
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 11 October 2021
Sec. Evolutionary and Population Genetics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.717621
 Tianzi Liu1,2†
Tianzi Liu1,2† Mohetaboer Momin3,4†Huiyue Zhou1,2
Mohetaboer Momin3,4†Huiyue Zhou1,2 Qiwen Zheng1,2
Qiwen Zheng1,2 Fangfang Fan3,4
Fangfang Fan3,4 Jia Jia3,4Mengyuan Liu3,4Minghui Bao3,4
Jia Jia3,4Mengyuan Liu3,4Minghui Bao3,4 Jianping Li3,4
Jianping Li3,4 Yong Huo3,4Jialin Liu1,2,5Yaning Zhang1,2,5Xuemei Mao6Xiao Han6
Yong Huo3,4Jialin Liu1,2,5Yaning Zhang1,2,5Xuemei Mao6Xiao Han6 Zhiyuan Hu6,7,8Changqing Zeng1,2,5
Zhiyuan Hu6,7,8Changqing Zeng1,2,5 Fan Liu1,2,5*Yan Zhang3,4*
Fan Liu1,2,5*Yan Zhang3,4*Plasma total homocysteine (tHCY) is a known risk factor of a wide range of complex diseases. No genome scans for tHCY have been conducted in East Asian populations. Here, we conducted an exome-wide association study (ExWAS) for tHCY in 5,175 individuals of Chinese Han origin, followed by a replication study in 668 Chinese individuals. The ExWAS identified two loci, 1p36.22 (lead single-nucleotide polymorphism (SNP) rs1801133, MTHFR C677T) and 16q24.3 (rs1126464, DPEP1), showing exome-wide significant association with tHCY (p < 5E−7); and both loci have been previously associated with tHCY in non-East Asian populations. Both SNPs were replicated in the replication study (p < 0.05). Conditioning on the genotype of C677T and rs1126464, we identified a novel East Asian-specific missense variant rs138189536 (C136T) of MTHFR (p = 6.53E−10), which was also significant in the replication study (p = 9.8E−3). The C136T and C677T variants affect tHCY in a compound heterozygote manner, where compound heterozygote and homozygote genotype carriers had on average 43.4% increased tHCY than had other genotypes. The frequency of the homozygote C677T genotype showed an inverse-U-shaped geospatial pattern globally with a pronounced frequency in northern China, which coincided with the high prevalence of hyperhomocysteinemia (HHCY) in northern China. A logistic regression model of HHCY status considering sex, age, and the genotypes of the three identified variants reached an area under the receiver operating characteristic curve (AUC) value of 0.74 in an independent validation cohort. These genetic observations provide new insights into the presence of multiple causal mutations at the MTHFR locus, highlight the role of genetics in HHCY epidemiology among different populations, and provide candidate loci for future functional studies.
Plasma total homocysteine (tHCY) is a known risk factor of a wide range of complex diseases, including cardiovascular and cerebrovascular diseases (Nygard et al., 1995; Refsum et al., 1998; Ganguly and Alam, 2015), type 2 diabetes (Wijekoon et al., 2007), age-related macular degeneration (Pinna et al., 2018), chronic kidney disease (Ji et al., 2019), Alzheimer’s disease (Mansoori et al., 2012), and cancers (Hasan et al., 2019).
Plasma tHCY variation has a known genetic component with a heritability estimated ranging from 47 to 70% in different populations (Jee et al., 2002; Kullo et al., 2006; Bathum et al., 2007; Siva et al., 2007; Nilsson et al., 2009). To date, 12 independent genome-wide association studies (GWASs) for tHCY have been conducted, among which nine were carried out in populations of European decent (Souto et al., 2005; Hazra et al., 2009; Malarstig et al., 2009; Pare et al., 2009; Tanaka et al., 2009; Wernimont et al., 2011; van Meurs et al., 2013; Williams et al., 2014; Shane et al., 2018), two in Africans (AFRs) (Kim et al., 2016; Raffield et al., 2018) and one in Filipinos (Lange et al., 2010). These studies collectively identified several genomic loci showing genome-wide significant association with tHCY levels. The one on chromosome 1p36.22 has been most extensively reported, where a single missense variant of the Methylenetetrahydrofolate Reductase gene (MTHFR C677T, rs1801133) showed an extraordinarily large effect on tHCY levels (Liew and Gupta, 2015; Moll and Varga, 2015); i.e., compared with wild-type carriers, individuals with homozygote T alleles had on average 31.08% increased tHCY levels (Shane et al., 2018). MTHFR is the rate-limiting enzyme in methionine metabolism, while the 677T variant results in a thermolabile enzyme that is ∼70% less effective during the conversion of 5,10-methyltetrahydrofolate (5,10-MTHF) to 5-methyltetrahydrofolate (5-MTHF) (Liew and Gupta, 2015). Since 5-MTHF acts as a cofactor in tHCY methylation to methionine, deficiency of MTHFR eventually leads to increased tHCY levels and a higher risk of multiple diseases (Goyette et al., 1994). Besides 1p36.22, DNA variants in or close to CBS on 21q22.3 (Pare et al., 2009; Lange et al., 2010; van Meurs et al., 2013; Williams et al., 2014), NOX4 on 11q14.3 (Pare et al., 2009; Lange et al., 2010), CPS1 on 2q34 (Lange et al., 2010; van Meurs et al., 2013; Williams et al., 2014), DPEP1 on 16q24.3 (Pare et al., 2009; Lange et al., 2010; van Meurs et al., 2013), and CUBN on 10p13 (Tanaka et al., 2009; van Meurs et al., 2013) were identified for association with tHCY by at least two independent GWASs, thus representing the most robust genetic findings.
China has a pronounced prevalence of hyperhomocysteinemia (HHCY), particularly in regions with low dietary folate intake (Hao et al., 2003; Yang et al., 2013; Yang et al., 2014), which also accounted for an increased risk of ischemic stroke in patients with hypertension in these regions (Clarke et al., 2002; Li et al., 2015). Surprisingly, no genome scans for tHCY have been conducted in Chinese populations nor in any East Asian populations. Therefore, the questions regarding the possibility of the presence of multiple causal mutations and East Asian (EAS)-specific alleles of tHCY, the genetic explanation for the geospatial pattern of HHCY prevalence in China, and the predictability of HHCY risk in Chinese population remained to be answered. To answer these questions, we conducted an exome-wide association study (ExWAS) of plasma tHCY level in a Chinese cohort including 5,175 individuals, followed by a replication study in 668 individuals of Chinese Han origin.
The discovery stage ExWAS included a total of 5,175 living residents of Beijing of Chinese Han origin. This cohort is characterized with a mid-aged distribution (mean age 57.14 ± 8.93 years), a pronounced proportion of females (62.17%), considerable proportions of current smokers (19.74%) and alcohol users (23.59%), and a relatively small proportion of vitamin B supplementation users (8.75%). The population structure analysis using northern (CHB) and southern (CHS) Chinese from the 1000 Genomes Project (McVean et al., 2012) as reference indicated that most of our discovery individuals are more likely to be northern Chinese (Supplementary Figure S1). The tHCY level (median 11.95 μmol/L, mean 14.09 μmol/L, sd 8.64 μmol/L) was on average higher than that reported by Schurks et al. in 44,147 Europeans (detailed sample characteristics was provided in Supplementary Table S1). In this study, age, sex, smoking, drinking, creatinine, vitamin B usage, and the first two principal components were adjusted for their potential confounding effects in association analyses.
A total of 89,131 quality-controlled, mostly coding autosomal DNA variants were tested for association with tHCY. These included those with minor allele count (MAC) >3 to detect rare coding variants with large effects. The distribution of genomic principal components (Supplementary Figure S2), the genomic inflation factor (λ = 1.03), and the distribution of the observed test statistics (Figure 1) did not reveal any evidence for the presence of population substructure. The ExWAS identified a total of 18 variants at two distinct loci showing exome-wide significant association with tHCY (p < 5.0E−7, Figure 1A; Table 1).
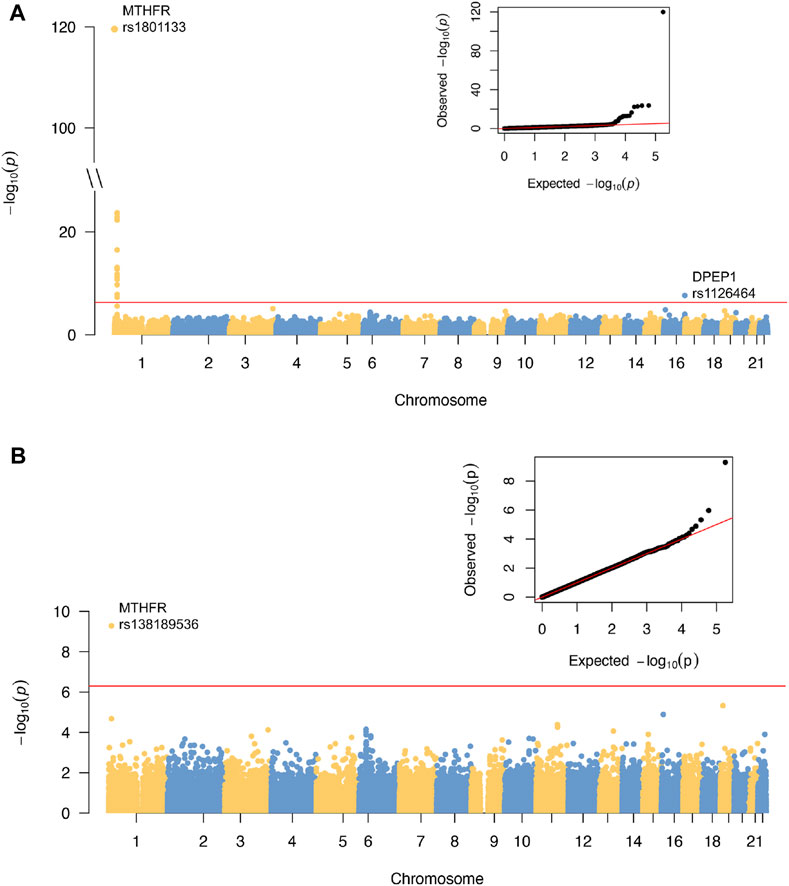
FIGURE 1. Results of ExWAS analysis and conditional analysis of tHCY in discovery cohort. (A) Manhattan plot and Q-Q plot of the ExWAS. (B) Manhattan plot and Q-Q plot of the conditional analysis. For Manhattan plots, the horizontal axes are genomic location (GRCh37.p13) of all the tested SNPs. The vertical axes are the negative log of p-values. Red lines indicate the threshold for exome-wide significance at 5E−7. ExWAS, exome-wide association study; tHCY, total homocysteine; SNP, single-nucleotide polymorphism.
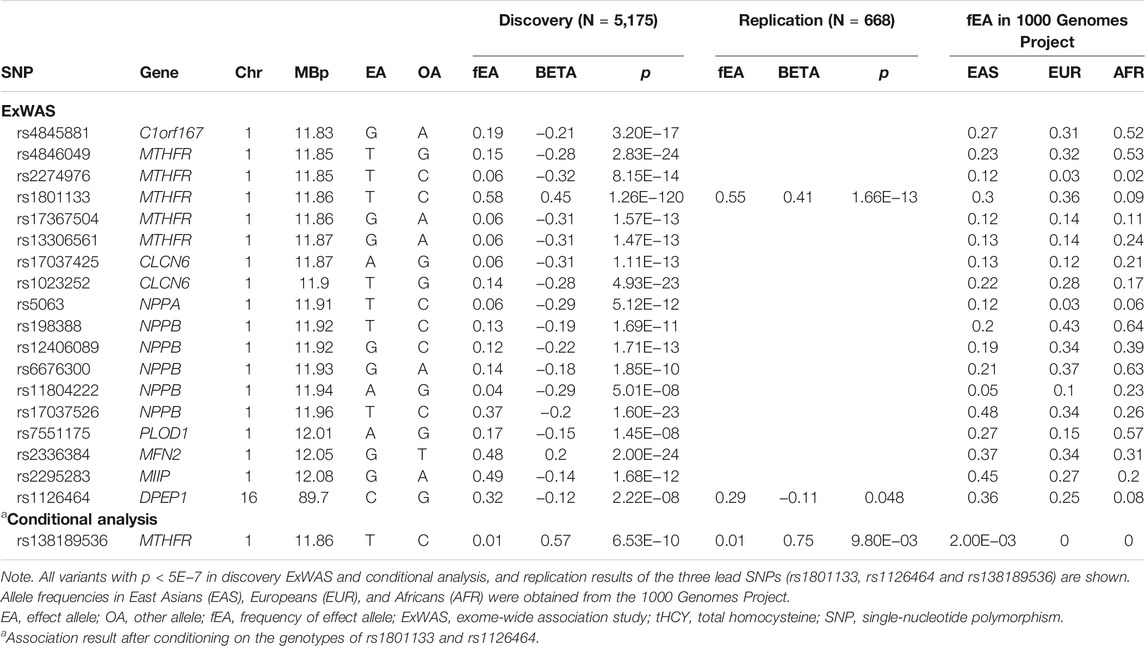
TABLE 1. Exome-wide significant variants associated with tHCY levels from ExWAS.
The first locus was on chromosome 1p36.22, consisting of 17 significant variants (Figure 2A; Table 1), and harbored eight known genes: C1orf167, MTHFR, CLCN6, NPPA, NPPB, PLOD1, MFN2, and MIIP. This locus included the well-known missense variant of MTHFR (rs1801133, C677T, β = 0.45, p = 1.3E−120, Table 1), which was also the lead single-nucleotide polymorphism (SNP) of this locus. In our sample, 18.5% had the CC genotype of MTHFR C677T, 46.7% had the CT genotype, and 34.8% had the TT genotype. Individuals with the TT genotype had an on average 46.22% higher tHCY level than those with CC genotypes (17.4 vs. 11.9 μmol/L, p = 6.8E−60, Supplementary Table S2), explaining 6.66% of the total phenotype variance and 9.95% of the covariate-adjusted phenotype variance. In the 1000 Genomes Project data (Li et al., 2019), the geospatial distribution of the frequency of the tHCY-increasing T allele showed a latitudinal inverse-U-shaped gradient in Europe and EAS continents (Supplementary Figure S3), with the highest frequency observed around latitude of 40° north (44–46%), i.e., southern Europe (Spain and Italy) and northern China. The frequency of homozygote TT genotype showed a similar geospatial distribution with a pronounced frequency (17–24%) around latitude 40° north (Figure 3). In China, the frequency of the TT genotype showed a remarkable difference between northern China around latitude 40° (22% in CHB from the 1000 Genomes Project) and southern China around latitude 20° (9% in CHS from the 1000 Genomes Project, Figure 3A), which largely coincides with the higher HHCY prevalence in northern China.
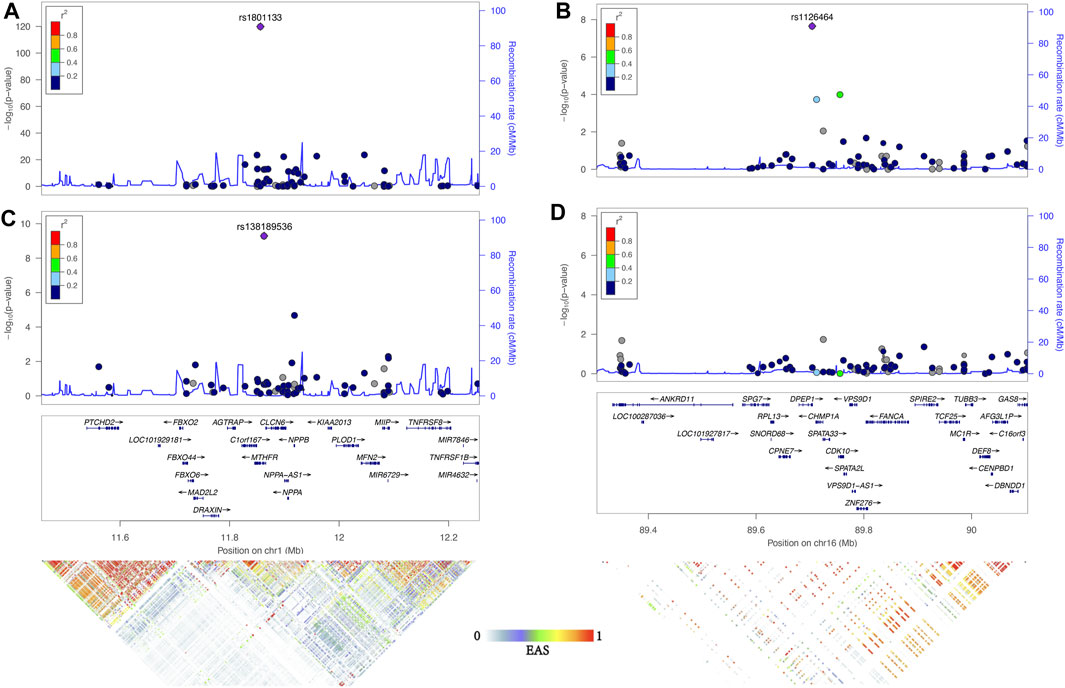
FIGURE 2. Association of two loci significantly associate with tHCY. Regional association plots were generated by Locuszoom of 1p36.22 and 16q24.3 of the results of ExWAS (A and B) and conditional analysis (C and D) using linkage disequilibrium information from the November 2014 release of the 1000 Genomes Project ASN samples. The index SNP in each region is shown as a purple diamond. Genes in the region and LD heatmap (r2) patterns according to the 1000 Genomes Project EAS dataset are aligned on the bottom. tHCY, total homocysteine; ExWAS, exome-wide association study; SNP, single-nucleotide polymorphism; LD, linkage disequilibrium.
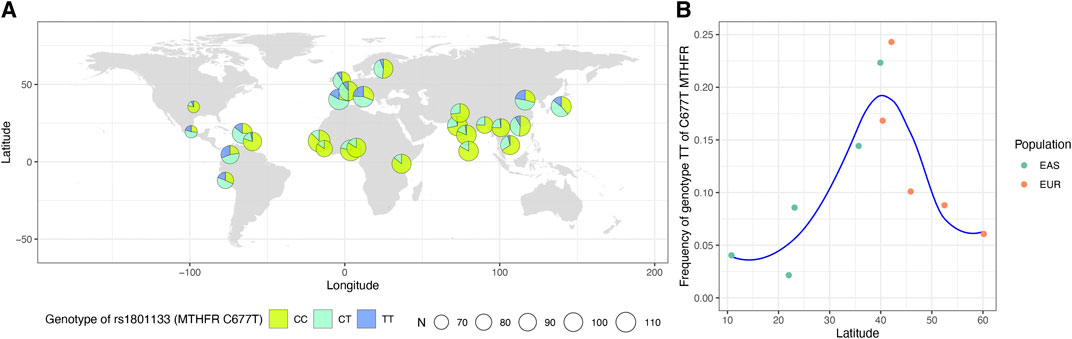
FIGURE 3. Worldwide prevalence patterns for genotypes of MTHFR C677T (rs1801133). (A) Map displaying the geospatial distribution of the prevalence patterns for genotypes of MTHFR C677T across the world. The map was drawn based on the genotypes of 2,504 subjects obtained from the 1000 Genomes Project datasets for this SNP. The pie denotes the sampling locations. (B) The frequency of TT genotype of MTHFR C677T based on the genotypes of East Asians and Europeans obtained from the 1000 Genomes Project datasets and the corresponding latitude of each population. SNP, single-nucleotide polymorphism.
The second locus was detected on chromosome 16q24.3 consisting of only one exome-wide significant signal, which was an exon variant of DPEP1 (rs1126464, β = −0.12, p = 2.2E−8, Figure 2B; Table 1). rs1126464 has been previously associated with tHCY in Europeans and Filipinos (Pare et al., 2009; Lange et al., 2010; van Meurs et al., 2013) with similar allele effects as observed in our sample. This SNP was also associated with waist circumference, hypertension, and osteoarthritis in previous studies. Individuals homozygous for the major allele G (mean tHCY = 14.1 μmol/L, Supplementary Table S2) had an on average 11.02% higher tHCY level than those with CC genotypes (mean tHCY = 12.7 μmol/L, p = 1.5E−6, Supplementary Table S2). The frequency of the tHCY decreasing C allele also showed large differences between major populations (36% in EASs, 25% in Europeans and 8% in AFRs, Table 1; Supplementary Figure S4). Besides, according to the GTEx database (GTEx Consortium, 2013), rs1126464 is a cis-regulated expression quantitative trait locus (eQTL) of CDK10, CHMP1A, FANCA, and VPS9D1 in multiple tissues, such as whole blood, tibial artery, skin, and thyroid (Supplementary Table S3; Supplementary Figure S5).
Next, we repeated the exome scan conditioning on the genotypes of MTHFR C677T and DPEP1 rs1126464, and we identified one novel rare missense variant showing exome-wide significant association with tHCY (rs138189536, MTHFR C136T, p = 2.18E−3 before conditional analysis, and p = 6.53E−10 after conditional analysis, Figures 1B, 2C, 2D; Table 1). This C136T variant was in weak linkage disequilibrium (LD) with the C677T variant (r2 = 0.014). A previous enzyme kinetics experiment has shown that the arginine to tryptophan substitution at C136T led to a reduced enzymatic activity of MTHFR, which provided direct evidence for a functional role of this variant, consistent with our observation of the presence of multiple causal mutations at this locus (Tan et al., 2021; Weile et al., 2021). A haplotype analysis further showed that these two alleles (677T and 136T) together affect tHCY in a compound heterozygote manner (Figure 4). The compound heterozygote of 677T and 136T and the homozygote 677T carriers had on average 43.4% increased tHCY than had the wild-type and any-heterozygote carriers (17.5 vs. 12.2 μmol/L, Figure 4), and carriers of compound heterozygote of C136T and C677T had a slightly (1.5%) increased tHCY compared with homozygote 677T and 136C carriers (17.7 vs. 17.5 μmol/L, Figure 4). Note that the homozygote 136T genotype was not observed by previous studies due to its low frequency in EASs and its absence in non-Asian populations, which also explained the less significant association observed for C136T relative to C677T. The 136T allele was rare in our sample (1% in both discovery and replication samples, Table 1) as well as in the EAS sample from the 1000 Genomes Project (0.2%, Table 1). This allele is absent in non-Asian populations, which may explain the failure of previous GWASs in detecting its effect (frequency of C136T was 0 in both EUR and AFR from the 1000 Genomes Project, Table 1).
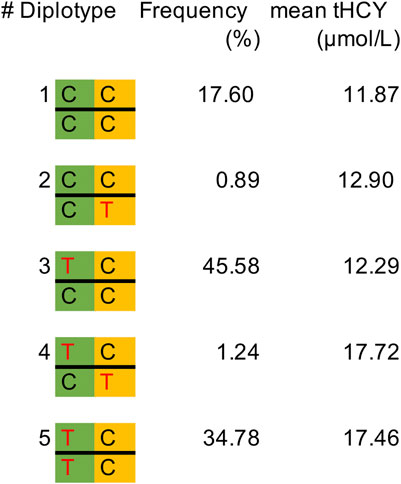
FIGURE 4. Frequency of diplotypes and the mean tHCY level in the discovery sample. MTHFR C677T is indicated in the green background, and MTHFR C136T is indicated in the orange background. Missense alleles 677T and 136T are indicated in red color. tHCY, total homocysteine.
A replication study was conducted in a total of 668 individuals of Chinese Han origin (Supplementary Table S4). As expected, MTHFR C677T was again highly significantly associated with tHCY with a similar effect size as observed in the discovery ExWAS (β = 0.41, p = 1.7E−13, Table 1). The frequency of homozygote 677TT genotype in replication sample (32%) was similar as observed in the discovery sample. The 16q24.3 rs1126464 in DPEP1 showed a nominally significant association with tHCY, and its allele effect was similar to that observed in the discovery ExWAS (β = −0.11, p = 0.048, Table 1). Conditioning on the genotype of MTHFR C677T and DPEP1 rs1126464, we successfully replicated the C136T with a similar effect size as observed in the discovery ExWAS (β = 0.75, p = 9.8E−3, Table 1).
A multiple regression analysis including sex, age, and the three abovementioned variants (MTHFR C677T, MTHFR C136T, and DPEP1 rs1126464) revealed that these variables together explained 18.6% of tHCY variance in the discovery individuals (Table 2). We then constructed linear and logistic models in the discovery sample by including sex and age as predictors with or without the three identified variants and used these models to predict the tHCY levels and the HHCY status (defined as tHCY >15 μmol/L) in the replication sample. The linear model consisting of sex, age, and the three variants provided a higher accuracy in explaining tHCY levels (R2 = 0.14) than did the model without the three variants (R2 = 0.05). The logistic model consisting of sex, age, and the three variants also provided a higher prediction accuracy (area under the receiver operating characteristic (ROC) curve (AUC) = 0.74, Figure 5A) in predicting HHCY status (defined with tHCY ≥15 μmol/L) than did the model without the three variants (AUC = 0.68, Figure 5A). Although the AUC values were lower than a clinically desired level (AUC = 0.85) for diagnosis, our prediction model consisting of sex, age, and the three variants provided fairly accurate prediction results for a good proportion of individuals, that is, individuals with predicted probabilities of HHCY <0.2 or >0.8 (34.9% < 0.2 and 5.8% > 0.8, Figure 5B; also see Supplementary Table S5). In practice, our model may provide an informative test for a total of 40.7% of these individuals with predicted probabilities of HHCY <0.2 or >0.8 but less informative for the rest.
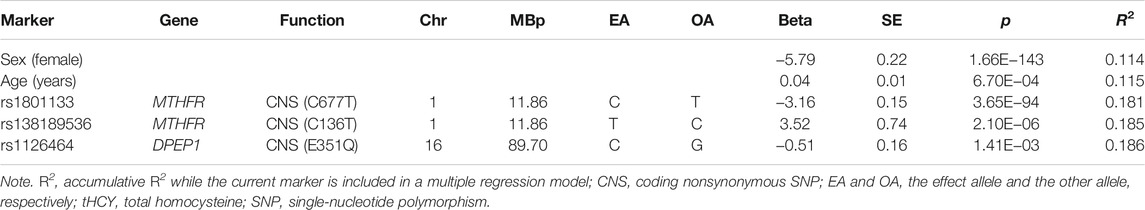
TABLE 2. Explained variance of tHCY by three identified SNPs.
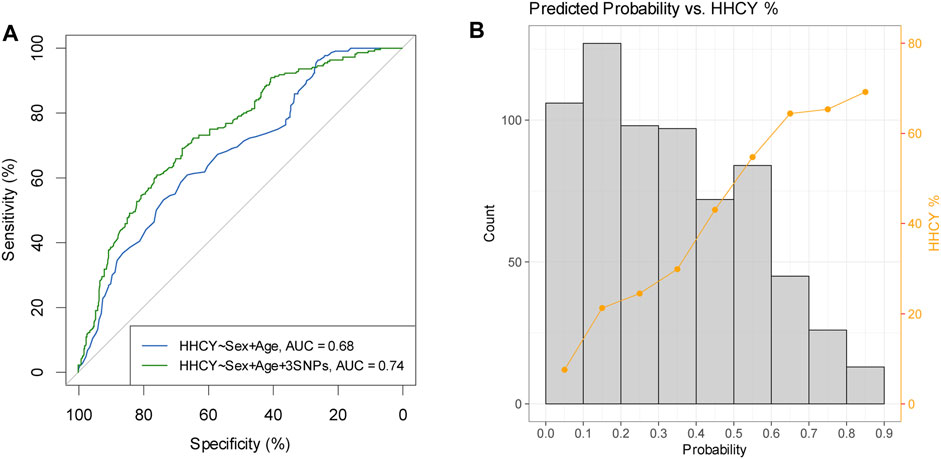
FIGURE 5. Prediction results of HHCY prediction in replication samples (N = 668). (A) Receiver operating characteristic curves for predicting HHCY status of prediction models considering sex and age as predictors with three identified SNPs (green line) or without three identified SNPs (blue line). (B) Histogram of predicted probability overplayed with percentage of HHCY in each probability bin (prediction model based on sex, age, and three identified variants as predictors). HHCY, hyperhomocysteinemia; SNP, single-nucleotide polymorphism.
This study represents the first genome scan of tHCY in a well-sized population of Chinese Han origin. The extraordinary large effect of MTHFR C677T on tHCY was confirmed in Chinese populations. The geospatial distribution of the frequency of the 677T homozygous genotype is consistent with the HHCY incidence in China, serving as a supplementary explanation for the hypothesis that folate inadequate intake accounts for the high prevalence of HHCY in northern China (Hao et al., 2003; Yang et al., 2013; Yang et al., 2014). Importantly, we detected the presence of multiple causal mutations at MTHFR by identifying a rare EAS-specific missense mutation (MTHFR C136T), which together with C677T affects tHCY in a compound heterozygote manner. This emphasized the need of sequence of the entire gene in genetic testing for HHCY. We also verified the effect of a known tHCY-associated exon variant of DPEP1 in Chinese populations. Using these three variants, we proposed a risk assessment model of HHCY, which may be informative for approximately 40% of Chinese individuals.
We observed the strongest evidences for association between MTHFR C677T and tHCY in both of the discovery and replication samples. This variant alone explained approximately 10.0% of the total covariate-adjusted variance of tHCY in the discovery sample. In our study, the tHCY level of the homozygous 677T carriers was on average 50% higher than the wild-type carriers, consistent with the findings from non-East Asian populations. Besides its effect on tHCY, the 677T allele has also been reported to affect vitamin B9 (folate) and B12 concentrations. MTHFR irreversibly reduces 5,10-MTHF to 5-MTHF, and 5-MTHF was converted to the tetrahydrofolate (THF) form of folate during the conversion of HCY to methionine. Further, function studies have revealed that MTHFR C677T results in ∼70% decrease of enzyme activity and thereby accompanied by decreased plasma folate level and increased tHCY level (Scott and Weir, 1981; Liew and Gupta, 2015).
C136T has been reported as a missense mutation of MTHFR, which may increase the occurrence and recurrence of pulmonary embolism (Tan et al., 2021). To our knowledge, this is the first genome scan revealing the influence of MTHFR C136T on tHCY. It is noticeable that C136T is only present in EASs, highlighting the importance of well-sized association study on tHCY in Chinese. C136T and C677T together influence tHCY in a compound heterozygote manner, consistent with the role as missense variants of both C136T and C677T in reducing the activity of MTHFR enzyme, supporting the previous findings of the existence of multiple causal alleles on MTHFR (Burda et al., 2015). Clearly, sequencing the entire gene is preferred for genetic testing involving MTHFR.
HHCY is a strong risk factor of cardiovascular and cerebrovascular diseases, type 2 diabetes, and cancers (Nygard et al., 1995; Refsum et al., 1998; Schurks et al., 2008; Mansoori et al., 2012; Ganguly and Alam, 2015; Pinna et al., 2018; Hasan et al., 2019; Ji et al., 2019). The prevalence of HHCY is very high in China, particularly in northern China (Yang et al., 2014), which has been hypothesized to account for an increased risk of multiple diseases in northern China. Previous epidemiological studies have revealed that the high prevalence of HHCY was likely due to the inadequate intake of folate. In this study, we observed that the frequency of homozygous MTHFR 677TT genotype is as high as 0.32 to 0.35 in northern China, compared with 0.09 in southern China. This observation may serve as an alternative or supplementary explanation of the large difference of HHCY prevalence between northern and southern China. The observation that 677T being a “bad” allele and at the same time a major allele in northern China suggested the presence of selective pressures. Previous population genetic studies have suggested a role of natural selection in shaping the latitudinal inverse U-shaped gradient of C677T, which may involve factors such as regional climate, UV radiation, and folate intake (Pepe et al., 1998; Bauduer and Lacombe, 2005; Wang et al., 2012; Li et al., 2015). Compared with low-latitudes people living under strong UV, people living in mid-latitudes are less likely to undergo photolysis of in vivo folate. Compared with people living at high latitudes with sunlight deficiency and less vegetable foods, people living in mid-latitudes can improve their body folate status through dietary intake. Under these favorable conditions, the 677TT genotype can be preserved to the maximum extent. However, the exact mechanism underlying the adaption of an increased level of tHCY remains elusive.
The current study has several strengths. First, our study is the first ExWAS of tHCY in a well-sized Chinese population, based on which we could confirm the effects of known alleles identified in other populations and identify novel EAS-specific variants explaining additional tHCY variance. Second, we performed a replication study in an independent sample and fully adjusted potential confounders to ensure true positive findings. Third, we used an exome-wide conditional analysis method, and our results emphasized its efficiency in detecting the presence of multiple causal mutations. This study also has several limitations. The coverage of our exome-genotyping data is much lower than that whole genome sequencing data, so we could not examine the effects of some previously reported tHCY-associated SNPs in our study. The participants in our study were mostly from northern China (Beijing); without southern Chinese samples, we could not directly examine the relationship between the geographic distribution of 677T and HHCY incidence in China. Future studies with a higher geographical coverage together with the folate intake information would help to refine the hypothesis of geographic distribution of HHCY incidence in China.
In conclusion, the ExWAS of tHCY in a Chinese population detected the presence of multiple causal mutations at the MTHFR locus by identifying an EAS-specific missense variant C163T, which together with the well-known 677T allele affects tHCY in a compound heterozygote manner. The geospatial distribution of homozygote 677T genotype coincides with that of HHCY incidence in China, supplementing the current hypothesis about the high HHCY risk in northern China due to insufficient folate intake. A logistic model consisting of sex, age, and three DNA variants constructed in the discovery sample could fairly accurately predict the HHCY risk in the testing sample.
The establishment of the discovery cohort has been described in details previously (Fan et al., 2016). In brief, 9,540 participants aged ≥40 years were recruited from the Gucheng and Pingguoyuan communities of Shijingshan District in Beijing of China. Baseline survey was investigated from December 2011 to April 2012 as described previously (Fan et al., 2016). Among them, a total of 6,480 subjects were genotyped by ExomeChip. To obtain high-quality genotypes, strict criteria were applied to filter out low-quality genotypes. We undertook plate-, individual-, and variant-level checks to exclude poor-quality genotype calls from the dataset. A total of 5,959 samples passed the above quality control. We excluded 702 participants based on the identity by descent (IBD) analysis conducted in our samples. We used pi-hat ≥0.35 as cutoff to remove higher than the first degree (based on the pi-hat distribution, the unbiased estimate is 0.5) of close relatives by keeping the sample with the highest call rate for each family group. We then excluded 82 participants who did not have tHCY data. Finally, 5,175 eligible participants of Chinese Han ancestry were included in this analysis.
Two cohorts, myocardial infarction (MI) cohort and Chinese Academy of Sciences (CAS) cohort, were included in the replication stage. Both cohorts were from Beijing, China. Participants of MI cohort were hospitalized with MI in Department of Cardiology, Peking University First Hospital, from February 2005 to May 2013. Initially, a total of 593 participants had their genotypes tested using customized ExomeChip. The details of this cohort, methods, and primary results have been reported elsewhere. After data quality controls and exclusion of those without tHCY and other missing covariates, a total of 410 eligible Chinese Han ancestry MI patients were included in this analysis. The CAS participants consisted of 273 employees of CAS who were invited to join in a personalized health management during their annually physical examination. tHCY measurements and epidemiological information were collected before the health management period. After data quality controls, a total of 258 eligible Chinese Han ancestry participants were included in this analysis.
Information, including demographic status, health behavior, disease history, and medical use, was collected using a standardized questionnaire in the discovery cohort and CAS cohort and extracted from in-hospital electronic medical records in the MI cohort.
Venous blood samples were obtained by venipuncture for all participants after overnight fasting. Plasma samples separated within 30 min and extracted DNA samples were stored at −80°C until measurement. For discovery sample, plasma tHCY was measured using an autobiochemical analyzer (AU480; Beckman Coulter, Brea, CA, United States) with the circulating enzymatic method in the core laboratory of the National Clinical Research Center for Kidney Disease of Nanfang Hospital in Guangzhou, China. The details of this method were subscribed in previous study (Momin et al., 2017). Serum creatinine (Scr) at baseline was measured on the Roche C8000 Automatic Analyzer (Roche Diagnostics, Basel, Switzerland) in the laboratory of the Chinese PLA General Hospital.
All subjects were genotyped using the Asian ExomeChip, a specially designed exome array with a custom content of 58,317 variants on top of the standard Infinium HumanExome BeadChip (Illumina, San Diego, CA, United States), which integrated a total of 302,218 variants. Details of the Asian ExomeChip design has been described in previous studies (Zhang et al., 2014; Tang et al., 2015). In brief, the original design of the exome array includes 242,901 markers, with the majority of over 200K coding variants identified from ∼12,000 sequenced genomes and exomes of primarily European ancestry. The underrepresentation of non-European genomes in the original design limited the coverage of low-frequency variants in Asian populations. To allow the comprehensive genotyping across the full allele frequency spectrum, a custom panel of ∼30K nonsense/missense variants were added based on three independent Asian sequencing datasets of ∼1,000 Chinese samples.
A total of 5,959 individuals passed the individual-based QC criteria for filtering, which included a call rate of <99%, sex mismatch, and excess heterozygosity. We also performed variant-level quality control by excluding variants that have <99% genotype call rate or deviation from the Hardy–Weinberg equilibrium (p < 1E−4). In total, 282,456 variants passed the quality control. Among them, 119,020 variants were polymorphic variants in our data. Finally, after exclusion of related samples using a greedy algorithm (Sun et al., 2021) and variants with MAC ≤3, a total of 5,175 unrelated individuals with tHCY data and 89,131 autosomal variants were retained in further analysis.
The population structure analysis inferred by ADMIXTURE software (Alexander et al., 2009) of the discovery samples were conducted with CHB and CHS from the 1000 Genomes Project as reference. We ran ADMIXTURE using an LD pruned marker set of 25,121 variants, assuming 2 (K = 2) genetic clusters. To normalize the plasma tHCY, residuals were obtained using linear regression after adjusting for age, sex, smoking, drinking, creatinine, vitamin usage, and the first two principal components. Then inverse normal transformation residuals were created for analysis. The ExWAS of residuals of tHCY was carried out in PLINK v1.9 (Purcell et al., 2007) using linear regression model, assuming an additive allele effect. The genomic control inflation factor was close to 1.0 (λ = 1.03) in ExWAS and was not further considered. The p-values equal to or smaller than 5E−7 based on Bonferroni correction of 89,131 autosomal variants were considered as exome-wide significant. Conditional analysis of discovery ExWAS result was performed using PLINK v1.9 (Purcell et al., 2007). Results of discovery ExWAS and conditional analysis were visualized by Manhattan and Q–Q plots generated by an R package qqman (Turner, 2018). Regional LD analysis was conducted using a self-written R script LDplot using EAS data from the 1000 Genomes Project. Regional Manhattan plots were created by LocusZoom (Pruim et al., 2010) with LD population according to the 1000 Genomes Project ASN dataset. We conducted linear regression analysis using each identified variant as the explanatory variable, and the fitness R2 from this model was considered as the phenotype variance explained by the variants.
Identified variants were annotated using ANNOVAR (Version 2017-07–17) (Wang et al., 2010) with respect to the hg19 genome build. The allele frequencies of candidate variants in different populations were examined in the 2,504 subjects of the 1000 Genomes Project and visualized using R. The significant expression eQTL effects of identified SNP in all available tissues were annotated using the data from The Genotype-Tissue Expression project (GTEx, http://www.gtexportal.org/home/, data source: GTEx Analysis Release V8 (dbGaP Accession phs000424.v8.p2) (GTEx Consortium, 2013). The nominal p-value of each variant–gene pair was calculated from the genome-wide empirical p-value and the beta distribution model of each gene. The variant–gene pairs with a p-value lower than the gene-level threshold (0.05 false discovery rate) were considered significant enough to be included in the list of variant–gene pairs. All the variants with exome-wide significant association in the discovery analysis and conditional analysis were selected for replication with a focus on the top-associated SNP per region. The replication was carried out separately in a total of 668 samples, using linear regression adjusted for age, sex, smoking, and drinking, assuming an additive allele effect. For replication study, the significance level was set to 0.017 based on Bonferroni correction of three significant variants. Explained phenotypic variance was derived for identified independent associated variants using backward stepwise linear regression analyses. In order to check if the compound heterozygote, rather than double heterozygotes, may indeed explain the identified association, we inferred haplotypes using the expectation maximization algorithm implemented in R library haplo.stat (Schaid et al., 2002).
Fine-tuned individual-level data analyses were conducted in 5,175 unrelated Chinese individuals from discovery stage. HHCY status was defined with tHCY >15 μmol/L in both discovery samples and replication samples. A multiple linear regression analysis was conducted to access the independent effect of three variants. The model considers three identified variants in discovery analysis and conditional analysis (MTHFR C677T, MTHFR C136T and DPEP1 rs1126464), together with sex and age as explanatory factors. We then applied multiple linear regression models including sex and age as predictors with or without the three variants developed in the discovery samples to the replication samples (N = 668) for tHCY level prediction. Prediction accuracy was estimated using R-squared correlation (R2) with tHCY phenotype. For HHCY risk prediction, multiple logistic regression models including sex and age as predictors with or without the three variants developed in the discovery samples were applied to the replication samples. Prediction accuracy was estimated using the AUC. AUC is the integral of ROC curves and ranges from 0.5 representing total lack of prediction (no better than flipping a coin) to 1.0 representing perfect prediction. Sensitivities and specificities were calculated using confusion matrices considering the predicted probability > t as the predicted shape type, where t optimized the sum of sensitivity and specificity.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: OMIX, China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences (https://ngdc.cncb.ac.cn/omix), accession OMIX499.
This study was approved by the ethics committee of Peking University First Hospital and Beijing Zhongguancun Hospital, and each participant provided written informed consent. This study has been performed following the ethical standards of the Declaration of Helsinki.
TL and MM: data curation, software, formal analysis, investigation, visualization, methodology, data acquisition, manuscript preparation and revision, and final approval. HZ, QZ, FF, JJ, ML, MB, JLL, YH, JPL, and YZ: data curation, formal analysis, investigation, data acquisition, and final approval. CZ, ZH, XM, and XH: resources, supervision, funding acquisition, and final approval. FL and YZ: conceptualization, resources, supervision, funding acquisition, methodology, data acquisition, article preparation and revision, and final approval.
Strategic Priority Research Program of Chinese Academy of Sciences (Grant No. XDB38010400) Science and Technology Service Network Initiative of Chinese Academy of Sciences (Grant No. KFJ-STS-ZDTP-079) Peking University First Hospital seed research fund (Grant No. 2018SF072) University of Michigan-Peking University Health Science Center (UM-PUHSC) joint institute for translational and clinical research (Grant No. BMU20110177) UMHS-PUHSC Joint Institute for Translational and Clinical Research and the Fundamental Research Funds for the Central Universities (Grant No. BMU20110177, BMU20160530) Beijing Municipal Science and Technology Project (Grant No. Z181100001618014) Open Project of Key Laboratory of Genomic and Precision Medicine, Chinese Academy of Sciences
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank the volunteer study participants for their contribution to this research.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.717621/full#supplementary-material
Alexander, D. H., Novembre, J., and Lange, K. (2009). Fast Model-Based Estimation of Ancestry in Unrelated Individuals. Genome Res. 19 (9), 1655–1664. doi:10.1101/gr.094052.109
Bathum, L., Petersen, I., Christiansen, L., Konieczna, A., Sørensen, T. I., and Kyvik, K. O. (2007). Genetic and Environmental Influences on Plasma Homocysteine: Results from a Danish Twin Study. Clin. Chem. 53 (5), 971–979. doi:10.1373/clinchem.2006.082149
Bauduer, F., and Lacombe, D. (2005). Factor V Leiden, Prothrombin 20210A, Methylenetetrahydrofolate Reductase 677T, and Population Genetics. Mol. Genet. Metab. 86 (1-2), 91–99. doi:10.1016/j.ymgme.2005.04.002
Burda, P., Schäfer, A., Suormala, T., Rummel, T., Bürer, C., Heuberger, D., et al. (2015). Insights into Severe 5,10-methylenetetrahydrofolate Reductase Deficiency: Molecular Genetic and Enzymatic Characterization of 76 Patients. Hum. Mutat. 36 (6), 611–621. doi:10.1002/humu.22779
Clarke, R., Collins, R., Lewington, S., Donald, A., Alfthan, G., Tuomilehto, J., et al. (2002). Homocysteine and Risk of Ischemic Heart Disease and Stroke: a Meta-Analysis. JAMA 288 (16), 2015–2022. doi:10.1001/jama.288.16.2015
Fan, F., Qi, L., Jia, J., Xu, X., Liu, Y., Yang, Y., et al. (2016). Noninvasive Central Systolic Blood Pressure Is More Strongly Related to Kidney Function Decline Than Peripheral Systolic Blood Pressure in a Chinese Community-Based Population. Hypertension 67 (6), 1166–1172. doi:10.1161/HYPERTENSIONAHA.115.07019
Ganguly, P., and Alam, S. F. (2015). Role of Homocysteine in the Development of Cardiovascular Disease. Nutr. J. 14, 6. doi:10.1186/1475-2891-14-6
Goyette, P., Sumner, J. S., Milos, R., Duncan, A. M. V., Rosenblatt, D. S., Matthews, R. G., et al. (1994). Human Methylenetetrahydrofolate Reductase: Isolation of cDNA, Mapping and Mutation Identification. Nat. Genet. 7 (2), 195–200. doi:10.1038/ng0694-195
GTEx Consortium (2013). The Genotype-Tissue Expression (GTEx) Project. Nat. Genet. 45 (6), 580–585. doi:10.1038/ng.2653
Hao, L., Ma, J., Stampfer, M. J., Ren, A., Tian, Y., Tang, Y., et al. (2003). Geographical, Seasonal and Gender Differences in Folate Status Among Chinese Adults. J. Nutr. 133 (11), 3630–3635. doi:10.1093/jn/133.11.3630
Hasan, T., Arora, R., Bansal, A. K., Bhattacharya, R., Sharma, G. S., and Singh, L. R. (2019). Disturbed Homocysteine Metabolism Is Associated with Cancer. Exp. Mol. Med. 51 (2), 1–13. doi:10.1038/s12276-019-0216-4
Hazra, A., Kraft, P., Lazarus, R., Chen, C., Chanock, S. J., Jacques, P., et al. (2009). Genome-wide Significant Predictors of Metabolites in the One-Carbon Metabolism Pathway. Hum. Mol. Genet. 18 (23), 4677–4687. doi:10.1093/hmg/ddp428
Jee, S., Song, K., Shim, W., Kim, H., Suh, I., Park, J., et al. (2002). Major Gene Evidence after MTHFR-Segregation Analysis of Serum Homocysteine in Families of Patients Undergoing Coronary Arteriography. Hum. Genet. 111 (2), 128–135. doi:10.1007/s00439-002-0757-8
Ji, A., Pan, C., Wang, H., Jin, Z., Lee, J. H., Wu, Q., et al. (2019). Prevalence and Associated Risk Factors of Chronic Kidney Disease in an Elderly Population from Eastern China. Int. J. Environ. Res. Public Health 16 (22), 4383. doi:10.3390/ijerph16224383
Kim, S., Nho, K., Ramanan, V. K., Lai, D., Foroud, T. M., Lane, K., et al. (2016). Genetic Influences on Plasma Homocysteine Levels in African Americans and Yoruba Nigerians. J. Alzheimers Dis. 49 (4), 991–1003. doi:10.3233/JAD-150651
Kullo, I. J., Ding, K., Boerwinkle, E., Turner, S. T., Mosley, T. H., Kardia, S. L. R., et al. (2006). Novel Genomic Loci Influencing Plasma Homocysteine Levels. Stroke 37 (7), 1703–1709. doi:10.1161/01.STR.0000225929.96190.b3
Lange, L. A., Croteau-Chonka, D. C., Marvelle, A. F., Qin, L., Gaulton, K. J., Kuzawa, C. W., et al. (2010). Genome-wide Association Study of Homocysteine Levels in Filipinos Provides Evidence for CPS1 in Women and a Stronger MTHFR Effect in Young Adults. Hum. Mol. Genet. 19 (10), 2050–2058. doi:10.1093/hmg/ddq062
Li, J., Jiang, S., Zhang, Y., Tang, G., Wang, Y., Mao, G., et al. (2015). H-type Hypertension and Risk of Stroke in Chinese Adults: A Prospective, Nested Case-Control Study. J. Transl Int. Med. 3 (4), 171–178. doi:10.1515/jtim-2015-0027
Li, Y., Zhao, W., Li, D., Tao, X., Xiong, Z., Liu, J., et al. (2019). RETRACTED ARTICLE: EDAR, LYPLAL1, PRDM16, PAX3, DKK1, TNFSF12, CACNA2D3, and SUPT3H Gene Variants Influence Facial Morphology in a Eurasian Population. Hum. Genet. 138 (6), 681–689. doi:10.1007/s00439-019-02023-7
Liew, S.-C., and Gupta, E. D. (2015). Methylenetetrahydrofolate Reductase (MTHFR) C677T Polymorphism: Epidemiology, Metabolism and the Associated Diseases. Eur. J. Med. Genet. 58 (1), 1–10. doi:10.1016/j.ejmg.2014.10.004
Mälarstig, A., Buil, A., Souto, J. C., Clarke, R., Blanco-Vaca, F., Fontcuberta, J., et al. (2009). Identification of ZNF366 and PTPRD as Novel Determinants of Plasma Homocysteine in a Family-Based Genome-wide Association Study. Blood 114 (7), 1417–1422. doi:10.1182/blood-2009-04-215269
Mansoori, N., Tripathi, M., Luthra, K., Alam, R., Lakshmy, R., Sharma, S., et al. (2012). MTHFR (677 and 1298) and IL-6-174 G/C Genes in Pathogenesis of Alzheimer's and Vascular Dementia and Their Epistatic Interaction. Neurobiol. Aging 33 (5), 1003. e1-8. doi:10.1016/j.neurobiolaging.2011.09.018
McVean, G. A., Altshuler, D. M., Durbin, R. M., Abecasis, G. R., Bentley, D. R., Chakravarti, A., et al. (2012). An Integrated Map of Genetic Variation from 1,092 Human Genomes. Nature 491 (7422), 56–65. doi:10.1038/nature11632
Moll, S., and Varga, E. A. (2015). Homocysteine and MTHFR Mutations. Circulation 132 (1), e6–9. doi:10.1161/CIRCULATIONAHA.114.013311
Momin, M., Fan, F., Li, J., Qin, X., Jia, J., Qi, L., et al. (2017). Associations of Plasma Homocysteine Levels with Peripheral Systolic Blood Pressure and Noninvasive central Systolic Blood Pressure in a Community-Based Chinese Population. Sci. Rep. 7 (1), 6316. doi:10.1038/s41598-017-06611-3
Nilsson, S. E., Read, S., Berg, S., and Johansson, B. (2009). Heritabilities for Fifteen Routine Biochemical Values: Findings in 215 Swedish Twin Pairs 82 Years of Age or Older. Scand. J. Clin. Lab. Invest. 69 (5), 562–569. doi:10.1080/00365510902814646
Nygård, O., Vollset, S. E., Refsum, H., Stensvold, I., Tverdal, A., Nordrehaug, J. E., et al. (1995). Total Plasma Homocysteine and Cardiovascular Risk Profile. JAMA 274 (19), 1526–1533. doi:10.1001/jama.1995.03530190040032
Paré, G., Chasman, D. I., Parker, A. N., Zee, R. R. Y., Mälarstig, A., Seedorf, U., et al. (2009). Novel Associations of CPS1, MUT, NOX4, and DPEP1 with Plasma Homocysteine in a Healthy Population. Circ. Cardiovasc. Genet. 2 (2), 142–150. doi:10.1161/CIRCGENETICS.108.829804
Pepe, G., Camacho Vanegas, O., Giusti, B., Brunelli, T., Marcucci, R., Attanasio, M., et al. (1998). Heterogeneity in World Distribution of the Thermolabile C677T Mutation in 5,10-methylenetetrahydrofolate Reductase. Am. J. Hum. Genet. 63 (3), 917–920. doi:10.1086/302015
Pinna, A., Zaccheddu, F., Boscia, F., Carru, C., and Solinas, G. (2018). Homocysteine and Risk of Age-Related Macular Degeneration: a Systematic Review and Meta-Analysis. Acta Ophthalmol. 96 (3), e269–e276. doi:10.1111/aos.13343
Pruim, R. J., Welch, R. P., Sanna, S., Teslovich, T. M., Chines, P. S., Gliedt, T. P., et al. (2010). LocusZoom: Regional Visualization of Genome-wide Association Scan Results. Bioinformatics 26 (18), 2336–2337. doi:10.1093/bioinformatics/btq419
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: a Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 81 (3), 559–575. doi:10.1086/519795
Raffield, L. M., Ellis, J., Olson, N. C., Duan, Q., Li, J., Durda, P., et al. (2018). Genome-wide Association Study of Homocysteine in African Americans from the Jackson Heart Study, the Multi-Ethnic Study of Atherosclerosis, and the Coronary Artery Risk in Young Adults Study. J. Hum. Genet. 63 (3), 327–337. doi:10.1038/s10038-017-0384-9
Refsum, Md, H., Ueland, Md, P. M., Nygård, Md, O., and Vollset, Md, Dr.Ph, S. E. (1998). Homocysteine and Cardiovascular Disease. Annu. Rev. Med. 49 (1), 31–62. doi:10.1146/annurev.med.49.1.31
Schaid, D. J., Rowland, C. M., Tines, D. E., Jacobson, R. M., and Poland, G. A. (2002). Score Tests for Association between Traits and Haplotypes when Linkage Phase Is Ambiguous. Am. J. Hum. Genet. 70 (2), 425–434. doi:10.1086/338688
Schurks, M., Zee, R. Y. L., Buring, J. E., and Kurth, T. (2008). Interrelationships Among the MTHFR 677C>T Polymorphism, Migraine, and Cardiovascular Disease. Neurology 71 (7), 505–513. doi:10.1212/01.wnl.0000316198.34558.e5
Scott, J., and Weir, D. G. (1981). THE METHYL FOLATE TRAP A Physiological Response in Man to Prevent Methyl Group Deficiency in Kwashiorkor (Methionine Deficiency) and an Explanation for Folic-Acid-Induced Exacerbation of Subacute Combined Degeneration in Pernicious Anaemia. The Lancet 318 (8242), 337–340. doi:10.1016/s0140-6736(81)90650-4
Shane, B., Pangilinan, F., Mills, J. L., Fan, R., Gong, T., Cropp, C. D., et al. (2018). The 677C→T Variant of MTHFR Is the Major Genetic Modifier of Biomarkers of Folate Status in a Young, Healthy Irish Population. Am. J. Clin. Nutr. 108 (6), 1334–1341. doi:10.1093/ajcn/nqy209
Siva, A., De Lange, M., Clayton, D., Monteith, S., Spector, T., and Brown, M. J. (2007). The Heritability of Plasma Homocysteine, and the Influence of Genetic Variation in the Homocysteine Methylation Pathway. QJM 100 (8), 495–499. doi:10.1093/qjmed/hcm054
Souto, J. C., Blanco-Vaca, F., Soria, J. M., Buil, A., Almasy, L., Ordoñez-Llanos, J., et al. (2005). A Genomewide Exploration Suggests a New Candidate Gene at Chromosome 11q23 as the Major Determinant of Plasma Homocysteine Levels: Results from the GAIT Project. Am. J. Hum. Genet. 76 (6), 925–933. doi:10.1086/430409
Sun, P., Zhou, W., Fu, Y., Cheung, C. Y. Y., Dong, Y., Yang, M.-L., et al. (2021). An Asian-specific MPL Genetic Variant Alters JAK-STAT Signaling and Influences Platelet Count in the Population. Hum. Mol. Genet. 30 (9), 836–842. doi:10.1093/hmg/ddab062
Tan, J.-S., Yan, X.-X., Wu, Y., Gao, X., Xu, X.-Q., Jiang, X., et al. (2021). Rare Variants in MTHFR Predispose to Occurrence and Recurrence of Pulmonary Embolism. Int. J. Cardiol. 331, 236–242. doi:10.1016/j.ijcard.2021.01.073
Tanaka, T., Scheet, P., Giusti, B., Bandinelli, S., Piras, M. G., Usala, G., et al. (2009). Genome-wide Association Study of Vitamin B6, Vitamin B12, Folate, and Homocysteine Blood Concentrations. Am. J. Hum. Genet. 84 (4), 477–482. doi:10.1016/j.ajhg.2009.02.011
Tang, C. S., Zhang, H., Cheung, C. Y. Y., Xu, M., Ho, J. C. Y., Zhou, W., et al. (2015). Exome-wide Association Analysis Reveals Novel Coding Sequence Variants Associated with Lipid Traits in Chinese. Nat. Commun. 6, 10206. doi:10.1038/ncomms10206
Turner, S. D. (2018). qqman: An R Package for Visualizing GWAS Results Using Q-Q and manhattan Plots. J. Open Source Softw. 3 (25), 731. doi:10.21105/joss.00731
van Meurs, J. B., Pare, G., Schwartz, S. M., Hazra, A., Tanaka, T., Vermeulen, S. H., et al. (2013). Common Genetic Loci Influencing Plasma Homocysteine Concentrations and Their Effect on Risk of Coronary Artery Disease. Am. J. Clin. Nutr. 98 (3), 668–676. doi:10.3945/ajcn.112.044545
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 38 (16), e164. doi:10.1093/nar/gkq603
Wang, Y., Pei, L., Wang, J., and Zheng, X. (2012). Is the Prevalence of MTHFR C677T Polymorphism Associated with Ultraviolet Radiation in Eurasia? J. Hum. Genet. 57 (12), 780–786. doi:10.1038/jhg.2012.113
Weile, J., Kishore, N., Sun, S., Maaieh, R., Verby, M., Li, R., et al. (2021). Shifting Landscapes of Human MTHFR Missense-Variant Effects. Am. J. Hum. Genet. 108 (7), 1283–1300. doi:10.1016/j.ajhg.2021.05.009
Wernimont, S. M., Clark, A. G., Stover, P. J., Wells, M. T., Litonjua, A. A., Weiss, S. T., et al. (2011). Folate Network Genetic Variation, Plasma Homocysteine, and Global Genomic Methylation Content: a Genetic Association Study. BMC Med. Genet. 12, 150. doi:10.1186/1471-2350-12-150
Wijekoon, E. P., Brosnan, M. E., and Brosnan, J. T. (2007). Homocysteine Metabolism in Diabetes. Biochem. Soc. Trans. 35 (Pt 5), 1175–1179. doi:10.1042/BST0351175
Williams, S. R., Yang, Q., Chen, F., Liu, X., Keene, K. L., Jacques, P., et al. (2014). Genome-wide Meta-Analysis of Homocysteine and Methionine Metabolism Identifies Five One Carbon Metabolism Loci and a Novel Association of ALDH1L1 with Ischemic Stroke. Plos Genet. 10 (3), e1004214. doi:10.1371/journal.pgen.1004214
Yang, B., Fan, S., Zhi, X., Wang, Y., Wang, Y., Zheng, Q., et al. (2014). Prevalence of Hyperhomocysteinemia in China: a Systematic Review and Meta-Analysis. Nutrients 7 (1), 74–90. doi:10.3390/nu7010074
Yang, B., Liu, Y., Li, Y., Fan, S., Zhi, X., Lu, X., et al. (2013). Geographical Distribution of MTHFR C677T, A1298C and MTRR A66G Gene Polymorphisms in China: Findings from 15357 Adults of Han Nationality. PLoS One 8 (3), e57917. doi:10.1371/journal.pone.0057917
Keywords: homocysteine, hyperhomocysteinemia, MTHFR C677T, MTHFR C136T, coding variants, population heterogeneity
Citation: Liu T, Momin M, Zhou H, Zheng Q, Fan F, Jia J, Liu M, Bao M, Li J, Huo Y, Liu J, Zhang Y, Mao X, Han X, Hu Z, Zeng C, Liu F and Zhang Y (2021) Exome-Wide Association Study Identifies East Asian-Specific Missense Variant MTHFR C136T Influencing Homocysteine Levels in Chinese Populations RH: ExWAS of tHCY in a Chinese Population. Front. Genet. 12:717621. doi: 10.3389/fgene.2021.717621
Received: 02 June 2021; Accepted: 02 September 2021;
Published: 11 October 2021.
Edited by:
Guo-Bo Chen, Zhejiang Provincial People’s Hospital, ChinaCopyright © 2021 Liu, Momin, Zhou, Zheng, Fan, Jia, Liu, Bao, Li, Huo, Liu, Zhang, Mao, Han, Hu, Zeng, Liu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fan Liu, bGl1ZmFuQGJpZy5hYy5jbg==; Yan Zhang, ZHJ6aHkxMTA4QDE2My5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.