
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 07 October 2021
Sec. Evolutionary and Population Genetics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.709419
This article is part of the Research Topic Association Between Individuals’ Genomic Ancestry and Variation in Disease Susceptibility View all 13 articles
 Vasily E. Ramensky1,2*
Vasily E. Ramensky1,2* Alexandra I. Ershova1
Alexandra I. Ershova1 Marija Zaicenoka3
Marija Zaicenoka3 Anna V. Kiseleva1Anastasia A. Zharikova1,2
Anna V. Kiseleva1Anastasia A. Zharikova1,2 Yuri V. Vyatkin1,4Evgeniia A. Sotnikova1Irina A. Efimova1
Yuri V. Vyatkin1,4Evgeniia A. Sotnikova1Irina A. Efimova1 Mikhail G. Divashuk1,5Olga V. Kurilova1Olga P. Skirko1
Mikhail G. Divashuk1,5Olga V. Kurilova1Olga P. Skirko1 Galina A. Muromtseva1Olga A. Belova6Svetlana A. Rachkova6Maria S. Pokrovskaya1Svetlana A. Shalnova1
Galina A. Muromtseva1Olga A. Belova6Svetlana A. Rachkova6Maria S. Pokrovskaya1Svetlana A. Shalnova1 Alexey N. Meshkov1†Oxana M. Drapkina1†
Alexey N. Meshkov1†Oxana M. Drapkina1†We performed a targeted sequencing of 242 clinically important genes mostly associated with cardiovascular diseases in a representative population sample of 1,658 individuals from the Ivanovo region northeast of Moscow. Approximately 11% of 11,876 detected variants were not found in the Single Nucleotide Polymorphism Database (dbSNP) or reported earlier in the Russian population. Most novel variants were singletons and doubletons in our sample, and virtually no novel alleles presumably specific for the Russian population were able to reach the frequencies above 0.1–0.2%. The overwhelming majority (99.3%) of variants detected in this study in three or more copies were shared with other populations. We found two dominant and seven recessive known pathogenic variants with allele frequencies significantly increased compared to those in the gnomAD non-Finnish Europeans. Of the 242 targeted genes, 28 were in the list of 59 genes for which the American College of Medical Genetics and Genomics (ACMG) recommended the reporting of incidental findings. Based on the number of variants detected in the sequenced subset of ACMG59 genes, we approximated the prevalence of known pathogenic and novel or rare protein-truncating variants in the complete set of ACMG59 genes in the Ivanovo population at 1.4 and 2.8%, respectively. We analyzed the available clinical data and observed the incomplete penetrance of known pathogenic variants in the 28 ACMG59 genes: only 1 individual out of 12 with such variants had the phenotype most likely related to the variant. When known pathogenic and novel or rare protein-truncating variants were considered together, the overall rate of confirmed phenotypes was about 19%, with maximum in the subset of novel protein-truncating variants. We report three novel protein truncating variants in APOB and one in MYH7 observed in individuals with hypobetalipoproteinemia and hypertrophic cardiomyopathy, respectively. Our results provide a valuable reference for the clinical interpretation of gene sequencing in Russian and other populations.
The next-generation sequencing projects of the last decade revealed the complicated spectrum of rare and ultra-rare allelic variation in most human genes (Tennessen et al., 2012; Lek et al., 2016; Van Hout et al., 2020). The sequencing of large population cohorts greatly enriched our understanding of the prevalence of presumably pathogenic variants in reportedly healthy people (Dorschner et al., 2013; Amendola et al., 2015; Dewey et al., 2016) and initiated studies aimed at reviewing the implied pathogenicity of genetic variants (Dewey et al., 2014; Shah et al., 2018) and penetrance of variants classified as pathogenic (Cooper et al., 2013; Chen et al., 2016; Wright et al., 2019; Van Rooij et al., 2020). This activity was shaped by the standards and guidelines for the classification of sequence variants using criteria informed by expert opinion developed by The American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015; Nykamp et al., 2017).
The ACMG also suggested the list of genes for which the reporting of known and expected pathogenic variants, also termed incidental findings, may be recommended (Green et al., 2013). The updated guidelines presented ACMG59, the list of the mostly dominant 59 genes associated with medically actionable disorders (Kalia et al., 2017). However, limited information on the penetrance of many variants even in this relatively small set of genes makes interpretation challenging. This uncertainty is gradually decreasing as more studies evaluate the carrier status of pathogenic gene variants in general populations (Amendola et al., 2016; Wright et al., 2019; Van Rooij et al., 2020). Numerous papers reported the prevalence of known and expected pathogenic variants in various populations, for example, Dutch (Haer-Wigman et al., 2019; Van Rooij et al., 2020), Qatari (Jain et al., 2018), Korean (Kwak et al., 2017), Australian (Lacaze et al., 2020), British (Van Hout et al., 2020), and Taiwanese (Kuo et al., 2020). The present study investigates the incidental findings of pathogenic variants in 28 genes from the ACMG59 list in the Russian population from the Ivanovo region.
Russia is one of the most ethnically diverse countries in the world; however, it is underrepresented in the large human genome sequencing projects (Lek et al., 2016; Van Hout et al., 2020). Earlier genome-wide studies of the Russian population were mostly based on the limited number of samples and focused on the genetic history and admixture patterns (Mallick et al., 2016; Wong et al., 2017). Recently, the whole-genome variation was analyzed for the 264 healthy adult participants of the Genome Russia Project (Zhernakova et al., 2020) demonstrating adaptive, clinical, and functional consequences. Approximately 3–4% of the called variants were classified as novel when compared to the dbSNP database (Sherry et al., 2001); in many cases, allele frequencies demonstrated divergence from the neighboring populations. Barbitoff et al. (2019) reported variant frequencies in a larger subset of 694 exomes from the northwest Russia. The results indicated that 9.3% of discovered variants were not present in dbSNP. Moreover, this whole-exome study demonstrated the overrepresentation of several disease-causing variants for Mendelian disorders, such as phenylketonuria (PAH, rs5030858), Wilson’s disease (ATP7B, rs76151636), factor VII deficiency (F7, rs36209567), and the kyphoscoliosis type of Ehlers–Danlos syndrome (FKBP14, rs542489955). For the Russian population, however, pathogenic variant frequencies were reported mostly for relatively small cohorts including patients and their families and targeted at specific genes and disorders, for example, familial hypercholesterolemia (Meshkov et al., 2021; Miroshnikova et al., 2021); cystic fibrosis, phenylketonuria, alpha-1 antitrypsin deficiency, and sensorineural hearing loss (Kiseleva et al., 2020; Petrova et al., 2020); cardiomyopathy (Marakhonov et al., 2019; Zaklyazminskaya et al., 2019; Kulikova et al., 2021; Shestak et al., 2021); and breast and ovarian cancer (Brovkina et al., 2018; Solodskikh et al., 2019).
The aim of this work was to take a step forward in the study of the genetic makeup of the Russian population, with the emphasis on rare variation, in particular, known and expected pathogenic variants in a subset of clinically important genes. We developed the gene panel which included 242 protein-coding genes associated with cardiovascular diseases and high risk of early or sudden cardiac death and used the population samples collected in the course of the ESSE-RF cardiovascular epidemiology project (Boitsov et al., 2013). Below, we report the results of the targeted sequencing of 1,685 unrelated participants from Ivanovo, one of the ESSE-RF regions. The availability of clinical data made it possible to evaluate the carriers of known and expected novel pathogenic variants for certain phenotypes.
The individuals for our study were selected from the study “Epidemiology of Cardiovascular Diseases and Risk Factors in Regions of the Russian Federation (ESSE-RF).” The ESSE-RF is a multicenter population-based study, conducted from 2012 to 2014, covering 13 regions of Russia, differing in climatic, geographic, economic, and demographic characteristics (Boitsov et al., 2013). About 1,600–1,900 people, aged 25–64, were randomly selected from every region, including Ivanovo, a region approximately 300 km northeast of Moscow with predominant Russian population. Please see more details regarding sample selection in the Supplementary Methods.
The sequenced set contained 1,685 individuals: 1,056 females with median age 52 at the moment of enrollment and 629 males with median age 44, respectively. We used PLINK v1.90 (Chang et al., 2015) to calculate identity by state (IBS) values and estimate the identity by descent (IBD) proportion (PI_HAT) for all possible pairs of individuals. To ensure that our dataset does not include closely related participants, we removed all pairs with PI_HAT > 0.33. The average PI_HAT value across all 1,418,770 pairs of 1,685 individuals was 0.027.
Clinical data were obtained from several major sources: (1) questionnaires administered face-to-face at the beginning of the ESSE-RF study (2012), (2) fasting venous blood samples, (3) electrocardiogram (ECG) records, (4) coronary artery disease (CAD) validation in 2013, and (5) endpoint data. Additionally, (6) the available medical records of 10 patients with variants of interest in the ACMG59 genes observed at the Ivanovo Cardiology dispensary were analyzed. Blood samples for genetic analysis were stored at the Biobank of the National Medical Research Center for Therapy and Preventive Medicine (Moscow, Russia) (Pokrovskaya et al., 2019; Anisimov et al., 2021). Demographic characteristics, information about the most common non-communicable diseases, ECG parameters, and multiple blood biochemical parameters including lipid profiles are included into analysis. Endpoints such as all-cause mortality, myocardial infarctions, strokes, new cases of coronary artery disease, and revascularization are collected annually in the ESSE-RF. Clinical and endpoint data were available for all patients. Coronary artery disease was verified for participants with positive Rose Angina Questionnaire or the patient’s positive answer to questions concerning previous diagnoses of either coronary artery disease or myocardial infarction. It is worth noting, however, that even with these data at hand, we were able to evaluate the individuals with known or expected pathogenic mutations for a limited number of phenotypes, for example, hypobetalipoproteinemia and prolonged QT interval.
The study was reviewed and approved by the Independent Ethic Committee of the National Medical Research Center for Therapy and Preventive Medicine (Protocol number 07-03/12 from 03.07.2012) and conducted according to the principles of the Helsinki Declaration. The participants provided their written informed consent to take part in this study.
We developed the targeted panel including coding exons of 242 protein-coding genes associated with cardiovascular diseases and high risk of early or sudden cardiac death, in particular, cardiomyopathy (for example, MYBPC3, MYH7, DSP, LMNA, and DES), channelopathy (KCNQ1, KCNH2, SCN5A, and others), and hypercholesterolemia and hypobetalipoproteinemia (APOB, PCSK9, LDLR, LDLRAP1, ANGPTL3, and others). Exon coordinates were expanded by 25 bp to include splice region variants. The sequenced target contained 4,311 exons with the total length of 1.04 Mbp. The following 28 genes from our panel are in the list of 59 well-characterized medically actionable disease genes recommended by the American College of Medical Genetics and Genomics for the return of incidental findings in clinical genomic sequencing (Kalia et al., 2017): ACTA2, ACTC1, APOB, DSC2, DSG2, DSP, GLA, KCNH2, KCNQ1, LDLR, LMNA, MYBPC3, MYH11, MYH7, MYL2, MYL3, PCSK9, PKP2, PRKAG2, RYR1, RYR2, SCN5A, SDHD, SMAD3, TMEM43, TNNI3, TNNT2, and TPM1. These genes comprise the cardiovascular disorder-related part of the ACMG59 panel. We refer to this subset of our target as ACMG59 genes. The full list of targeted genes with phenotypes is shown in Supplementary Table 1.
Target sequencing data processing and quality control evaluation were performed with the custom- designed pipeline based on GATK 3.8 and generally following the Broad institute best practices for variant calling with both standard GATK hard filters and VQSR (details in the Supplementary Methods). Paired-end reads were aligned to the hg19 genome sequence which is the major human reference in this project. Since the VQSR quality evaluation is more effective for the hg38, we also ran the pipeline with this reference and used the resulting filters independently: a variant was accepted for downstream analysis only if both hg37 and hg38 GATK filters flagged it as PASS and all non-reference genotype quality values were maximal (GQ = 99). This filtering retained 11,876 (84.3%) variants of the initial set of 14,087 variants and is a conservative approach aimed at minimizing the false positive variant calls.
Single-nucleotide variants and short indels were annotated with ENSEMBL Variant Effect Predictor (VEP) v.100 (McLaren et al., 2016) and cross-checked against the standalone versions of ClinVar (2021/01/10) and gnomAD (v2.1.1) databases that accumulate information on variant clinical significance and population frequencies (Landrum et al., 2016; Karczewski et al., 2020). We used ClinVar as the major source of known pathogenic and likely pathogenic variants (KP). VEP identified known variants present in the dbSNP database (Sherry et al., 2001). For each annotated variant, VEP aggregates maximal alternative allele frequency across 1000 Genomes, ESP and ExAC/gnomAD, and reports in the MAX_AF field. Allele frequencies in our dataset were also compared, where available, to those in the Russian northwest population (NWR) (Barbitoff et al., 2019).
We used LOFTEE (Karczewski et al., 2020), SIFT (Ng and Henikoff, 2006), and PolyPhen-2 (Adzhubei et al., 2010) VEP plugins to flag low-confidence protein-truncating variants (PTVs, high- impact variants in VEP) and predict potentially damaging missense variants, respectively. Missense variants simultaneously predicted as deleterious by SIFT and damaging by PolyPhen-2 are referred to as strictly damaging. We used VarSome (Kopanos et al., 2019) to perform automated variant classification according to the ACMG guidelines (Richards et al., 2015).
We explored the spectrum of genome variation in the Russian population by sequencing 242 clinically important protein-coding genes in 1,658 unrelated individuals from the Ivanovo region. The strict quality control and filtering procedure described in the section “Materials and Methods” filtered out 2,209 variants out of the initial 14,087 keeping 11,423 SNVs and 453 short indels in the targeted regions. The observed transition/transversion Ti/Tv ratio was 3.10 for all SNVs in the exons and flanking regions and 3.48 for missense and synonymous variants suggesting the paucity of false positive variants after filtering.
Missense variants comprise the largest group by annotation (40.8%), followed by synonymous (27.2%) and intron variants (21.3%). We split the bulk of the observed variation into five groups based on their annotation and potential impact on gene function: protein-truncating variants (PTVs), strictly damaging missense variants (predicted to be damaging both by SIFT and PolyPhen-2), other missense variants, in-frame indels, and other variants, including synonymous, UTR, intron, and other variants with the lowest expected impact (Table 1 and Figure 1). The fraction of strictly damaging predictions is maximal among singleton missense variants (31.9%) and gradually reduces with increasing frequency, reaching 15.1% for missense variants with allele frequency in our sample AF > 1%. This confirms the observation of a negative correlation between the alternative allele frequency and the ratio of missense variants predicted as damaging in large cohorts (Tennessen et al., 2012). Out of the total 11,876 high-quality SNV and short indels discovered in our dataset, 7,582 (63.8%) were singletons or doubletons (allele count AC < 3) with estimated alternative allele frequency not exceeding approximately 0.1%. The overwhelming majority (99.3%) of variants detected in our study in three or more copies are reported in the dbSNP and shared with other populations. The allele frequencies of these variants are very close to those in the European non-Finnish (NFE) gnomAD exomes with Pearson’s correlation coefficient R = 0.997 (Figure 2).

Table 1. Overview of variants in the 242 targeted genes in the Ivanovo population.
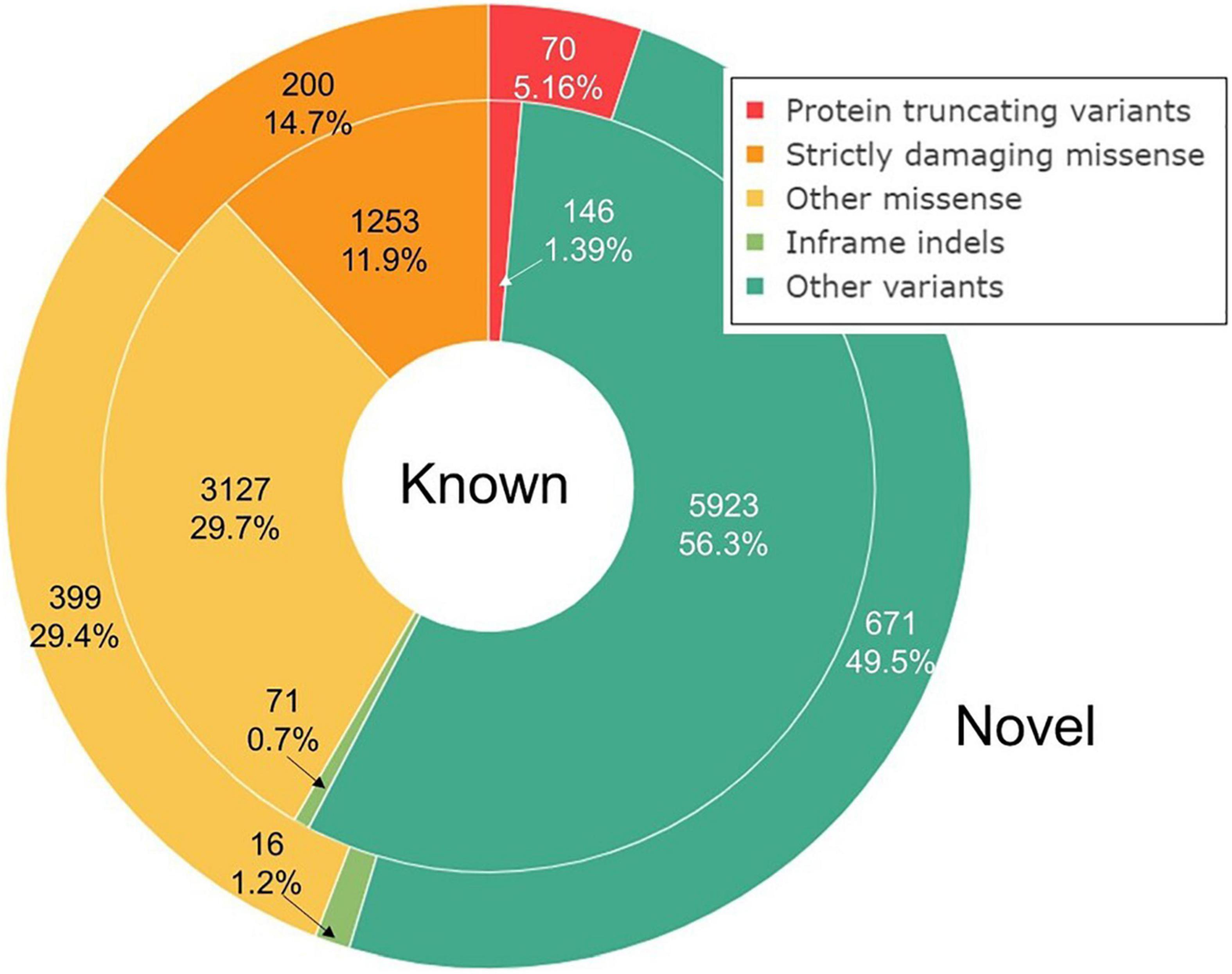
Figure 1. Variant annotation. Known and novel variants are represented by inner and outer rings, respectively. Variant annotation was performed by ENSEMBL VEP. Strictly damaging variants are those predicted as damaging both by SIFT and PolyPhen-2.
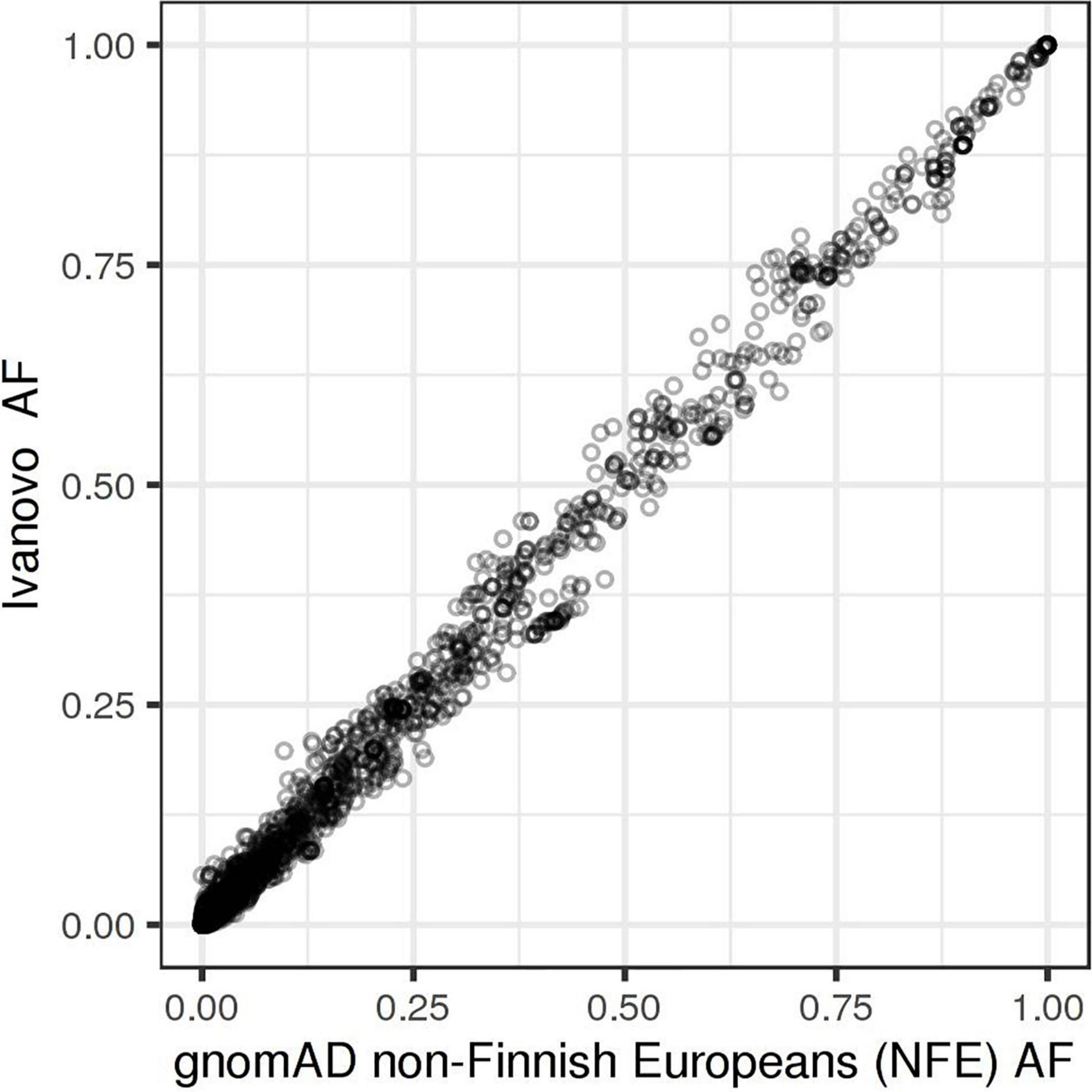
Figure 2. Allele frequencies in the Ivanovo population (y-axis) compared to gnomAD non-Finnish Europeans (x-axis).
A total of 1,356 (11.4%) variants out of 11,876 were novel, that is, not present in dbSNP 153. The fraction of novel variants was highest among the protein-truncating ones (32.4%) apparently due to the purifying selection acting on specific types of variants enriched with deleterious alleles (Lek et al., 2016). In-frame indels were not that abundant but include the second largest fraction of novel variants (18.4%). The majority (95.9%) of novel variants were not reported earlier in the survey of 694 exomes from the northwest Russia (NWR) (Barbitoff et al., 2019), the largest report on the whole exome sequencing in the Russian population to date. This is not surprising, taking in view the fact that our set is 2.4 times larger and thus allows the surveying of a wider spectrum of rare variation.
The novel variants in the Ivanovo population were found mostly among singletons and doubletons with only 26 variants with three-to-seven copies of alternative alleles (Supplementary Table 2). There were no novel protein-truncating variants other than singletons or doubletons. In Table 2, we report 12 selected novel variants with three or more alleles in our dataset and that were annotated as moderate impact by VEP. All these variants were missense except for a complex non-frameshifting variant in TRIM63 that was reported by GATK as three adjacent short indels. This complex haplotype was confirmed by the visual inspection of read alignments with IGV software (Robinson et al., 2011) and reported in Table 2 as a single in-frame indel spanning protein residues 98–109. Seven missense variants were predicted as damaging both by SIFT and PolyPhen-2 and were denoted with asterisk in Table 2. The remarkably large SYNE2 gene harbored two such novel strictly damaging variants, Thr3804Asn and Glu4972Lys, each observed in three distinct individuals. Only two variants (Ser291Phe in DMD and Lys4037Glu in SYNE1) were found earlier in NWR exomes. The relatively high prevalence of these variants in the Russian population should be taken into account in the course of exome or genome sequencing in the clinical context (Richards et al., 2015).
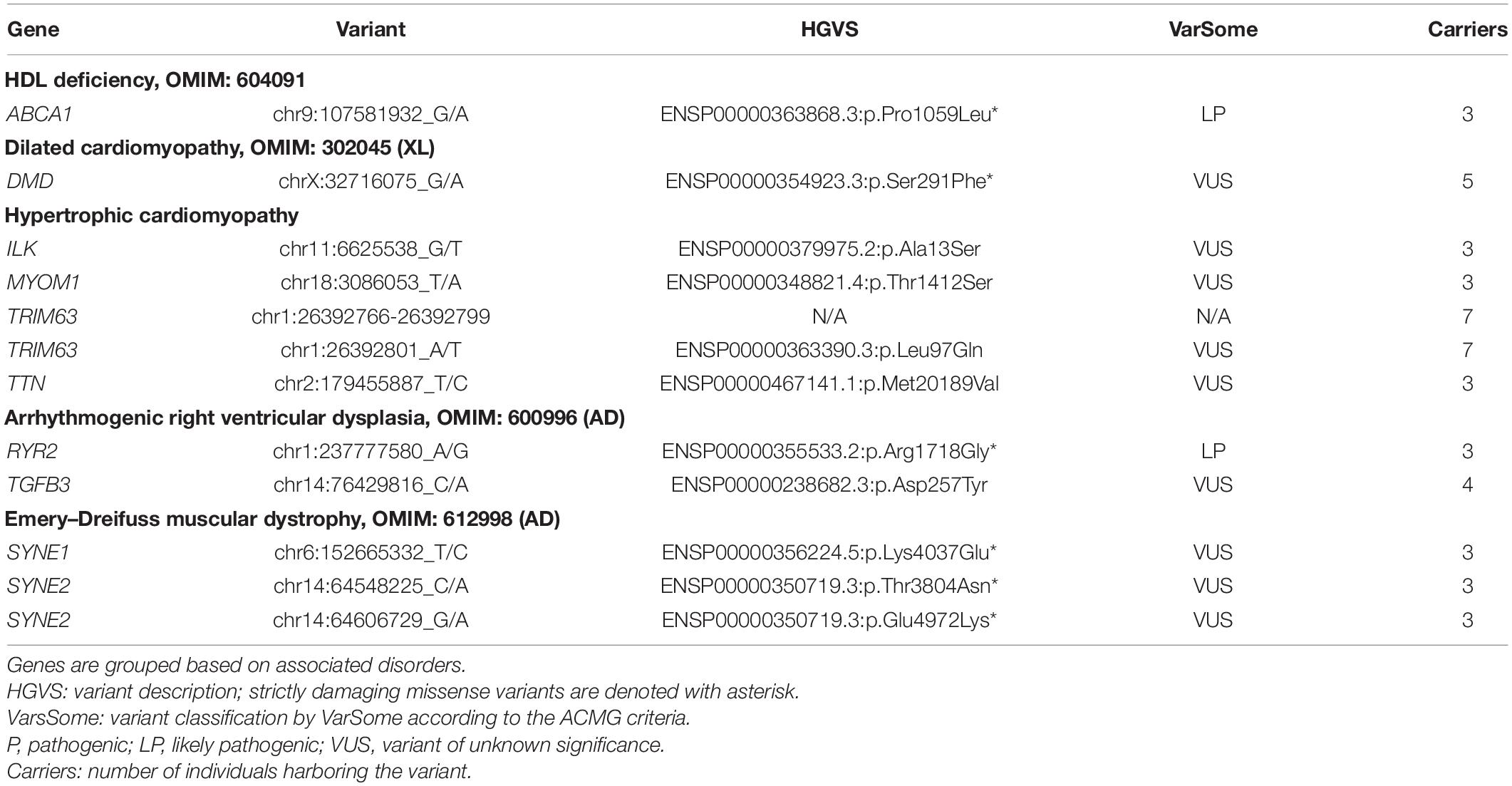
Table 2. Novel variants with three or more alleles in our dataset annotated by VEP as high or moderate impact.
The observed variant counts by type are tabulated for each gene in Supplementary Table 1. We found two genes that were significantly enriched with novel variants: MIB1 associated with left ventricular non-compaction (OMIM: 615092), with 13 novel and 21 known variants, and PHKA1 associated with muscle glycogenosis (OMIM:300559), with 12 novel and 17 known variants. The enrichment was validated by Fisher’s exact test that compared the numbers of novel and known variants in a particular gene vs. all genes taken together, with 1,356 novel and 10,520 known variants. The P-values for MIB1 and PHKA1 were 5.2 × 10–5 and 4.0 × 10–5, respectively. The next hit with excess of novel variants is LAMA2 with 27 novel and 107 known variants; however, unlike the first two hits, its resulting P = 0.002 apparently does not pass any reasonable threshold even after the most lenient multiple test correction. Taking into account only rare known variants in comparison instead of all known variants gives similar results. The observed relative excess of rare variants may be a signature of population-specific demographic events at certain loci.
Our sequencing target included 28 of the 59 genes known as the ACMG59 genes, for which the reporting of known pathogenic variants was recommended by the American College of Medical Genetics and Genomics (Kalia et al., 2017). Numerous studies reported the prevalence estimates for known and expected pathogenic variants in these genes (Amendola et al., 2015; Haer-Wigman et al., 2019; Kuo et al., 2020; Lacaze et al., 2020; Van Hout et al., 2020).
In the Ivanovo sample of 1,685 individuals, six genes (DSP, KCNQ1, MYBPC3, MYH7, RYR2, TMEM43) harbored eight known pathogenic or likely pathogenic variants (KP). Besides, nine novel high confidence protein-truncating variants were found in seven genes: APOB, DSP, KCNQ1, MYH7, MYH11, PCSK9, and RYR2 (Table 3). All observed genotypes involving these variants were heterozygous, and none of the 17 variants were observed in the NWR exomes.
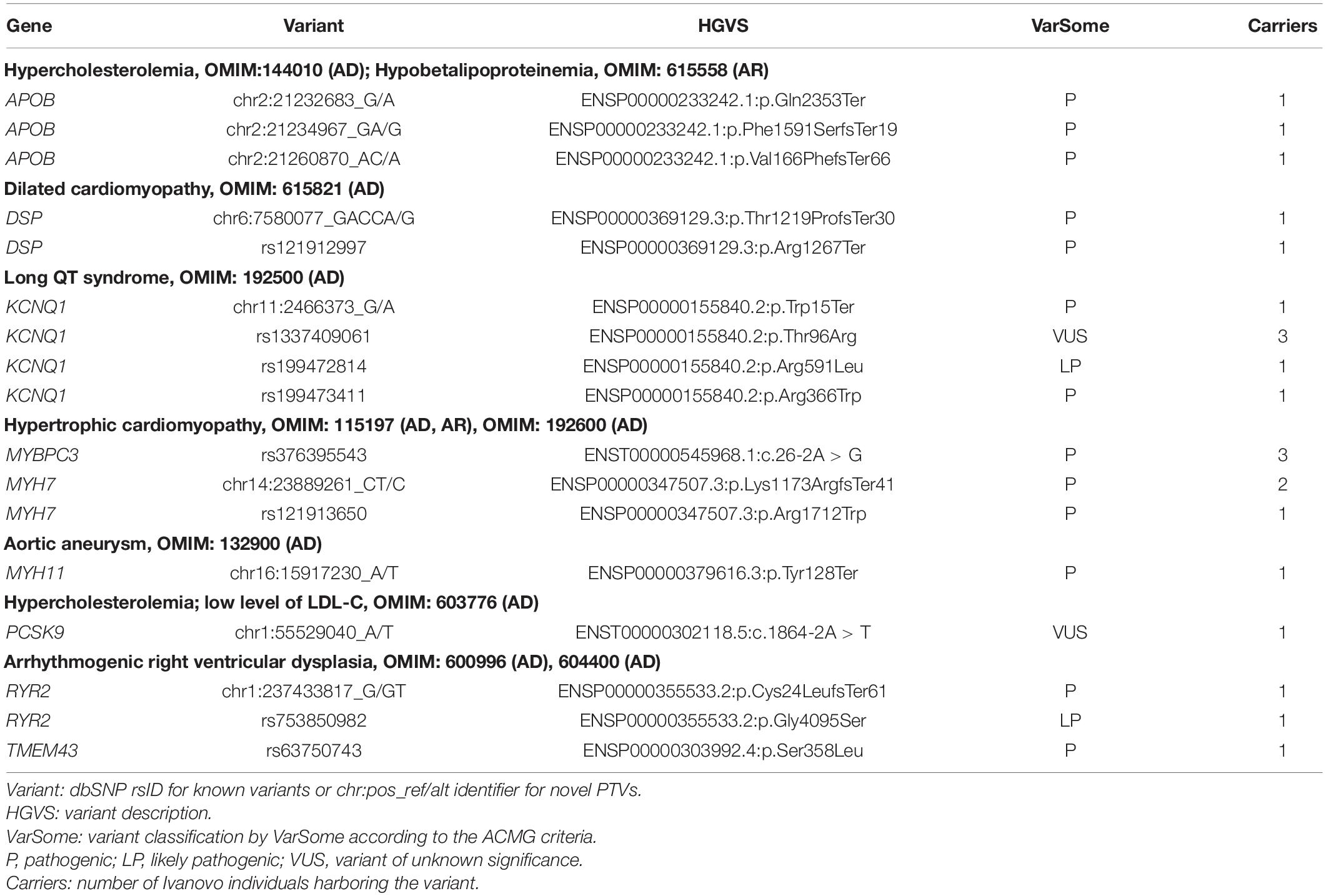
Table 3. Known pathogenic or likely pathogenic and novel protein-truncating variants in the 28 genes from the ACMG59 list.
We used VarSome (Kopanos et al., 2019) to classify KP and novel PTVs according to the ACMG criteria (Richards et al., 2015). All such variants in the ACMG59 genes were evaluated as pathogenic or likely pathogenic with two exceptions classified as variants of unknown significance (VUS). The first one is the missense substitution Thr96Arg in KCNQ1 reported by one submitter as likely pathogenic without any supporting evidence and predicted as benign/tolerated by SIFT and PolyPhen-2. The second one is the novel splice acceptor variant in the PCSK9 gene (ENST00000302118.5:c.1864-2A > T), considered high quality by LOFTEE and predicted deleterious by multiple tools. The LDL-C value of the carrier is 2.64 mmol/l which is in the normal range. In both cases, variants were classified by VarSome as VUS but not excluded from our analysis.
For all seven genes with novel PTVs, protein truncation is known to be an established disease-causing mechanism: in particular, no benign PTVs were reported in ClinVar for KCNQ1, MYH11, and MYH7 and only one instance of benign PTV in DSP. In PCSK9, truncating mutations are associated with low plasma levels of low-density lipoprotein cholesterol (LDL-C) (Cohen et al., 2005). Six of eight known KPs were observed as singletons and 2 in 3 individuals each, giving 12 individuals in total which comprises 0.71% of the total sample of 1,685 participants. The initial set of variant calls included a KP variant in RYR2 (rs794728721), but it was filtered out by both hg37 and hg38 GATK filters and was not included in the filtered variant set. Eight novel truncating variants were singletons, and one was observed in two participants, which gave the extra 10 participants harboring novel protein-truncating variants. The genes also harbored nine rare PTVs with worldwide maximal allele frequency below 0.1% as reported by VEP and the total of 14 carriers in the Ivanovo population (Supplementary Table 3). The 24 participants with novel or rare PTVs gave an extra 1.4% of the total sample of 1,685 participants. Taking in view our gene sequencing target included approximately one-half of the current list of 56 dominant ACMG59 genes (excluding ATP7B, MUTYH, and PMS2), one may approximate the prevalence of dominant KP variants and novel or rare PTVs in the Ivanovo population at 1.4 and 2.8%, respectively.
Evaluating the complete set of 242 sequenced genes, we identified 85 pathogenic or likely pathogenic ClinVar variants in 44 genes: 36 protein-truncating, 47 missense with 36 of them predicted to be strictly damaging, and two intron variants (Supplementary Table 3). This set of KP variants included eight variants in the ACMG59 genes described above. All observed genotypes involving known pathogenic or novel variants discussed in this section were heterozygous. Among the 77 known KP in non-ACMG59 genes, 9 and 63 were associated with dominant (AD) and recessive (AR) diseases, respectively (Table 4).
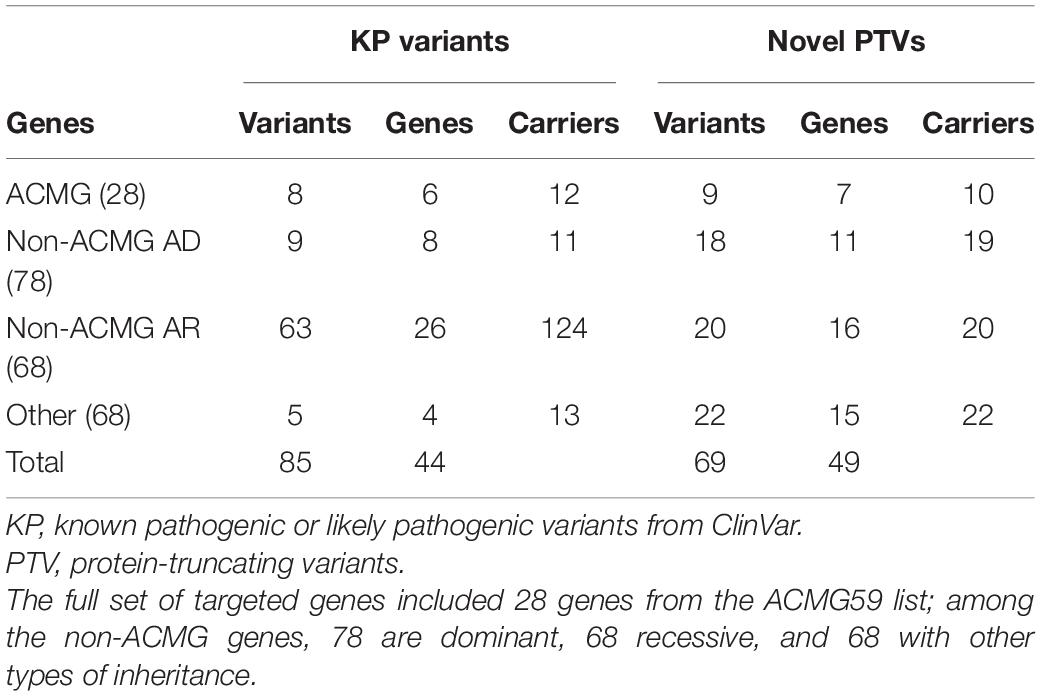
Table 4. Known pathogenic or likely pathogenic and novel protein-truncating variants in the full set of 242 targeted genes.
Known variants in dominant non-ACMG genes were mostly present in one participant, except for the missense Pro279Leu variant in the MEF2A gene (rs121918529) observed in three participants and associated with CAD/myocardial infarction. As expected, known disease variants in recessive genes were more prevalent, with 12 recessive variants observed in three or more participants. Among them were two variants in GAA (rs375470378) and PMM2 (rs28936415), found in eight participants each. One protein-truncating variant in APOC3 without any assigned specific inheritance mode (rs138326449) was classified as pathogenic and associated with coronary artery disease and hyperalphalipoproteinemia by a single submitter in ClinVar (VCV000139560.3) and found in nine individuals.
The full set of 242 targeted genes also contained 69 novel PTVs in 48 genes (Table 4 and Supplementary Table 3), all being singletons, except for the two doubleton cases: the frameshift variant ENSP00000347507.3:p.Lys1173ArgfsTer41 in the ACMG59 gene MYH7 and the stop gain variant ENSP00000364979.4:p.Arg1063Ter in the non-ACMG gene COL4A1. Unlike known pathogenic variants, novel PTVs did not exhibit any detectable enrichment or depletion in the dominant or recessive genes, suggesting that their functional role may require thorough evaluation.
We used VarSome (Kopanos et al., 2019) to classify KP and novel PTVs according to the ACMG criteria (Richards et al., 2015). All variants were evaluated as pathogenic or likely pathogenic with few exceptions: five KP variants and six novel PTVs were classified by VarSome as VUS, and one KP variant as likely benign (Supplementary Table 3). The latter is the missense Ala296Thr variant rs80356462 in the SIX5 gene, classified by ClinVar as pathogenic, with no assertion criteria provided (ClinVar id: VCV000008599.1) and predicted benign by multiple computational tools, including SIFT and PolyPhen-2.
With novel PTVs observed in 48 genes, every fifth of the targeted genes in the Ivanovo sample contained a novel heterozygous PTV, although only in one or two individuals from the studied population. These genes included eight genes which are confidently depleted for PTV variation in gnomAD (Karczewski et al., 2020): DSP, RYR2, COL4A1, EYA1, FBN2, MYH11, NNT, and SVEP1. The most PTV-depleted gene in this list is the collagen type IV alpha 1 chain COL4A1 with only six PTVs observed in gnomAD. The novel protein-truncating variant Arg1063Ter in this gene is observed in two participants.
We also detected 82 protein-truncating variants with worldwide maximal allele frequency below 0.1% as reported by VEP and refer to them as rare PTVs (Supplementary Table 3). Twenty-three of them (28%) are reported in ClinVar as VUS or variants with conflicting interpretation. Most of the rare PTV alleles are rare in the Ivanovo population as well, with only six seen in 3–6 individuals and one (rs201068740) in 14 individuals, all heterozygous.
For the 16 KP variants harbored by three or more individuals, we compared the allele frequencies in our population with gnomAD v.2.1.1 non-Finnish Europeans (NFE). Since three or more copies of an allele correspond to population frequencies roughly equal or exceeding 0.1% in our sample with 1,685 individuals, we expected that KP variants observed in the Ivanovo population more frequently than in NFE would be present among such variants. Indeed, we found two dominant and eight recessive pathogenic variants with allele frequencies 6.7–67.2 times greater than in the NFE aggregated genomes and exomes allele counts (Table 5). In each case, the significance of the observed frequency difference was validated with the Fisher’s test on direct allele counts. The variants were accepted if the corresponding P-values did not exceed 0.05 after Benjamini–Hochberg correction (Supplementary Table 4).
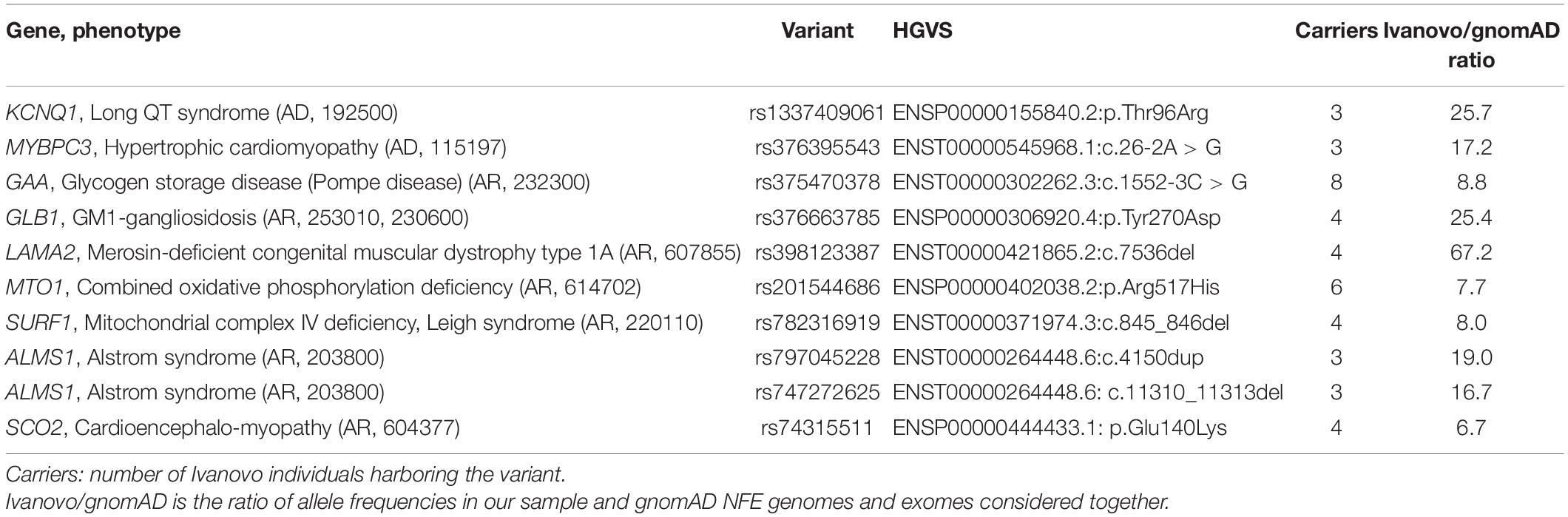
Table 5. KP variants with frequencies in the Ivanovo population significantly exceeding the NFE.
Missense substitution Thr96Arg in KCNQ1 (dbSNP: rs1337409061) was recently submitted to ClinVar as likely pathogenic and associated with long QT syndrome (ClinVar id: VCV000983021.1). This allele was observed in four copies only in non-Finnish Europeans with the frequency of 3.4 × 10–5 in that population. With three harboring participants, the frequency in the Ivanovo population equals 8.9 × 10–4 with lower and upper 95% confidence interval (CI) margins 2.0 × 10–4 and 2.6 × 10–3, respectively. The Thr96Arg substitution, however, was classified by VarSome as VUS, had no supporting evidence for pathogenicity in ClinVar, and was not observed among the variants discovered in 9 and 53 unrelated Russian families with the long QT syndrome (Polyak et al., 2016; Maltese et al., 2017). The presence of this variant in the general Russian population without any prevalence in patients may suggest that its pathogenicity needs to be confirmed by future independent submissions.
Pathogenic splice acceptor variant c.26-2A > G in MYBPC3 (rs376395543) is associated with hypertrophic cardiomyopathy (VCV000042644.10) and is observed in six copies in non-Finnish Europeans (frequency 5.2 × 10–5) and one allele copy in Latino Americans. This variant was observed in three individuals in our set, with population frequency estimated as 8.9 × 10–4 (CI from 2.0 × 10–4 to 2.6 × 10–3), not observed in NWR exomes and classified as pathogenic by VarSome.
Table 5 also contains eight pathogenic recessive variants which were significantly overrepresented in the Ivanovo population compared to gnomAD NFE. The most frequent one harbored by eight participants in our study was the acid alpha-glucosidase GAA intronic variant c.1552-3C > G (rs375470378) associated with glycogen storage disease type II, also known as Pompe disease. This intronic variant is reported in ClinVar as pathogenic or likely pathogenic by multiple submitters (VCV000419722.9) and likely pathogenic by VarSome. The observed frequency in the Ivanovo population (0.24%) significantly exceeds the NFE (0.03%) and other population frequencies, with the exception of the Estonian population where the observed frequency is 3/4480 (0.07%) with marginally significant difference from the Ivanovo population (Fisher’s test P = 0.045) likely due to modest study sample sizes. The northwest Russia frequency is estimated as 0.14%, suggesting that this variant is mostly prevalent in Northern Europe. Glycogen storage disease type II (Pompe disease) is an autosomal recessive metabolic disorder which damages muscle and nerve cells and results from the accumulation of glycogen in the lysosome due to the deficiency of the alpha-glucosidase enzyme. The worldwide prevalence of this disease is in the range 1:14,000 to 1:300,000 (Van der Ploeg and Reuser, 2008), with no estimates for Russia available (Semyachkina et al., 2014). It was also hypothesized that the overwhelming majority of Pompe disease cases in Russia may not be diagnosed (Nikitin, 2016). By combining allele counts in our study and the NWR, we calculated the frequency of homozygous carriers of the disease-causing GAA variant rs375470378 as 1:226,000 which is the lower estimate of Pompe disease prevalence in Russia due to the existence of other pathogenic alleles.
The largest frequency difference between NFE and our sample was observed for the alpha-2 laminin LAMA2 frameshift deletion c.7536delC (rs398123387) associated with merosin-deficient congenital muscular dystrophy type 1A (MDC1A) and segregating in the Ivanovo population in four copies (0.11%), which is approximately 67 times greater than the NFE frequency of 0.0017%. This variant was reported earlier in a single Russian patient (Dadali et al., 2010) and later in 21% of all affected chromosomes in the sample of 29 unrelated MDC1A patients, suggesting that this is the most abundant disease-causing LAMA2 allele in the Russian population (Milovidova et al., 2018). Based on the analysis of four microsatellite markers in the LAMA2 region of chromosome 6, the authors suggested that this mutation belongs to a founder haplotype spanning at least 3.2 cM. Our data seem to confirm this assumption and suggest the frequency range (0.11% with CI from 0.03 to 0.30%) for this variant in the unaffected population.
The frameshift variant c.845_846del in the Cytochrome C oxidase assembly Factor SURF1 (rs782316919) is associated with Leigh syndrome due to mitochondrial complex IV deficiency (VCV000012770.12) and has been reported earlier as the most prevalent SURF1 disease allele among Russian, Ukrainian, and Polish patients (Piekutowska-Abramczuk et al., 2009; Tsygankova et al., 2010). Our frequency estimate of 0.11% is comparable with those given earlier for the NWR and Poland (0.22 and 0.28%, respectively) and is by order of magnitude higher than that in the NFE (0.014%), confirming the hypothesis of the Eastern European origin of this variant (Piekutowska-Abramczuk et al., 2009).
The missense variant Arg517His in the MTO1 gene associated with combined oxidative phosphorylation deficiency (rs201544686, VCV000089037.4) and c.4150dup duplication in the ALMS1 gene associated with the Alstrom syndrome (rs797045228, VCV000210127.8) were reported earlier among 14 known pathogenic variants most prevalent in NWR (Barbitoff et al., 2019). It is noteworthy that we discovered another pathogenic variant in ALMS1 (rs747272625, VCV000550797.4) detected in three other participants and overrepresented in the Ivanovo population compared to NFE (0.09% vs. 0.005%). The missense Tyr270Asp variant in GLB1 is associated with GM1-gangliosidosis (rs376663785, VCV000284172.5) and was observed in three copies in the Ivanovo population. Finally, the missense substitution Glu140Lys in SCO2 is associated with cardioencephalomyopathy, also known as mitochondrial complex IV deficiency (rs74315511, VCV000005681.11). The observed Ivanovo frequency for this allele (0.0012) is 6.7 greater than the overall NFE frequency (0.00017) and most close to that of the Estonian subset of NFE (0.0008) and NWR (0.0007), suggesting the putative geographical origin of this variant.
We analyzed the available clinical data of participants with detected pathogenic/likely pathogenic variants or novel protein-truncating variants in 28 of ACMG59 genes and other genes with autosomal dominant inheritance. For the ACMG59 genes, we also evaluated phenotypes in the carriers of rare protein-truncating variants (VEP MAX_AF < 0.1%). Variants subject to carrier evaluation are marked in Supplementary Table 3; the results are presented in Table 6. For the first group that contained 17 KP variants in 23 carriers, we found five individuals with matching phenotypes: prolonged QTc interval (Arg366Trp in KCNQ1 and Arg858His in CACNA1C), hypertriglyceridemia (Gln313Ter in APOA5), CAD (Pro279Leu in MEF2A), and cerebral infarction (Arg153Cys in NOTCH3).

Table 6. Variants with confirmed phenotypes.
The second group, novel PTVs, comprised 27 variants in 27 carriers and yielded four individuals: three with hypobetalipoproteinemia, all with various novel truncating mutations in APOB, and one individual with hypertrophic cardiomyopathy and frameshift deletion in MYH7 (Table 6). Various truncated forms of APOB have been found earlier to segregate with the familial hypobetalipoproteinemia phenotype (OMIM:615558). Missense mutations are the most prevalent cardiomyopathy-related pathogenic mechanism in MYH7; however, ClinVar reports 20 cases of pathogenic and likely pathogenic truncating variants with various degrees of confidence.
Finally, nine rare PTVs in the ACMG59 genes were harbored by 14 carriers, two of which displayed expected phenotypes: myopathy (frameshift deletion in RYR1) and prolonged QTc interval (frameshift insertion in KCNQ1). We also checked 17 carriers of 18 novel truncating variants in non-ACMG59 genes and did not discover any remarkable genotype–phenotype associations.
Out of the total 11 genotype–phenotype associations described above, 7 are in the ACMG59 genes: 1 among KP variants, 4 among novel PTVs, and 2 among the rare PTVs. Taking in view that the total numbers of carriers checked in these three groups were 12, 10, and 14, respectively (Supplementary Table 3), we estimated the overall success rate of phenotype evaluation in the ACMG59 genes as 19% (=7/36). It is worth noting that the highest phenotype confirmation rate (40%) was observed in the subset of novel PTVs, thus emphasizing the importance of studying this class of variation by deeper sequencing the individuals from the Russian population.
In the course of the analysis of the study endpoints, we found a female participant who died of heart failure due to dilated cardiomyopathy. The manual review of rare candidate variants harbored by this individual discovered the singleton missense variant Thr891Met in the FLNC gene (rs766023596). This substitution was submitted to ClinVar (VCV000855393.2) by a single submitter as VUS associated with this type of cardiomyopathy (Cui et al., 2018). Our observation provides evidence suggesting the pathogenicity of this variant.
The overwhelming majority of variants detected in our study in three or more copies were known and shared with other populations. The alternative allele frequencies of these variants were highly correlated with non-Finnish European population. This is in agreement with the earlier observation that in the case of a smaller sample set of 694 individuals from the northwest region of Russia, the majority of disease alleles were shared between Russian and European populations (Barbitoff et al., 2019). However, with the sample size of 1,685 individuals, we detected a considerable fraction (11.4%) of novel variants not present in the dbSNP and not reported earlier in the Russian population. Most such variants were singletons and doubletons in our sample, with the exceptions discussed above: only 26 novel alleles were observed in three-to-seven copies, with no protein-truncating variants among them. Our results therefore suggest that virtually no “private” alleles specific for the Russian population reach the frequencies above 0.1–0.2%
The fraction of novel variants was highest among the protein-truncating ones: out of 218 PTVs discovered in our dataset, 70 are novel and, with one exception, not reported before in the Russian population. This effect can be explained by the purifying selection acting on specific classes of variants enriched with deleterious alleles. Since rare and novel protein-truncating variants are of special interest in the context of clinical genome interpretation (DeBoever et al., 2018), our study emphasizes the importance of deeper sequencing, taking in view the size and ethnic diversity of the Russian population.
Approximately 18% of 242 targeted genes harbored known pathogenic or likely pathogenic variants reported earlier in ClinVar. Similar to the novel variants, most of disease-causing alleles were singletons or doubletons in our sample. We report nine variants with disease alleles significantly more frequent in the Ivanovo population compared to the non-Finnish Europeans. The largest frequency difference between NFE and our sample (>67x) was observed for the alpha-2 laminin LAMA2 frameshift deletion c.7536delC (rs398123387) associated with merosin-deficient congenital muscular dystrophy type 1A (MDC1A). It was suggested earlier that this deletion is the most abundant disease-causing LAMA2 mutation in the Russian population residing on the founder haplotype spanning at least 3.2 cM (Milovidova et al., 2018). We estimated the frequency range for this variant in the unaffected population as 0.11 with 95% CI range from 0.03 to 0.30%.
To the best of our knowledge, this is the first report of the spectrum of known and putative pathogenic variants in the ACMG59 genes in the Russian population. We approximated the prevalence of known dominant pathogenic variants as 1.4% which is in agreement with the range of 1.3−2.7% suggested recently for other European populations (Haer-Wigman et al., 2019; Kuo et al., 2020; Lacaze et al., 2020; Van Hout et al., 2020). Since our target included only 28 genes of the ACMG59 list, further studies may provide more precise estimates of KP variant prevalence in these genes in the Russian population. Our results also emphasize the importance of sequencing representative cohorts (>1,000 individuals) to uncover the population-specific genetic variation of clinical relevance.
We observed the incomplete penetrance of known pathogenic variants in the ACMG59 genes: only 1 individual out of 12 with KP variants in these genes had the phenotype most likely related to the variant. When known pathogenic and novel or rare protein-truncating variants were considered together, the overall rate of confirmed phenotypes was about 19%, with maximum in the subset of novel PTVs. This is in overall agreement with the other studies: in the population cohort from the Rotterdam Study, there were 13% of the carriers of known pathogenic variants in the ACMG59 genes who had phenotype correlated with genotype (Van Rooij et al., 2020). The Rotterdam study was based on a larger sample size of 2,628 individuals and involved an in-depth analysis of the phenotypes. The Rotterdam study included persons of age 55 and above, while in our study, the age of participants was above 25, which enabled the identification of individuals with earlier disease onset.
Hou et al. (2020) reported results of the whole-genome sequencing of 1,190 self-referred volunteers with a median age of 54 years (range 20–89+ years, 70.6% European) and showed that deeper phenotyping (metabolomics, advanced imaging, and clinical laboratory tests in addition to family/medical history) significantly increased the concordance between genetic and phenotypic data. Genotype–phenotype associations were identified in 66.5% of the participants with pathogenic or likely pathogenic variants, not restricted to the ACMG59 genes. Overall, 44.5% of the cases were revealed through genomics and metabolomics analysis and had phenotype manifestations affecting serum metabolite levels. We hypothesize that the volume of the available clinical information is the limiting factor for our ability to observe better concordance between genotype and phenotype. In particular, results of imaging tests, which are essential components of cardiomyopathy diagnostics, were not available for all Ivanovo participants. The medical records, which usually contain more detailed clinical information about a patient, were very useful in our analysis and enabled identification of two genotype–phenotype associations in 10 variant carriers with available medical records. In order to develop a procedure for the return of secondary findings in the ACMG59 genes, we are designing an in-depth clinical examination targeted at all study participants with detected known and expected pathogenic variants in these genes and the genetic cascade screening of their families.
This work presents the analysis of targeted sequencing of the largest sample of the general Russian population to date. The observed spectrum of genetic variants in a region with predominantly Russian population is close in genetic composition to European populations, with the exception of a rare variation. We believe that our results provide a valuable reference for the clinical interpretation of genome sequencing and are the first step toward creating a comprehensive reference of genetic variability observed in the Russian Federation.
Aggregated variant frequency information can be requested from the authors. Individual genotype information cannot be made available in order to protect participant privacy.
The studies involving human participants were reviewed and approved by the independent Ethics Committee of the National Medical Research Center for Therapy and Preventive Medicine (Protocol numbers 07-03/12 from 03.07.2012). The patients/participants provided their written informed consent to participate in this study.
OD, AM, AE, SS, and VR conceived and designed the study. VR, AE, AZ, MZ, AK, and YV analyzed the data and wrote the manuscript. IE, GM, OB, SR, and MP carried out participant and clinical data management. AK, ES, OK, OS, and MD performed the target design and sequencing. All authors contributed to the article, revised the manuscript, and approved the submitted version.
This study was supported by the State assignment no. 121021100127-8. VR acknowledges support by the RFBR and DFG joint research project no 20-54-12008.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors are grateful to the participants from Ivanovo for their contributions and support of this research.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.709419/full#supplementary-material
Adzhubei, I. A., Schmidt, S., Peshkin, L., Ramensky, V. E., Gerasimova, A., Bork, P., et al. (2010). A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249. doi: 10.1038/nmeth0410-248
Amendola, L. M., Dorschner, M. O., Robertson, P. D., Salama, J. S., Hart, R., Shirts, B. H., et al. (2015). Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome Res. 25, 305–315. doi: 10.1101/gr.183483.114
Amendola, L. M., Jarvik, G. P., Leo, M. C., McLaughlin, H. M., Akkari, Y., Amaral, M. D., et al. (2016). Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the clinical sequencing exploratory research consortium. Am. J. Hum. Genet. 98, 1067–1076. doi: 10.1016/j.ajhg.2016.03.024
Anisimov, S. V., Meshkov, A. N., Glotov, A. S., Borisova, A. L., Balanovsky, O. P., Belyaev, V. E., et al. (2021). National association of biobanks and biobanking specialists: new community for promoting biobanking ideas and projects in Russia. Biopreserv. Biobank. 19, 73–82. doi: 10.1089/bio.2020.0049
Barbitoff, Y. A., Skitchenko, R. K., Poleshchuk, O. I., Shikov, A. E., Serebryakova, E. A., Nasykhova, Y. A., et al. (2019). Whole-exome sequencing provides insights into monogenic disease prevalence in Northwest Russia. Mol. Genet. Genomic Med. 7:e964. doi: 10.1002/mgg3.964
Boitsov, S. A., Chazov, E. I., Shlyakhto, E. V., Shalnova, S. A., Konradi, A. O., Karpov, Y. A., et al. (2013). Epidemiology of cardiovascular diseases in different regions of Russia (ESSE-RF). The rationale for and design of the study. Profilakticheskaya Med. 16, 25–34.
Brovkina, O. I., Shigapova, L., Chudakova, D. A., Gordiev, M. G., Enikeev, R. F., Druzhkov, M. O., et al. (2018). The ethnic-specific spectrum of germline nucleotide variants in DNA damage response and repair genes in hereditary breast and ovarian cancer patients of tatar descent. Front. Oncol. 8:421. doi: 10.3389/fonc.2018.00421
Chang, C. C., Chow, C. C., Tellier, L. C., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4:7. doi: 10.1186/s13742-015-0047-8
Chen, R., Shi, L., Hakenberg, J., Naughton, B., Sklar, P., Zhang, J., et al. (2016). Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat. Biotechnol. 34, 531–538. doi: 10.1038/nbt.3514
Cohen, J., Pertsemlidis, A., Kotowski, I. K., Graham, R., Garcia, C. K., and Hobbs, H. H. (2005). Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 37, 161–165. doi: 10.1038/ng1509
Cooper, D. N., Krawczak, M., Polychronakos, C., Tyler-Smith, C., and Kehrer-Sawatzki, H. (2013). Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum. Genet. 132, 1077–1130. doi: 10.1007/s00439-013-1331-2
Cui, H., Wang, J., Zhang, C., Wu, G., Zhu, C., Tang, B., et al. (2018). Mutation profile of FLNC gene and its prognostic relevance in patients with hypertrophic cardiomyopathy. Mol. Genet. Genomic Med. 6, 1104–1113. doi: 10.1002/mgg3.488
Dadali, E. L., Rudenskaia, G. E., Shchagina, O. A., Tiburkova, T. B., Sukhorukov, V. S., Kharlamov, D. A., et al. (2010). [Merosin-deficient congenital muscular dystrophy]. Z. Nevrol. Psikhiatrii 110, 83–89.
DeBoever, C., Tanigawa, Y., Lindholm, M. E., McInnes, G., Lavertu, A., Ingelsson, E., et al. (2018). Medical relevance of protein-truncating variants across 337,205 individuals in the UK Biobank study. Nat. Commun. 9:1612. doi: 10.1038/s41467-018-03910-9
Dewey, F. E., Grove, M. E., Pan, C., Goldstein, B. A., Bernstein, J. A., Chaib, H., et al. (2014). Clinical interpretation and implications of whole-genome sequencing. JAMA 311:1035. doi: 10.1001/jama.2014.1717
Dewey, F. E., Murray, M. F., Overton, J. D., Habegger, L., Leader, J. B., Fetterolf, S. N., et al. (2016). Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 354:aaf6814. doi: 10.1126/science.aaf6814
Dorschner, M. O., Amendola, L. M., Turner, E. H., Robertson, P. D., Shirts, B. H., Gallego, C. J., et al. (2013). Actionable, pathogenic incidental findings in 1,000 participants’ exomes. Am. J. Hum. Genet. 93, 631–640. doi: 10.1016/j.ajhg.2013.08.006
Green, R. C., Berg, J. S., Grody, W. W., Kalia, S. S., Korf, B. R., Martin, C. L., et al. (2013). ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 15, 565–574. doi: 10.1038/gim.2013.73
Haer-Wigman, L., van der Schoot, V., Feenstra, I., Vulto-van Silfhout, A. T., Gilissen, C., Brunner, H. G., et al. (2019). 1 in 38 individuals at risk of a dominant medically actionable disease. Eur. J. Hum. Genet. 27, 325–330. doi: 10.1038/s41431-018-0284-2
Hou, Y.-C. C., Yu, H.-C., Martin, R., Cirulli, E. T., Schenker-Ahmed, N. M., Hicks, M., et al. (2020). Precision medicine integrating whole-genome sequencing, comprehensive metabolomics, and advanced imaging. Proc. Natl. Acad. Sci. U.S.A. 117, 3053–3062. doi: 10.1073/pnas.1909378117
Jain, A., Gandhi, S., Koshy, R., and Scaria, V. (2018). Incidental and clinically actionable genetic variants in 1005 whole exomes and genomes from Qatar. Mol. Genet. Genomics 293, 919–929. doi: 10.1007/s00438-018-1431-8
Kalia, S. S., Adelman, K., Bale, S. J., Chung, W. K., Eng, C., Evans, J. P., et al. (2017). Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 19, 249–255. doi: 10.1038/gim.2016.190
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443. doi: 10.1038/s41586-020-2308-7
Kiseleva, A. V., Klimushina, M. V., Sotnikova, E. A., Divashuk, M. G., Ershova, A. I., Skirko, O. P., et al. (2020). A data-driven approach to carrier screening for common recessive diseases. J. Pers. Med. 10:140. doi: 10.3390/jpm10030140
Kopanos, C., Tsiolkas, V., Kouris, A., Chapple, C. E., Albarca Aguilera, M., Meyer, R., et al. (2019). VarSome: the human genomic variant search engine. Bioinformatics 35, 1978–1980. doi: 10.1093/bioinformatics/bty897
Kulikova, O., Brodehl, A., Kiseleva, A., Myasnikov, R., Meshkov, A., Stanasiuk, C., et al. (2021). The desmin (DES) mutation p.A337P is associated with left-ventricular non-compaction cardiomyopathy. Genes 12:121. doi: 10.3390/genes12010121
Kuo, C., Hwu, W., Chien, Y., Hsu, C., Hung, M., Lin, I., et al. (2020). Frequency and spectrum of actionable pathogenic secondary findings in Taiwanese exomes. Mol. Genet. Genomic Med. 8:e1455. doi: 10.1002/mgg3.1455
Kwak, S. H., Chae, J., Choi, S., Kim, M. J., Choi, M., Chae, J.-H., et al. (2017). Findings of a 1303 Korean whole-exome sequencing study. Exp. Mol. Med. 49:e356. doi: 10.1038/emm.2017.142
Lacaze, P., Sebra, R., Riaz, M., Tiller, J., Revote, J., Phung, J., et al. (2020). Medically actionable pathogenic variants in a population of 13,131 healthy elderly individuals. Genet. Med. 22, 1883–1886. doi: 10.1038/s41436-020-0881-7
Landrum, M. J., Lee, J. M., Benson, M., Brown, G., Chao, C., Chitipiralla, S., et al. (2016). ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–D868. doi: 10.1093/nar/gkv1222
Lek, M., Karczewski, K. J., Minikel, E. V., Samocha, K. E., Banks, E., Fennell, T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536, 285–291. doi: 10.1038/nature19057
Mallick, S., Li, H., Lipson, M., Mathieson, I., Gymrek, M., Racimo, F., et al. (2016). The simons genome diversity project: 300 genomes from 142 diverse populations. Nature 538, 201–206. doi: 10.1038/nature18964
Maltese, P. E., Orlova, N., Krasikova, E., Emelyanchik, E., Cheremisina, A., Kuscaeva, A., et al. (2017). Gene-targeted analysis of clinically diagnosed long QT Russian families. Int. Heart J. 58, 81–87. doi: 10.1536/ihj.16-133
Marakhonov, A. V., Brodehl, A., Myasnikov, R. P., Sparber, P. A., Kiseleva, A. V., Kulikova, O. V., et al. (2019). Noncompaction cardiomyopathy is caused by a novel in-frame desmin (DES) deletion mutation within the 1A coiled-coil rod segment leading to a severe filament assembly defect. Hum. Mutat. 40, 734–741. doi: 10.1002/humu.23747
McLaren, W., Gil, L., Hunt, S. E., Riat, H. S., Ritchie, G. R. S., Thormann, A., et al. (2016). The ensembl variant effect predictor. Genome Biol. 17:122. doi: 10.1186/s13059-016-0974-4
Meshkov, A., Ershova, A., Kiseleva, A., Zotova, E., Sotnikova, E., Petukhova, A., et al. (2021). The LDLR, APOB, and PCSK9 variants of index patients with familial hypercholesterolemia in Russia. Genes 12:66. doi: 10.3390/genes12010066
Milovidova, T. B., Bulach, M. V., Schagina, O. A., and Polyakov, A. V. (2018). Molecular genetic analysis of congenital merozin-negative muscular dystrophy in Russia. Med. Genet. 17, 38–45.
Miroshnikova, V., Romanova, O., Ivanova, O., Fedyakov, M., Panteleeva, A., Barbitoff, Y., et al. (2021). Identification of novel variants in the LDLR gene in Russian patients with familial hypercholesterolemia using targeted sequencing. Biomed. Rep. 14:15. doi: 10.3892/br.2020.1391
Ng, P. C., and Henikoff, S. (2006). Predicting the effects of amino acid substitutions on protein function. Annu. Rev. Genomics Hum. Genet. 7, 61–80. doi: 10.1146/annurev.genom.7.080505.115630
Nikitin, S. S. (2016). Resolution of the 3rd Russian panel of experts in diagnostics and treatment of Pompe disease. Neuromuscul. Dis. 6, 89–90.
Nykamp, K., Anderson, M., Powers, M., Garcia, J., Herrera, B., Ho, Y.-Y., et al. (2017). Sherloc: a comprehensive refinement of the ACMG–AMP variant classification criteria. Genet. Med. 19, 1105–1117. doi: 10.1038/gim.2017.37
Petrova, N. V., Kashirskaya, N. Y., Vasilyeva, T. A., Kondratyeva, E. I., Zhekaite, E. K., Voronkova, A. Y., et al. (2020). Analysis of CFTR mutation spectrum in ethnic Russian cystic fibrosis patients. Genes 11:554. doi: 10.3390/genes11050554
Piekutowska-Abramczuk, D., Popowska, E., Pronicki, M., Karczmarewicz, E., Tylek-Lemanska, D., Sykut-Cegielska, J., et al. (2009). High prevalence of SURF1 c.845_846delCT mutation in Polish Leigh patients. Eur. J. Paediatr. Neurol. 13, 146–153. doi: 10.1016/j.ejpn.2008.03.009
Pokrovskaya, M. S., Sivakova, O. V., Efimova, I. A., Meshkov, A. N., Metelskaya, V. A., Shalnova, S. A., et al. (2019). Biobanking as a necessary tool for research in the field of personalized medicine in the scientific medical center. Per. Med. 16, 501–509. doi: 10.2217/pme-2019-0049
Polyak, M. E., Ivanova, E. A., Polyakov, A. V., and Zaklyazminskaya, E. V. (2016). Mutation spectrum of the gene KCNQ1 in russian patients with long QT syndrome. Russ. J. Cardiol. 10, 15–20. doi: 10.15829/1560-4071-2016-10-15-20
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–423. doi: 10.1038/gim.2015.30
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi: 10.1038/nbt.1754
Semyachkina, A. N., Sukhorukov, V. S., Bukina, T. M., Yablonskaya, M. I., Merkuryeva, E. S., Kharabadze, M. N., et al. (2014). Glycogen storage disease type II (Pompe disease) in children. Ross. Vestn. Perinatol. Pediatr. Russian Bull. Perinatol. Pediatr. 59, 48–55.
Shah, N., Hou, Y.-C. C., Yu, H.-C., Sainger, R., Caskey, C. T., Venter, J. C., et al. (2018). Identification of misclassified clinvar variants via disease population prevalence. Am. J. Hum. Genet. 102, 609–619. doi: 10.1016/j.ajhg.2018.02.019
Sherry, S. T., Ward, M. H., Kholodov, M., Baker, J., Phan, L., Smigielski, E. M., et al. (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 29, 308–311. doi: 10.1093/nar/29.1.308
Shestak, A. G., Bukaeva, A. A., Saber, S., and Zaklyazminskaya, E. V. (2021). Allelic dropout is a common phenomenon that reduces the diagnostic yield of PCR-based sequencing of targeted gene panels. Front. Genet. 12:337. doi: 10.3389/fgene.2021.620337
Solodskikh, S. A., Panevina, A. V., Gryaznova, M. V., Gureev, A. P., Serzhantova, O. V., Mikhailov, A. A., et al. (2019). Targeted sequencing to discover germline variants in the BRCA1 and BRCA2 genes in a Russian population and their association with breast cancer risk. Mutat. Res. Mol. Mech. Mutagen. 813, 51–57. doi: 10.1016/j.mrfmmm.2018.12.005
Tennessen, J. A., Bigham, A. W., O′Connor, T. D., Fu, W., Kenny, E. E., Gravel, S., et al. (2012). Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337, 64–69. doi: 10.1126/science.1219240
Tsygankova, P. G., Mikhaĭlova, S. V., Zakharova, E. I., Pichkur, N. A., Il′ina, E. S., Nikolaeva, E. A., et al. (2010). Syndrome Leigh caused by mutations in the SURF1 gene: clinical and molecular-genetic characteristics. Z. Nevrol. Psikhiatrii 110, 25–32.
Van der Ploeg, A. T., and Reuser, A. J. (2008). Pompe’s disease. Lancet 372, 1342–1353. doi: 10.1016/S0140-6736(08)61555-X
Van Hout, C. V., Tachmazidou, I., Backman, J. D., Hoffman, J. D., Liu, D., Pandey, A. K., et al. (2020). Exome sequencing and characterization of 49,960 individuals in the UK Biobank. Nature 586, 749–756. doi: 10.1038/s41586-020-2853-0
Van Rooij, J., Arp, P., Broer, L., Verlouw, J., van Rooij, F., Kraaij, R., et al. (2020). Reduced penetrance of pathogenic ACMG variants in a deeply phenotyped cohort study and evaluation of ClinVar classification over time. Genet. Med. 22, 1812–1820. doi: 10.1038/s41436-020-0900-8
Wong, E. H. M., Khrunin, A., Nichols, L., Pushkarev, D., Khokhrin, D., Verbenko, D., et al. (2017). Reconstructing genetic history of Siberian and Northeastern European populations. Genome Res. 27, 1–14. doi: 10.1101/gr.202945.115
Wright, C. F., West, B., Tuke, M., Jones, S. E., Patel, K., Laver, T. W., et al. (2019). Assessing the pathogenicity, penetrance, and expressivity of putative disease-causing variants in a population setting. Am. J. Hum. Genet. 104, 275–286. doi: 10.1016/j.ajhg.2018.12.015
Zaklyazminskaya, E., Mikhailov, V., Bukaeva, A., Kotlukova, N., Povolotskaya, I., Kaimonov, V., et al. (2019). Low mutation rate in the TTN gene in paediatric patients with dilated cardiomyopathy – a pilot study. Sci. Rep. 9:16409. doi: 10.1038/s41598-019-52911-1
Keywords: genetic testing, rare variants, secondary findings, pathogenic variants, penetrance
Citation: Ramensky VE, Ershova AI, Zaicenoka M, Kiseleva AV, Zharikova AA, Vyatkin YV, Sotnikova EA, Efimova IA, Divashuk MG, Kurilova OV, Skirko OP, Muromtseva GA, Belova OA, Rachkova SA, Pokrovskaya MS, Shalnova SA, Meshkov AN and Drapkina OM (2021) Targeted Sequencing of 242 Clinically Important Genes in the Russian Population From the Ivanovo Region. Front. Genet. 12:709419. doi: 10.3389/fgene.2021.709419
Received: 13 May 2021; Accepted: 31 August 2021;
Published: 07 October 2021.
Edited by:
Tatiana V. Tatarinova, University of La Verne, United StatesReviewed by:
Meng Wu, Editas Medicine, Inc., United StatesCopyright © 2021 Ramensky, Ershova, Zaicenoka, Kiseleva, Zharikova, Vyatkin, Sotnikova, Efimova, Divashuk, Kurilova, Skirko, Muromtseva, Belova, Rachkova, Pokrovskaya, Shalnova, Meshkov and Drapkina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vasily E. Ramensky, cmFtZW5za3lAZ21haWwuY29t
†These authors have contributed equally to this work and share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.