
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 01 September 2021
Sec. Livestock Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.720081
This article is part of the Research Topic Genetic Diversity and Selection Signatures in Composite Breeds View all 16 articles
 Zhong Xu1
Zhong Xu1 Shuqi Mei1Jiawei Zhou1Yu Zhang1Mu Qiao1Hua Sun1Zipeng Li1Lianghua Li1
Shuqi Mei1Jiawei Zhou1Yu Zhang1Mu Qiao1Hua Sun1Zipeng Li1Lianghua Li1 Binke Dong1
Binke Dong1 Favour Oluwapelumi Oyelami2Junjing Wu1*Xianwen Peng1*
Favour Oluwapelumi Oyelami2Junjing Wu1*Xianwen Peng1*The primary purpose of the current study was to assess the genetic diversity, runs of homozygosity (ROH) and ROH islands in a Chinese composite pig and explore hotspot regions for traces of selection. First, we estimated the length, number, and frequency of ROH in 262 Xidu black pigs using the Porcine SNP50 BeadChip and compared the estimates of inbreeding coefficients, which were calculated based on ROHs (FROH) and homozygosity (FHOM). Our result shows that a total of 7,248 ROH exceeding 1Mb were detected in 262 pigs. In addition, Sus scrofa chromosome (SSC) 8 and SSC10, respectively, has the highest and lowest chromosome coverage by ROH. These results suggest that inbreeding estimation based on total ROH may be a useful method, especially for crossbreed or composite populations. We also calculated an inbreeding coefficient of 0.077 from the total ROH. Eight ROH islands were found in this study. These ROH islands harbored genes associated with fat deposition, muscular development, reproduction, ear shape, and adaptation, such as TRAF7, IGFBP7, XPO1, SLC26A8, PPARD, and OR1F1. These findings may help to understand the effects of environmental and artificial selection on the genome structure of composite pigs. Our results provide a basis for subsequent genomic selection (GS), and provides a reference for the hybrid utilization of other pig breeds.
Crossbreeding is a common strategy to improve livestock production because it can explore complementarities of additive genetic effects as well as heterosis caused by non-additive genetic effects (Howard et al., 2016). The Enshi black pig, a typical native black breed in China, mainly lives in mountainous areas of southwest China at an average altitude of more than 800m. It is well-known for its adaptability to a mountainous environment, excellent meat quality, fat storage ability, and cold-wet tolerance (China National Commission of Animal Genetic Resources, 2011). Since the 1990s, under the impact of exotic germplasm with high growth rate and lean meat rate, the Enshi black pig has been facing extinction due to its low growth rate. To overcome these deficiencies and conserve the Enshi black pig, crossbreeding programs have been implemented to increase productivity, and the Xidu black pig is a new composite breed that has been developed for this situation. Crossbreeding combines the cold-wet environmental adaptation of the Enshi black pig with the high fertility of the Meishan pig and the fast growth rate of the Hubei white pig. When the three-way crossbreeding [Hubei white×(Meishan×Enshi black)] was formed, they were inter-se mating and selected to become the Xidu black pig breed, which having about 50% Hubei white, 25% Meishan, and 25% Enshi black pig inheritance. It is worth noting that the Xidu black pig is now well established and can be used as a purebred without the need for any ongoing crossbreeding programs.
Generally, crossbred offspring can be mated among themselves in each generation, and selection for specific traits and genetic improvement can be applied during this process. Therefore, it is essential to manage genetic diversity by avoiding high inbreeding rates in composite breed, which will retain high levels of heterozygosity and heterosis (Peripolli et al., 2020). It is difficult to assess genetic diversity using pedigree data mostly because the genealogical relationships between parental breeds that are used in crossbreeding cannot be established. However, the analysis of genomic data could solve this problem (Ganteil et al., 2020). There are several methods used in estimating genetic diversity from genomic data. Some of these methods include the use of observed and expected heterozygosity, runs of homozygosity (ROH), and linkage disequilibrium (LD).
Runs of homozygosity are long continuous homozygous segments in the genome that are formed in an individual by the combination of two identical haplotypes from a common ancestor (Ceballos et al., 2018). As an important genome feature, ROH provides an essential reference for the study of the genome structure. Besides, in animal genetics, the presence of homozygous segments in the genome can be influenced by intensive selection, population history, and consanguinity levels (Peripolli et al., 2017). Also, inbreeding estimates based on ROH is usually considered to be more accurate for estimating individual inbreeding levels when compared with other existing methods (Keller et al., 2011). In addition, ROH hotspots are known to be non-randomly distributed across the genome, and can reveal selection pressure events since selection is one of the main causes of homozygous stretches on the genome. Recently, ROH has been mostly employed in estimating the genomic inbreeding and selection signatures of many livestock populations (Zhang et al., 2018b; Xu et al., 2019; Shi et al., 2020), but less commonly used in crossbred or composite populations.
The LD analysis is also an efficient approach for determining the level of genetic diversity within a studied population. Generally, LD can be defined as the non-random genetic relationship between two loci in a population (Saravanan et al., 2020). Thus, exploring the pattern and extent of LD in the genome can provide essential insights for guiding genome-wide association studies (GWAS) and genome selection (GS; Mokry et al., 2014).
The main objectives of this study were: (1) to investigate the characteristics of ROH on the genome of the Xidu black pig, and also identify the genomic regions with high ROH frequency; and (2) to estimate genetic diversity parameters, such as inbreeding rates and LD in this composite pig population.
All experimental procedures were approved by the Institutional Animal Care and Use Committee of the Hubei Academy of Agriculture Sciences, and all methods involved pigs were in accordance with the agreement of Institutional Animal Care and Use Committee of the Hubei Academy of Agriculture Sciences (Permit number: 36/2016).
The animal genomics dataset used in the current study were gotten from pigs raised in a composite swine breeding farm located in Enshi, Hubei, China. In order to make the sample representative, we chose individuals from the core group to avoid full siblings. At last, there were 262 individuals consisting of approximately three generations. The 262 animals were genotyped using the Porcine Single nucleotide polymorphism SNP50 BeadChip (Illumina, United States), consisting of 51,315 Single nucleotide polymorphism (SNPs) evenly distributed along the pig genome. Genotype quality control was carried out using PLINK v1.90 (Chang et al., 2015) software based on the following filtering criteria: (1) the call rate of SNPs and individuals were higher than 0.9; (2) the minor allele frequency (MAF) was greater than 0.01; and (3) only SNPs mapped to autosomes were included. The latest version of the pig genome, Sus scrofa 11.1 was used in this study.
We identified ROH in individuals using PLINK v1.90 software, which uses a sliding window approach to detect autozygous segments. The algorithm is as follows: take a window of X SNPs and slide them across the genome. Determine at each window position whether the window looks sufficiently “homogeneous” (yes/no). Then, for each SNP, calculate the proportion of the “homozygous” window that overlaps that position. Call segmentation based on this metric, such as a threshold based on the average value. To define a ROH, the criterion and thresholds were as follows: (1) a minimum ROH length of 1Mb; (2) at least 50 homozygous SNPs included in a ROH; (3) a minimum density of a SNP in 100Kb; (4) a sliding window of 50 SNPs across the genome that moves one SNP at a time; and (5) up to one heterozygous SNP and five missing SNPs were allowed in a sliding window. Detected ROHs were later classified into three different classes based on their length: 1–5, 5–10, and >10Mb. The total number and length of ROHs were counted for all individuals.
We identified the genomic regions that were mostly associated with ROHs, by calculating the proportion of SNPs in ROH. This was done by counting the number of times the SNP was detected in those ROH across individuals. Afterward, we selected the top 1% of SNPs that were commonly observed in ROHs. Adjacent SNPs that were above this threshold were finally merged into genomic regions which are called ROH islands, which is characterized by being shared by a majority of individuals in the population (Dixit et al., 2020). We used the database provided by NCBI to annotate the genes in the ROH island. Through a large number of accurate literature searches, the biological function of each annotated gene in the ROH island was inferred.
In this study, two types of genomic inbreeding coefficients were calculated, one based on ROH (FROH) and the other based on excess of homozygosity (FHOM). Genomic inbreeding coefficients (FROH) were computed for all individuals by the following formula, as proposed by McQuillan et al. (2008):
The LD was measured using the r2, which was calculated for each pair of SNPs per chromosome according to Hill and Robertson (1968). The pairwise LD (r2) were calculated using the parameters “--ld-window 99,999 --ld-window-kb 1,000 --ld-window-r2 0” in PLINK v1.90. To visualize the decline of LD, the physical distances between SNPs were divided into 100-Kb intervals, and the average of r2 in each group was then estimated.
After quality control, 262 pigs and 38,275 SNPs were retained. The average observed (Ho) and expected (He) heterozygosity estimates were 0.37 and 0.35, respectively, and the average MAF was 0.26. We observed that the Ho was somewhat higher than the He.
In this study, a total of 7,248 ROHs were identified in the 262 animals with an average of 27.66 ROH per animal. The average ROH length was 6.32Mb, and the longest fragment in Sus scrofa chromosome 8 (SSC8) was 57.84Mb (1,000 SNPs). Table 1 summarizes the descriptive statistics of the ROH number and length by classes. The total ROH number of the composite Xidu Black pigs was mainly composed of shorter segments (1–5Mb), which accounted for about 56.16% of all the detected ROH. The genome coverage of long segments (>10Mb) accounted for 37.11% of the total ROH. Short ROH reflects ancestral inbreeding history, while long ROH segments are usually formed by recent inbreeding. This indicated that both ancient and recent inbreeding events might have affected this population, but recent inbreeding or selection pressures have mainly influenced the genome of the Xidu Black pig population.
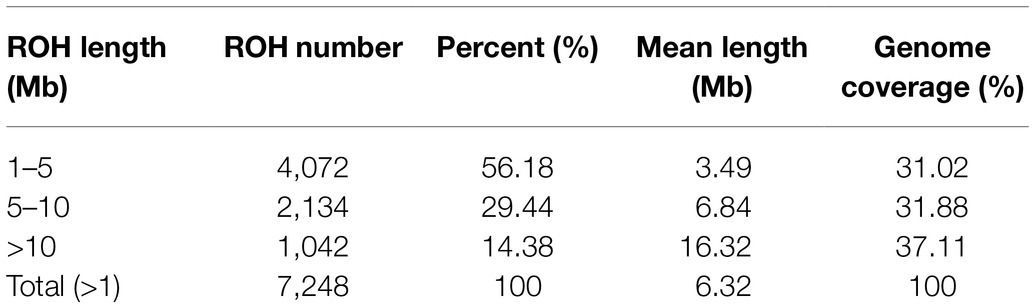
Table 1. Descriptive statistics of runs of homozygosity (ROH) number and length (in Mb) by ROH length class (ROH 1–5Mb, ROH 5–10Mb, ROH>10Mb, and total).
The relationship between the total ROH number and the total length of the genome covered by ROH in each individual is shown in Figure 1, and is greatly different among animals. In this population, the most extreme animal with long ROHs had a length of 777.06Mb (34.41% of the pig genome). The variability of the total number and length of ROH among individuals was high. Similar distributions were also observed in other pigs (Xu et al., 2019; Shi et al., 2020) and livestock species, such as sheep (Mastrangelo et al., 2017) and cattle (Peripolli et al., 2018).
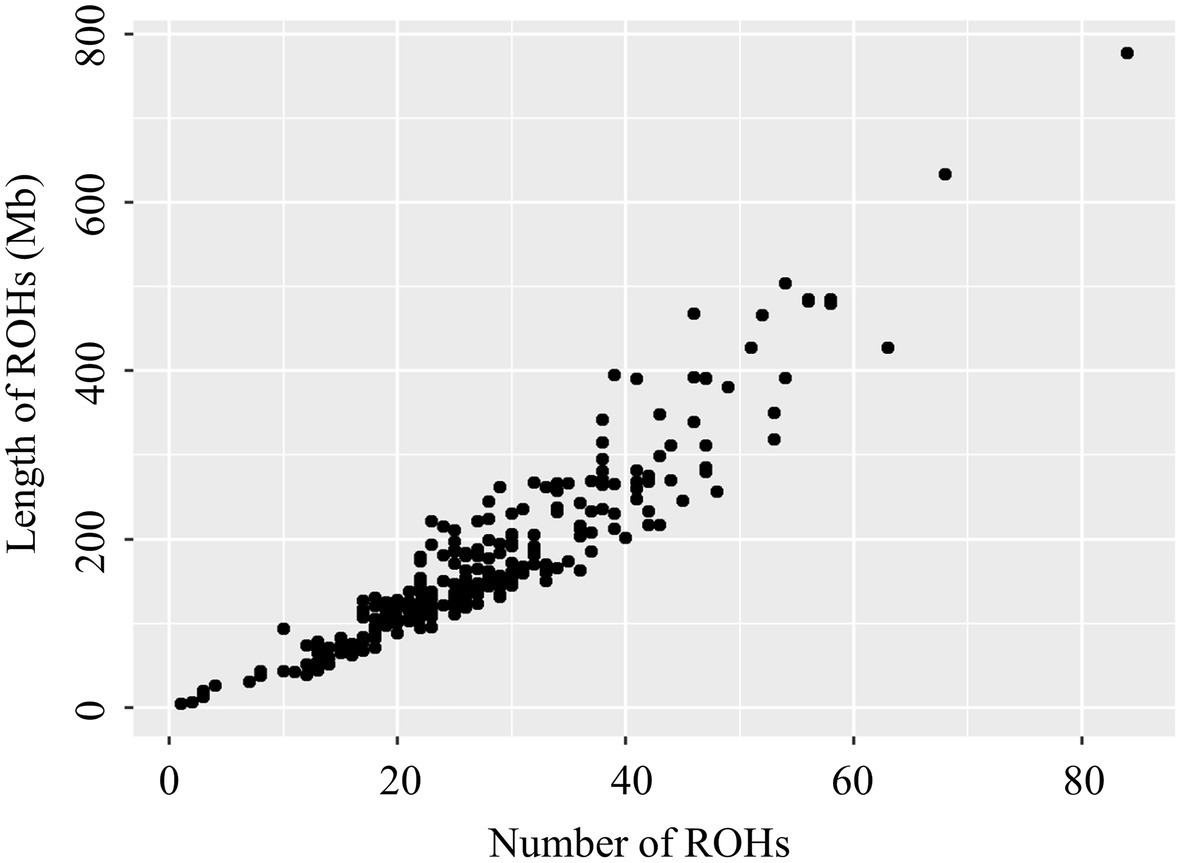
Figure 1. Relationship between the total number of ROH segments (x-axis) and the total length (Mb) of the genome in ROH (y-axis) for all individuals. Each dot represents an individual.
For chromosomes, the distribution of total number of ROHs in each chromosome and percentage coverage per chromosome are presented in Figure 2. The number of ROHs per chromosome was greater on SSC1 (710 segments), while the smallest number of ROHs was on SSC17 (118 segments). Previous studies on pigs (Xu et al., 2019; Zhan et al., 2020) have also reported the highest number of ROH on SSC1, possibly because SSC1 is the largest chromosome in the pig genome, and has more markers than other chromosomes. The highest ROH coverage was observed on SSC8 (3.26%), whereas the lowest was on SSC10 (1.19%). Our result suggests that the chromosomes with high ROH coverage might have been influenced by positive selection, which consequently increases the accumulation of advantageous alleles on the chromosome. According to our results, some genomic regions, with the highest ROH coverage, on the SSC4 require more consideration.
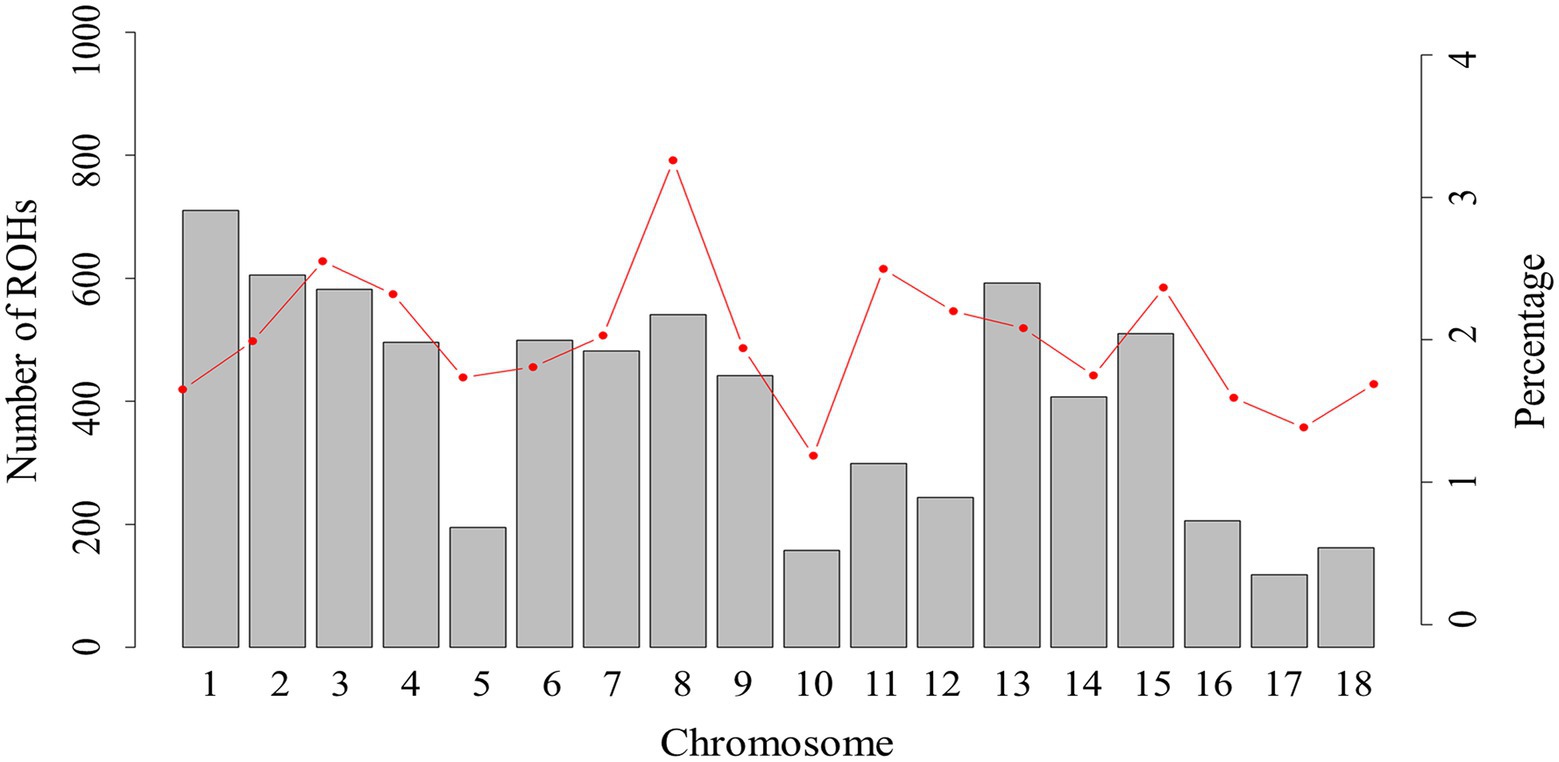
Figure 2. The number of ROHs and percentage coverage per chromosome in Xiduhei pig population. The vertical bars show the total number of ROHs per chromosome and the line shows the percentage of chromosome covered with ROH.
Here, we calculated the genomic inbreeding coefficients (FROH and FHOM) using the genotype data of 262 individuals. The mean value of the inbreeding coefficient based on total observed ROHs (FROH_total) was 0.077 and ranged from 0.002 to 0.344. The estimated FHOM inbreeding coefficients was −0.054 with a range from −0.199 to 0.251 in this population (negative values correspond to individuals with lower-than-average homozygosity). Traditionally, the inbreeding coefficient was estimated based on pedigree data. However, when it comes to cross-bred individuals, things get complicated because the genealogical relationships between parental breeds cannot be established. Moreover, in reality, pedigree information might be incorrect and incomplete, and does not usually take into account the various stochastic events of recombination, which might have occurred during meiosis (Marras et al., 2015). Thus, the inbreeding coefficient value estimated based on pedigree data could not totally show the actual relatedness among individuals within a population. In this study, we used genomic data to estimate inbreeding coefficient.
To further investigate the inbreeding coefficients which were obtained by different estimation methods, we conducted a pairwise comparisons between FHOM and FROH. The pairwise correlations among five types of inbreeding coefficients were shown in Figure 3. Among all pairwise correlations, the highest correlation was 0.94 between FROH_total and FROH>10Mb. This result showed that long ROH segments (>10Mb) were the main source of FROH_total. The inbreeding coefficients obtained by different categories of ROHs with FHOM ranged from 0.63 to 0.83, with the highest correlation found between FROH_total and FHOM. These results are in line with previous research in other pig populations (Xu et al., 2019; Shi et al., 2020) and cattle (Mastrangelo et al., 2016; Biscarini et al., 2020). Furthermore, we found that many individuals had a negative FHOM value, which might be because FHOM was sensitive to allele frequency for populations with a higher level of heterozygosity compared to FROH estimators (Zhang et al., 2015). Zhang et al. (2015) found a negative FHOM value for Danish Red Cattle (RDC), a composite breed, which is likely due to the admixture present in RDC. A similar result was obtained for a crossbred cattle in Vrindavani (Chhotaray et al., 2021). Therefore, the results of this study suggest that the inbreeding level based on total ROH may be a useful method, especially for crossbreed or composed populations.
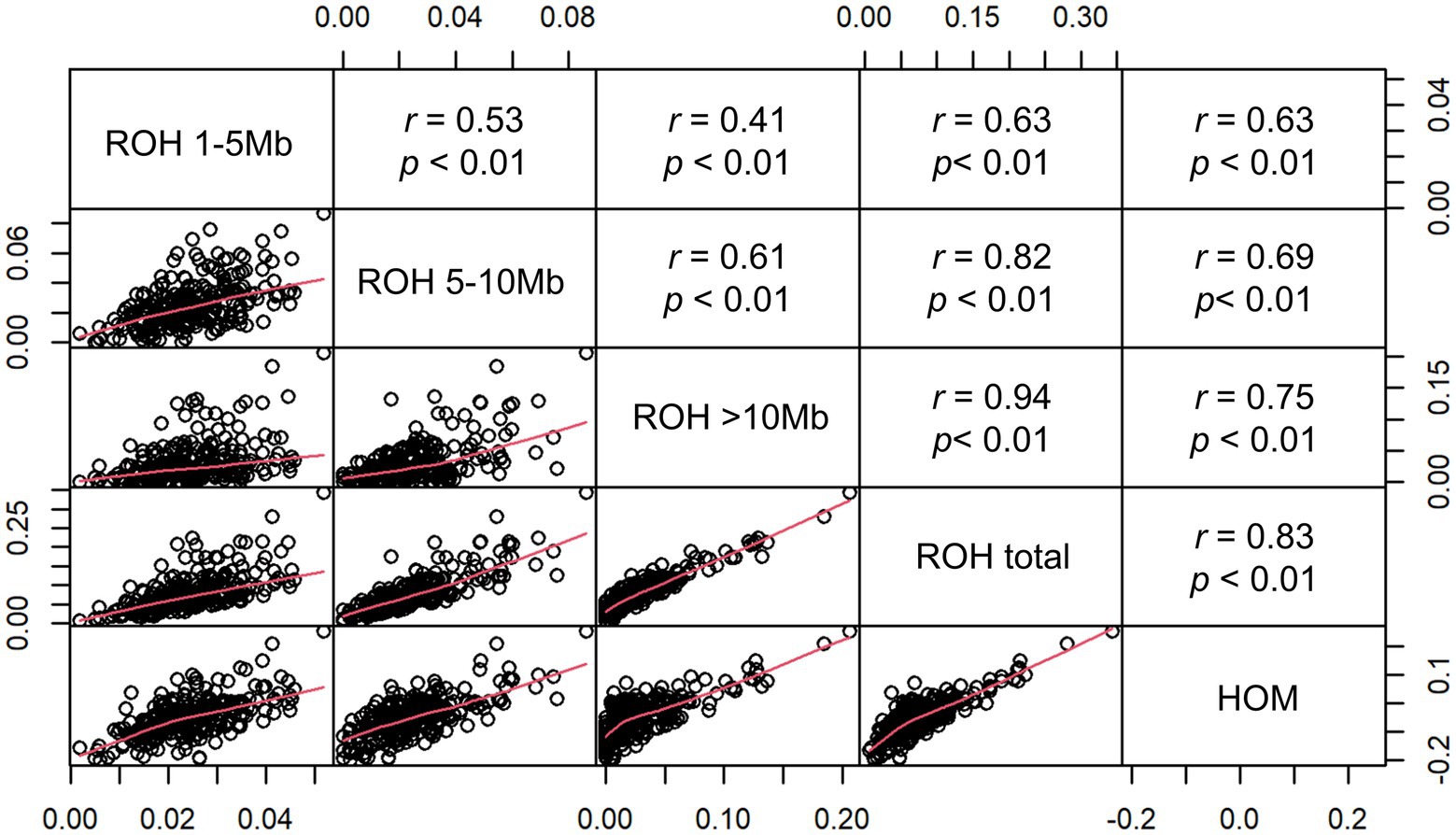
Figure 3. Scatterplots (lower panel) and Pearson’s correlations (upper panel) of the genomic inbreeding coefficients FROH (FROH 1~5 Mb, FROH5~10 Mb, FROH>10 Mb, and FROH_total) and FHOM.
Furthermore, we plotted the percentage of SNPs in ROHs against their respective positions along the chromosomes (Figure 4). The result shows a non-uniformity in the frequency of different SNPs within the ROH regions across the genome. The most frequent SNP in ROH (121 occurrences, 46.18%) was mapped at ∼35Mb on SSC11, and the closest gene to this SNP was the U6 gene. Regions of the genome with high homozygosity around the ROH islands may contain positively selected targets and might be under strong selection pressure (Pemberton et al., 2012). To identify the genomic regions that were mostly associated with ROH in all individuals, we considered the top 1% of SNPs with the highest occurrences (over 30.15% of the samples) in the ROH as candidate SNPs (Figure 4). We identified a total of eight ROH island regions (Table 2), with length ranging from 0.804Mb on SSC2 to 3.188Mb on SSC11.
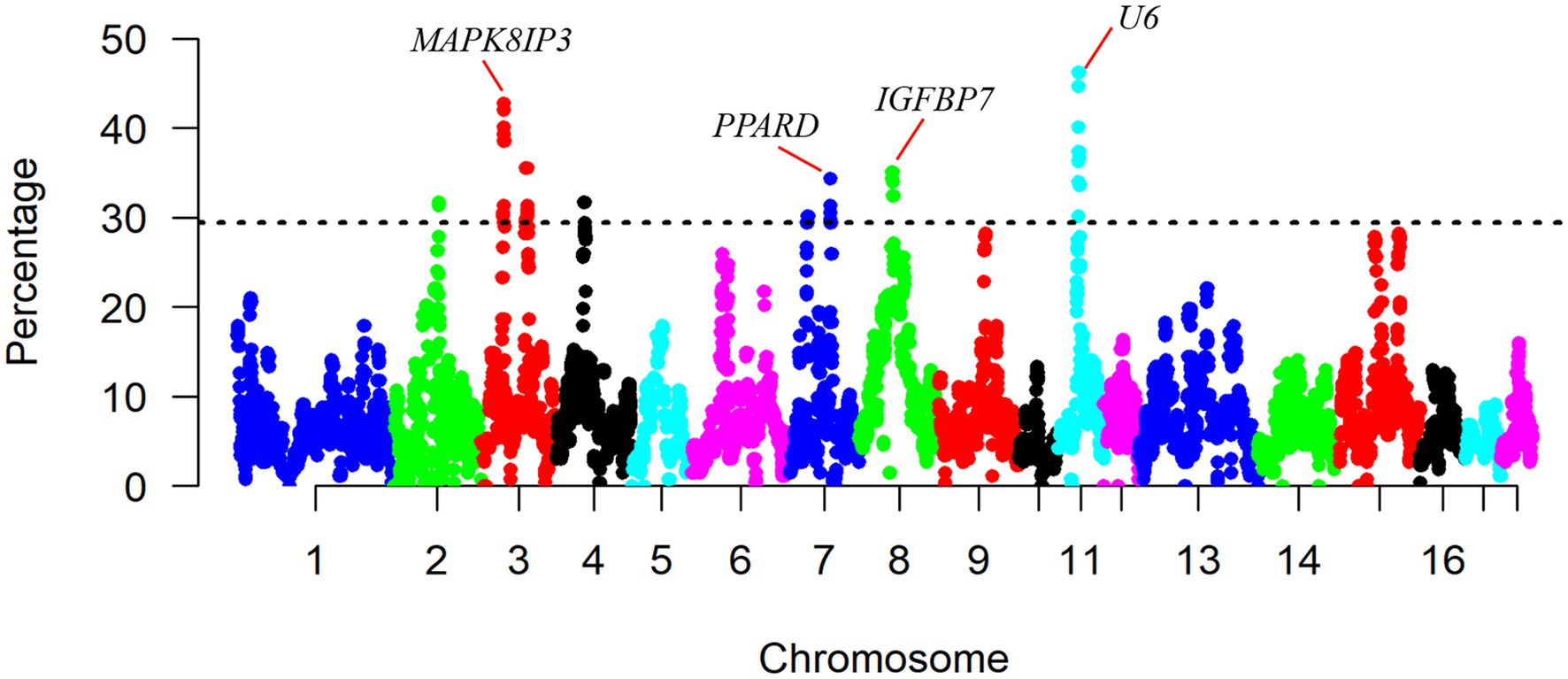
Figure 4. Manhattan plot of occurrences (%) of a SNP in ROHs across individuals.
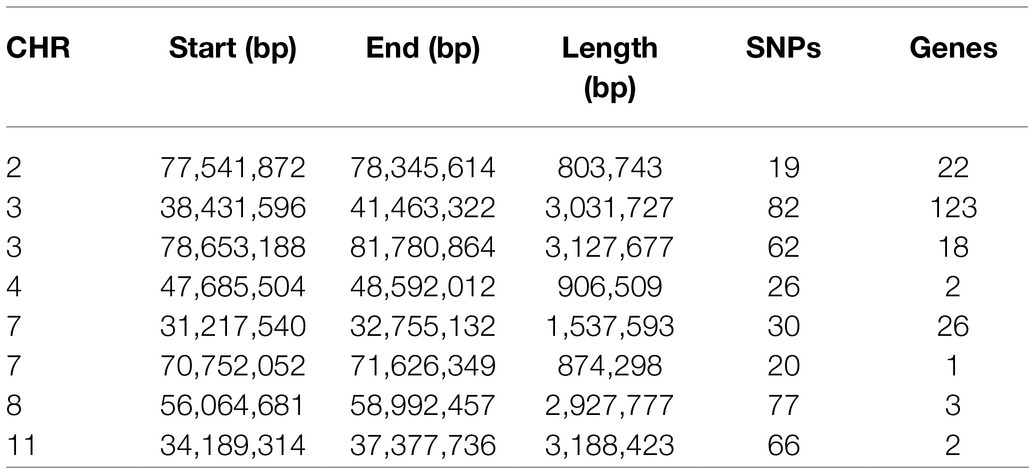
Table 2. List of genomic regions of extended homozygosity detected in Xidu black pigs.
Chromosome position, the start and end position of ROH, ROH length, number of SNPs, and the number of genes within the ROH islands were reported in Table 2. To evaluate the potential functional importance of the detected ROH islands, we analyzed the gene content of the identified regions. In summary, we annotated a total of 199 genes that were detected within these ROH islands. The chromosome position, start and end, gene name and Ensembl Gene ID were provided in Supplementary Table S1.
In this study, we focus on the genes related to some specific livestock-traits which are also important in breeding. We identified numerous candidate genes associated with muscular development and fat deposition. Among these genes is the TRAF7 gene, a MyoD1 transcriptional target that can regulate NF-κB activity during myogenesis. Studies have shown that missense mutations in TRAF7 causes developmental delay or skeletal dysplasia (Tsikitis et al., 2010; Tokita et al., 2018). IGFBP7 can promote lipid accumulation and triglyceride production in mature adipocytes and plays an important regulatory role in the differentiation of preadipocyte cells that can affect fat deposition (Hu et al., 2021). PRSS33 was related to lipid transport and metabolism, and was also detected in reported selection signature regions in Enshi black pigs, one of the founder breeds. Since Xidu black pigs are characterized for meat quality, we also detected some important genes that are associated with specific meat quality traits: TRAP1 and CREBBP are associated with pork meat pH in Finnish Yorkshire pigs (Verardo et al., 2017); SLC9A3R2 gene has been shown to be differentially expressed in longissimus muscle tissues of Meishan and Large White pigs (Jiugang et al., 2011), and was considered to be the candidate genes for meat quality (Wu et al., 2020). OTX1 is a novel regulator of proliferation, migration, invasion, and apoptosis in lung adenocarcinoma (Yang et al., 2020), and were involved in the battle between foot-and-mouth disease virus and the host (Zhang et al., 2018a). We identified several candidate genes related to reproduction traits: XPO1, a nuclear transport receptor, plays an essential role in meiotic resumption in porcine full-grown and growing oocytes (Onuma et al., 2018); SLC26A8, also known as testis anion transporter 1, is required for sperm terminal differentiation and male fertility in the mouse (Toure et al., 2007).
OR1F1, an olfactory receptor, was demonstrated to function in odor perception activation (Li et al., 2013). CLDN9 played an essential role in maintaining barrier function in airway epithelial cells (Gon et al., 2017), and E4F1 is essential for skin homeostasis (Lacroix et al., 2010). Notably, the Xidu black pigs reside in a subtropical region, which is characterized by high temperature and humidity. Therefore, we considered that OR1F1, CLDN9, and E4F1 as key factors for the environmental adaptability of Xidu black pigs. The most interesting candidate gene in this population seems to be PPARD gene, which was shown to affect the shape of the external ear and fat deposition in pigs (Meidtner et al., 2009; Ren et al., 2011). Due to the hybrid parents (Hubei white pig, Meishan pig, and Enshi black pig) have different ear shapes, the ear shapes of the base population had erected, forward sloping and drooping phenotype. Therefore, during the breeding process of Xidu black pigs, the forward sloping and slightly drooping ear shape was constantly selected. This gene may play a key role in ear shape in this population and needs more attention.
A total of 38,275 SNPs were used to calculate the average LD between all adjacent SNPs, with a distance less than 100Kb. The average and SD of estimated r2 was 0.289±0.316, which was similar to the result that (Joaquim et al., 2019) estimated in a crossbred Landrace pig population. It was observed that the LD value decreased with the increase of the distance between markers (Figure 5). When the distance was greater than 1,000Kb, the average r2 was about 0.15. The LD extent (r2=0.3) in Xidu black pigs was about 25Kb. This value was less than 79.54Kb (r2=0.3) for Meishan pigs (one of the original parental breed) obtained by the same method (Liu et al., 2020). The effectiveness of GWAS and genomic selection (GS) relies on the LD between the markers. According to the literature, a mean r2 value above 0.30 is considered as a strong LD sufficient for QTL mapping (Shifman et al., 2003). However, an average r2 of 0.20 is considered enough to achieve an accuracy of 0.85 for genomic estimated breeding value (GEBV; Meuwissen et al., 2001). In this study, we found that moderate LD (r2≥0.2) extended up to 150–200Kb. Assuming that the total length of the pig genome is 2.5Gb, this suggests that at least 12,500–16,667SNPs would be required for effective GWAS in this breed. This result could be particularly useful when designing breed-specific SNP array panels in future genomic study and selection programs for this composite pig breed.
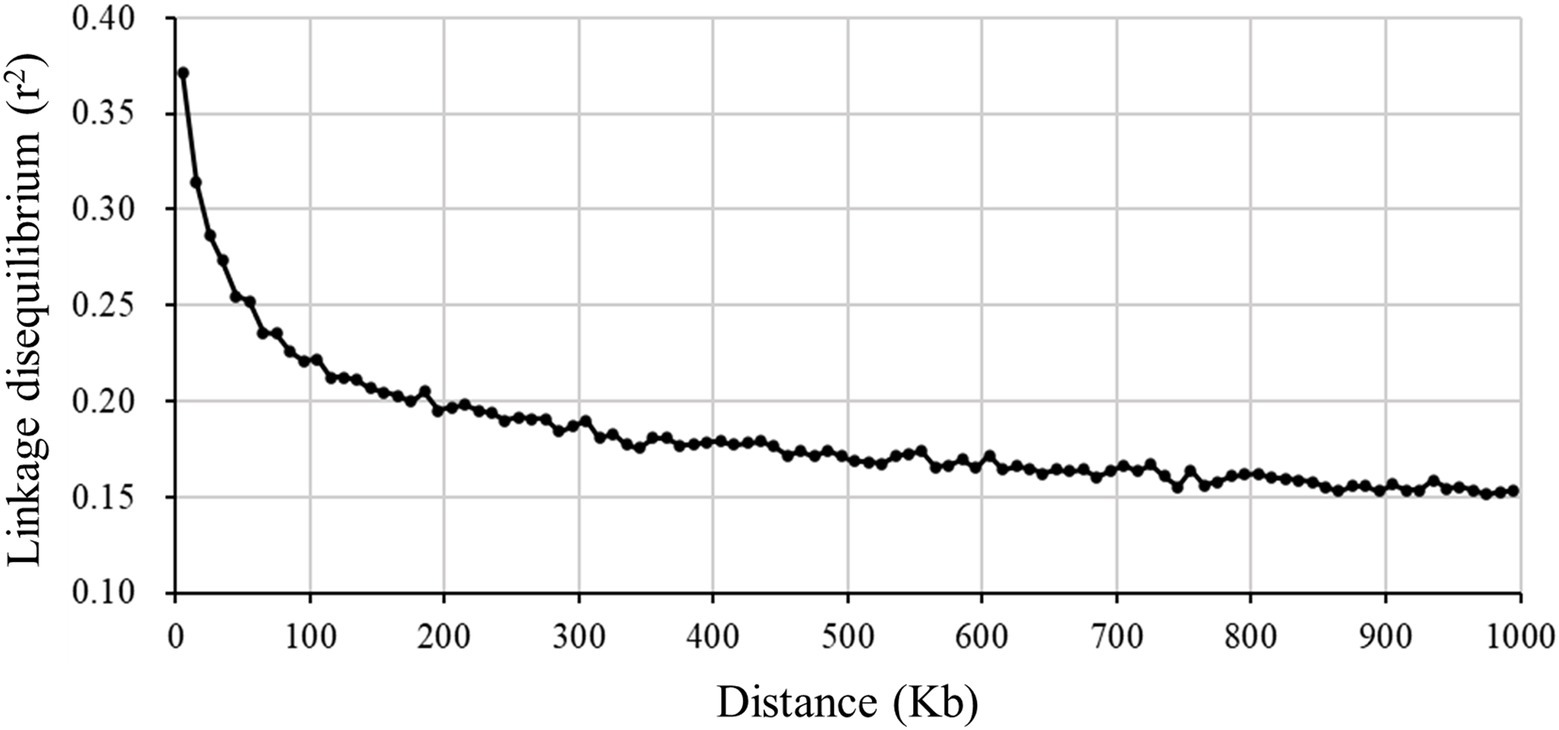
Figure 5. Decay of average linkage disequilibrium (r2) over distance (Kb) between markers for a composite pig breed.
In summary, in this study, we investigated the patterns of ROH, inbreeding coefficients and LD in the Xidu black pigs. To our knowledge, this is the first study about ROH patterns and autozygosity islands in a composite pig breed. The results of this study suggest that inbreeding based on total ROH may be a useful method, especially for crossbred or composite populations. The detected ROH patterns in this population suggests recent inbreeding events, agreeing with the newest developments in this composite pig breed. Besides, the reported genes within the identified ROH islands point to phenotypic characteristics related to reproduction, fat deposition, ear shape, and environmental adaptation. We believe that these findings will further assist in genome-wide association studies, GS, as well as the design and implementation of breed improvement and conservation programs.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://figshare.com/articles/dataset/Genome-wide_assessment_of_runs_of_homozygosity_and_estimates_of_genomic_inbreeding_in_a_Chinese_composite_pig_breed/14904567.
All experimental procedures were approved by the Institutional Animal Care and Use Committee of the Hubei Academy of Agriculture Sciences, and all methods involved pigs were in accordance with the agreement of Institutional Animal Care and Use Committee of the Hubei Academy of Agriculture Sciences (Permit number: 36/2016).
SM and XP: data curation and supervision. SM: funding acquisition. JW: project administration. ZX: writing – original draft. SM, XP, and FO: writing – review and editing. JZ, YZ, MQ, HS, ZL, LL, and BD: data analysis. All authors contributed to the article and approved the submitted version.
This work was financially supported by the Hubei Natural Science Foundation (2020BBB069), China Agriculture Research System of MOF and MARA, the Innovation Team of the Hubei Agricultural Science and Technology Innovation Center (2019-620-000-001-18), and Hubei Academy of Agricultural Sciences Youth Science Fund (2020NKYJJ10).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.720081/full#supplementary-material
Biscarini, F., Mastrangelo, S., Catillo, G., Senczuk, G., and Ciampolini, R. (2020). Insights into genetic diversity, runs of homozygosity and heterozygosity-rich regions in maremmana semi-feral cattle using pedigree and genomic data. Animals 10:2285. doi: 10.3390/ani10122285
Ceballos, F. C., Joshi, P. K., Clark, D. W., Ramsay, M., and Wilson, J. F. (2018). Runs of homozygosity: windows into population history and trait architecture. Nat. Rev. Genet. 19, 220–234. doi: 10.1038/nrg.2017.109
Chang, C. C., Chow, C. C., Tellier, L. C., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4:7. doi: 10.1186/s13742-015-0047-8
Chhotaray, S., Panigrahi, M., Pal, D., Ahmad, S. F., Bhanuprakash, V., Kumar, H., et al. (2021). Genome-wide estimation of inbreeding coefficient, effective population size and haplotype blocks in Vrindavani crossbred cattle strain of India. Biol. Rhythm. Res. 52, 666–679. doi: 10.1080/09291016.2019.1600266
China National Commission of Animal Genetic Resources (2011). Animal Genetic Resources in China Pigs. Beijing: Chinese Agriculture Press.
Dixit, S. P., Singh, S., Ganguly, I., Bhatia, A. K., Sharma, A., Kumar, N. A., et al. (2020). Genome-wide runs of homozygosity revealed selection signatures in bos indicus. Front. Genet. 11:92. doi: 10.3389/fgene.2020.00092
Ganteil, A., Rodriguez-Ramilo, S. T., Ligonesche, B., and Larzul, C. (2020). Characterization of autozygosity in pigs in three-way crossbreeding. Front. Genet. 11:584556. doi: 10.3389/fgene.2020.584556
Gon, Y., Maruoka, S., Kishi, H., Kozu, Y., Kazumichi, K., Nomura, Y., et al. (2017). NDRG1 is important to maintain the integrity of airway epithelial barrier through claudin-9 expression. Cell Biol. Int. 41, 716–725. doi: 10.1002/cbin.10741
Hill, W. G., and Robertson, A. (1968). Linkage disequilibrium in finite populations. Theor. Appl. Genet. 38, 226–231. doi: 10.1007/BF01245622
Howard, J. T., Tiezzi, F., Huang, Y., Gray, K. A., and Maltecca, C. (2016). Characterization and management of long runs of homozygosity in parental nucleus lines and their associated crossbred progeny. Genet. Sel. Evol. 48:91. doi: 10.1186/s12711-016-0269-y
Hu, Z., Wu, J., Qin, L., Jin, H., Cao, Y., and Zhao, Y. (2021). IGFBP7 downregulation or overexpression effect on bovine preadipocyte differentiation. Anim. Biotechnol. 32, 21–30. doi: 10.1080/10495398.2019.1642906
Jiugang, Z., Jing, L., and Yonggang, L. (2011). A novel porcine gene--SLC9A3R2, differentially expressed in the longissimus muscle tissues from meishan and large white pigs. Folia Biol. 57, 20–25.
Joaquim, L. B., Chud, T. C. S., Marchesi, J. A. P., Savegnago, R. P., Buzanskas, M. E., Zanella, R., et al. (2019). Genomic structure of a crossbred landrace pig population. PLoS One 14:e0212266. doi: 10.1371/journal.pone.0212266
Keller, M. C., Visscher, P. M., and Goddard, M. E. (2011). Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data. Genetics 189, 237–249. doi: 10.1534/genetics.111.130922
Lacroix, M., Caramel, J., Goguet-Rubio, P., Linares, L. K., Estrach, S., Hatchi, E., et al. (2010). Transcription factor E4F1 is essential for epidermal stem cell maintenance and skin homeostasis. Proc. Natl. Acad. Sci. U. S. A. 107, 21076–21081. doi: 10.1073/pnas.1010167107
Li, M., Tian, S., Jin, L., Zhou, G., Li, Y., Zhang, Y., et al. (2013). Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat. Genet. 45, 1431–1438. doi: 10.1038/ng.2811
Liu, C., Li, P., Zhou, W., Ma, X., Wang, X., Xu, Y., et al. (2020). Genome data uncover conservation status, historical relatedness and candidate genes under selection in Chinese indigenous pigs in the Taihu Lake region. Front. Genet. 11:591. doi: 10.3389/fgene.2020.00591
Marras, G., Gaspa, G., Sorbolini, S., Dimauro, C., Ajmone-Marsan, P., Valentini, A., et al. (2015). Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim. Genet. 46, 110–121. doi: 10.1111/age.12259
Mastrangelo, S., Tolone, M., Di Gerlando, R., Fontanesi, L., Sardina, M. T., and Portolano, B. (2016). Genomic inbreeding estimation in small populations: evaluation of runs of homozygosity in three local dairy cattle breeds. Animal 10, 746–754. doi: 10.1017/S1751731115002943
Mastrangelo, S., Tolone, M., Sardina, M. T., Sottile, G., Sutera, A. M., Di Gerlando, R., et al. (2017). Genome-wide scan for runs of homozygosity identifies potential candidate genes associated with local adaptation in Valle del Belice sheep. Genet. Sel. Evol. 49:84. doi: 10.1186/s12711-017-0360-z
McQuillan, R., Leutenegger, A. L., Abdel-Rahman, R., Franklin, C. S., Pericic, M., Barac-Lauc, L., et al. (2008). Runs of homozygosity in European populations. Am. J. Hum. Genet. 83, 359–372. doi: 10.1016/j.ajhg.2008.08.007
Meidtner, K., Schwarzenbacher, H., Scharfe, M., Severitt, S., Blocker, H., and Fries, R. (2009). Haplotypes of the porcine peroxisome proliferator-activated receptor delta gene are associated with backfat thickness. BMC Genet. 10:76. doi: 10.1186/1471-2156-10-76
Meuwissen, T. H., Hayes, B. J., and Goddard, M. E. (2001). Prediction of total genetic value using genome-wide dense marker maps. Genetics 157, 1819–1829. doi: 10.1093/genetics/157.4.1819
Mokry, F. B., Buzanskas, M. E., de Alvarenga Mudadu, M., do Amaral Grossi, D., Higa, R. H., Ventura, R. V., et al. (2014). Linkage disequilibrium and haplotype block structure in a composite beef cattle breed. BMC Genomics 15(Suppl. 7):S6. doi: 10.1186/1471-2164-15-S7-S6
Onuma, A., Fujioka, Y. A., Fujii, W., Sugiura, K., and Naito, K. (2018). Effects of exportin 1 on nuclear transport and meiotic resumption in porcine full-grown and growing oocytes. Biol. Reprod. 98, 501–509. doi: 10.1093/biolre/iox168
Pemberton, T. J., Absher, D., Feldman, M. W., Myers, R. M., Rosenberg, N. A., and Li, J. Z. (2012). Genomic patterns of homozygosity in worldwide human populations. Am. J. Hum. Genet. 91, 275–292. doi: 10.1016/j.ajhg.2012.06.014
Peripolli, E., Munari, D. P., Silva, M., Lima, A. L. F., Irgang, R., and Baldi, F. (2017). Runs of homozygosity: current knowledge and applications in livestock. Anim. Genet. 48, 255–271. doi: 10.1111/age.12526
Peripolli, E., Stafuzza, N. B., Amorim, S. T., de Lemos, M. V. A., Grigoletto, L., Kluska, S., et al. (2020). Genome-wide scan for runs of homozygosity in the composite Montana Tropical((R)) beef cattle. J. Anim. Breed. Genet. 137, 155–165. doi: 10.1111/jbg.12428
Peripolli, E., Stafuzza, N. B., Munari, D. P., Lima, A. L. F., Irgang, R., Machado, M. A., et al. (2018). Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr (Bos indicus) dairy cattle. BMC Genomics 19:34. doi: 10.1186/s12864-017-4365-3
Ren, J., Duan, Y., Qiao, R., Yao, F., Zhang, Z., Yang, B., et al. (2011). A missense mutation in PPARD causes a major QTL effect on ear size in pigs. PLoS Genet. 7:e1002043. doi: 10.1371/journal.pgen.1002043
Saravanan, K. A., Panigrahi, M., Kumar, H., Parida, S., Bhushan, B., Gaur, G. K., et al. (2020). Genome-wide assessment of genetic diversity, linkage disequilibrium and haplotype block structure in Tharparkar cattle breed of India. Anim. Biotechnol. 30, 1–15. doi: 10.1080/10495398.2020.1796696
Shi, L., Wang, L., Liu, J., Deng, T., Yan, H., Zhang, L., et al. (2020). Estimation of inbreeding and identification of regions under heavy selection based on runs of homozygosity in a large white pig population. J. Anim. Sci. Biotechnol. 11:46. doi: 10.1186/s40104-020-00447-0
Shifman, S., Kuypers, J., Kokoris, M., Yakir, B., and Darvasi, A. (2003). Linkage disequilibrium patterns of the human genome across populations. Hum. Mol. Genet. 12, 771–776. doi: 10.1093/hmg/ddg088
Tokita, M. J., Chen, C. A., Chitayat, D., Macnamara, E., Rosenfeld, J. A., Hanchard, N., et al. (2018). De novo missense variants in TRAF7 cause developmental delay, congenital anomalies, and dysmorphic features. Am. J. Hum. Genet. 103, 154–162. doi: 10.1016/j.ajhg.2018.06.005
Toure, A., Lhuillier, P., Gossen, J. A., Kuil, C. W., Lhote, D., Jegou, B., et al. (2007). The testis anion transporter 1 (Slc26a8) is required for sperm terminal differentiation and male fertility in the mouse. Hum. Mol. Genet. 16, 1783–1793. doi: 10.1093/hmg/ddm117
Tsikitis, M., Acosta-Alvear, D., Blais, A., Campos, E. I., Lane, W. S., Sanchez, I., et al. (2010). Traf7, a MyoD1 transcriptional target, regulates nuclear factor-kappaB activity during myogenesis. EMBO Rep. 11, 969–976. doi: 10.1038/embor.2010.154
Verardo, L. L., Sevon-Aimonen, M. L., Serenius, T., Hietakangas, V., and Uimari, P. (2017). Whole-genome association analysis of pork meat pH revealed three significant regions and several potential genes in Finnish Yorkshire pigs. BMC Genet. 18:13. doi: 10.1186/s12863-017-0482-x
Wu, F., Sun, H., Lu, S., Gou, X., Yan, D., Xu, Z., et al. (2020). Genetic diversity and selection signatures within diannan small-ear pigs revealed by next-generation sequencing. Front. Genet. 11:733. doi: 10.3389/fgene.2020.00733
Xu, Z., Sun, H., Zhang, Z., Zhao, Q., Olasege, B. S., Li, Q., et al. (2019). Assessment of autozygosity derived from runs of homozygosity in Jinhua pigs disclosed by sequencing data. Front. Genet. 10:274. doi: 10.3389/fgene.2019.00274
Yang, C. Y., Wang, L., Mu, D. C., Li, F. F., Ran, P. Z., Shen, H., et al. (2020). OTX1 is a novel regulator of proliferation, migration, invasion and apoptosis in lung adenocarcinoma. Eur. Rev. Med. Pharmacol. Sci. 24, 9497–9510. doi: 10.26355/eurrev_202009_23035
Zhan, H., Zhang, S., Zhang, K., Peng, X., Xie, S., Li, X., et al. (2020). Genome-wide patterns of homozygosity and relevant characterizations on the population structure in Pietrain pigs. Genes (Basel) 11:577. doi: 10.3390/genes11050577
Zhang, Q., Calus, M. P., Guldbrandtsen, B., Lund, M. S., and Sahana, G. (2015). Estimation of inbreeding using pedigree, 50k SNP chip genotypes and full sequence data in three cattle breeds. BMC Genet. 16:88. doi: 10.1186/s12863-015-0227-7
Zhang, T., Chen, H., Qi, L., Zhang, J., Wu, R., Zhang, Y., et al. (2018a). Transcript profiling identifies early response genes against FMDV infection in PK-15 cells. Viruses 10:364. doi: 10.3390/v10070364
Keywords: pig, crossbreed, runs of homozygosity, inbreeding, linkage disequilibrium
Citation: Xu Z, Mei S, Zhou J, Zhang Y, Qiao M, Sun H, Li Z, Li L, Dong B, Oyelami FO, Wu J and Peng X (2021) Genome-Wide Assessment of Runs of Homozygosity and Estimates of Genomic Inbreeding in a Chinese Composite Pig Breed. Front. Genet. 12:720081. doi: 10.3389/fgene.2021.720081
Edited by:
Tiago do Prado Paim, Goiano Federal Institute (IFGOIANO), BrazilReviewed by:
Ran Li, Northwest A and F University, ChinaCopyright © 2021 Xu, Mei, Zhou, Zhang, Qiao, Sun, Li, Li, Dong, Oyelami, Wu and Peng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junjing Wu, amVhbm5lMTEwNkAxMjYuY29t; Xianwen Peng, cHh3cGFsQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.