
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 12 August 2021
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.708864
This article is part of the Research Topic Novel Insights into the Genetics of Growth Disorders View all 10 articles
 Valentina Mancioppi1
Valentina Mancioppi1 Flavia Prodam1,2,3*
Flavia Prodam1,2,3* Simona Mellone4
Simona Mellone4 Roberta Ricotti1
Roberta Ricotti1 Enza Giglione1
Enza Giglione1 Nicolino Grasso1
Nicolino Grasso1 Denise Vurchio4
Denise Vurchio4 Antonella Petri1
Antonella Petri1 Ivana Rabbone1
Ivana Rabbone1 Mara Giordano4,5
Mara Giordano4,5 Simonetta Bellone1,3
Simonetta Bellone1,3Short stature is a frequent disorder in the pediatric population and can be caused by multiple factors. In the last few years, the introduction of Next Generation Sequencing (NGS) in the molecular diagnostic workflow led to the discovery of mutations in novel genes causing short stature including heterozygous mutations in ACAN gene. It encodes for aggrecan, a primary proteoglycan component specific for the structure of the cartilage growth plate, articular and intervertebral disc. We report a novel ACAN heterozygous pathogenic variant in a family with idiopathic short stature, early-onset osteoarthritis and osteoarthritis dissecans (SSOAOD). We also performed a literature review summarizing the clinical characteristic of ACAN's patients. The probands are two Caucasian sisters with a family history of short stature and osteoarthritis dissecans. They showed dysmorphic features such as mild midface hypoplasia, brachydactyly and broad thumbs, especially the great toes. The same phenotype was presented in the mother who had had short stature and suffered from intervertebral disc disease. DNA sequencing identified a heterozygous pathogenic variation (c.4390delG p.Val1464Ter) in the sisters, with a maternal inheritance. The nonsense mutation, located on exon 12, results in premature truncation and presumed loss of protein function. In terms of treatment, our patients underwent recombinant human growth hormone replacement therapy, associated with gonadotropin releasing hormone therapy, in order to block early growth cessation and therefore reach a better final height. Our case suggests that SSOAOD ACAN related should be considered in the differential diagnosis of children with autosomal dominant short stature and family history of joints disease.
Short stature is defined as a height of at least two standard deviations below the average observed in age and sex control population (Hauer et al., 2017). It's a frequent disorder affecting 3% of the pediatric population and one of the most common causes of pediatric endocrinologist evaluation (Stavber et al., 2020). According to its etiology, short stature can be classified into primary growth disorder, secondary growth disorder and idiopathic short stature (ISS) (Xu et al., 2018). In addition to forms determined by common variants with polygenic inheritance, recent studies have highlighted that monogenic defects could be the cause of the growth disorder observed in non-syndromic children (Grunauer and Jorge, 2018; Vasques et al., 2019). More than 700 genes are associated with growth failure, with SHOX haploinsufficiency as the most frequent form, estimated to cause 2–3% of idiopathic short stature (ISS) cases (Fukami et al., 2016).
The introduction of NGS in the molecular diagnostic work-flow led to the discovery of mutations in novel genes causing short stature including heterozygous mutations in ACAN gene (ACAN, OMIM:*155760) (Sentchordi-Montané et al., 2018). ACAN is located on chromosome 15q26 and consists of 19 exons, ranging in size from 77 to 4224 bp. It encodes for aggrecan, a primary proteoglycan component specific for the structure of the cartilage growth plate, articular and intervertebral disc (Uchida et al., 2020). Aggrecan is fundamental in articular cartilage, providing the hydrated gel structure necessary for the load-bearing properties of joints (Dateki, 2017) as well as chondrocyte and bone morphogenesis (Quintos et al., 2015). The core protein consists of three globular domains (G1, G2, and G3), interglobular domain (IGD), and centrally located glycosaminoglycan attachment region (GAG), consisting of chondroitin sulfate attachment domains (CS1 and CS2) and keratan sulfate attachment domain (KS) (Figure 1). G1 domain forms interactions with hyaluronan, G2 biological function is still not known whereas the G3 domain binds to different extracellular proteoglycans (i.e., tenascin and fibulin) via its C-type lectin repeat domain (CLD) (Gkourogianni et al., 2017). ACAN biallelic mutations are related to spondyloepimetaphyseal dysplasia, aggrecan type (SEMD, OMIM#612813) (Tompson et al., 2009), while mutations at the heterozygous state can cause spondyloepiphyseal dysplasia, Kimberley type (SEDK, OMIM#608361) (Gleghorn et al., 2005), short stature associated with or without accelerated bone age (BA), early-onset osteoarthritis (OA) and/or osteoarthritis dissecans (SSOAOD, OMIM#165800) and various idiopathic short stature phenotypes (Stattin et al., 2010; Quintos et al., 2015; Tatsi et al., 2017; Van der Steen et al., 2017).
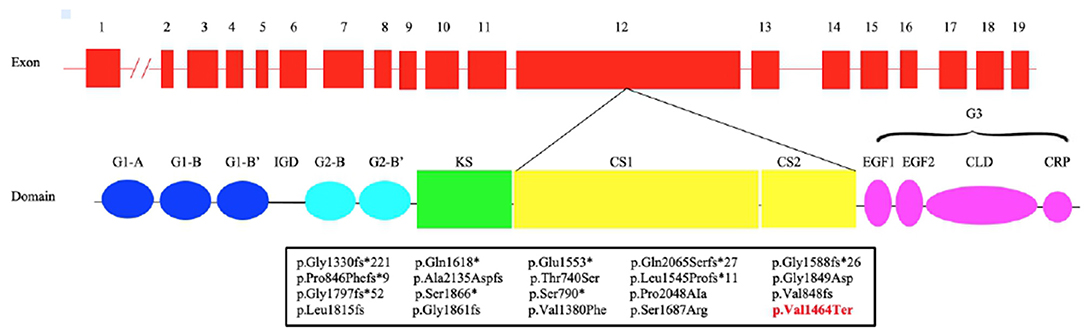
Figure 1. Structure of ACAN gene and aggrecan protein. Pathogenic variants in exon 12 (bottom) with respective predicted changes in the amino acid sequence are shown. The mutations identified in previous studies are shown in black, while our is in red. CLD, C-type lectin domain; CRP, complement regulatory protein-like domain; CS1, chondroitin sulfate 1; CS2, chondroitin sulfate 2; EGF1, 2, epidermal growth factor–like domain 1, 2; G1, globular domain 1; G2, globular domain 2; G3, globular domain 3; IGD, interglobular domain; KS, keratan sulfate.
Here we report two sisters from a family with idiopathic short stature, early-onset osteoarthritis and osteoarthritis dissecans caused by novel heterozygous ACAN variant.
The probands are two caucasian sisters (III-1 and III-2, Figures 2A,B), previously referred to another hospital for persistent short stature.
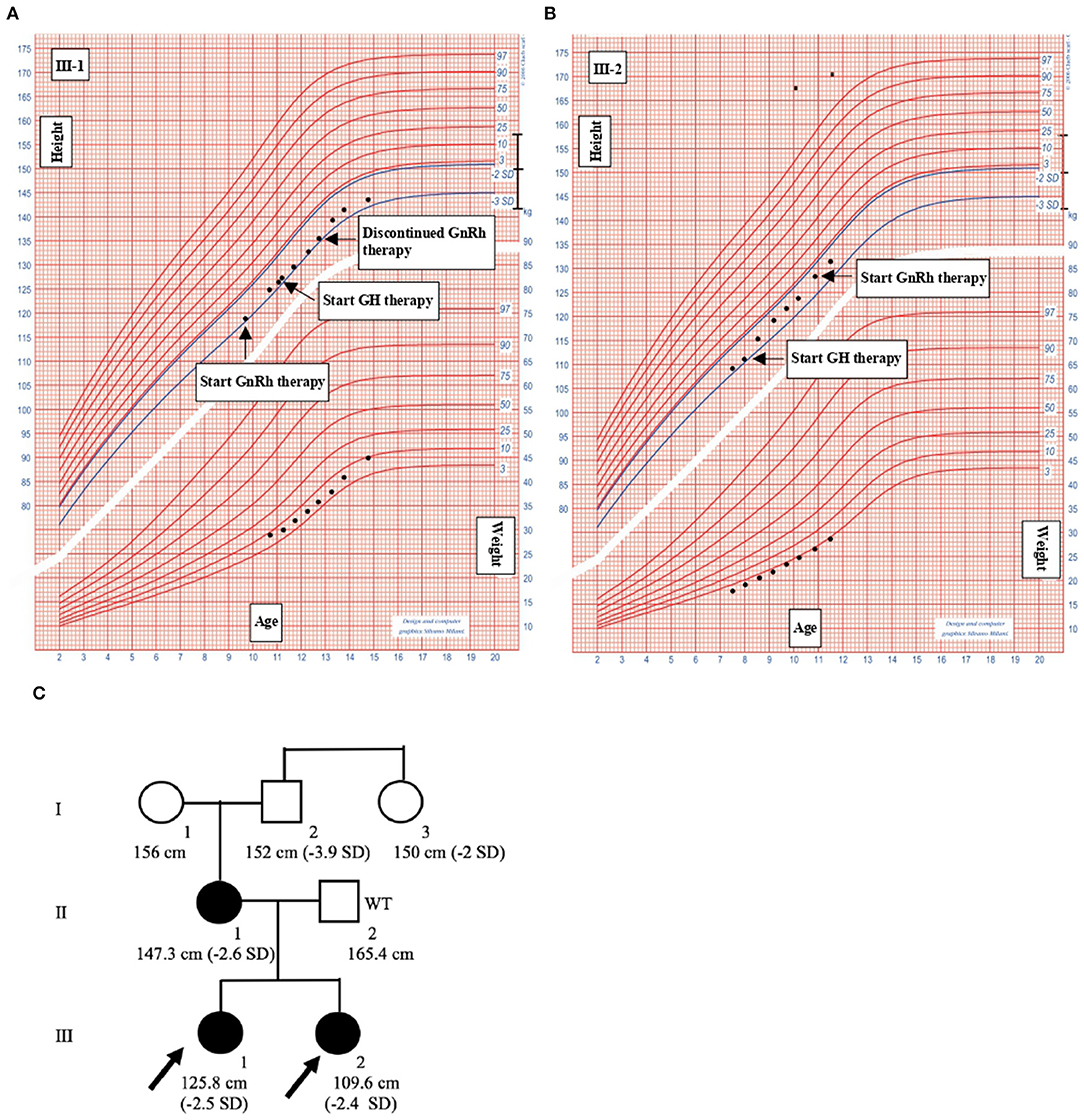
Figure 2. Clinical informations of the family members. (A) III-1. (B) III-2. Growth charts of the probands. Heights and weights are shown as black points. GH, Growth hormone; GnRH, gonadotropin-releasing hormone. (C) Pedigree of the family with familiar short stature. Shaded symbols indicate patients with stature below 2 SD; empty symbols with “WT” (Wild Type) are family members without mutations; empty symbols indicate family members who have not undergone ACAN sequencing.
III-1 was born full-term after an uncomplicated pregnancy and delivery. At birth, her weight was 3.3 kg (−0.05 SD), length of 46 cm (−2.12 SD), head circumference of 36 cm (+1.68 SD) and she presented normal psychomotor development (Table 1). Growth rate has been below normal during most of her childhood. At our first evaluation, she was 10.8 years old, her height was 125.8 cm (−2.5 SD) with a genetic target of 150.4 cm (−2 SD). The previous year her physical examination revealed bilateral breast development (Tanner stage B2) and gonadotropin releasing hormone stimulation test was performed (peak FSH value of 18.6 mU/ml, peak PH 7.3 mU/ml). She started Gonadotropin-releasing hormone (GnRH) treatment for precocious puberty with Triptorelin 11.25 mg i.m. every 4 weeks. The father's height (II-2) was 165.4 cm (−1.84 SD) while maternal height (II-1) was 147.3 cm (−2.6 SD). Maternal family history of short stature was described: the grandfather's height (I-2) was 152 cm (−3.9 SD), the grandmother's height (I-1) was 156 cm (−1.12 SD) and the height of the grandfather's sister (I-3) was 150 cm (−2 SD) (Figure 2C). They had all the same phenotype characterized by short stature, macrocephaly, short neck, barrel chest, muscle hypertrophy and severe osteoarthritis. III-1 showed dysmorphic features such as macrocephaly, frontal bossing, low-set rotated ears, flat nasal filter, brachydactyly and broad thumbs, especially the great toes. Growth hormone (GH) stimulation test (arginine) produced a peak GH value of 25.6 ng/ml. Additional laboratory and endocrine assessment revealed normal complete blood count, chemistry panel, inflammatory markers, celiac screening, epatic-renal-thyroid function test, insulin-like growth factor-1 (IGF-1) (Table 1). The hand and wrist radiography showed no characteristic signs of skeletal dysplasia, other than shortening of the 1st distal phalanx. According to the Greulich and Pyle method, her bone age (BA) was 0.6 years advanced relative to the calendar age (CA). She started recombinant human GH (rhGH) replacement therapy at a dose of 0.25 mg/kg/week when she was 11 year old. At 12.6 years of age, the treatment with GnRH was discontinued and menarche occurred at 14.5 years of age. Moreover in the last two years, she had a history of persistent pain in both knees: x-ray and magnetic resonance imaging detected multiple osteochondral lesions consistent with osteochondritis dissecans and she underwent a surgical operation. At her latest visit (age 14.7 years) her height was 143 cm (−2.3 SD) with a growth velocity of 2 cm/year (+0.6 SD).
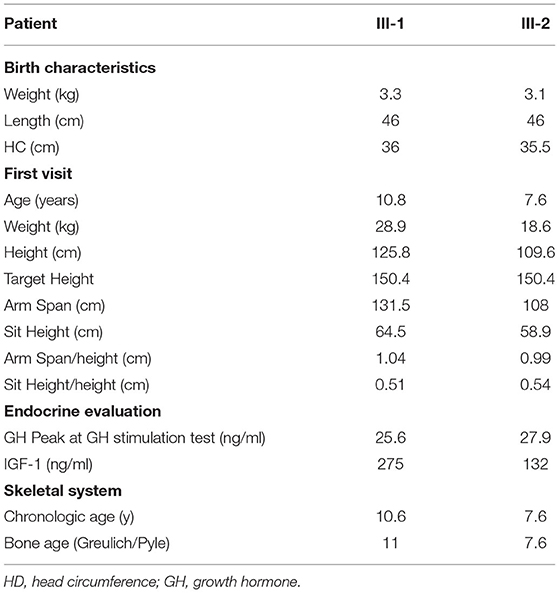
Table 1. Phenotype of our two patients with ACAN mutations.
The younger sister (III-2) was born full-term after an uncomplicated pregnancy with a birth weight of 3.1 kg (−0.85 SD), birth length of 46 cm (−2.34 SD), head circumference of 35.5 cm (+0.97), normal mental and motor development (Table 1). Short stature developed in early childhood, with appropriate weight for height. At our first visit, she was 7.6 years old, her height was 109.6 cm (−2.4 SD) and she was prepubertal. She had the same dysmorphic features as the older sister, bone age corresponding to chronological age, no signs of skeletal dysplasia. Growth hormone (GH) stimulation test (arginine) produced a peak GH value of 27.9 ng/ml. She had normal screening laboratory evaluation including complete blood count, inflammatory markers, celiac screening, epatic-renal-thyroid function test, insulin-like growth factor-1 (IGF-1) or other systemic explanation for her short stature (Table 1). At 8.2 years of age, she started GH therapy at a dose of 0.27 mg/kg/week. Thelarche occurred at approximately 11 years of age (height-−2.3 SD) and she was started on GnRH agonist (Triptorelin 11.25 mg i.m. every 4 weeks, still ongoing). She also suffered from persistent ankles' pain (especially the right), with the magnetic resonance imaging revealing mild osteochondritis dissecans. She didn't need to perform any surgical procedure but had to use bilateral ankle braces. At her latest visit (age 11.6 years) her height was 131.1 cm (−2.1 SD) with a growth rate of 5.3 cm/year (+1.4 SD).
After starting GH's therapy, both sisters were seen every 6 months for monitoring their growth and pubertal development but also for undergoing laboratory and endocrine assessment for glucose and lipid metabolism, epatic-renal-thyroid and corticotrope function test. All the evaluations were always normal; the dose adjustment for GH therapy was based on IGF-I levels. No side effects were registered for both GH and GnRH therapy. Clinical, anthropometric, and metabolic features during GH treatment are summarized in Table 2.
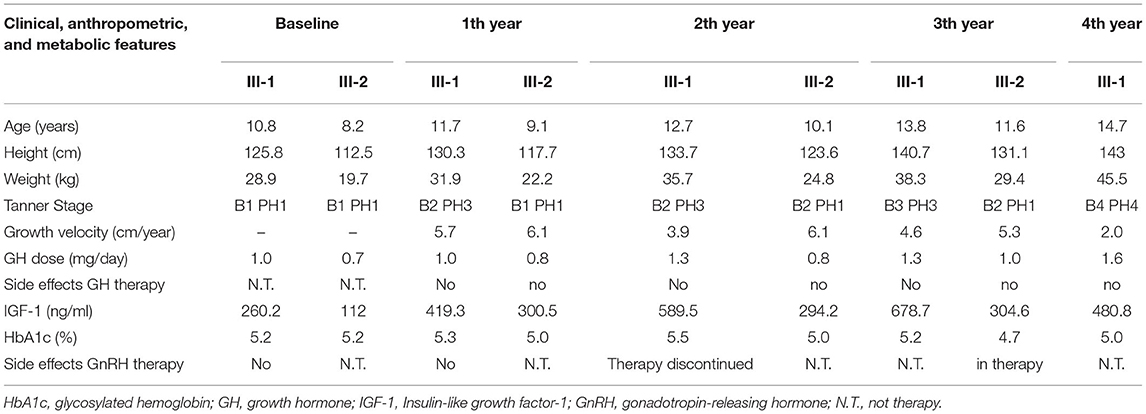
Table 2. Clinical, anthropometric, and metabolic features at baseline and during GH treatment.
The mother had the same phenotype, characterized by short stature, macrocephaly, short neck, frontal bossing, flat nasal filter, brachydactyly and broad thumbs, especially the great toes. Despite having a stature < 2.6 SD, she was never referred to an endocrinologist or didn't undergo any endocrine assessment. From 35–40 years, she started suffering from severe intervertebral disc disease but didn't need to perform any surgical procedure.
Following written informed consent, the genomic DNA of the patients was extracted from peripheral blood through the ReliaPrep Blood gDNA Miniprep System (Promega), according to the manufacturer's recommendation. The GenetiSure Dx Postnatal Array 4x180K + SNP (Agilent Technologies, USA) was used following standard protocols.
The entire coding region of the SHOX gene (exon 1–exon 6a/6b) and intron–exon boundaries were amplified by PCR and sequenced by Big Dye Terminator Kit (Applied Biosystems, Foster City, CA) through the automatic sequencer ABI PRISM 3100 Genetic Analyzer (Applied Biosystems). Search for deletions/duplications was performed by an MLPA assay using an MLPA Commercial Kit (SALSA MLPA Kit P018-G1 SHOX; MRC-Holland, Amsterdam, Netherlands) following the manufacturer's instructions.
A custom-designed NGS panel including 67 genes [A2ML1, ACAN, ANKRD11, BRAF, CBL, CHD7, COL2A1, COMP, CUL7, FGD1, FGF8, FGFR1, FGFR3, FLNB GH1, GHR, GHRH, GHRHR, GHSR, GLI2, GLI3, GNAS, HDAC6, HESX1, HMGA2, HRAS, IGF1, IGF1R, IGFALS, IHH, KAT6B, KDM6A, KRAS, LHX3, LHX4, LIG4, LZTR1, MATN3, MEK1, MEK2, NF1, NPPC, NPR2, NRAS, OTX2, PCNT, PDE3A, PDE4D, POC1A, POU1F1, PROK2, PROKR2, PROP1, PTPN11, RAF1, RIT1, SHH, SHOC2, SLC26A2, SOS1, SOS2, SOX2, SOX3, SOX9, SPRED1, STAT5B, TRIM37] involved in bone growth plate development, hypothalamic-pituitary IGF-1 axis and reported mutated in syndromic short stature was used to investigate the presence of pathogenic variants in the two sibs. An Agilent SureSelectQXT target capture method was utilized with a panel size corresponding to 1.3 Mb. The sequencing probes were designed to cover all coding exons of the 67 genes and the 20 bp flanking sequence from the intron-exon boundary. Sequencing reaction was performed using an Illumina MiSeq and MiSeq v2 300 Cycle Reagent Kits. Variant calling was performed with the SureCall v3.5 software (Agilent Technologies) and VCF files were annotated with the wANNOVAR tool. Finally post aligned, annotated, and categorized sequence data was analyzed using a personalized bioinformatics pipeline: variants were filtered by frequency, excluding those with a MAF ≥0.01 in the public databases: 1000 Genome project (https://www.internationalgenome.org/) and gnomAD (https://gnomad.broadinstitute.org/). We prioritized variants causing frameshift, stop codon, splicing or aminoacidic changes predicted as pathogenic by at least four in silico prediction software (SIFT, Polyphen-2, Mutation Taster, CADD). Classification of variants was evaluated using the consensus guidelines as set out by the American College of Medical Genetics and Genomics (ACMG) guidelines (https://www.acmg.net/ACMG). Sanger sequencing was performed to confirm the presence or absence of these mutations in all the family members.
In the two sisters (III-1 and III-2) the aCGH revealed a normal female Karyotype and SHOX molecular analysis excluded any form of SHOX-haploinsufficiency. A targeted NGS panel containing 67 genes involved in syndromic and non-syndromic short stature detected in both sibs the presence of a heterozygous pathogenic variation in exon 12, namely c.4390delG that causes the changing of the reading frame from GTA encoding Valine at position 464 to stop codon (p.Val1464Ter). The variant, validated by Sanger sequencing, was inherited from the short stature mother (Figure 3).
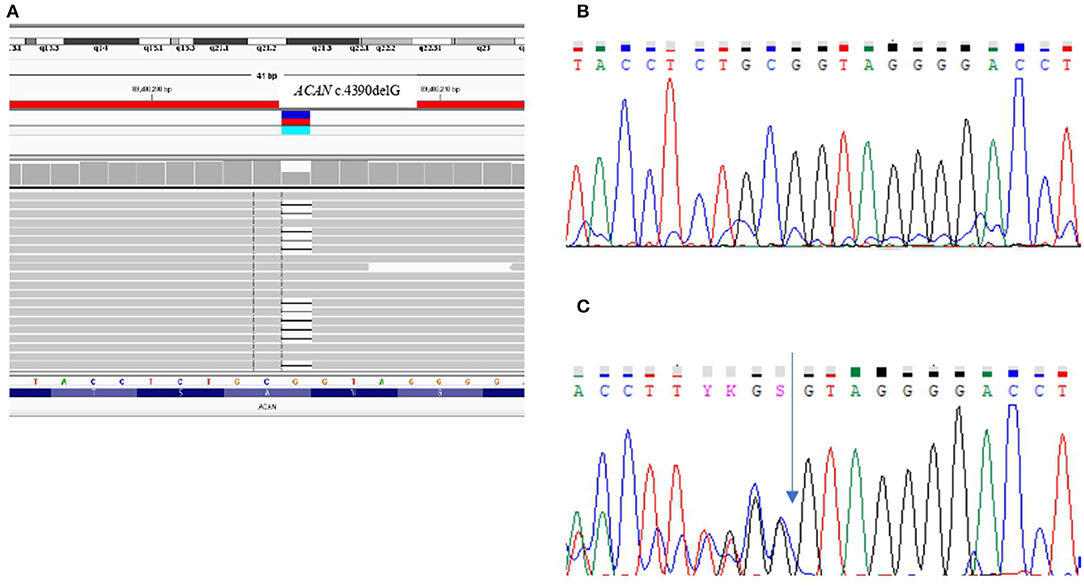
Figure 3. Genetic analysis. (A) NGS file and (B,C) Sanger sequencing chromatograms reporting the wild-type (B) and the c.4390delG variant identified in the patients (C) in exon 12 (NM_013227) of the ACAN gene.
The etiology of short stature is complex and can be related to multiple factors such as GH deficiency, ISS, genetic inheritance, malnutrition, chronic disease (such as hypothyroidism, chronic liver and kidney diseases), constitutional delay of growth and puberty, intrauterine growth retardation, skeletal dysplasia, psychosocial stress and environmental factors (Liang et al., 2020; Wang et al., 2020). Based on new gene sequencing techniques, it is now possible to identify the cause of some short stature cases previously diagnosed as ISS.
We report a novel heterozygous mutation in ACAN (c.4390delG; p.Val1464Ter), located in exon 12, in a family with short stature associated with early-onset osteoarthritis and osteoarthritis dissecans (SSOAOD). Nilsson et al. (2014) reported for the first time that heterozygous ACAN mutations could be the cause of short stature and accelerated bone age (Nilsson et al., 2014). Since than an increasing number of ACAN pathogenic variants have been identified. To date, 93 mutations in this gene have been reported in HGMD (http://www.hgmd.cf.ac.uk/ac/index.php), with the majority of the variants (81/93) associated with ISS. Among them, 87.9% were inherited whereas the remaining arose de novo. Up to now 18 different mutations in exon 12 have been described (Figure 1; Table 3) (Gleghorn et al., 2005; Quintos et al., 2015; Gkourogianni et al., 2017; Hattori et al., 2017; Hauer et al., 2017; Tatsi et al., 2017; Van der Steen et al., 2017; Sentchordi-Montané et al., 2018; Zeng et al., 2018; Fukuhara et al., 2019; Uchida et al., 2020; Lin et al., 2021). This represents the exon with the highest number of mutations, accounting for the 19% of ACAN alterations (Liang et al., 2020; Lin et al., 2021). Exon 12 encodes for the chondroitin sulfate attachment domains (CS1 and CS2) and along with exon 11, for the keratan sulfate attachment domain (KS). Thanks to the sulfation of these domains (KS, CS1 and CS2) and the aggregation with hyaluronan, a fixed negative charge is generated (Dateki, 2017). This process is fundamental to cartilage because it attracts ions and water molecules, letting the cartilage bear the high mechanical load found in the skeletal joint (Gibson and Briggs, 2016). All heterozygous aggrecan mutations known so far cause short stature of similar severity, indicating that they similarly affect growth plate cartilage. Nevertheless, not all of them cause involve the articular cartilage and, therefore, determine early-onset osteoarthritis. Early-frameshift or splice-site mutations are likely to cause early truncation of the protein and may therefore functionally represent haploinsufficiency of ACAN. Patients with these types of mutations have short stature but no evidence of early-onset osteoarthritis, suggesting that ACAN haploinsufficiency primarily affects growth plate cartilage. Previously reported familial osteochondritis dissecans family and our family have missense or non-sence mutations in the C-type lectin domain and articular disease, suggesting that the presence of aggrecan protein with a dysfunctional C-type lectin domain causes a more severe phenotype affecting both growth plate and articular cartilage function (Nilsson et al., 2014). The nonsense mutation found in the present patients results in premature truncation and presumed loss of protein function. Ten frameshift variants, four missense variants and four nonsense variants (Table 3) had been previously identified in exon 12, causing a premature termination codon and the loss of the C-terminal globular domains (G3) (Figure 1; Table 3). G3 is fundamental for the proper protein function, contributing to the interaction between the aggrecan molecule and various components of the extracellular matrix (ECM) (Aspberg, 2012; Quintos et al., 2015). Nevertheless, numerous authors reiterate the importance of performing additional studies to determine whether these variants cause mRNA nonsense-mediated decay (NMD) or allow translation of a truncated aggrecan protein into the cartilage extracellular matrix (Gibson and Briggs, 2016; Stavber et al., 2020). To date, the few functional studies on the consequences of mutations in the G3 domain conducted showed the secretion of a truncated aggrecan, ruining cartilage structure and function with a dominant-negative effect (Tompson et al., 2009; Stattin et al., 2010; Gibson and Briggs, 2016).
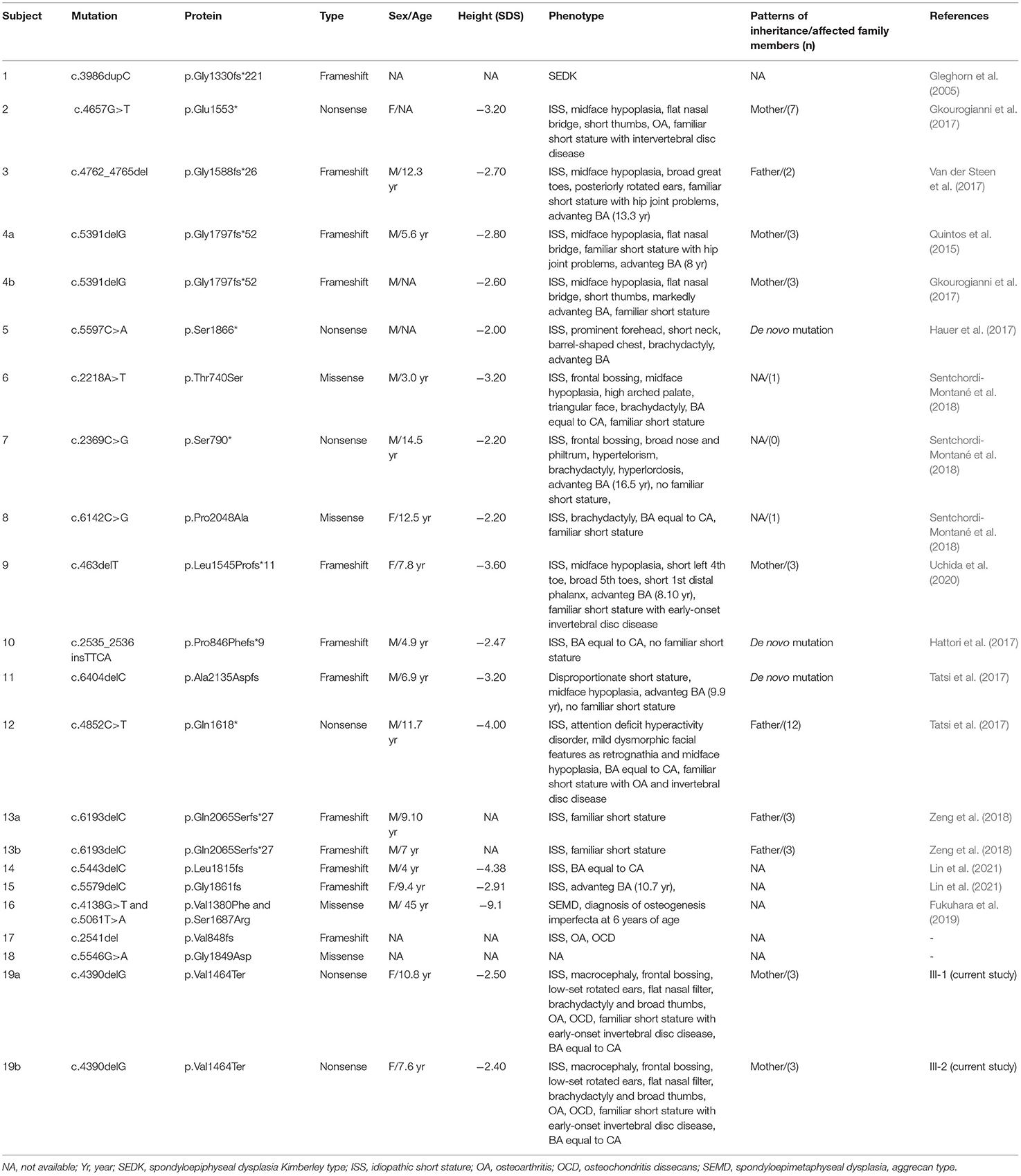
Table 3. Clinical findings of patients with ACAN mutation in exon 12 in this study and the literatures.
The heterozygous variant detected in III-1 and III-2 was maternally inherited and their similar phenotype is consistent with that described in previous reports (Table 3), with short stature below −2.4/−2.5 SD, prognathia, flat nasal bridge, midfacial hypoplasia, posteriorly rotated ears, broad forehead, relative macrocephaly, brachydactyly, broad great toes, short thumbs, lordosis, genu valgum, joint problems (Dateki, 2017; Dateki et al., 2017; Hu et al., 2017; Van der Steen et al., 2017; Kim et al., 2020; Nilsson, 2020). The same characteristics can be detected, also, among patients carrying ACAN mutations located on other domains, underlying the absence of a genotype-phenotype correlation. The three most frequent features described among all ACAN's patients were short neck, mild midface hypoplasia (frontal bossing, flat nasal bridge, and long philtrum), and brachydactyly, secondly osteoarthritis, spine malformation, short limbs, thoracic deformity (Liang et al., 2020). Both the sibs developed early-onset osteoarthritis (II-1 in the knee, while the III-2 in the ankle) and, subsequently, osteochondritis dissecans. While these clinical presentations are relatively common in patients carrying ACAN mutations, in the other cases with truncating mutations in the G3 domain there were only two cases of early-onset osteoarthritis (Gleghorn et al., 2005; Gkourogianni et al., 2017) but no other osteochondritis dissecans. The two sibs presented joint problems at a young age (between 10–12 years), in contrast to previous reports where the common start was late adolescence or even later (Gkourogianni et al., 2017). Their affected mother (II-1) suffered from severe intervertebral disc disease, starting at 35–40 years, indicating wide phenotype variability within this family.
In terms of treatment, although the two sisters didn't develop precocious puberty, they underwent gonadotropin releasing hormone (GnRH) therapy, to block early growth cessation and therefore reached a better final height. Several studies evaluated the effectiveness of combined GH and GnRH analog treatment for achieving a proper final height in patients with ACAN mutations (Dateki, 2017; Van der Steen et al., 2017; Zeng et al., 2018; Stavber et al., 2020). Gkourogianni et al. (2017) found that the mean height SD for ACAN adult patients previously treated with GH was −2.5, while that of untreated adult individuals was −3.0 (Gkourogianni et al., 2017). Furthermore, Van der Steen et al. (2017) reported that GH-treated adult individuals, who received in addition GnRH analog treatment for two years from puberty (mean age of GnRH treatment's start between 10–12 years, with normal timing of puberty), gain 5–8 cm at their adult height compared to their family members, with the same sex and ACAN mutation (Van der Steen et al., 2017). Stavber et al. (2020) described the cases of three children who, in addition to GH treatment, received treatment with GnRHa during puberty after 10 years of age. During the combined therapy there was a significant improvement in SD height and growth velocity increased significantly. Nevertheless, the author underlines that it's not possible to establish the efficacy of the therapy since none of the patients have already achieved the final height at the time of publication (Stavber et al., 2020). Therefore, it is still controversial if ACAN patients need to undergo GH therapy with or without GnRHa to achieve a proper final height and further studies are needed (Dateki, 2017).
In conclusion we identified a novel heterozygous ACAN mutation causing SSOAOD in the family. SSOAOD ACAN related should be considered in the differential diagnosis of children with autosomal dominant short stature and family history of joints disease, even in patients in which BA is equal to CA. Diagnosis of the condition is difficult, and genetic testing may help. As for treatment, growth therapies could be beneficial for improving affected individuals' adult height.
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
VM collected the anamnestic as well as the biochemical data for the patients. VM performed the literature search, reviewed and extracted data from the papers. MG and SM performed the genetic analysis. VM performed the figures and table designing and the manuscript writing with the assistance of MG, SB, and FP. All authors discussed the results and contributed to the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank the patients for their participation in this work.
Aspberg, A. (2012). The different roles of aggrecan interaction domains. J. Histochem. Cytochem. 60, 987–996. doi: 10.1369/0022155412464376
Dateki, S. (2017). ACAN mutations as a cause of familial short stature. Clin. Pediatr. Endocrinol. 26, 119–125. doi: 10.1297/cpe.26.119
Dateki, S., Nakatomi, A., Watanabe, S., Shimizu, H., Inoue, Y., Baba, H., et al. (2017). Identification of a novel heterozygous mutation of the Aggrecan gene in a family with idiopathic short stature and multiple intervertebral disc herniation. J. Hum. Genet. 62, 717–721. doi: 10.1038/jhg.2017.33
Fukami, M., Seki, A., and Ogata, T. (2016). SHOX Haploinsufficiency as a cause of syndromic and nonsyndromic short stature. Mol. Syndromol. 7, 3–11. doi: 10.1159/000444596
Fukuhara, Y., Cho, S. Y., Miyazaki, O., Hattori, A., Seo, J. H., Mashima, R., et al. (2019). The second report on spondyloepimetaphyseal dysplasia, aggrecan type: a milder phenotype than originally reported. Clin. Dysmorphol. 28, 26–29. doi: 10.1097/MCD.0000000000000241
Gibson, B. G., and Briggs, M. D. (2016). The aggrecanopathies; an evolving phenotypic spectrum of human genetic skeletal diseases. Orphanet J. Rare Dis. 11:86. doi: 10.1186/s13023-016-0459-2
Gkourogianni, A., Andrew, M., Tyzinski, L., Crocker, M., Douglas, J., Dunbar, N., et al. (2017). Clinical characterization of patients with autosomal dominant short stature due to aggrecan mutations. J. Clin. Endocrinol. Metab. 102, 460–469. doi: 10.1210/jc.2016-3313
Gleghorn, L., Ramesar, R., Beighton, P., and Wallis, G. (2005). A mutation in the variable repeat region of the aggrecan gene (AGC1) causes a form of spondyloepiphyseal dysplasia associated with severe, premature osteoarthritis. Am. J. Hum. Genet. 77, 484–490. doi: 10.1086/444401
Grunauer, M., and Jorge, A. A. L. (2018). Genetic short stature. Growth Horm. IGF Res. 38, 29–33. doi: 10.1016/j.ghir.2017.12.003
Hattori, A., Katoh-Fukui, Y., Nakamura, A., Matsubara, K., Kamimaki, T., Tanaka, H., et al. (2017). Next generation sequencing-based mutation screening of 86 patients with idiopathic short stature. Endocr. J. 64(10), 947–954. doi: 10.1507/endocrj.EJ17-0150
Hauer, N. N., Sticht, H., Boppudi, S., Büttner, C., Kraus, C., Trautmann, U., et al. (2017). Genetic screening confirms heterozygous mutations in ACAN as a major cause of idiopathic short stature. Sci Rep. 22:12225. doi: 10.1038/s41598-017-12465-6
Hu, X., Gui, B., Su, J., Li, H., Li, N., Yu, T., et al. (2017). Novel pathogenic ACAN variants in non-syndromic short stature patients. Clin. Chim. Acta 469, 126–129. doi: 10.1016/j.cca.2017.04.004
Kim, Y. T., Jang, K. M., Keum, C. W., Oh, S. H., and Chung, W. Y. (2020). Identification of a heterozygous ACAN mutation in a 15-year-old boy with short stature who presented with advanced bone age: a case report and review of the literature. Ann. Pediatr. Endocrinol. Metabol. 25, 272–276. doi: 10.6065/apem.1938198.099
Liang, H., Miao, H., Pan, H., Yang, H., Gong, F., Duan, L., et al. (2020). Growth-promoting therapies may be useful in short stature patients with nonspecific skeletal abnormalities caused by Acan heterozygous mutations: six Chinese cases and literature review. Endocr. Pract. 26, 1255–1268. doi: 10.4158/EP-2019-0518
Lin, L., Li, M., Luo, J., Li, P., Zhou, S., Yang, Y., et al. (2021). A high proportion of novel ACAN mutations and their prevalence in a large cohort of Chinese short stature children. J. Clin. Endocrinol. Metab. 106(7):e2711–e2719. doi: 10.1210/clinem/dgab088
Nilsson, O. (2020). Aggrecanopathies highlight the need for genetic evaluation of ISS children. Eur J. Endocrinol. 183:C9–C10. doi: 10.1530/EJE-20-0420
Nilsson, O., Guo, M. H., Dunbar, N., Popovic, J., Flynn, D., Jacobsen, C., et al. (2014). Short stature, accelerated bone maturation, and early growth cessation due to heterozygous aggrecan mutations. J. Clin. Endocrinol. Metab. 99, E1510–E1518. doi: 10.1210/jc.2014-1332
Quintos, J. B., Guo, M. H., and Dauber, A. (2015). Idiopathic short stature due to novel heterozygous mutation of the aggrecan gene. J. Pediatr. Endocrinol. Metab. 28, 927–932. doi: 10.1515/jpem-2014-0450
Sentchordi-Montané, L., Aza-Carmona, M., Benito-Sanz, S., Barreda-Bonis, A. C., Sánchez-Garre, C., Prieto-Matos, P., et al. (2018). Heterozygous aggrecan variants are associated with short stature and brachydactyly: description of 16 probands and a review of the literature. Clin. Endocrinol. (Oxf). 88, 820–829. doi: 10.1111/cen.13581
Stattin, E. L., Wiklund, F., Lindblom, K., Onnerfjord, P., Jonsson, B. A., Tegner, Y., et al. (2010). A missense mutation in the aggrecan C-type lectin domain disrupts extracellular matrix interactions and causes dominant familial osteochondritis dissecans. Am. J. Hum. Genet. 86, 126–137. doi: 10.1016/j.ajhg.2009.12.018
Stavber, L., Hovnik, T., Kotnik, P., Lovrečić, L., Kovač, J., Tesovnik, T., et al. (2020). High frequency of pathogenic ACAN variants including an intragenic deletion in selected individuals with short stature. Eur. J. Endocrinol. 182, 243–253. doi: 10.1530/EJE-19-0771
Tatsi, C., Gkourogianni, A., Mohnike, K., DeArment, D., Witchel, S., Andrade, A. C., et al. (2017). Aggrecan mutations in nonfamilial short stature and short stature without accelerated skeletal maturation. J. Endocr. Soc. 1, 1006–1011. doi: 10.1210/js.2017-00229
Tompson, S. W., Merriman, B., Funari, V. A., Fresquet, M., Lachman, R. S., Rimoin, D. L., et al. (2009). A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C-type lectin domain of aggrecan. Am. J. Hum. Genet. 84, 72–79. doi: 10.1016/j.ajhg.2008.12.001
Uchida, N., Shibata, H., Nishimura, G., and Hasegawa, T. (2020). A novel mutation in the ACAN gene in a family with autosomal dominant short stature and intervertebral disc disease. Hum. Genome Var. 7:44. doi: 10.1038/s41439-020-00132-8
Van der Steen, M., Pfundt, R., Maas, S. J. W. H., Bakker-van Waarde, W. M., Odink, R. J., and Hokken-Koelega, A. C. S. (2017). ACAN gene mutations in short children born SGA and response to growth hormone treatment. J. Clin. Endocrinol. Metab. 102, 1458–1467. doi: 10.1210/jc.2016-2941
Vasques, G. A., Andrade, N. L. M., and Jorge, A. A. L. (2019). Genetic causes of isolated short stature. Arch. Endocrinol. Metab. 63, 70–78. doi: 10.20945/2359-3997000000105
Wang, Y., Ge, J., Ma, J., Qiao, L., and Li, T. (2020). Short stature with precocious puberty caused by aggrecan gene mutation: a case report. Medicine (Baltimore) 99:e21635. doi: 10.1097/MD.0000000000021635
Xu, D., Sun, C., Zhou, Z., Wu, B., Yang, L., Chang, Z., et al. (2018). Novel aggrecan variant, p. Gln2364Pro, causes severe familial nonsyndromic adult short stature and poor growth hormone response in Chinese children. BMC Med. Genet. 19:79. doi: 10.1186/s12881-018-0591-z
Keywords: short stature, ACAN, mutation, aggrecans, osteoarthritis dissecans
Citation: Mancioppi V, Prodam F, Mellone S, Ricotti R, Giglione E, Grasso N, Vurchio D, Petri A, Rabbone I, Giordano M and Bellone S (2021) Retrospective Diagnosis of a Novel ACAN Pathogenic Variant in a Family With Short Stature: A Case Report and Review of the Literature. Front. Genet. 12:708864. doi: 10.3389/fgene.2021.708864
Received: 12 May 2021; Accepted: 19 July 2021;
Published: 12 August 2021.
Edited by:
Massimo Zeviani, University of Padua, ItalyReviewed by:
Saadullah Khan, Kohat University of Science and Technology, PakistanCopyright © 2021 Mancioppi, Prodam, Mellone, Ricotti, Giglione, Grasso, Vurchio, Petri, Rabbone, Giordano and Bellone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Flavia Prodam, ZmxhdmlhLnByb2RhbUBtZWQudW5pdXBvLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.