 Katalin Komlosi1*
Katalin Komlosi1* Olivier Claris2,3†
Olivier Claris2,3† Sophie Collardeau-Frachon4,5†
Sophie Collardeau-Frachon4,5† Julia Kopp1
Julia Kopp1 Ingrid Hausser6Juliette Mazereeuw-Hautier7
Ingrid Hausser6Juliette Mazereeuw-Hautier7 Nathalie Jonca8,9Andreas D. Zimmer1
Nathalie Jonca8,9Andreas D. Zimmer1 Damien Sanlaville10,11
Damien Sanlaville10,11 Judith Fischer1
Judith Fischer1- 1Institute of Human Genetics, Medical Center, Faculty of Medicine, University of Freiburg, Freiburg, Germany
- 2Department of Neonatology, Hospices Civils de Lyon, Hôpital Femme Mère Enfant, Bron, France
- 3Claude Bernard University, Lyon, France
- 4Hospices Civils de Lyon, Hôpital Femme Mère Enfant, Institut de Pathologie, Bron, France
- 5Faculté de médecine Lyon Est, Claude Bernard University, Lyon, France
- 6Institute of Pathology, Heidelberg University Hospital, Heidelberg, Germany
- 7Dermatology Department, Reference Center for Rare Skin Diseases, CHU Larrey, Université Paul Sabatier, Toulouse, France
- 8Infinity, CNRS, Inserm, UPS, Université Toulouse, Toulouse, France
- 9CHU Toulouse, Hôpital Purpan, Laboratoire de Biologie Cellulaire et Cytologie, Institut Fédératif de Biologie, Toulouse, France
- 10Hospices Civils de Lyon, Hôpital Femme Mère Enfant, Service de Génétique, Bron, France
- 11Institut Neuromyogène, Université de Lyon, Lyon, France
Neonatal collodion baby or ichthyosis can pose a diagnostic challenge, and in many cases, only additional organ involvement or the course of the disease will help differentiate between non-syndromic and syndromic forms. Skin abnormalities are described in about 20% of the congenital disorders of glycosylation (CDG). Among those, some rare CDG forms constitute a special group among the syndromic ichthyoses and can initially misdirect the diagnosis towards non-syndromic genodermatosis. DOLK-CDG is such a rare subtype, resulting from a defect in dolichol kinase, in which the congenital skin phenotype (often ichthyosis) is later associated with variable extracutaneous features such as dilatative cardiomyopathy, epilepsy, microcephaly, visual impairment, and hypoglycemia and may lead to a fatal course. We report two neonatal cases of lethal ichthyosis from the same family, with distal digital constrictions and a progressive course leading to multi-organ failure and death. Postmortem trio whole-exome sequencing revealed the compound heterozygous variants NM_014908.3: c.1342G>A, p.(Gly448Arg) and NM_014908.3: c.1558A>G, p.(Thr520Ala) in the DOLK gene in the first affected child, which were confirmed in the affected sibling. Reduced staining with anti-α-Dystroglycan antibody was observed in frozen heart tissue of the second child as an expression of reduced O-mannosylation due to the dolichol kinase deficiency. In addition to the detailed dermatopathological changes, both cases presented hepatic and extrahepatic hemosiderosis on histological examination. Our patients represent an early and fatal form of DOLK-CDG with a striking presentation at birth resembling severe collodion baby. Both cases emphasize the phenotypic variability of glycosylation disorders and the importance to broaden the differential diagnosis of ichthyosis and to actively search for organ involvement in neonates with ichthyosis.
Introduction
Pediatric disorders leading to perinatal death still present a challenge for genetic counseling when rapid whole exome/whole genome sequencing (WES/WGS) in the neonatal setting is not available. Affected children often die before a genetic diagnosis can be established and phenotypic descriptions are often insufficient. Complete and rigorous autopsy with multiple tissue sampling of affected fetuses or children is of great importance for detecting all anomalies that can indicate a potential diagnosis, and providing frozen tissue for genetic and functional analysis. A definitive diagnosis always requires a multidisciplinary approach including clinical data, autopsy findings, and genetic results, especially for correct interpretation of the pathogenicity of the identified DNA variants. A confirmed genetic defect is the requirement for targeted carrier testing in parents and prenatal or preimplantation genetic diagnosis in further pregnancies.
Inherited ichthyoses are classified as Mendelian disorders of cornification (MEDOC), which are further defined on the basis of clinical and genetic features and can be divided into non-syndromic and syndromic forms. To date, mutations in more than 50 genes are known to result in various types of ichthyoses (Fischer and Bourrat, 2020). The syndromic ichthyoses are generally very rare and are classified based on the mode of inheritance, and can be further subdivided according to the predominant symptoms (Oji et al., 2010; Fischer and Bourrat, 2020).
Rare forms of congenital disorders of glycosylation (CDG) are among the inborn errors of metabolism presenting at birth with ichthyosis. CDG are due to deficient glycosylation of proteins and lipids and are characterized by multi-organ symptoms with a wide range of clinical severity, from mild symptoms to severe multisystem dysfunction, and even a fatal course (Francisco et al., 2019). The spectrum of clinical manifestations comprises psychomotor delay and intellectual disability, muscle hypotonia, seizures, endocrine and coagulation abnormalities, ophthalmologic anomalies, failure to thrive, and variable dysmorphic features. Skin abnormalities are described in only about 20% of the different CDG forms (Rymen et al., 2012; Haijes et al., 2020; Komlosi et al., 2020). Generally, the skin manifestations represent only a single feature within a much broader phenotype and include orange peel skin, ichthyosis, increased skin laxity, hypo/hyperpigmentation, tumoral calcinosis, aplasia cutis congenita, hypohidrosis, hyperthermia, lipodystrophy, and psoriasis (Rymen et al., 2012; Kouwenberg et al., 2014; Alsubhi et al., 2017; Van Damme et al., 2017; Haijes et al., 2020; Komlosi et al., 2020). CDG linked to ichthyosis or ichthyosiform dry skin with variable neurologic and multi-organ involvement are due to deficiencies within the dolichol (DOLK-, SRD5A3-CDG) and the GPI (glycosylphosphatidylinositol) anchor biosynthesis pathway (PIGL-CDG, MPDU1-CDG) (Schenk et al., 2001; Kranz et al., 2007; Kahrizi et al., 2011; Lefeber et al., 2011; Kapusta et al., 2013; Jaeken et al., 2014; Rymen and Jaeken, 2014; Rush et al., 2017; Thiel et al., 2018; Hall et al., 2020; Ng et al., 2021). Recently, mild manifestations were also observed in a patient with COG5-CDG (Rymen et al., 2012). We recently described a family with two affected children with COG6-CDG in whom very dry, tight, and rigid skin with hyperkeratosis and scaling was the prominent skin feature at birth (Komlosi et al., 2020).
Construction of the complex N-linked glycan chains on proteins starts with production of the lipid carrier dolichyl phosphate, which serves to anchor the nascent oligosaccharide to the endoplasmic reticulum. It is known that dolichol and dolichyl phosphate metabolites are expressed ubiquitously in humans and are localized to the endoplasmic reticulum but also present in other organelles such as the Golgi apparatus, mitochondria, and lysosomes. In addition to their critical role in glycan synthesis, dolichols have a number of other proposed functions including modulating physiochemical properties of lipid bilayers and shielding of cellular lipids from oxidative damage (Cantagrel et al., 2010; Buczkowska et al., 2015). Dolichol kinase deficiency (DOLK-CDG, OMIM #610768) is an autosomal recessive disorder that results from deficiency of the enzyme responsible for the terminal step in the synthesis of dolichol phosphate. The range of symptoms and age at presentation are highly variable in the patients described so far (Kranz et al., 2007; Lefeber et al., 2011; Kapusta et al., 2013; Rush et al., 2017; Hall et al., 2020). The phenotypic spectrum encompasses three major forms with (1) neurological abnormalities with hypotonia, microcephaly, seizures, and visual impairment with or without ichthyosis; (2) isolated cardiomyopathy; and (3) multiorgan involvement, which can also include ichthyosis (Kranz et al., 2007; Lefeber et al., 2011; Helander et al., 2013; Kapusta et al., 2013; Lieu et al., 2013; Rush et al., 2017; Hall et al., 2020; Paprocka et al., 2021). A few cases with biallelic pathogenic variants in DOLK (OMIM *610746) with severe multi-organ involvement encompassing profound muscular hypotonia, ichthyosiform skin, nystagmus, epilepsy, and pulmonary infections leading to death within the first months of life have also been described (Kranz et al., 2007; Lefeber et al., 2011; Lieu et al., 2013; Rush et al., 2017; Hall et al., 2020).
We present two children of a non-consanguineous French couple with compound heterozygous variants in the DOLK gene presenting with severe ichthyosis, distal digital constrictions, cardiomegaly, thrombocytopenia, diffuse coagulation defect, and progressive multi-organ failure, which resulted in death in the neonatal period. They represent two additional cases of the early and fatal form of DOLK-CDG with a striking presentation at birth resembling autosomal recessive congenital ichthyosis with progressive multi-organ failure.
Materials and Methods
Clinical Report
Patient 1
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Ethics Committee of the University of Freiburg (ethical code number: 436/17). The parents of the patients gave written informed consent to the publication of the clinical information and the pictures.
Patient 1 was male and the first child of healthy French European parents (Figure 1A). The mother was a 30-year-old with a history of two spontaneous abortions. Pregnancy was complicated by oligohydramnios and intrauterine growth retardation. Prenatal ultrasound examinations did not show any malformation and the couple did not wish further prenatal genetic diagnosis. Pregnancy was marked by rupture of the amniotic membrane at 31 + 2 weeks of gestation and administration of antenatal corticosteroids. The infant was born prematurely at 35 + 1 weeks of gestation by vaginal delivery with a birth weight of 1,840 g (3rd percentile), length of 42.5 cm (3rd percentile), and occipitofrontal circumference (OFC) of 31.2 cm (10th percentile). Apgar scores were 10/10/10/10 at 1, 3, 5, and 10 min, respectively. Physical examination showed a congenital anomaly of all four extremities resembling amniotic bands with extremity edema and tendinous retractions at the level of the joints (Figure 1B), underdeveloped thenar, rigid fingers, signs of oligohydramnios with low-set ears, hypertelorism, broad forehead and flat nose, a narrow chest, and arthrogryposis (Supplementary Figure S1A). The most prominent feature was the very dry, nonelastic, and hard skin. Chest x-ray showed a narrow thorax and lung hypoplasia as a consequence of the oligohydramnios and cardiomegaly with right deviation of the heart. There was no sign of hepatosplenomegaly on physical examination, with no abdominal ultrasound performed. After an initial stable cardiac and respiratory state, the neonate developed progressive respiratory failure at 12 h of age and was transferred to a level III Neonatal Intensive Care Unit. He was difficult to ventilate adequately, and developed pneumomediastinum and pneumothorax requiring exsufflation. He was subsequently switched from conventional ventilation to high-frequency oscillatory ventilation. Sequential echocardiographs were performed showing severe persistent pulmonary hypertension, a large right-to-left shunting patent ductus arteriosus, and a mild but significant left ventricle hypokinesia with mild mitral and aortic insufficiency. Despite administration of dobutamine and norepinephrine, hypotension persisted and the child developed multi-organ failure after 48 h, with anuria, severe metabolic acidosis (pH 6.97), elevated lactate (15 mmol/L), thrombocytopenia (64 G/L), elevated transaminases, necrosis of the digits (Figure 1B), and a progressive respiratory failure. He died on postnatal day 3. The immediate cause of death was hemodynamic instability, ventricular fibrillation, and severe metabolic acidosis.
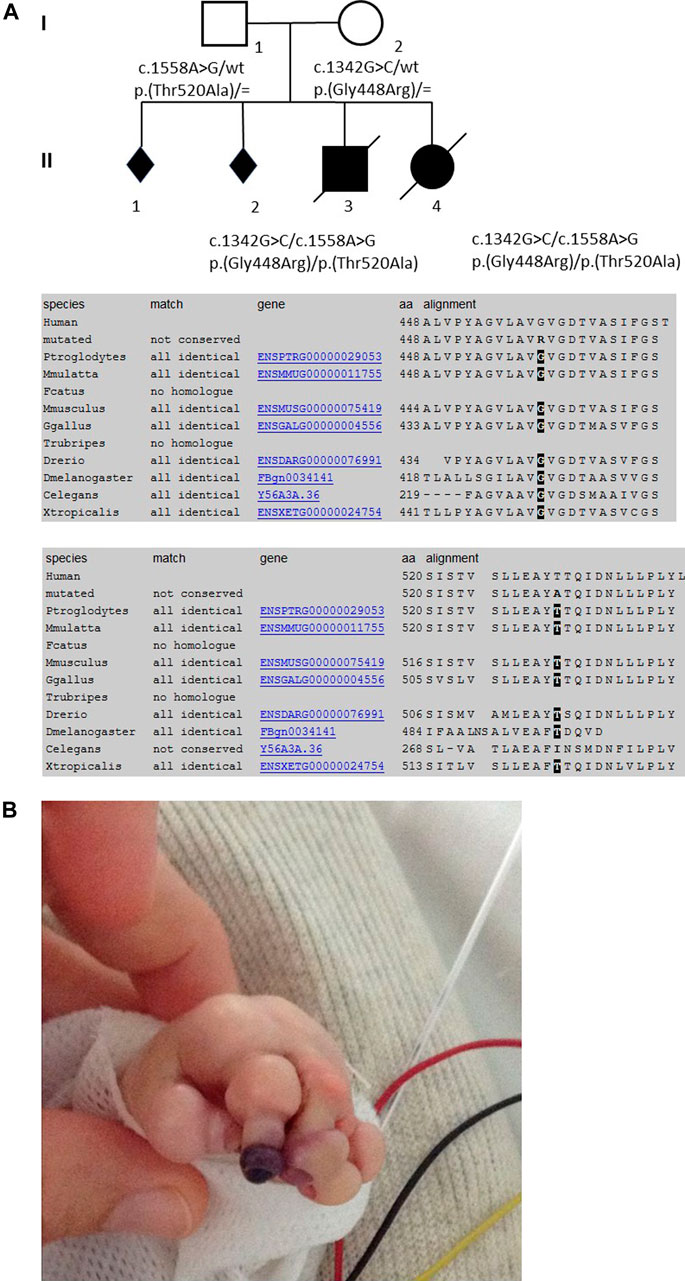
FIGURE 1. Pedigree of the family, conservation of the AAs among the diverse species (from MutationTaster, www.mutationtaster.org), and skin manifestations of the first child in the neonatal period.
Patient 2
Patient 2 was female, born to the same parents 20 months after patient 1 (Figure 1A). The pregnancy was complicated by prenatal ultrasound findings of hyperechogenic intestines in gestational week 23. Conventional karyotyping and array CGH analysis from amniotic fluid was normal; CFTR screening revealed a heterozygous paternal mutation. After premature rupture of the amniotic membrane and chorioamnionitis, the infant was born prematurely at 32 + 4 weeks of gestation by vaginal delivery with a birth weight of 1,800 g (25th–50th percentile), a length of 44.5 cm (50th–75th percentile), and OFC of 29 cm (25th percentile). Apgar scores were 9/8/8/10 at 1, 3, 5, and 10 min, respectively. Physical exam showed collodion membrane with tight and hard skin with fissures, no ectropion or eclabion, and edema of the extremities (Supplementary Figures S1A,B). The fingers and toes were fixed in rigid volar/plantar flexion (Supplementary Figure S1B) and there was a blackish appearance of some fingers, raising the possibility of necrosis. Relieving incisions of the fingers (fingers II to V of both hands) were performed on day 4. There was no evidence of respiratory distress syndrome and the child was maintained on nasal CPAP (continuous positive airway pressure) until she developed apneic episodes and intubation was indicated. Although initially stable hemodynamically, on postnatal day 5, she developed several bradycardic episodes. There was no sign of hepatosplenomegaly and abdominal ultrasound was normal. Laboratory investigations revealed a diffuse coagulation defect (thrombocyte count: 201 G/L; total coagulation activity: 5.29xT, normal range: 0.8–1.2; Fibrinogen 1.8 g/L; Factor II: 9%, Factor V: 7%, Factor VII: 8%, Factor X: 16%) and plasma transfusion was administered. Ophthalmological examination showed extensive retinal hemorrhage of the right eye. On postnatal day 7, following episodes of crying and recurrent vomiting, the child developed a sudden cardiopulmonary arrest leading to death.
Molecular Genetic Analyses
Since there was no confirmed genetic diagnosis during the neonatal period in either of the siblings, postmortem genetic analyses were performed. With the clinical suspicion of congenital syndromic ichthyosis, a multi-gene panel was initially performed from DNA extracted from a postmortem liver sample of the second child of the family. Mutation analysis of 66 genes associated with non-syndromic and syndromic ichthyosis (in-house designed HaloPlex Custom Kit, Agilent Technologies, Inc. Santa Clara, CA, United States) detected no pathogenic alterations.
For further analysis, trio whole-exome sequencing of the first child and the parents was performed using DNA extracted from a postmortem thymus sample and from peripheral blood of the healthy parents. Target enrichment of all coding genomic regions (exome) was performed with a Twist Human Core Exome Kit (Twist Bioscience) and sequencing was run on an Illumina platform (NextSeq500 System, San Diego, United States) with 150-bp paired-end reads. Candidate causative variants were validated by Sanger sequencing.
Histopathological Analyses
Histopathological examination of the skin (H&E staining) was performed from the abdominal wall along the incision that was performed for internal examination in the first child (Figure 5A) and from the right inner thigh and the scalp of the second affected child (Figures 5B,C).
To assess glycogen storage in the liver and heart tissue of the first and second child, Periodic Acid–Schiff (PAS) and Periodic Acid–Schiff–diastase (PAS diastase) stainings were performed on FFPE tissues. For the detection of hemosiderosis, Perls staining was performed on FFPE of liver, spleen, pancreas, renal, thyroid, and thymus samples.
O-mannosylation was assessed by immunohistochemistry on frozen heart tissue sample of the second child with anti-α-Dystroglycan antibody (clone VIA4-1, Sigma-Aldrich Merck, Germany) directed against the O-mannosyl glycans of alpha-dystroglycan. As a control, a normal heart tissue sample from a healthy proband of the same age was used. To determine whether the antibody worked, a tissue sample from adult skeletal muscle was used.
Results
Molecular Genetic Results
Trio whole-exome sequencing revealed the compound heterozygous variants NM_014908.3:c.1342G>C, p.(Gly448Arg) and NM_014908.3:c.1558A>G, p.(Thr520Ala) in the DOLK gene in the affected first child. Sanger sequencing confirmed both variants in the DNA of the second child (Figure 1A). The variant NM_014908.3:c.1342G > C, p. (Gly448Arg), which is maternally derived, has not been described in the literature so far, nor is it listed in clinical databases (ClinVar or HGMD® Professional 2020.3). In the population database gnomAD (the Genome Aggregation Database v2.1.1; http://gnomad.broadinstitute.org/), it is listed in heterozygous state in one individual (heterozygous carriers 1/251,402, allele frequency 0.0004%). Several in silico algorithms predict a deleterious effect of the variant and even an effect on splicing is predicted. Residue 448 is highly conserved among eukaryotes (Figure 1A). According to the ACMG guidelines (Richards et al., 2015) this variant was classified as likely pathogenic (class 4). The second variant NM_014908.3:c.1558A > G, p. (Thr520Ala), which is paternally derived, was already described (Retterer et al., 2016; Rush et al., 2017 and HGMD CM1618868) as pathogenic and results in a non-conservative amino acid substitution of a polar, hydrophilic threonine with a nonpolar, hydrophobic alanine residue. The Threonine-520 residue contributes to a highly conserved cytoplasmic domain (Rush et al., 2017, Figure 1A). Data from gnomAD suggest that this variant is rare, with an allele frequency of 0.004% (heterozygous carriers 9/251,490).
No further causative or possibly causative variants were identified in the trio analysis that would explain the striking phenotype in the siblings. Biallelic pathogenic variants in the DOLK gene are known to be responsible for DOLK-CDG (OMIM #610768).
Results of the Macroscopic and Histopathological Analyses
In addition to the clinical anomalies previously described in Patient 1 (dry skin, pterygium, arthrogryposis of joints of all limbs, IUGR, cardiomegaly, and pulmonary hypoplasia, Supplementary Figure S1A), autopsy revealed a hypotrophic liver with diffuse steatosis (Figure 2, arrow) and hemosiderosis (Figure 3). There was no evidence of glycogen storage on PAS diastase staining in the liver (Figure 2). The heart weight was increased (18 g, normal value for age = 9.6 ± 2.4 g) without malformation, but ventricle wall measurement did not reveal hypertrophy and no steatosis or glycogen storage was seen on microscopic examination (Figure 2). Extrahepatic epithelial hemosiderosis was observed on Perls stainings in pancreatic acini and thyroid follicles (Figure 3, lower panel).
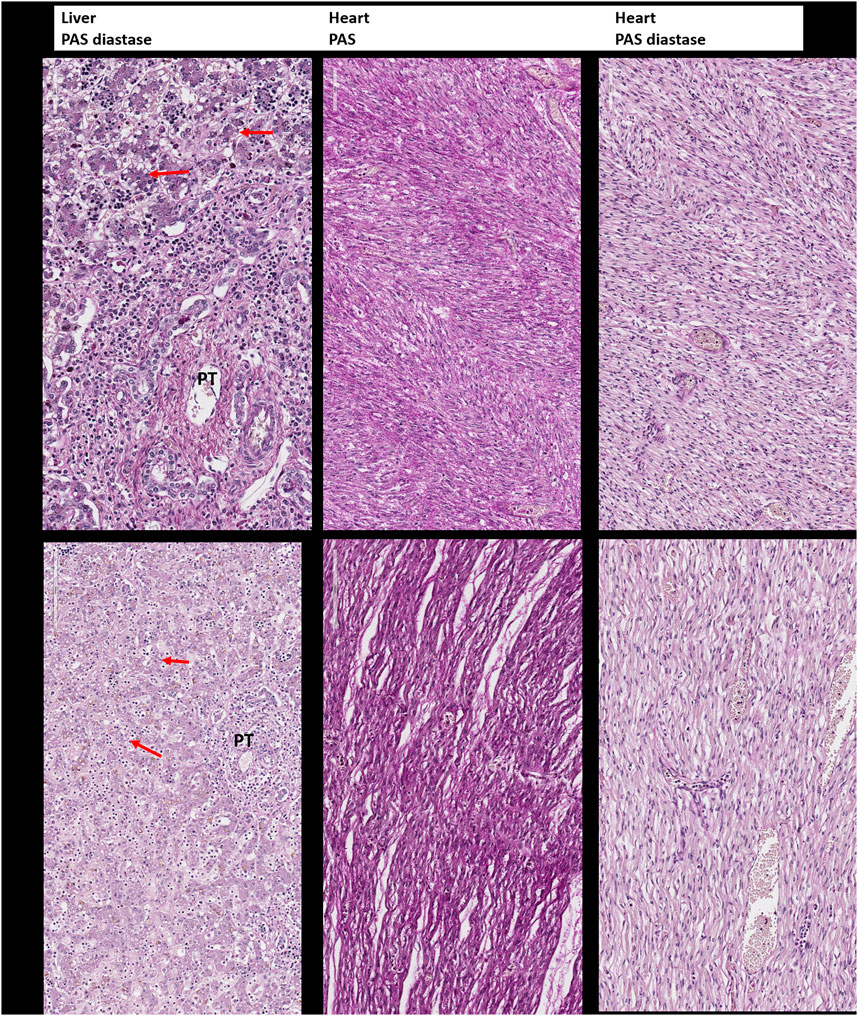
FIGURE 2. Periodic Acid–Schiff (PAS) and Periodic Acid–Schiff–diastase (PAS-D, PAS diastase) staining for the detection of glycogen. Upper panel: Liver and heart tissue of the first child: no evidence of glycogen storage and diffuse small vacuoles of steatosis in the hepatocytes (arrow). PT: portal tract. Lower panel: Liver and heart tissue of the second child with the same findings as in the brother.
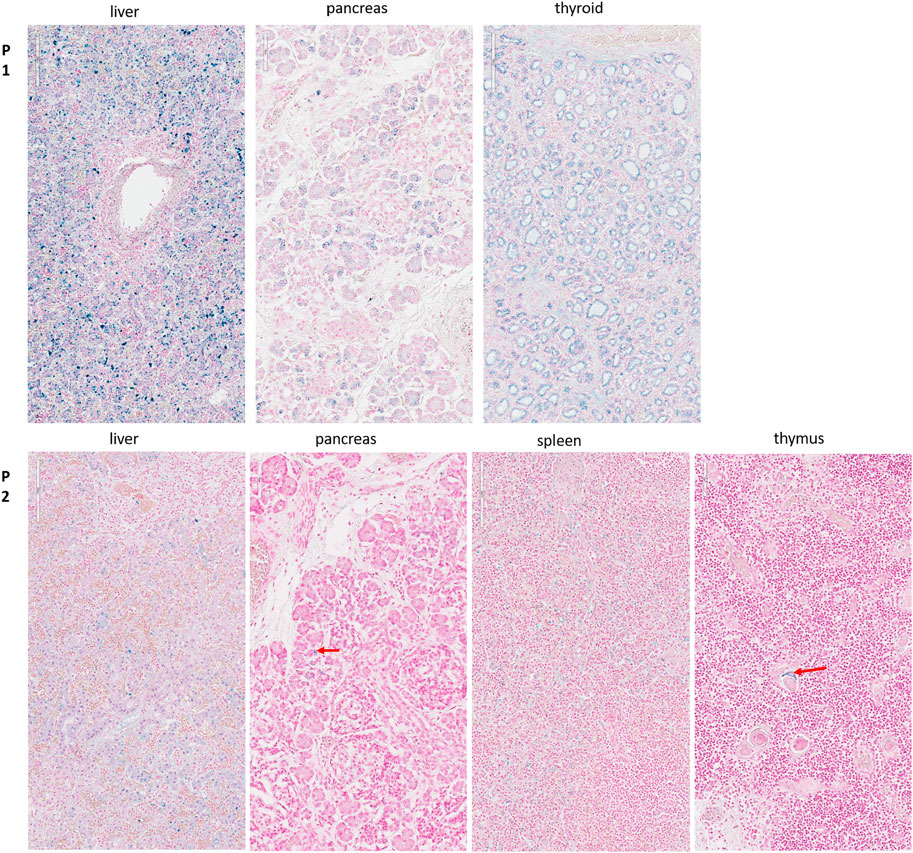
FIGURE 3. Perls staining for the detection of hemosiderosis. Upper panel (P1): Diffuse hemosiderosis in liver parenchyma, thyroid follicles, and pancreatic acini of the first child. Lower panel (P2): Liver: relatively diffuse hemosiderosis in liver parenchyma, less intense than in her brother; Pancreas: focal hemosiderosis in a few acini (arrow); Spleen: diffuse hemosiderosis in macrophages; Thymus: focal hemosiderosis in a few Hassal corpuscles (arrow).
On postmortem examination of the second child, there was no IUGR, but pterygium and arthrogryposis of all limbs and joints, as well as skin anomalies described at birth (Supplementary Figures S1B,C). In addition, autopsy highlighted cardiomegaly: the heart weighed 24 g (normal = 12.6 ± 2.9 g) with mildly increased right ventricular thickness (5 mm, normal = 3.2 ± 1 mm) without microscopic anomalies. No glycogen storage was seen on PAS and PAS diastase stainings (Figure 2). Immunohistochemistry with α-Dystroglycan antibody on frozen heart tissue showed a reduced expression compared to an age-matched control (Figure 4).
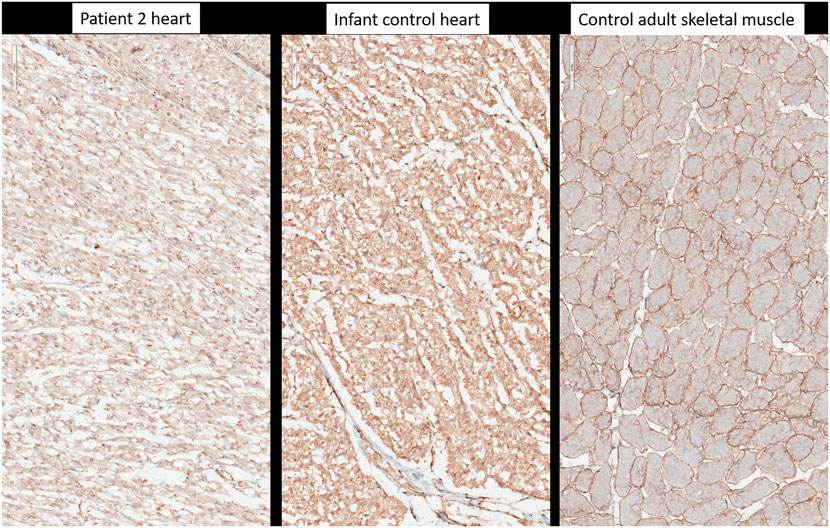
FIGURE 4. Immunohistochemistry with anti-alpha-dystroglycan (clone VIA4-1, Sigma-Aldrich-Merck, Germany). Heart tissue of patient 2, heart tissue of a healthy age-matched control, control from adult skeletal muscle.
Liver weight in the second child was normal but diffuse steatosis (Figure 2) and hemosiderosis, less intense than in the brother, were present (Figure 3). Extrahepatic hemosiderosis was also seen on Perls staining in macrophages of spleen (diffuse hemosiderosis), while there was focal hemosiderosis in the thymus, in a few Hassal corpuscles (arrow), and in the pancreatic acini (Figure 3).
On histopathological examination of the skin from the abdominal wall (Figure 5A) of Patient 1, the epidermis exhibited global compact orthohyperkeratosis with moderate to severe hyperplasia of the stratum corneum (Figure 5A, arrow). The granular layer encompassed one cellular layer, and there was acanthosis and no epidermal or sub-epidermal detachment (no bullae) and no inflammatory infiltrate with leucocytes or erythrocyte (no signs of erythroderma). Histopathology from the right inner thigh of the second child was similar to those of her brother (Figure 5B); from the scalp, it showed pronounced follicular plugging with onion scale-like keratin deposits in hair follicle orifices (Figure 5C, arrow).
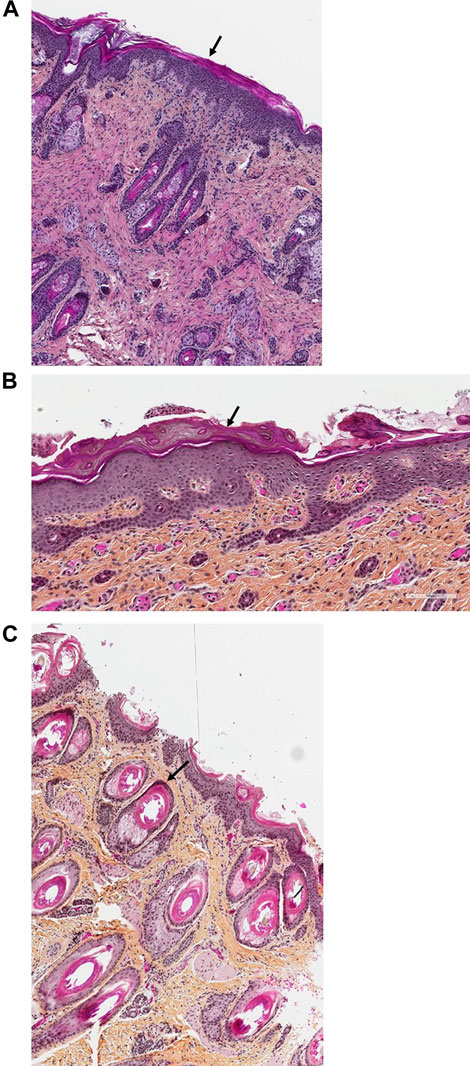
FIGURE 5. (A–C). Skin histology of Patients 1 and 2. In the histopathological examination of the skin (H&E staining) from the abdominal wall along the incision that was performed for internal examination in the first child (Figure 5A, magnification ×40) and from the right inner thigh of the second affected child (Figure 5B, magnification ×100), the epidermis exhibited global compact orthohyperkeratosis with moderate to severe hyperplasia of the stratum corneum (arrows). The granular layer encompassed one cellular layer; there was acanthosis and no epidermal or sub-epidermal detachment (no bullae) and no inflammatory infiltrate with leucocytes or erythrocyte. Pronounced follicular plugging with onion scale-like keratin deposits were seen in the hair follicle orifices from the second child (Figure 5C, magnification ×100, arrow).
Discussion
Here, we present two new cases of a lethal neonatal phenotype of DOLK-CDG whose severe ichthyosis at birth initially misdirected diagnostic efforts towards an autosomal recessive congenital ichthyosis. However, the combination of severe skin phenotype, distal digital constrictions, cardiomegaly, thrombocytopenia, coagulation defect, and multi-organ failure led to reconsideration and suspicion of an inborn error of metabolism. If CDG is suspected or considered early in the differential diagnosis of severely ill neonates, transferrin isoelectric focusing can be performed to screen for N-linked glycosylation defects as a rapid biochemical first-tier investigation. Due to the rapid fatal course in both of our patients, unfortunately, no isoelectric focusing of serum transferrin was performed. However, we attempted to show the effects of the compound heterozygous mutations of the dolichol kinase gene on reduced O-mannosylation in the second child. Dolichol-phosphate is converted to dolichol-phosphate-mannose, the monosaccharide donor for N-glycosylation inside the ER lumen and for O-mannosylation of alpha-dystroglycan. Immunohistochemistry with anti-α-Dystroglycan antibody directed against the O-mannosyl glycans of α-Dystroglycan on frozen heart tissue sample of the second child showed a reduced expression as an evidence of deficient protein mannosylation.
Dolichol kinase deficiency (DK) can present with a wide range and severity of symptoms (Kranz et al., 2007; Lefeber et al., 2011; Lieu et al., 2013; Rush et al., 2017; Hall et al., 2020). It can often be fatal, particularly when manifesting with cardiomyopathy. At the most severe end of the spectrum, two independent families with two and four affected children each, presenting with a lethal neonatal course including severe ichthyosis, digit skin constriction rings, dilated cardiomyopathy/cardiomegaly, hepatosplenomegaly, thrombocytopenia, hypoglycemia, and hypothyroidism, have been reported (Rush et al., 2017; Hall et al., 2020). Review of those most severely affected cases shows symptoms partly overlapping with those of our patients (Table 1). Besides presenting with striking congenital ichthyosis, both of our patients had a narrow chest and evidence of pulmonary hypoplasia on autopsy. In both children, evidence of hepatic and extrahepatic (spleen, thymus, thyroid, and pancreas) hemosiderosis was seen, which has only been described in a few cases of CDG so far (Agarwal et al., 2007; Léticée et al., 2010) and can lead to hematological, endocrinological, and immunological abnormalities. Endocrinopathy has already been described in other DOLK-CDG cases. Both of our patients had cardiomegaly; however, due to the rapid fatal course, they did not yet manifest signs of a dilatative cardiomyopathy. Deficient O-mannosylation of α-Dystroglycan in the heart tissue also points to a functional defect of the heart muscle in our second patient. Since altered glycosylation of certain coagulation factors can alter their plasma levels and thus influence the coagulation profile (Hansson and Stenflo, 2005), diffuse coagulopathies are often observed in CDG. The first child of the family showed necrosis of the digits already on the second day of his life probably due to thrombosis possibly caused by deficient glycosylation of clotting factors, e.g., Protein C and Protein S. The second child showed a severe diffuse coagulation defect with strongly decreased coagulation factor activities as an expression of a glycosylation defect and consequent intraretinal hemorrhage and required plasma transfusion.
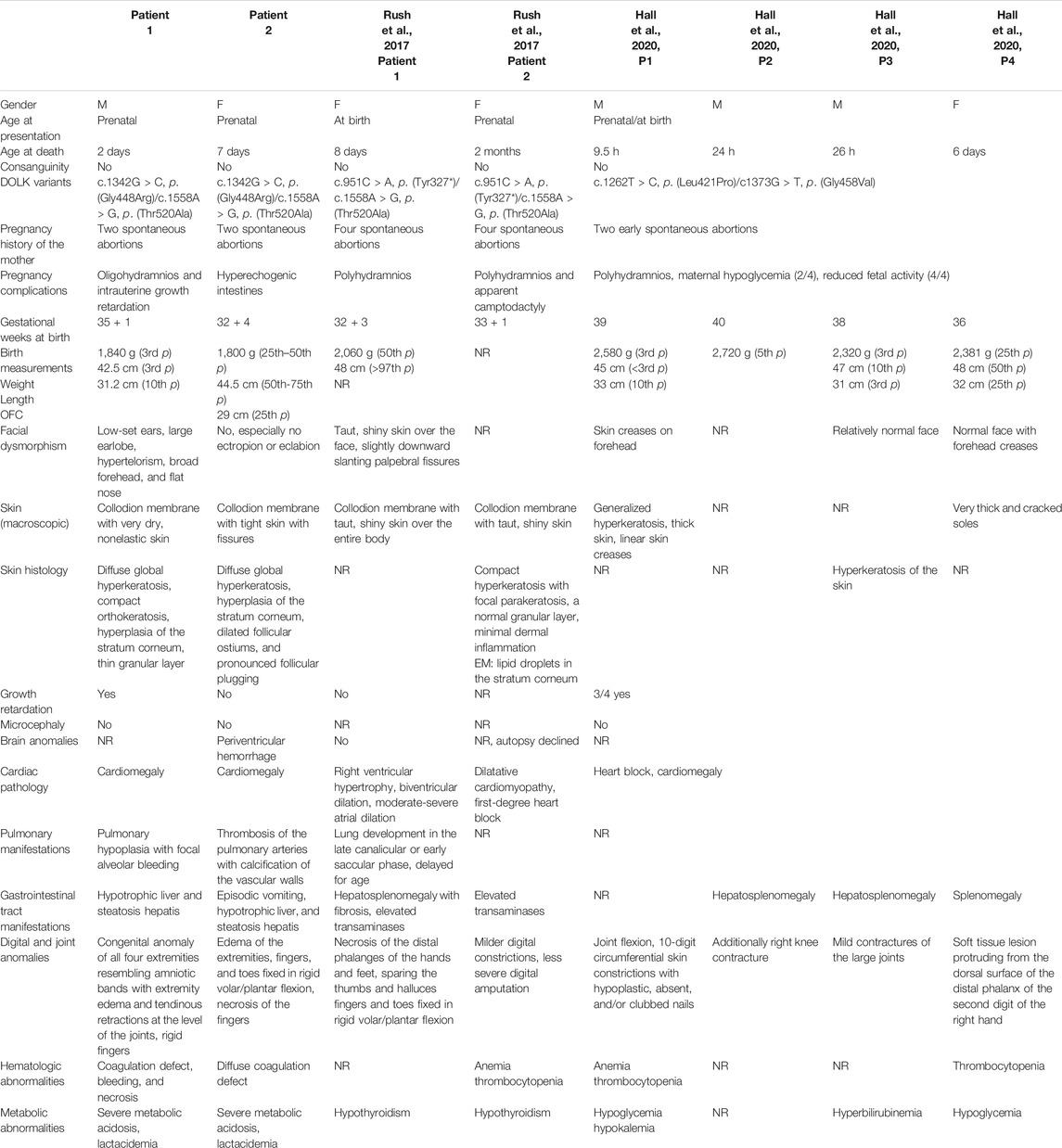
TABLE 1. Clinical and laboratory comparison of eight DOLK-CDG cases with fatal neonatal or early infantile course.
It is interesting to note that the mothers in all three families reported with severe neonatal DOLK-CDG children had previous early miscarriages (Rush et al., 2017; Hall et al., 2020 and our family). In our family, no additional genetic cause was detected in the trio-exom sequencing, raising the possibility of a contribution of the DOLK mutation. Other prenatal manifestations in the fatal neonatal cases were IUGR (2/8), oligohydramnios (2/8), polyhydramnios (5/8), hyperechogenic intestines (in our case), and unexplained hypoglycemia of the mother during the pregnancy (2/8) (Rush et al., 2017; Hall et al., 2020 and Table 1).
Skin abnormalities have been described in about 20% of the different CDG forms (Rymen et al., 2012; Haijes et al., 2020; Komlosi et al., 2020). Recently, some rare forms have also been classified among the syndromic ichthyosis due to their characteristic skin manifestations (Fischer and Bourrat, 2020). Both siblings described here showed a striking congenital ichthyosis as previously described in this disease by Rush et al. (2017) and mentioned by Kranz et al. (2007) while in the four siblings with fatal neonatal DOLK-CDG recently described by Hall et al. (2020), less severe ichthyosis was seen. The pathomechanisms for the skin involvement in dolichol kinase deficiency are still not fully understood and there is only one previous report describing the detailed dermatopathological changes in this disease (Rush et al., 2017) while Hall et al. shows only the hyperkeratosis in one of the four siblings (Hall et al., 2020). Most theories explaining skin involvement are based on the assumption of the instability of key glycoproteins in skin. Since dolichol biosynthesis follows the sterol pathway up to the formation of farnesyl-PP, it has also been proposed that accumulation of toxic sterol precursors could contribute to the phenotype in patients with SRD5A3-and DOLK-CDG (Gründahl et al., 2012; Rymen and Jaeken, 2014). Since the diagnosis of a CDG disorder was established in our cases on postmortem samples, 3 years after the death of the second child in the family, no fresh tissue sample was available to assess the accumulation of a possible toxic metabolite precursor. Glycogen storage was not observed in the liver and heart tissue of both children on microscopic examination. One missense mutation found in our patients [DOLK NM_014908.3:c.1558A>G, p.(Thr520Ala)] was also found in a patient published by Rush et al. (2017), and the reported dermatopathological changes in this patient concurred with ours as well. Histopathology in our cases was typical for ichthyosis with global compact orthohyperkeratosis, thin granular layer, and follicular plugging (Figures 5A–C). Unfortunately, no skin specimen was available anymore for further ultrastructural analysis in our case. In the previous report by Denecke and Kranz (2009), electron microscopy of a skin biopsy sample revealed hyperkeratosis (Kranz et al., 2007), while Rush et al. showed evidence of abnormal lipid droplet accumulation in the stratum corneum and keratinocytes (Rush et al., 2017). With increasing awareness for rare metabolic disorders including CDG and with the expanding possibilities of rapid genetic diagnoses even in the neonatal setting, it will be very important to further elucidate the pathomechanism of skin and organ involvement in future confirmed DOLK-CDG cases.
Conclusion
Our two cases presenting a detailed histopathology besides the clinical data expand the prenatal and postnatal phenotype of DOLK-CDG. They illustrate the broad differential diagnosis of neonatal syndromic ichthyosis and emphasize the importance to actively search for organ involvement in neonates with ichthyosis and to consider the congenital disorders of glycosylation among the possible diagnoses. Congenital ichthyosis with cardiac involvement and distal digital constrictions in combination with multi-organ failure and coagulation defects should prompt strong consideration of DOLK-CDG. In case of suspicion, a rapid first-tier screening using transferrin isoelectric focusing can be performed, followed by genetic analyses. Trio exome sequencing in children with severe disease course and early perinatal death can be valuable in informing the reproductive options of the affected families and allowing them access to prenatal and preimplantation genetic diagnosis.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of the University of Freiburg (ethical code number: 436/17). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author Contributions
JF conceived the manuscript. OC performed the clinical diagnosis and treatment of the neonate, and OC, JM-H, and NJ initiated the genetic diagnostics and DS carried out the genetic counseling and further genetic diagnostics of the proband and their parents; SC-F carried out the macroscopic and microscopic pathological examinations; JK and JF carried out the molecular analyses and reviewed the molecular genetic results; AZ performed the bioinformatic analyses; KK and JF planned the manuscript; KK, JF, and IH interpreted the results and researched the literature; KK and JF prepared the manuscript. OC, SC-F, DS, IH, JK, AZ, and JF edited and reviewed the manuscript; JF contributed substantially to the conception, design, and critical revision of the work for important intellectual content. KK drafted the paper and coordinated writing of the manuscript. All authors discussed, read, and approved the manuscript. All authors approve the version to be published and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding
This work was partially supported by the German Research Foundation, Grant/Award Number: FI1767/3‐1. The authors confirm independence from the sponsors; the content of the article has not been influenced by the sponsors in any way.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We wish to thank the family for their consent to the publication. We also thank the team of the molecular genetic laboratory of the Institute of Human Genetics Freiburg, Germany, and the team of Institute of Pathology of the Hôpital Femme Mère Enfant, Hospices Civils de Lyon, France for technical assistance in the analyses.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.719624/full#supplementary-material
References
Agarwal, B., Ahmed, A., Rushing, E. J., Bloom, M., Kadom, N., Vezina, G., et al. (2007). Congenital Disorder of Glycosylation-X: Clinicopathologic Study of an Autopsy Case with Distinct Neuropathologic Features. Hum. Pathol. 38, 1714–1719. doi:10.1016/j.humpath.2007.05.028
Alsubhi, S., Alhashem, A., Faqeih, E., Alfadhel, M., Alfaifi, A., Altuwaijri, W., et al. (2017). Congenital Disorders of Glycosylation: The Saudi Experience. Am. J. Med. Genet. 173, 2614–2621. doi:10.1002/ajmg.a.38358
Buczkowska, A., Swiezewska, E., and Lefeber, D. J. (2015). Genetic Defects in Dolichol Metabolism. J. Inherit. Metab. Dis. 38, 157–169. doi:10.1007/s10545-014-9760-1
Cantagrel, V., Lefeber, D. J., Ng, B. G., Guan, Z., Silhavy, J. L., Bielas, S. L., et al. (2010). SRD5A3 Is Required for Converting Polyprenol to Dolichol and Is Mutated in a Congenital Glycosylation Disorder. Cell 142, 203–217. doi:10.1016/j.cell.2010.06.001
Denecke, J., and Kranz, C. (2009). Hypoglycosylation Due to Dolichol Metabolism Defects. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1792, 888–895. doi:10.1016/j.bbadis.2009.01.013
Fischer, J., and Bourrat, E. (2020). Genetics of Inherited Ichthyoses and Related Diseases. Acta Derm. Venereol. 100, adv00096. doi:10.2340/00015555-3432
Francisco, R., Marques-da-Silva, D., Brasil, S., Pascoal, C., Dos Reis Ferreira, V., Morava, E., et al. (2019). The challenge of CDG Diagnosis. Mol. Genet. Metab. 126, 1–5. doi:10.1016/j.ymgme.2018.11.003
Gründahl, J. E. H., Guan, Z., Rust, S., Reunert, J., Müller, B., Du Chesne, I., et al. (2012). Life with Too Much Polyprenol: Polyprenol Reductase Deficiency. Mol. Genet. Metab. 105, 642–651. doi:10.1016/j.ymgme.2011.12.017
Haijes, H. A., Jaeken, J., and Hasselt, P. M. (2020). Hypothesis: Determining Phenotypic Specificity Facilitates Understanding of Pathophysiology in Rare Genetic Disorders. Jrnl Inher Metab. Disea 43, 701–711. doi:10.1002/jimd.12201
Hall, B. D., Stevenson, R. E., and Jones, J. R. (2020). Fatal Hyperkeratosis Syndrome in Four Siblings Due to Dolichol Kinase Deficiency. Am. J. Med. Genet. 182, 1421–1425. doi:10.1002/ajmg.a.61574
Hansson, K., and Stenflo, J. (2005). Post-translational Modifications in Proteins Involved in Blood Coagulation. J. Thromb. Haemost. 3, 2633–2648. doi:10.1111/j.1538-7836.2005.01478.x
Helander, A., Stödberg, T., Jaeken, J., Matthijs, G., Eriksson, M., and Eggertsen, G. (2013). Dolichol Kinase Deficiency (DOLK-CDG) with a Purely Neurological Presentation Caused by a Novel Mutation. Mol. Genet. Metab. 110, 342–344. doi:10.1016/j.ymgme.2013.07.002
Jaeken, J., Rymen, D., and Matthijs, G. (2014). Congenital Disorders of Glycosylation: Other Causes of Ichthyosis. Eur. J. Hum. Genet. 22, 444. doi:10.1038/ejhg.2013.168
Kahrizi, K., Hu, C. H., Garshasbi, M., Abedini, S. S. S., Ghadami, S., Kariminejad, R., et al. (2011). Next Generation Sequencing in a Family with Autosomal Recessive Kahrizi Syndrome (OMIM 612713) Reveals a Homozygous Frameshift Mutation in SRD5A3. Eur. J. Hum. Genet. 19, 115–117. doi:10.1038/ejhg.2010.132
Kapusta, L., Zucker, N., Frenckel, G., Medalion, B., Gal, T. B., Birk, E., et al. (2013). From Discrete Dilated Cardiomyopathy to Successful Cardiac Transplantation in Congenital Disorders of Glycosylation Due to Dolichol Kinase Deficiency (DK1-CDG). Heart Fail. Rev. 18, 187–196. doi:10.1007/s10741-012-9302-6
Komlosi, K., Gläser, S., Kopp, J., Hotz, A., Alter, S., Zimmer, A. D., et al. (2020). Neonatal Presentation of COG6‐CDG with Prominent Skin Phenotype. JIMD Rep. 55, 51–58. doi:10.1002/jmd2.12154
Kouwenberg, D., Gardeitchik, T., Mohamed, M., Lefeber, D. J., and Morava, E. (2014). Wrinkled Skin and Fat Pads in Patients with ALG8-CDG: Revisiting Skin Manifestations in Congenital Disorders of Glycosylation. Pediatr. Dermatol. 31, e1–e5. doi:10.1111/pde.12233
Kranz, C., Jungeblut, C., Denecke, J., Erlekotte, A., Sohlbach, C., Debus, V., et al. (2007). A Defect in Dolichol Phosphate Biosynthesis Causes a New Inherited Disorder with Death in Early Infancy. Am. J. Hum. Genet. 80, 433–440. doi:10.1086/512130
Lefeber, D. J., de Brouwer, A. P. M., Morava, E., Riemersma, M., Schuurs-Hoeijmakers, J. H. M., Absmanner, B., et al. (2011). Autosomal Recessive Dilated Cardiomyopathy Due to DOLK Mutations Results from Abnormal Dystroglycan O-Mannosylation. Plos Genet. 7, e1002427. doi:10.1371/journal.pgen.1002427
Léticée, N., Bessières-Grattagliano, B., Dupré, T., Vuillaumier-Barrot, S., de Lonlay, P., Razavi, F., et al. (2010). Should PMM2-Deficiency (CDG Ia) Be Searched in Every Case of Unexplained Hydrops Fetalis? Mol. Genet. Metab. 101, 253–257. doi:10.1016/j.ymgme.2010.06.009
Lieu, M. T., Ng, B. G., Rush, J. S., Wood, T., Basehore, M. J., Hegde, M., et al. (2013). Severe, Fatal Multisystem Manifestations in a Patient with Dolichol Kinase-Congenital Disorder of Glycosylation. Mol. Genet. Metab. 110, 484–489. doi:10.1016/j.ymgme.2013.09.016
Ng, B. G., Hackmann, K., Jones, M. A., Eroshkin, A. M., He, P., Wiliams, R., et al. (2021). Mutations in the Glycosylphosphatidylinositol Gene PIGL Cause CHIME Syndrome. Am. J. Hum. Genet. 90, 685–688. doi:10.1016/j.ajhg.2012.02.010
Oji, V., Tadini, G., Akiyama, M., Blanchet Bardon, C., Bodemer, C., Bourrat, E., et al. (2010). Revised Nomenclature and Classification of Inherited Ichthyoses: Results of the First Ichthyosis Consensus Conference in Sorèze 2009. J. Am. Acad. Dermatol. 63, 607–641. doi:10.1016/j.jaad.2009.11.020
Paprocka, J., Jezela-Stanek, A., Tylki-Szymańska, A., and Grunewald, S. (2021). Congenital Disorders of Glycosylation from a Neurological Perspective. Brain Sci. 11, 88. doi:10.3390/brainsci11010088
Retterer, K., Juusola, J., Cho, M. T., Vitazka, P., Millan, F., Gibellini, F., et al. (2016). Clinical Application of Whole-Exome Sequencing across Clinical Indications. Genet. Med. 18, 696–704. doi:10.1038/gim.2015.148
Richards, S., Aziz, N., Aziz, N., Bale, S., Bick, D., Das, S., et al. (2015). Standards and Guidelines for the Interpretation of Sequence Variants: a Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423. doi:10.1038/gim.2015.30
Rush, E. T., Baker, C. V., and Rizzo, W. B. (2017). Dolichol Kinase Deficiency (DOLK-CDG): Two New Cases and Expansion of Phenotype. Am. J. Med. Genet. 173, 2428–2434. doi:10.1002/ajmg.a.38287
Rymen, D., and Jaeken, J. (2014). Skin Manifestations in CDG. J. Inherit. Metab. Dis. 37, 699–708. doi:10.1007/s10545-014-9678-7
Rymen, D., Keldermans, L., Race, V., Régal, L., Deconinck, N., Dionisi-Vici, C., et al. (2012). COG5-CDG: Expanding the Clinical Spectrum. Orphanet. J. Rare. Dis. 7, 94. doi:10.1186/1750-1172-7-94
Schenk, B., Imbach, T., Frank, C. G., Grubenmann, C. E., Raymond, G. V., Hurvitz, H., et al. (2001). MPDU1 Mutations Underlie a Novel Human Congenital Disorder of Glycosylation, Designated Type if. J. Clin. Invest. 108, 1687–1695. doi:10.1172/jci200113419
Thiel, C., Wortmann, S., Riedhammer, K., Alhaddad, B., Mayatepek, E., Prokisch, H., et al. (2018). Severe Ichthyosis in MPDU1-CDG. J. Inherit. Metab. Dis. 41, 1293–1294. doi:10.1007/s10545-018-0189-9
Keywords: DOLK, congenital disorders of glycosylation, whole exome sequencing, Mendelian disorders of cornification, syndromic ichthyosis
Citation: Komlosi K, Claris O, Collardeau-Frachon S, Kopp J, Hausser I, Mazereeuw-Hautier J, Jonca N, Zimmer AD, Sanlaville D and Fischer J (2021) Fatal Neonatal DOLK-CDG as a Rare Form of Syndromic Ichthyosis. Front. Genet. 12:719624. doi: 10.3389/fgene.2021.719624
Received: 02 June 2021; Accepted: 02 November 2021;
Published: 08 December 2021.
Edited by:
Anna Tylki-Szymańska, Children’s Memorial Health Institute (IPCZD), PolandReviewed by:
Julien H. Park, University of Münster, GermanyHasan Orhan Akman, Columbia University Irving Medical Center, United States
Daisy Rymen, University Hospitals Leuven, Belgium
Copyright © 2021 Komlosi, Claris, Collardeau-Frachon, Kopp, Hausser, Mazereeuw-Hautier, Jonca, Zimmer, Sanlaville and Fischer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katalin Komlosi, a2F0YWxpbi5rb21sb3NpQHVuaWtsaW5pay1mcmVpYnVyZy5kZQ==
†These authors have contributed equally to this work