 Yang Gao1,2,3†
Yang Gao1,2,3† Chuan Xu1,2,3†Qing Tan1,4†Qunshan Shen1,2,3Huan Wu1,2,3
Chuan Xu1,2,3†Qing Tan1,4†Qunshan Shen1,2,3Huan Wu1,2,3 Mingrong Lv1,2,5Kuokuo Li1,2,5Dongdong Tang1,2,3Bing Song1,2,3
Mingrong Lv1,2,5Kuokuo Li1,2,5Dongdong Tang1,2,3Bing Song1,2,3 Yuping Xu1,2,3Ping Zhou1,2,3Zhaolian Wei1,2,3Fangbiao Tao2,3
Yuping Xu1,2,3Ping Zhou1,2,3Zhaolian Wei1,2,3Fangbiao Tao2,3 Yunxia Cao1,2,3*
Yunxia Cao1,2,3* Xiaojin He1,2,3*
Xiaojin He1,2,3*- 1Department of Obstetrics and Gynecology, Reproductive Medicine Center, The First Affiliated Hospital of Anhui Medical University, Hefei, China
- 2NHC Key Laboratory of Study on Abnormal Gametes and Reproductive Tract, Anhui Medical University, Hefei, China
- 3Key Laboratory of Population Health Across Life Cycle, Anhui Medical University, Ministry of Education of the People's Republic of China, Hefei, China
- 4Anhui Provincial Human Sperm Bank, The First Affiliated Hospital of Anhui Medical University, Hefei, China
- 5Anhui Province Key Laboratory of Reproductive Health and Genetics, Anhui Medical University, Hefei, China
Primary ciliary dyskinesia (PCD) is a clinically and genetically heterogeneous ciliopathy affecting the cilia and sperm flagella. Mutations in genes related to the structural and functional defects of respiratory ciliary axoneme have been reported to be the predominant cause of this symptom; however, evidence regarding male infertility and genotype–phenotype associations between some of these genes and flagellar axoneme remains unclear. Here, we reported a male patient from a non-consanguineous Chinese family who exhibited left/right body asymmetry and oligoasthenoterazoospermia factor infertility. Novel compound heterozygous mutations in ARMC4 (NM:018076: c.2095C>T: p. Gln699*; c.1679C>T: p. Ala560Val) were identified in this patient, and his parents were a heterozygous carrier for the mutations. Morphological and ultrastructural analysis of the spermatozoa from the man showed aberrant sperm flagella with axonemal disorganization and outer dynein arm (ODA) loss. In addition, immunofluorescence analysis of the spermatozoa from the proband and a control man revealed a significant lower expression of ARMC4 protein due to pathogenic mutations. Therefore, our findings help to expand the spectrum of ARMC4 pathogenic mutations and linked biallelic ARMC4 mutations to male infertility for the first time.
Introduction
Primary ciliary dyskinesia (PCD, MIM 244400) is a genetically heterogeneous syndrome (Fliegauf et al., 2007), clinically characterized by the presence of chronic airway symptoms, obstructive lung disease, defects in laterality, and infertility (Goutaki et al., 2016). In human, a wide spectrum of recessive inherited genes, predominantly encoding ciliary/flagellar components, has linked PCD to various axonemal ultrastructural abnormalities. To date, more than 40 genes have been reported to cause this syndrome (Lucas et al., 2020). However, most genetic research studies of PCD are based on a cohort of patients with typical symptoms diagnosed at the early stage of life, which consequently ignore the genetic relationship between male infertility and PCD (Sironen et al., 2019; Guo et al., 2020).
In this study, we identified novel biallelic ARMC4 mutations in a Chinese male patient with PCD and infertility associated with oligoasthenoterazoospermia. The ARMC4 gene encodes a component of the outer dynein arm-docking complex (ODA-DC) that mediates ODA targeting and/or anchoring onto the doublet microtubule. Mutations in ARMC4 gene have been reported to cause PCD in human, mice, and zebrafish (Hjeij et al., 2013). Moreover, Hjeij et al. and Onoufriadis et al. both demonstrated that ARMC4-related PCD showed defects of the left–right axis and distal ODAs of the respiratory cilia (Hjeij et al., 2013; Onoufriadis et al., 2014). However, no sperm-related examinations were performed in these cases, and whether ARMC4 mutations have effects on sperm flagella structure remains unclear. This is the first report of ARMC4 mutations that associates with oligoasthenoterazoospermia in human and expands the spectrum of known ARMC4 mutations.
Patients and Methods
The proband was a 30-year-old man from a non-consanguineous Han Chinese family. He was recruited to identify the genetic risk factors at the Reproductive Center of the First Affiliated Hospital of Anhui Medical University due to his 3–4 years of primary infertility combined with dexiocardia symptom in 2020. The routine clinical examinations excluded the usual causes for male infertility including developmental effects, hormone levels, chromosomal aberration, and Y chromosome microdeletion. However, semen analyses showed oligoasthenospermia, which is clinically considered as a risk factor for infertility. Moreover, chest X-ray revealed left/right body asymmetry of the proband, but the parents are normal. Based on these findings, whole-exome sequencing (WES) was performed on the proband to identify genetic causes as previously described (He et al., 2020). Sanger sequencing was further used to validated pathogenic variants in his family (Supplementary Table 2). Immunofluorescence and transmission electron microscopy (TEM) analysis were performed to detect changes of the spermatozoa of the proband. All subjects were enrolled in accordance with the protocol approved by the Ethics Review Committee of the First Affiliated Hospital of Anhui Medical University and signed the informed consent.
Results
Identification of Novel Biallelic Mutations in ARMC4
Whole-exome sequencing was performed on the man, and compound heterozygous variants affecting exonic regions or splice sites were screened based on a recessive disease model. First, according to our filtering criteria, variants present in the 1000G, ExAC, and gnomAD databases with allele frequency >0.01 were removed. Second, we screened the variants predicted to be deleterious by more than three of the four biological systems (SIFT, PolyPhen-2, MutationTaster, CADD). The variant with CADD score higher than 20 was defined as deleterious. Third, genes associated with the ciliary phenotype were retained for further verification.
Based on the criteria, we found novel putative compound heterozygous variants in the ARMC4 gene (Table 1). Sanger sequencing confirmed the presence of the variants in the man, whereas his parents were heterozygous carriers (Figure 1A). Both of the variants were located in a conserved ARM domain of the ARMC4 protein, and the affected residues were highly conserved among different species (Figure 1B).
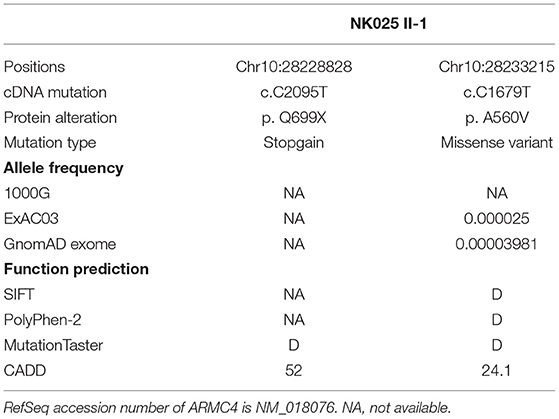
Table 1. Overview of the identified ARMC4 mutations.
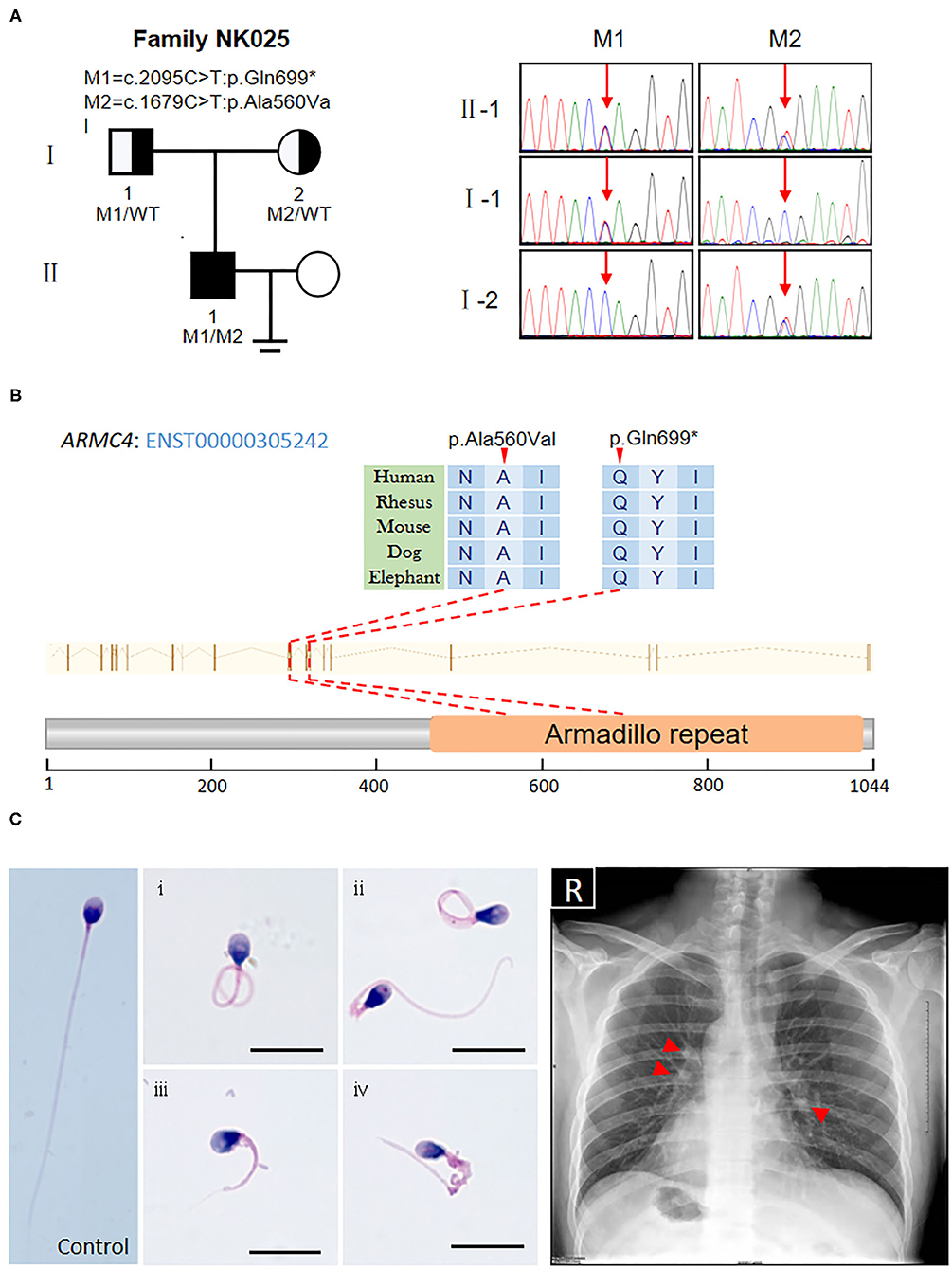
Figure 1. Identification of novel biallelic ARMC4 mutations in a Chinese man with left/right body asymmetry and oligoasthenospermia-associated male infertility. (A) The pedigree of the investigated men affected by biallelic mutations in ARMC4. Sanger sequencing results are shown on the right. Red arrows indicated the mutated positions. (B) Schematic of the positions of mutations identified in the ARMC4 genome and protein sequence. One of the mutations introduces a stop codon which leads to a premature stop and the other is a missense mutation exchanging alanine with valine at position 560. Both mutated residues are located in the evolutionarily conserved region. (C) Morphology of the spermatozoa from ARMC4-mutated men shows the MMAF phenotype, defined by coiled (i), bent (ii), short (iii), and irregular-caliber flagella (iv). Scale bar = 10μm. Chest X-ray of ARMC4-mutated men shows randomization of situs inversus (dextrocardia, gastric bubble right, liver on the left) and paracardial opacities with cystic lesions, suggesting atelectasis of the middle lobe and bronchiectasis (red arrows).
Clinical Description and Good Intracytoplasmic Sperm Injection Outcome of the ARMC4-Mutated Patient
Routine semen analysis of NK025 II-1 showed oligoasthenospermia (sperm concentration: 7.5 × 106/ml, progressive rate: 20%) (Supplementary Table 1) (Cooper et al., 2010). Morphological analysis of the spermatozoa from NK025 II-1 revealed multiple morphological abnormalities of the sperm flagella (MMAF) phenotype (Figure 1C), of which only 20.5% (41/200) of the spermatozoa exhibited normal tail morphologies, while coiled flagella, short flagella, angled flagella, and absent flagella of the spermatozoa were frequently observed (38, 20, 11.5, and 8%, respectively) (Supplementary Table 1) (Auger et al., 2016).
Next, we performed WES analysis on the spouse of the patient to exclude pathogenic ARMC4 variants before choosing intracytoplasmic sperm injection (ICSI) treatment. During the ICSI process, she received ovarian stimulation by the GnRH antagonist protocol. The rFSH was administered at a dose of 187.5 IU on day 2 of the menstrual cycle and 187.5 IU per day for 10 days. Subsequently, 18 meiosis II oocytes were injected with spermatozoa from NK025 II-1 and 15 two-pronuclear zygotes were observed on day 1. All the zygotes underwent normal cleavage. We finally acquired nine good quality embryos and frozen these on day 5 and day 6. One viable embryo was transferred in a frozen-thawed embryo cycle, and clinical pregnancy was diagnosed by transvaginal ultrasonography 30 days after transplantation fortunately (Table 2).
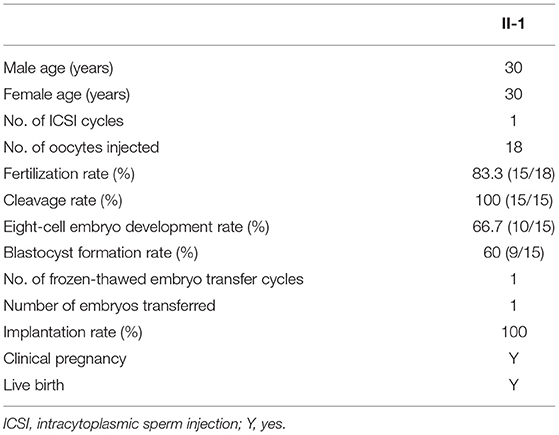
Table 2. ICSI outcomes of the ARMC4-mutated patient.
The MMAF Phenotype Was Linked to Lower ARMC4 Protein Expression and Ultrastructural Defects in the Spermatozoa From ARMC4-Mutated Men
The electron microscopy was used to evaluate the ultrastructural changes of the spermatozoa from NK025 II-1. High-resolution morphological analysis using scanning electron microscopy (SEM) confirmed that the spermatozoa of the patient displayed multiple sperm flagella abnormalities mainly with coiled and short flagella (≥58%) (Supplementary Figure 1). Moreover, TEM found aberrant axonemal cross-sectional structures of the spermatozoa of the patient (loss of the ODAs and defects of the central pair microtubules) compared with the spermatozoa from normal control (Figure 2A). To better understand the pathogenicity of ARMC4-mutated residues at the molecular level, we performed immunofluorescence for spermatozoa from NK025 II-1 and unaffected individuals. In normal spermatozoa, immunostaining of the ARMC4 protein was located along the length of the sperm flagella. However, ARMC4 immunostaining is markedly reduced along the full length of the flagella in the spermatozoa from NK025 II-1, which was consistent with the ODA defects observed (Figure 2B).
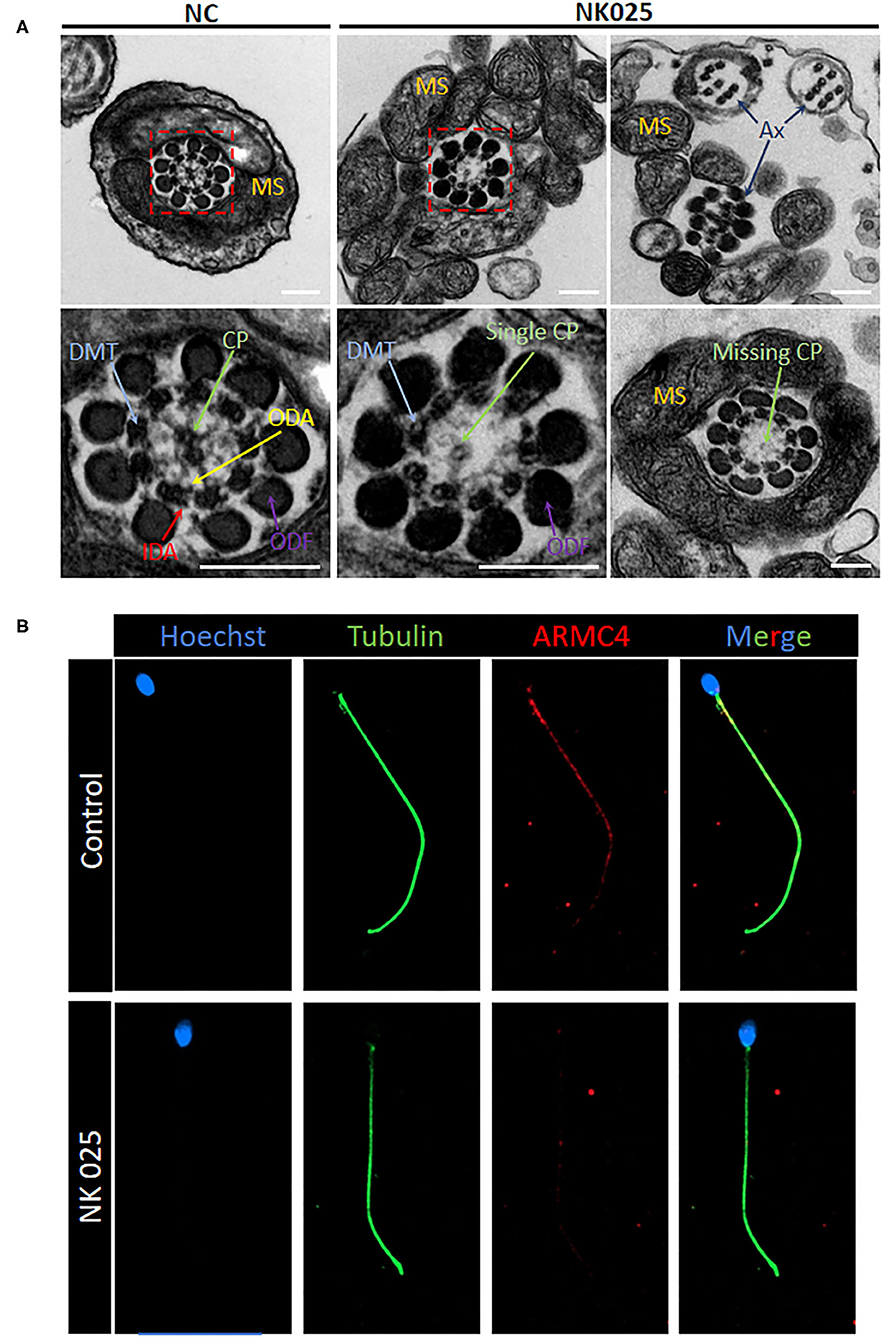
Figure 2. The ARMC4-mutated man exhibits lower ARMC4 protein expression and ultrastructural defects in axonemal structure. (A) Cross-sections of sperm flagella from the control man and the ARMC4-mutated men. Compared with those in normal sperm flagella, the axoneme of ARMC4-mutated men exhibited multiple defects of the axoneme, including absent of outer/inner dynein arms, CP defects (“9+0” “9+1”), and disorder of axoneme. Scale bars = 200 nm. (B) ARMC4 staining was present mainly in the midpiece and principal piece of spermatozoa flagella from the control man, but absent or had a significant decrease in the spermatozoa from the ARMC4-mutated men. CP, central microtubule pair (green arrows); ODA, outer dynein arm (yellow arrows); IDA, inner dynein arm (red arrows); MS, mitochondrial sheath; DMT, (blue arrows); ODF, outer dense fiber (purple arrows).
Discussion
Dysfunction of the cilia will lead to recurrent respiratory tract infections from early infancy, chronic rhinosinusitis, bronchiectasis, and otitis media (Mirra et al., 2017). Moreover, half of the patients had situs inversus, which is thought to be due to nodal cilia defects during embryonic development. Although advances in genetic etiology have identified pathogenic mutations in more than 40 flagella and motile cilia-associated genes, little is known about the fertility of such patients at their reproductive age not to mention the detailed sperm phenotypes of male patients (Goutaki et al., 2016).
In this study, we identified novel biallelic mutations in the PCD-related gene ARMC4 that might be responsible for human oligoasthenoterazoospermia factor infertility. As early as 2013, two main research studies have described the genotype–phenotype association between PCD and ARMC4 gene in a species conserved model. Hjeij et al. found that the absence of ARMC4 localization would lead to a significant reduction of ODAs in the respiratory cilia axonemes from patients with truncating mutations (Hjeij et al., 2013). Besides, Onoufriadis et al. found that ARMC4 transcript levels were upregulated significantly after ciliogenesis but undetectable in non-ciliated human bronchial epithelial cells (Onoufriadis et al., 2014). These findings suggested that the ARMC4 protein is required for a late step in proper targeting and anchoring of ODAs.
Motile cilia and sperm flagella are evolutionarily conserved and related organelles across species. In humans, respiratory motile cilia and sperm flagella share a common axoneme structure, consisting of a central microtubule pair surrounded by nine peripheral microtubule doublets (Lehti and Sironen, 2017). In addition, a variety of microtubular protein complexes, including radial spokes, nexin–dynein regulatory complexes, and inner and ODAs, are attached along the entire length of the axoneme (Toure et al., 2020). Although a role for ARMC4 in sperm flagella function has not been reported, the RNA expression profile has been inferred to be involved in sperm cells (Sironen et al., 2019).
The C-terminal of ARMC4 protein contains the well-known armadillo repeat motifs (ARMs). It is reported that tandem ARM-repeat sequence fold together as a superhelix, forming a conserved three-dimensional structure to interact with binding partners (Coates, 2003). Notably, the ARMs containing proteins are known to be involved in various processes, including signal transduction and cytoskeletal regulation (Coates, 2003; Cheng et al., 2013). In this present study, the identified novel biallelic ARMC4 mutations were both located in this region of the protein, consistent with the mutations found in previous studies (Hjeij et al., 2013; Onoufriadis et al., 2014). It is therefore highly suggested that the mutations may influence the protein-binding capabilities of ARMC4.
To verify the impact of mutations in the spermatozoa from this ARMC4-mutated man, we performed immunofluorescence analysis by using rabbit polyclonal anti-ARMC4 antibodies (HPA037829, Atlas Antibodies, Stockholm, Sweden). Immunofluorescence microscopy revealed that the ARMC4 protein localized mainly to the midpiece and principal piece of the sperm flagella in control sperm cells, and ARMC4-mutated sperm cells showed obviously a lower expression. We also performed cross-sectional analysis of the sperm flagella in an unaffected individual and ARMC4-mutated male patient by TEM. We found ODA abnormalities in most of the analyzed cross-sections of the patient, indicating that ARMC4 mutations affect ODA attachment along the entire axonemal length. In addition, previous studies have reported that men with PCD who were unable to reproduce naturally may have poorer outcomes with ICSI treatment (Yildirim et al., 2009; McLachlan et al., 2012). However, the specific mutations of these PCD patients were unknown. Here, we reported successful ICSI outcome and consequent live birth was achieved within first transplantation with sperm from an ARMC4-mutated PCD patient. We believe that it is of great importance to investigate the specific gene mutations on PCD patients and their fertility status or treatment outcomes to improve counseling and treatment of infertility in PCD patients.
Besides, we noticed that semen analysis of the ARMC4-mutated man also showed oligospermia (sperm count <15 million per ml); however, no experimental information was obtained to explain this phenomenon. Based on the observation that the ARMC4 protein is clearly expressed in the epididymal epithelium (The Human Protein Atlas: http://www.proteinatlas.org/), we speculated here that ARMC4-mutated men may have deficiency in epididymal cilia function (Ichioka et al., 2006). However, the proper mechanism of each gene and the effects of their abnormalities on male infertility are yet to be explored.
In conclusion, we identified novel pathogenic mutations in ARMC4 leading to low expression of ARMC4 protein in sperm flagella, which might cause male infertility with oligoasthenoterazoospermia and impact the ODA ultrastructure of mutated spermatozoa. Moreover, our clinical and genetic findings help to expand the spectrum of the ARMC4 gene and provide a deeper insight to our knowledge of the pathogenic effect of ARMC4 mutations in the reproductive system.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by the Ethics Review Committee of the First Affiliated Hospital of Anhui Medical University. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author Contributions
XH and YC designed the overall study concept. YG, XH, and YC wrote the manuscript. YG, CX, and QT performed the experiments. ML, KL, QS, HW, and DT analyzed the genetic data. BS, YX, FT, PZ, and ZW provided clinical data and supplied biological materials. All authors edited and reviewed the manuscript.
Funding
This work was supported by the National Key R&D Program of China (grant numbers 2019YFC1005106), the National Natural Science Foundation of China (grant numbers 81901541 and 81971441), Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (grant number 2019PT310002), National Major Scientific and Technological Special Project (2020ZX09201014), and Key R&D Program of Anhui Province (202004j07020032).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank the family who participated in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.715339/full#supplementary-material
References
Auger, J., Jouannet, P., and Eustache, F. (2016). Another look at human sperm morphology. Hum. Reprod. 31, 10–23. doi: 10.1093/humrep/dev251
Cheng, W., Ip, Y. T., and Xu, Z. (2013). Gudu, an Armadillo repeat-containing protein, is required for spermatogenesis in Drosophila. Gene 531, 294–300. doi: 10.1016/j.gene.2013.08.080
Coates, J. (2003). Armadillo repeat proteins: beyond the animal kingdom. Trends Cell Biol. 13, 463–471. doi: 10.1016/S0962-8924(03)00167-3
Cooper, T. G., Noonan, E., von Eckardstein, S., Auger, J., Baker, H. W. G., Behre, H. M., et al. (2010). World Health Organization reference values for human semen characteristics. Hum. Reprod. Update 16, 231–245. doi: 10.1093/humupd/dmp048
Fliegauf, M., Benzing, T., and Omran, H. (2007). When cilia go bad: cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 8, 880–893. doi: 10.1038/nrm2278
Goutaki, M., Meier, A. B., Halbeisen, F. S., Lucas, J. S., Dell, S. D., Maurer, E., et al. (2016). Clinical manifestations in primary ciliary dyskinesia: systematic review and meta-analysis. Eur. Respir. J. 48, 1081–1095. doi: 10.1183/13993003.00736-2016
Guo, Z., Chen, W., Wang, L., and Qian, L. (2020). Clinical and genetic spectrum of children with primary ciliary dyskinesia in China. J. Pediatr. 225, 157.e5–165.e5. doi: 10.1016/j.jpeds.2020.05.052
He, X., Liu, C., Yang, X., Lv, M., Ni, X., Li, Q., et al. (2020). Bi-allelic loss-of-function variants in CFAP58 cause flagellar axoneme and mitochondrial sheath defects and asthenoteratozoospermia in humans and mice. Am. J. Hum. Genet. 107, 514–526. doi: 10.1016/j.ajhg.2020.07.010
Hjeij, R., Lindstrand, A., Francis, R., Zariwala, M. A., Liu, X., Li, Y., et al. (2013). ARMC4 mutations cause primary ciliary dyskinesia with randomization of left/right body asymmetry. Am. J. Hum. Genet. 93, 357–367. doi: 10.1016/j.ajhg.2013.06.009
Ichioka, K., Kohei, N., Okubo, K., Nishiyama, H., and Terai, A. (2006). Obstructive azoospermia associated with chronic sinopulmonary infection and situs inversus totalis. Urology 68, 204.e5–207.e5. doi: 10.1016/j.urology.2006.01.072
Lehti, M. S., and Sironen, A. (2017). Formation and function of sperm tail structures in association with sperm motility defects. Biol. Reprod. 97, 522–536. doi: 10.1093/biolre/iox096
Lucas, J. S., Davis, S. D., Omran, H., and Shoemark, A. (2020). Primary ciliary dyskinesia in the genomics age. Lancet Respir. Med. 8, 202–216. doi: 10.1016/S2213-2600(19)30374-1
McLachlan, R. I., Ishikawa, T., Osianlis, T., Robinson, P., Merriner, D. J., Healy, D., et al. (2012). Normal live birth after testicular sperm extraction and intracytoplasmic sperm injection in variant primary ciliary dyskinesia with completely immotile sperm and structurally abnormal sperm tails. Fertil. Steril. 97, 313–318. doi: 10.1016/j.fertnstert.2011.11.003
Mirra, V., Werner, C., and Santamaria, F. (2017). Primary ciliary dyskinesia: an update on clinical aspects, genetics, diagnosis, and future treatment strategies. Front Pediatr 5:135. doi: 10.3389/fped.2017.00135
Onoufriadis, A., Shoemark, A., Munye, M. M., James, C. T., Schmidts, M., Patel, M., et al. (2014). Combined exome and whole-genome sequencing identifies mutations in ARMC4 as a cause of primary ciliary dyskinesia with defects in the outer dynein arm. J. Med. Genet. 51, 61–67. doi: 10.1136/jmedgenet-2013-101938
Sironen, A., Shoemark, A., Patel, M., Loebinger, M. R., and Mitchison, H. M. (2019). Sperm defects in primary ciliary dyskinesia and related causes of male infertility. Cell. Mol. Life Sci. doi: 10.1007/s00018-019-03389-7
Toure, A., Martinez, G., Kherraf, Z. E., Cazin, C., Beurois, J., Arnoult, C., et al. (2020). The genetic architecture of morphological abnormalities of the sperm tail. Hum. Genet. doi: 10.1007/s00439-020-02113-x
Keywords: male infertility, primary ciliary dyskinesia (Kartagener syndrome), ARMC4, oligoasthenoterazoospermia, ICSI
Citation: Gao Y, Xu C, Tan Q, Shen Q, Wu H, Lv M, Li K, Tang D, Song B, Xu Y, Zhou P, Wei Z, Tao F, Cao Y and He X (2021) Case Report: Novel Biallelic Mutations in ARMC4 Cause Primary Ciliary Dyskinesia and Male Infertility in a Chinese Family. Front. Genet. 12:715339. doi: 10.3389/fgene.2021.715339
Received: 02 June 2021; Accepted: 28 June 2021;
Published: 30 July 2021.
Edited by:
Lingqian Wu, Central South University, ChinaReviewed by:
Mingxi Liu, Nanjing Medical University, ChinaSinem Firtina, University of Istinye, Turkey
Copyright © 2021 Gao, Xu, Tan, Shen, Wu, Lv, Li, Tang, Song, Xu, Zhou, Wei, Tao, Cao and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yunxia Cao, Y2FveXVueGlhNiYjeDAwMDQwOzEyNi5jb20=; Xiaojin He, aHhqMDExNyYjeDAwMDQwOzEyNi5jb20=
†These authors have contributed equally to this work