 Przemyslaw Kosinski1,2*†
Przemyslaw Kosinski1,2*† Malgorzata Kedzia3†
Malgorzata Kedzia3† Adrianna Mostowska4Pawel Gutaj2,3Michal Lipa1,2
Adrianna Mostowska4Pawel Gutaj2,3Michal Lipa1,2 Ewa Wender-Ozegowska3Adriana Rozy5
Ewa Wender-Ozegowska3Adriana Rozy5 Joanna Chorostowska-Wynimko5*Miroslaw Wielgos1
Joanna Chorostowska-Wynimko5*Miroslaw Wielgos1 Aleksandra Jezela-Stanek5
Aleksandra Jezela-Stanek5- 1First Department of Obstetrics and Gynecology, Medical University of Warsaw, Warsaw, Poland
- 2“Club 35”, Scientific Group of Polish Society of Obstetricians and Gynaecologists, Warsaw, Poland
- 3Division of Reproduction, Department of Obstetrics, Gynecology, and Gynecologic Oncology, Poznan University of Medical Sciences, Poznań, Poland
- 4Department of Biochemistry and Molecular Biology, Poznan University of Medical Sciences, Poznań, Poland
- 5Department of Genetics and Clinical Immunology, National Institute of Tuberculosis and Lung Diseases, Warsaw, Poland
Background: Intrahepatic cholestasis of pregnancy (ICP; prevalence 0.2–15.6%) is the most common pregnancy-related liver disorder. It may have serious consequences for a pregnancy, including increased risk of preterm delivery, meconium staining of amniotic fluid, fetal bradycardia, distress, and fetal demise. In cases of high bile acids (>100μmol/L), patients have 10-fold increase in the risk of stillbirth. Biophysical methods of fetal monitoring, such as cardiotocography, ultrasonography, or Doppler have been proven unreliable for risk prediction in the course of intrahepatic cholestasis. Therefore, we believe extensive research for more specific, especially early, markers should be carried out. By analogy with cholestasis in children with inherited alpha-1 antitrypsin deficiency (AATD), we hypothesized the SERPINA1 Z pathogenic variant might be related to a higher risk of cholestasis in pregnancy. This study aimed to investigate the most common AATD variants (Z and S SERPINA1 alleles) in a group of cholestatic pregnant women.
Results: The Z carrier frequency was calculated to be 6.8%, which is much higher compared to the general population [2.3%; the Chi-squared test with Yates correction is 6.8774 (p=0.008)].
Conclusion: Increased prevalence of SERPINA1 PI*Z variant in a group of women with intrahepatic cholestasis may suggest a possible genetic origin of a higher risk of intrahepatic cholestasis in pregnancy.
Introduction
The SERPINA1 (Serpin Peptidase Inhibitor, Clade A, Member 1, MIM *107400) gene encodes alpha-1 antitrypsin [AAT, A1AT, formerly known as a protease inhibitor (PI)], a major plasma serine PI (Ferrarotti et al., 2012). The SERPINA1 gene is highly polymorphic, with more than 100 clinically significant variants. The most prevalent pathogenic alleles are PI*Z (c.1096G>A, p.Glu366Lys) and PI*S (c.863A>T, p.Glu288Val) identified in 95% of severe alpha-1 antitrypsin deficient (AATD) patients (McElvaney et al., 1997). Both result in a quantitative and functional alpha-1 antitrypsin deficiency (AATD). The serum levels of alpha-1-antitrypsin have also been recently found to change during pregnancy according to differential DNA methylation of SERPINA1 gene (Rotondo et al., 2020). It underlines that AATD might also be potentially due to epigenetic modifications other than to SERPINA1 variants. AATD in adults is characterized by a variety of clinical presentations (Stoller et al., 1993), while in children, the most common manifestation is neonatal cholestasis, also called “cholestatic hepatitis.”
Intrahepatic cholestasis of pregnancy (ICP) is the most common pregnancy-related liver disorder with incidence of 0.1–1.5% in Central/Western Europe and North America and up to 1.5–4% in Chile and Bolivia (Brouwers et al., 2015). ICP usually ensues in the second or third trimester and spontaneously withdraws within 2–3weeks post-delivery. It commonly reoccurs in subsequent pregnancies (45–70%; Lee et al., 2008). The pathomechanism of ICP is not fully understood. It is thought to be multifactorial, with both environmental, hormonal as well as genetic factors involved. As ICP may have serious consequences for the course of pregnancy, including increased risk of preterm delivery, meconium staining of amniotic fluid, fetal bradycardia, distress, and fetal demise (Rook et al., 2012), it may be clinically beneficial to search for additional diagnostic markers or predictors that would allow earlier identification of patients at higher risk of ICP. Given the link between the deficient variants of the SERPINA1 gene and pediatric intrahepatic cholestasis as a manifestation of AATD (Chen et al., 2018; Comba et al., 2018; Lin et al., 2019), we aimed to assess the presence and frequency of the two most common SERPINA1 nucleotide variants – PI*Z and PI*S – in a group of 103 women with a history of ICP.
Materials and Methods
The study group consisted of 103 pregnant females with ICP diagnosed according to current guidelines (Manzotti et al., 2019), i.e., peak serum bile acid concentration above 10μmol/L. The characteristics of the study group are presented in Table 1. All pregnant women were otherwise healthy and in particular co-morbidities such as non-alcoholic fatty liver disease, cystic fibrosis, COPD, or alcohol abuse were excluded. The average gestational age (GA) at the time of occurance of cholestasis was 33weeks. After the diagnosis, all patients were treated with ursodeoxycholic acid (UDCA) starting with a dose of 250mg three times a day. During the treatment the patients were hospitalized and fetal and maternal well-being were monitored. The UDCA dose was modified by experienced physician according to the therapeutic effect and intensified in case of progressive increase of total bile acids (TBA). The maximal applied dose did not exceed 1,500mg/day. UDCA was administered until the day of delivery. The TBA and aminotransferases concentrations were monitored after fasting period regularly: twice a week or even daily in selected cases. Written informed consent was obtained from all participating individuals. The study procedure was approved by the Local Ethics Committees of Poznan University of Medical Sciences (no. 197/18) and performed in accordance with the code of ethics of the Declaration of Helsinki.

Table 1. Characteristics of patients with cholestasis of pregnancy [the data is presented as median (range)].
Methods
Genotyping of PI*S and PI*Z AAT Alleles by Real-Time PCR
Genomic DNA was isolated from peripheral blood lymphocytes with the salting-out method. In this DNA extraction technique initially described by Miller et al. (1988), following cell lysis and proteinase K treatment, the cell debris and proteins are precipitated using a high-concentration salt solution. The DNA is then precipitated using ethanol and redissolved in sterile H2O. The quality and quantity of DNA were checked spectrophotometrically (NanoDrop One spectrophotometer, Thermo Fisher Scientific, United States). AAT genotyping was performed in the LightCycler 480 II machine (Roche Diagnostics Ltd., Switzerland) using a set of specific oligonucleotide primers and minor groove binding (MGB) hydrolysis probes for PI*S and PI*Z alleles (sequences available upon request; Struniawski et al., 2013). Each assay included an S/Z heterozygote, Z/Z homozygote. Two duplex real-time PCR reactions were performed simultaneously using fluorescent hydrolysis probes for the detection of both wild-type and variant alleles. The assembled reaction mixtures were amplified in 96-well microplates (Roche Applied Science) using the following cycling conditions: 10min incubation at 95°C followed by 40cycles at 95°C for 20s, and at 60°C for 60s. Fluorescence emissions of PI*S and PI*Z probes were detected in the FAM channel and VIC channel, respectively, during the 60°C annealing step of each PCR cycle. The amplification results were interpreted according to the conventional endpoint genotyping principles using the LightCycler 480 Software, version 1.5 (Roche Applied Science). A two-tailed Chi-squared test was employed to evaluate significance of difference in the rate of the SERPINA1 Z pathogenic variant between studied group and general population (Chorostowska-Wynimko et al., submitted manuscript). A post hoc analysis revealed that for the observed difference in the frequency in the rate of the SERPINA1 Z pathogenic variant between studied group and general population our study had 74.1% power (alpha 0.05) to detect significant difference.
Results
Final results were obtained from 103 DNA samples. The SERPINA1 variants of interest were detected in seven (PI*Z, 6.8%) and 1 (PI*S, 0.9%) patients with ICP. All identified individuals were heterozygous, PI*MZ or PI*MS, respectively. Their demographic and clinical details are presented in Table 2A. Clinical characteristics of neonates born to PI*MZ and PI*MS heterozygous mothers are presented in Table 2B. The Chi-squared test for the rate of the SERPINA1 Z pathogenic variant between studied group and general population with Yates correction is 6.8774 (p=0.008).
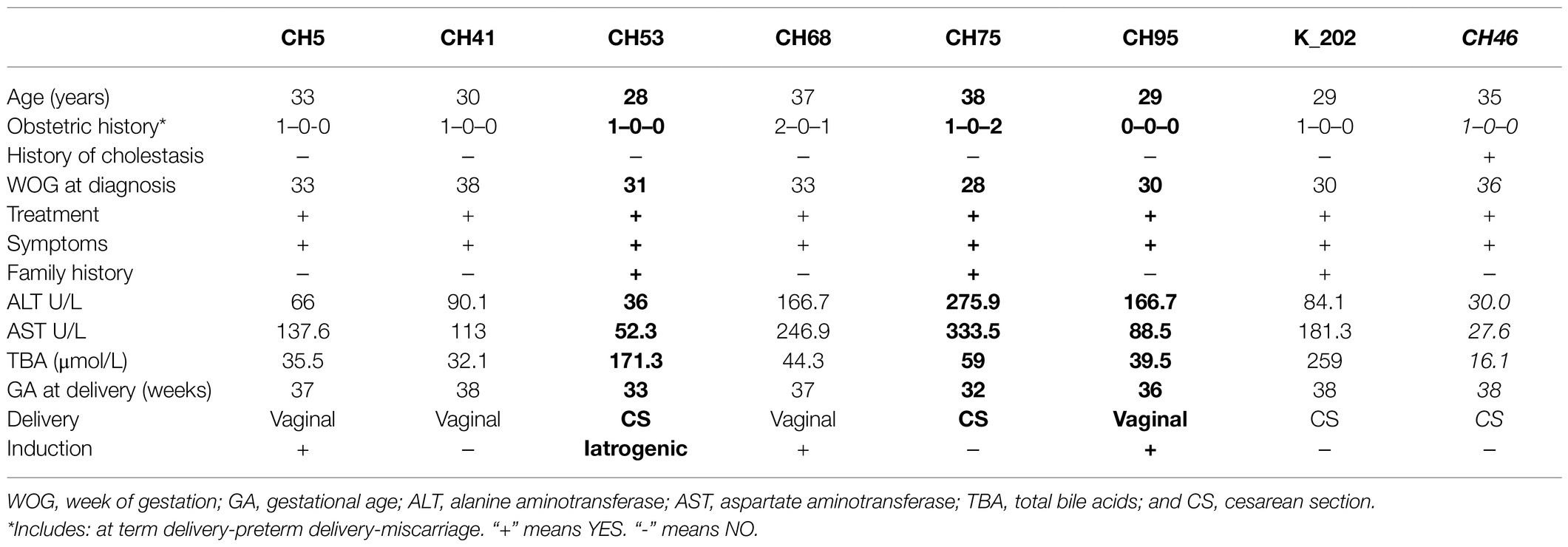
Table 2A. Demographic and clinical characteristics of PI*MZ heterozygotes (n=7) and PI*MS (n=1, highlighted in italics) identified in the group of 103 patients with intrahepatic cholestasis of pregnancy (ICP; premature births marked in bold).

Table 2B. Clinical characteristics of neonates born to PI*MZ and PI*MS (highlighted in italics) heterozygous mothers with recent history of ICP (see Table 2A).
After completed the study, we have retrospectively analyzed the medical files of women diagnosed as alpha-1 antitrysin deficiency carriers. No abnormalities suggesting liver dysfunctions [i.e., increased alanine aminotransferase (ALT), aspartate aminotransferase (AST), or bilirubin] were noted in any individuals. Family histories were also non-remarkable as far as lung diseases are concerned.
A detailed health assessment is planned, as well as further monitoring in regards to liver dysfunctions. All women will receive genetic counseling to inform of the potential risks, management and identification of indications for genetic testing toward AAT deficiency/carriership in close family members (siblings and offspring).
Discussion
Intrahepatic cholestasis of pregnancy is associated with an increased risk of fetal morbidity and mortality. Elevated TBA level, resulting in ICP, is also a risk factor for an adverse perinatal outcome, including spontaneous preterm birth, meconium-stained amniotic fluid, and neonatal unit admission (Glantz et al., 2008; Herrera et al., 2018; Chappell et al., 2019); Based on recent meta-analyses, the risk of stillbirth is increased in women with ICP and singleton pregnancies when serum bile acids concentrations are of 100μmol/L or more (Ovadia et al., 2019). Also other researchers confirmed that patients with serum bile acids >100μmol/L present an approximately 10-fold higher probability of stillbirth (Kawakita et al., 2015; Ovadia et al., 2019). The diagnosis is based on clinical manifestations, mainly pruritus, as well as abnormal TBA levels (Manzotti et al., 2019). Commonly available biophysical methods of fetal monitoring, such as cardiotocography, ultrasonography, or Doppler ultrasonography, have been proven unreliable for stillbirth risk assessment (Oztas et al., 2015). Therefore, identification of high-risk group for ICP would be of great value in the scientific and potentially also in the clinical setting. As for instance functional genetic variants are considered as biomarkers in oncology, our findings may help to establish new biomarkers for ICP. By analogy with cholestasis observed in children with inherited AATD, we hypothesized the SERPINA1 PI*Z deficiency variant might be linked to a higher risk of cholestasis in pregnancy.
According to the Online Mendelian Inheritance in Man’s “Gene-Phenotype Relationships,” certain variants of SERPINA1 gene are associated with: (1) Emphysema due to AAT deficiency (# 613490); (2) Emphysema-cirrhosis, due to AAT deficiency (# 613490); and (3) Hemorrhagic diathesis due to antithrombin Pittsburgh (# 613490; https://www.omim.org/entry/107400, accessed 10, 2020). The clinical manifestations observed in severe inherited AATD are, however, more diverse, and apart from those listed in Table 3, include bronchial asthma, bronchiectasis, panniculitis, and granulomatosis with polyangiitis (GPA; Stoller et al., 1993).

Table 3. SERPINA1 clinical synopses (adapted from OMIM).
In children, neonatal cholestasis is the most common presentation of severe AAT deficiency resulting from PI*ZZ genotypes. It affects ~10% of newborns and infants and was first mentioned by Aagenaes et al. (1972), who document five individuals (Bouchecareilh, 2020). There are more than 50 known pathogenic variants of the SERPINA1 gene (ClinVar https://www.ncbi.nlm.nih.gov/clinvar, accessed 10, 2020). The PI*Z, a glutamate-to-lysine substitution at position 342, and the PI*S, a glutamate-to-valine substitution at position 264, are considered the most clinically SERPINA1 variants (Schneider et al., 2020). The frequency of SERPINA1 Z and S alleles in the Polish population was preliminarily established in a newborn population screening of 658 subjects performed using dry blood spots (DBS) collected in Warsaw between September and December 2011 (Chorostowska-Wynimko et al., 2013). Main deficiency variants were detected in 28 individuals – the PI*Z allele in 18 (2.7%) and the PI*S allele in 10 (1.5%). More recently, in a group of 4,185 unselected neonates, the pathogenic variants were identified in 186 newborns, specifically the PI*Z allele in 96 and the PI*S allele in 90 individuals, corresponding to the estimated prevalence of accordingly 2.3 and 2.1% in the Polish population (Chorostowska-Wynimko et al., submitted manuscript). Meanwhile, the calculated frequency of the PI*Z variant in the ICP was 6.8%, thus considerably higher than expected for the general population. The obvious gender imbalance in the study group is of no clinical importance, as frequencies of SERPINA1 pathogenic variants are similar in both sexes.
The risk of cholestasis in pregnancy has been associated with genetic variants in ABCB4, ABCB11, ATP8B1, ABCC2, and TJP2 genes (Chen et al., 2018). Heterozygotes mutation in the ABCB4 (ATP-Binding Cassette, Subfamily B, Member 4) gene on chromosome 7q21 accounts for about 15% of ICP cases (Ziol et al., 2008). The gene is responsible for intrahepatic cholestasis of pregnancy 3 (ICP3, OMIM 614972). Heterozygotes variants in the ATP8B1 gene on chromosome 18q21, except intrahepatic cholestasis of pregnancy 1 (ICP1, OMIM 147480), can also cause progressive familial intrahepatic cholestasis 1 (PFIC1, OMIM 211600) and benign recurrent intrahepatic cholestasis 1 (BRIC1, OMIM 243300).1 ABCC2, encoded by the ATP-Binding Cassette, Subfamily C, Member 2 (ABCC2 gene), is an integral membrane glycoprotein expressed mainly in the canalicular (apical) membrane liver cells. It belongs to the ATP-binding cassette transporter superfamily and transports endogenous and exogenous anionic conjugates from hepatocytes to bile. The TJP2 gene, localized on 9q21.11, encodes tight junction protein-2, which belongs to a family of membrane-associated guanylate kinase (MAGUK) homologs involved in the organization of epithelial and endothelial intercellular junctions. Its pathogenic variants have been identified in patients with progressive familial intrahepatic cholestasis-4 (PFIC4, OMIM 615878; Sambrotta et al., 2014).
Here, we provided an insight into the possible significance of the PI*Z SERPINA1 and suggest its potential role as a risk factor for ICP. Such a relationship seems warranted, especially in light of the PI*Z disease-modifying role in liver diseases, i.e., in triggering hepatic dysfunction in cystic fibrosis (Bartlett et al., 2009; Elborn, 2016), as a disease modifier in alcoholic (AFLD) and nonalcoholic (NAFLD) fatty liver disease (Abul-Husn et al., 2018; Strnad et al., 2019), or due to its link to higher serum ALT and AST levels (Prins et al., 2017). Moreover, PI*Z carriers are over-represented among patients with end-stage liver disease, in individuals with cryptogenic cirrhosis (Graziadei et al., 1998), or patients referred for liver transplantation due to cirrhosis (Schaefer et al., 2018). There have never been any data published, nor do we provide evidence suggesting an association between PI*S polymorphic variant and clinically relevant risk of liver injury. This is in line with results published by others (Abul-Husn et al., 2018; Strnad et al., 2019).
The exact risk of liver disease in PI*MZ adults is currently unknown, and symptomatic liver dysfunction is not commonly observed in PI*ZZ individuals. Yet, an odds ratio for developing chronic liver disease by adult PI*Z heterozygotes varies between 1.8 and 3.1 (Fra et al., 2016). Clinical symptoms might be augmented by coexisting conditions, such as alcohol abuse, non-alcoholic fatty liver disease, or cystic fibrosis (CFTR variants; Strnad et al., 2020) as well as being significantly affected by age. PI*Z carriers become symptomatic for liver disease later in life – on average PI*ZZ at the age of 58years, PI*MZ at 73, and PI*SZ at 66years of age (Irving et al., 2014). The mechanisms behind AATD-mediated liver disease are not fully understood. The retention of Z AAT protein results in the formation of Z aggregates within the endoplasmic reticulum (ER) and contributes to hepatocyte damage as the key process, due to the fact that the aggregates have a significant gain-of-function hepatotoxic effect (Teckman, 2013; Bouchecareilh, 2020). Accordingly, the inclusion bodies (IB) are the histopathological hallmark of AATD liver disease. In a mouse model of AT deficiency-associated liver disease, the norursodeoxycholic acid (norUDCA), a side-shortened homolog of UDCA, has been shown to induce autophagy and, therefore, reduce the burden of Z AAT protein deposits (Hidvegi et al., 2010; Tang et al., 2018). Both UDCA and norUDCA are used in clinical practice to limit Z AAT induced liver damage.
Ursodeoxycholic acid is also commonly used for the treatment of ICP. Its effects are thought to occur by improving biliary flow, enhancing the protective mechanisms, and protecting the liver from bile acid-induced apoptosis (Pusl et al., 2008; Bicocca et al., 2018). Studies have demonstrated that UDCA treatment is associated with a reduction of pruritus (Kong et al., 2016). Unfortunately, clear evidence for the clinical benefit of UDCA in reducing serious pregnancy complications is not extensive. Most studies suggest the fetal stillbirth in patients with ICP is caused by the toxic influence of bile acids on fetal heart cells (Williamson et al., 2001; Ozel et al., 2020). Therefore, according to some authors, UDCA treatment may reduce the impact of the abovementioned pathological and toxic mechanisms that are implicated in the etiology of stillbirth in ICP, such as fetal arrhythmia (Miragoli et al., 2011).
Conclusion
The prevalence of SERPINA1 PI*Z variant in a group of women with intrahepatic cholestasis is higher compared to general population. Our data provide the first evidence for association between SERPINA1 Z variant and ICP.
Limitations
Given the small size of the analyzed group, these results should be interpreted with caution and further verified in ICP patients. This hypothesis should be also tested with two different control groups: pregnant women without ICP and non-pregnant women. In case this is confirmed, screening for SERPINA1 PI*Z might be used to emerge a group of women who might be more prone to ICP. It may also provide an inspiration for new treatment options or schemes dedicated to pregnant PI*Z carriers.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committees of Poznan University of Medical Sciences (no. 197/18). The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
PK, MK, PG, ML, JC-W, MW, and AJ-S: conception or design of the work. PK, MK, PG, AR, JC-W, and AJ-S: data collection. PK, MK, AM, PG, ML, EW-O, AR, JC-W, MW, and AJ-S: data analysis and interpretation. PK, MW, JC-W, and AJ-S: drafting the article. MK, AM, AR, EW-O, MW, and AJ-S: critical revision of the article. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ICP, Intrahepatic cholestasis of pregnancy; AATD, Alpha-1 antitrypsin deficiency; UDCA, Ursodeoxycholic acid; GA, Gestational age; WOG, Week of gestation; ALT, Alanine aminotransferase; AST, Aspartate aminotransferase; TBA, Total bile acids; CS, Cesarean section; DBS, Dry blood spots; ER, Endoplasmic reticulum; IB, Inclusion bodies.
Footnotes
References
Aagenaes, O., Matlary, A., Elgjo, K., Munthe, E., and Fagerhol, M. (1972). Neonatal cholestasis in alpha-1-antitrypsin deficient children. Clinical, genetic, histological and immunohistochemical findings. Acta Paediatr. Scand. 61, 632–642. doi: 10.1111/j.1651-2227.1972.tb15960.x
Abul-Husn, N. S., Cheng, X., Li, A. H., Xin, Y., Schurmann, C., Stevis, P., et al. (2018). A protein-truncating HSD17B13 variant and protection from chronic liver disease. N. Engl. J. Med. 378, 1096–1106. doi: 10.1056/NEJMoa1712191
Bartlett, J. R., Friedman, K. J., Ling, S. C., Pace, R. G., Bell, S. C., Bourke, B., et al. (2009). Genetic modifiers of liver disease in cystic fibrosis. JAMA 302, 1076–1083. doi: 10.1001/jama.2009.1295
Bicocca, M. J., Sperling, J. D., and Chauhan, S. P. (2018). Intrahepatic cholestasis of pregnancy: review of six national and regional guidelines. Eur. J. Obstet. Gynecol. Reprod. Biol. 231, 180–187. doi: 10.1016/j.ejogrb.2018.10.041
Bouchecareilh, M. (2020). Alpha-1 antitrypsin deficiency-mediated liver toxicity: why do some patients do poorly? What do we know so far? Chronic Obstr. Pulm. Dis. 7, 172–181. doi: 10.15326/jcopdf.7.3.2019.0148
Brouwers, L., Koster, M. P., Page-Christiaens, G. C., Kemperman, H., Boon, J., Evers, I. M., et al. (2015). Intrahepatic cholestasis of pregnancy: maternal and fetal outcomes associated with elevated bile acid levels. Am. J. Obstet. Gynecol. 212, 100.e101–107.e101. doi: 10.1016/j.ajog.2014.07.026
Chappell, L. C., Bell, J. L., Smith, A., Linsell, L., Juszczak, E., Dixon, P. H., et al. (2019). Ursodeoxycholic acid versus placebo in women with intrahepatic cholestasis of pregnancy (PITCHES): a randomised controlled trial. Lancet 394, 849–860. doi: 10.1016/S0140-6736(19)31270-X
Chen, H. L., Wu, S. H., Hsu, S. H., Liou, B. Y., Chen, H. L., and Chang, M. H. (2018). Jaundice revisited: recent advances in the diagnosis and treatment of inherited cholestatic liver diseases. J. Biomed. Sci. 25:75. doi: 10.1186/s12929-018-0475-8
Chorostowska-Wynimko, J., Struniawski, R., Poplawska, B., and Borszewska-Kornacka, J. (2012). Incidence of alpha-1 antitrypsin Z and S alleles in patients with granulomatosis with polyangiitis—pilot study. Pneumonol. Alergol. Pol. 81, 319–322.
Comba, A., Demirbas, F., Caltepe, G., Eren, E., and Kalayci, A. G. (2018). Retrospective analysis of children with alpha-1 antitrypsin deficiency. Eur. J. Gastroenterol. Hepatol. 30, 774–778. doi: 10.1097/MEG.0000000000001108
Ferrarotti, I., Thun, G. A., Zorzetto, M., Ottaviani, S., Imboden, M., Schindler, C., et al. (2012). Serum levels and genotype distribution of alpha1-antitrypsin in the general population. Thorax 67, 669–674. doi: 10.1136/thoraxjnl-2011-201321
Fra, A., Cosmi, F., Ordonez, A., Berardelli, R., Perez, J., Guadagno, N. A., et al. (2016). Polymers of Z alpha1-antitrypsin are secreted in cell models of disease. Eur. Respir. J. 47, 1005–1009. doi: 10.1183/13993003.00940-2015
Glantz, A., Reilly, S. J., Benthin, L., Lammert, F., Mattsson, L. A., and Marschall, H. U. (2008). Intrahepatic cholestasis of pregnancy: amelioration of pruritus by UDCA is associated with decreased progesterone disulphates in urine. Hepatology 47, 544–551. doi: 10.1002/hep.21987
Graziadei, I. W., Joseph, J. J., Wiesner, R. H., Therneau, T. M., Batts, K. P., and Porayko, M. K. (1998). Increased risk of chronic liver failure in adults with heterozygous alpha1-antitrypsin deficiency. Hepatology 28, 1058–1063. doi: 10.1002/hep.510280421
Herrera, C. A., Manuck, T. A., Stoddard, G. J., Varner, M. W., Esplin, S., Clark, E. A. S., et al. (2018). Perinatal outcomes associated with intrahepatic cholestasis of pregnancy. J. Matern. Fetal Neonatal Med. 31, 1913–1920. doi: 10.1080/14767058.2017.1332036
Hidvegi, T., Ewing, M., Hale, P., Dippold, C., Beckett, C., Kemp, C., et al. (2010). An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science 329, 229–232. doi: 10.1126/science.1190354
Irving, J. A., Haq, I., Dickens, J. A., Faull, S. V., and Lomas, D. A. (2014). Altered native stability is the dominant basis for susceptibility of alpha1-antitrypsin mutants to polymerization. Biochem. J. 460, 103–115. doi: 10.1042/BJ20131650
Kawakita, T., Parikh, L. I., Ramsey, P. S., Huang, C. C., Zeymo, A., Fernandez, M., et al. (2015). Predictors of adverse neonatal outcomes in intrahepatic cholestasis of pregnancy. Am. J. Obstet. Gynecol. 213, 570.e1–578.e1. doi: 10.1016/j.ajog.2015.06.021
Kong, X., Kong, Y., Zhang, F., Wang, T., and Yan, J. (2016). Evaluating the effectiveness and safety of ursodeoxycholic acid in treatment of intrahepatic cholestasis of pregnancy: a meta-analysis (a prisma-compliant study). Medicine 95:e4949. doi: 10.1097/MD.0000000000004949
Lee, R. H., Kwok, K. M., Ingles, S., Wilson, M. L., Mullin, P., Incerpi, M., et al. (2008). Pregnancy outcomes during an era of aggressive management for intrahepatic cholestasis of pregnancy. Am. J. Perinatol. 25, 341–345. doi: 10.1055/s-2008-1078756
Lin, H. C., Kasi, N., and Quiros, J. A. (2019). Alpha1-antitrypsin deficiency: transition of care for the child with AAT deficiency into adulthood. Curr. Pediatr. Rev. 15, 53–61. doi: 10.2174/1573396314666181113094517
Manzotti, C., Casazza, G., Stimac, T., Nikolova, D., and Gluud, C. (2019). Total serum bile acids or serum bile acid profile, or both, for the diagnosis of intrahepatic cholestasis of pregnancy. Cochrane Database Syst. Rev. 7:CD012546. doi: 10.1002/14651858.CD012546.pub2
McElvaney, N. G., Stoller, J. K., Buist, A. S., Prakash, U. B., Brantly, M. L., Schluchter, M. D., et al. (1997). Baseline characteristics of enrollees in the national heart, lung and blood institute registry of alpha 1-antitrypsin deficiency. Alpha 1-antitrypsin deficiency registry study group. Chest 111, 394–403. doi: 10.1378/chest.111.2.394
Miller, S. A., Dykes, D. D., and Polesky, H. F. (1988). A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 16:1215. doi: 10.1093/nar/16.3.1215
Miragoli, M., Kadir, S. H., Sheppard, M. N., Salvarani, N., Virta, M., Wells, S., et al. (2011). A protective antiarrhythmic role of ursodeoxycholic acid in an in vitro rat model of the cholestatic fetal heart. Hepatology 54, 1282–1292. doi: 10.1002/hep.24492
Ovadia, C., Seed, P. T., Sklavounos, A., Geenes, V., Di Ilio, C., Chambers, J., et al. (2019). Association of adverse perinatal outcomes of intrahepatic cholestasis of pregnancy with biochemical markers: results of aggregate and individual patient data meta-analyses. Lancet 393, 899–909. doi: 10.1016/S0140-6736(18)31877-4
Ozel, A., Alici Davutoglu, E., Eric Ozdemir, M., Oztunc, F., and Madazli, R. (2020). Assessment of fetal left ventricular modified myocardial performance index and its prognostic significance for adverse perinatal outcome in intrahepatic cholestasis of pregnancy. J. Matern. Fetal Neonatal Med. 33, 2000–2005. doi: 10.1080/14767058.2018.1535588
Oztas, E., Erkenekli, K., Ozler, S., Ersoy, A. O., Kurt, M., Oztas, E., et al. (2015). Can routine laboratory parameters predict adverse pregnancy outcomes in intrahepatic cholestasis of pregnancy? J. Perinat. Med. 43, 667–674. doi: 10.1515/jpm-2014-0207
Prins, B. P., Kuchenbaecker, K. B., Bao, Y., Smart, M., Zabaneh, D., Fatemifar, G., et al. (2017). Genome-wide analysis of health-related biomarkers in the UK household longitudinal study reveals novel associations. Sci. Rep. 7:11008. doi: 10.1038/s41598-017-10812-1
Pusl, T., Vennegeerts, T., Wimmer, R., Denk, G. U., Beuers, U., and Rust, C. (2008). Tauroursodeoxycholic acid reduces bile acid-induced apoptosis by modulation of AP-1. Biochem. Biophys. Res. Commun. 367, 208–212. doi: 10.1016/j.bbrc.2007.12.122
Rook, M., Vargas, J., Caughey, A., Bacchetti, P., Rosenthal, P., and Bull, L. (2012). Fetal outcomes in pregnancies complicated by intrahepatic cholestasis of pregnancy in a northern California cohort. PLoS One 7:e28343. doi: 10.1371/journal.pone.0028343
Rotondo, J. C., Oton-Gonzalez, L., Selvatici, R., Rizzo, P., Pavasini, R., Campo, G. C., et al. (2020). SERPINA1 gene promoter is differentially methylated in peripheral blood mononuclear cells of pregnant women. Front. Cell Dev. Biol. 8:550543. doi: 10.3389/fcell.2020.550543
Sambrotta, M., Strautnieks, S., Papouli, E., Rushton, P., Clark, B. E., Parry, D. A., et al. (2014). Mutations in TJP2 cause progressive cholestatic liver disease. Nat. Genet. 46, 326–328. doi: 10.1038/ng.2918
Schaefer, B., Mandorfer, M., Viveiros, A., Finkenstedt, A., Ferenci, P., Schneeberger, S., et al. (2018). Heterozygosity for the alpha-1-antitrypsin Z allele in cirrhosis is associated with more advanced disease. Liver Transpl. 24, 744–751. doi: 10.1002/lt.25057
Schneider, C. V., Hamesch, K., Gross, A., Mandorfer, M., Moeller, L. S., Pereira, V., et al. (2020). Liver phenotypes of European adults heterozygous or homozygous for pi*Z variant of AAT (pi*MZ vs pi*ZZ genotype) and noncarriers. Gastroenterology 159, 534.e511–548.e511. doi: 10.1053/j.gastro.2020.04.058
Stoller, J. K., Hupertz, V., and Aboussouan, L. S. (1993). “Alpha-1 antitrypsin deficiency,” in GeneReviews®. eds. M. P. Adam, H. H. Ardinger, R. A. Pagon, S. E. Wallace, L. J. H. Bean, and G. Mirzaa, et al. (Seattle: University of Washington).
Strnad, P., Buch, S., Hamesch, K., Fischer, J., Rosendahl, J., Schmelz, R., et al. (2019). Heterozygous carriage of the alpha1-antitrypsin pi*Z variant increases the risk to develop liver cirrhosis. Gut 68, 1099–1107. doi: 10.1136/gutjnl-2018-316228
Strnad, P., McElvaney, N. G., and Lomas, D. A. (2020). Alpha1-antitrypsin deficiency. N. Engl. J. Med. 382, 1443–1455. doi: 10.1056/NEJMra1910234
Struniawski, R., Szpechcinski, A., Poplawska, B., Skronski, M., and Chorostowska-Wynimko, J. (2013). Rapid DNA extraction protocol for detection of alpha-1 antitrypsin deficiency from dried blood spots by real-time PCR. Adv. Exp. Med. Biol. 756, 29–37. doi: 10.1007/978-94-007-4549-0_5
Tang, Y., Blomenkamp, K. S., Fickert, P., Trauner, M., and Teckman, J. H. (2018). NorUDCA promotes degradation of alpha1-antitrypsin mutant Z protein by inducing autophagy through AMPK/ULK1 pathway. PLoS One 13:e0200897. doi: 10.1371/journal.pone.0200897
Teckman, J. H. (2013). Liver disease in alpha-1 antitrypsin deficiency: current understanding and future therapy. COPD 10(Suppl. 1), 35–43. doi: 10.3109/15412555.2013.765839
Williamson, C., Gorelik, J., Eaton, B. M., Lab, M., de Swiet, M., and Korchev, Y. (2001). The bile acid taurocholate impairs rat cardiomyocyte function: a proposed mechanism for intra-uterine fetal death in obstetric cholestasis. Clin. Sci. 100, 363–369. doi: 10.1042/CS20000164
Keywords: ursodeoxycholic acid, alpha-1 antitrypsin deficiency, intrahepatic cholestasis of pregnancy, cholestasis, pregnancy
Citation: Kosinski P, Kedzia M, Mostowska A, Gutaj P, Lipa M, Wender-Ozegowska E, Rozy A, Chorostowska-Wynimko J, Wielgos M and Jezela-Stanek A (2021) Alpha-1 Antitrypsin Z Variant (AAT PI*Z) as a Risk Factor for Intrahepatic Cholestasis of Pregnancy. Front. Genet. 12:720465. doi: 10.3389/fgene.2021.720465
Edited by:
Ramcés Falfán-Valencia, Instituto Nacional de Enfermedades Respiratorias-México (INER), MexicoReviewed by:
Nancy Monroy-Jaramillo, National Institute of Neurology and Neurosurgery, MexicoDanielius Serapinas, Lithuanian University of Health Sciences, Lithuania
Ben Woolbright, University of Kansas Medical Center, United States
Copyright © 2021 Kosinski, Kedzia, Mostowska, Gutaj, Lipa, Wender-Ozegowska, Rozy, Chorostowska-Wynimko, Wielgos and Jezela-Stanek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Przemyslaw Kosinski, cGtvc2luc2tpQHd1bS5lZHUucGw=; Joanna Chorostowska-Wynimko, ai5jaG9yb3N0b3dza2FAaWdpY2hwLmVkdS5wbA==
†These authors have contributed equally to this work