 Liam Shaw†
Liam Shaw† Kevin J. Hamill
Kevin J. Hamill
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Genet., 12 August 2021
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.707087
This article is part of the Research TopicMolecular Mechanisms of Heritable Connective Tissue DisordersView all 9 articles
The laminins (LM) are a family of basement membranes glycoproteins with essential structural roles in supporting epithelia, endothelia, nerves and muscle adhesion, and signaling roles in regulating cell migration, proliferation, stem cell maintenance and differentiation. Laminins are obligate heterotrimers comprised of α, β and γ chains that assemble intracellularly. However, extracellularly these heterotrimers then assemble into higher-order networks via interaction between their laminin N-terminal (LN) domains. In vitro protein studies have identified assembly kinetics and the structural motifs involved in binding of adjacent LN domains. The physiological importance of these interactions has been identified through the study of pathogenic point mutations in LN domains that lead to syndromic disorders presenting with phenotypes dependent on which laminin gene is mutated. Genotype-phenotype comparison between knockout and LN domain missense mutations of the same laminin allows inferences to be drawn about the roles of laminin network assembly in terms of tissue function. In this review, we will discuss these comparisons in terms of laminin disorders, and the therapeutic options that understanding these processes have allowed. We will also discuss recent findings of non-laminin mediators of laminin network assembly and their implications in terms of basement membrane structure and function.
Basement membranes (BMs) are flexible 40–120 nm sheets that separates cells from underlying connective tissue and regulate important cell behaviors such as cell polarity and migration, metabolism, and in inducing differentiation (Paulsson, 1992). Most BMs consist of two layers; an electron-lucent layer, lamina lucida comprised predominantly of laminins (LMs) and nidogens, and an electron dense layer, lamina densa of type IV collagen (col IV) and perlecan (Paulsson, 1992). BMs assemble through a multistep process, with the LM network assembling first (Kalb and Engel, 1991; Smyth et al., 1998; Li et al., 2002, 2005; McKee et al., 2007, 2009) via anchoring of the LMs cell surface receptors (Kalb and Engel, 1991; Smyth et al., 1998; Li et al., 2002, 2005; McKee et al., 2007, 2009). Anchorage increases local LM concentration, allows polymerization and recruitment of other components to the LM scaffold.
Laminins are an obligatory feature of every BM. Each LM is an αβγ heterotrimer comprised of one of five α chains (encoded by LAMA1-5), one of four α chains (LAMB1-4) and one of three γ (LAMC1-3) chains (Aumailley et al., 2005; Domogatskaya et al., 2012; Aumailley, 2013). Use of alternative promoters in LAMA3 gives rise to either the short LMα3A or the longer LMα3B form, which are functionally and structurally distinct (Ryan et al., 1994; Ferrigno et al., 1997). Restrictions in heterotrimerization potential means that only 16 αβγchain combinations are possible (Paulsson et al., 1985; Engel et al., 1991; Nissinen et al., 1991; Utani et al., 1994). These are differentially expressed and play context specific roles. For example, α1β1γ1 (LM111) is essential for embryonic development and knockout of any of those genes is not compatible with life, whereas α3Aβ3γ2 (LM332) is highly expressed in mature epithelial tissues and loss of function leads to epidermal fragility.
The LM family has arisen by gene duplication and rearrangement, and common structural motifs are shared between members (Figure 1A). Archetypal LM chains consists of a laminin N-terminal domain (LN domain) followed by rod-like stretches of epidermal growth factor-like repeats (LE domains) interspersed with globular domains (L4 or LF domains) and followed by a coiled coil domain (LCC domain) through which αβγ heterotrimers form (Paulsson et al., 1985; Engel et al., 1991; Nissinen et al., 1991; Zimmerman and Blanco, 2007). In α chains, the LCC domain is followed by five LM globular domains (LG1-5), which harbor the highest affinity cell surface receptor sites (Timpl et al., 2000). While this architecture holds true for most LMs, not all chains contain all domains. Importantly, the α3A, α4, and γ2 chains contain shorter amino terminal arms lacking LN domain (Aumailley et al., 2005; Hamill et al., 2009).
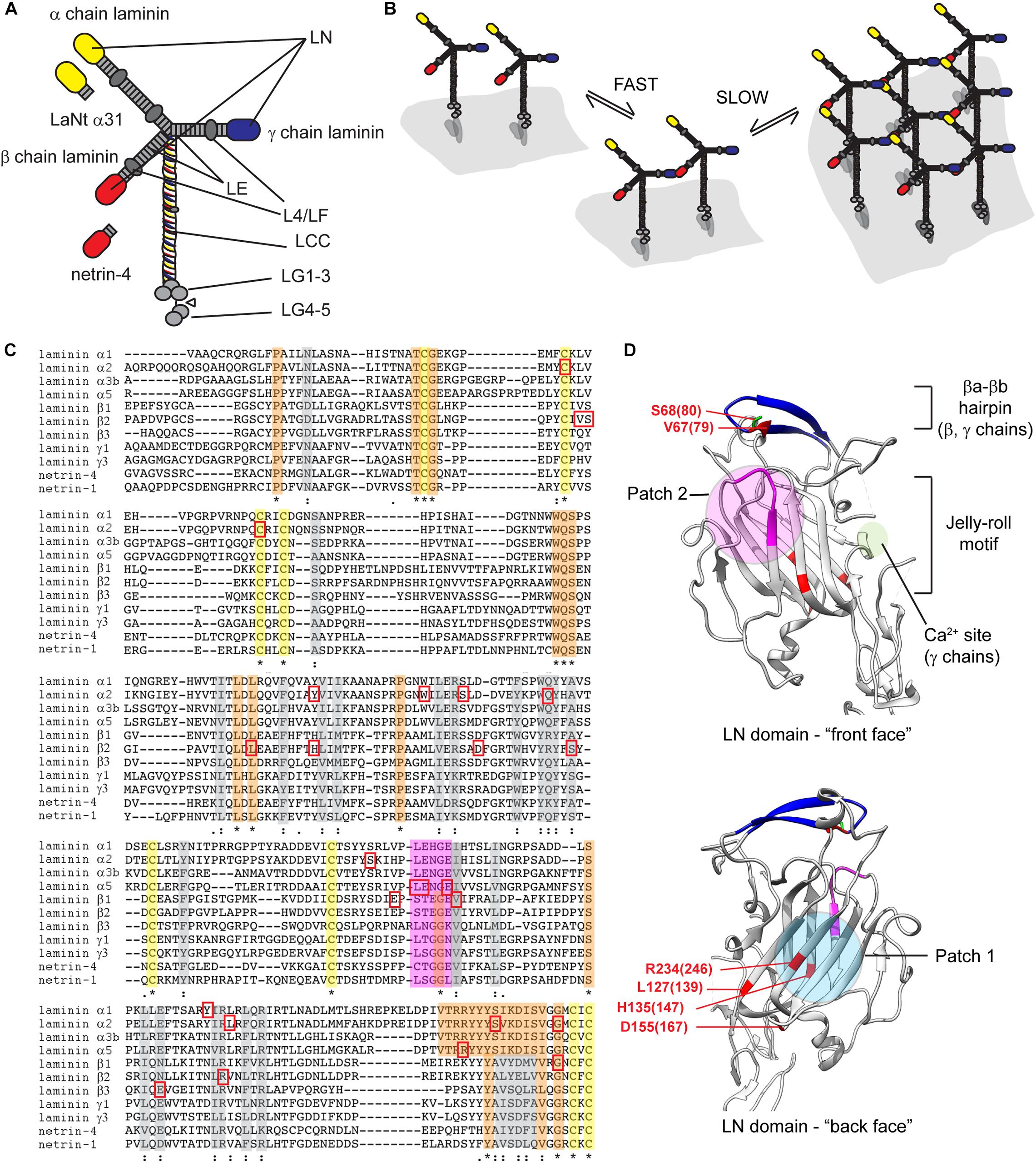
Figure 1. (A) Diagrammatic representation of archetypal LM structure. Yellow ovals αLN domains, red = βLN domains and blue = γLN domains. LE, laminin-type EGF-like repeat; LCC, laminin coiled coil domain; LG, laminin globular domains. (B) Two-step laminin network assembly. Secreted LMs associated with cell surface receptors, predominantly via their LG domain regions, β−γ LN domain interactions are then facilitated in a reaction with rapid on/off kinetics, then in the propagation step β−γ interactions are stabilized via incorporation of an α-chain leading to a stable cell-associated network. (C) Sequence alignments of LN domains from human laminin chains, netrin-1 and netrin-4. Yellow, orange, and gray highlights indicated conserved cysteines, fully conserved residues, or partially conserved residues. Magenta highlight identifies LENGE region. Red squares indicate pathogenic missense mutations. (D) Crystal structures of LN domains. View of the front and back face of the β1 chain is shown with features of conserved patches involved in LN-interaction (patch 2) and required for domain folding (patch 1). Amino acids associated Pierson syndrome mutations are indicated numbered based on LMβ1 with LMβ2 equivalent in parenthesis. Crystal structures derived from Hussain et al. (2011) and Carafoli et al. (2012) rendered using UCSF chimera (Pettersen et al., 2004). * = conserved residue, : = conservation of residues with strongly similar properties,. = conservation of residues with weakly similar properties.
Laminin N-terminal domain are 252–264 amino acid globular domains found not only in LMs but also other ECM proteins including netrins. 14% of all residues in LN domains are conserved with strict conservation of six key cysteines. There is 72% homology between LMα1 and LMα2, 77% between LMα3B and LMα5, 72% between LMβ1 and LMβ2, and 64% between LMγ1 and LMγ3 LN domains. Lowest conservation is between LMβ3 and the β1 and β2 LN domains (38 and 42%, respectively) (Garbe et al., 2002).
Laminins interactions have been studied over many years with important early work establishing a “three-arm” model; polymerization only occurs when all the constituent chains contain LN domains (Yurchenco and Cheng, 1994; Hohenester and Yurchenco, 2013). Moreover, interactions must be heterotypic, involving an α, β and γ LN domain (McKee et al., 2007). The assembly process is divided into a temperature-dependent oligomerization step and a calcium-dependent polymerization step. In vitro analyses have further shown that the αβγ ternary node assembly involves rapid but unstable formation of βγ pairs that are then stabilized through integration of an αLN domain (Hussain et al., 2011; Figure 1B). In line with the three-arm model, LMs that lack one or more LN domain cannot polymerize independently (Hohenester and Yurchenco, 2013). These include LM332 and LM411, which are abundantly expressed in many epithelial and endothelial BMs. For these LMs, alternative methods of interaction with other LM isoforms may be required for BM assembly. For LM332, incorporation into skin BM can be partly explained by an interaction between LM332’s β3LN domain with the LE domain of α3 in LM311. These LM dimers could then self-associate into higher order networks (Champliaud et al., 1996; Rousselle and Beck, 2013). Non-network forming LM BM incorporation likely also depends on compensatory interactions with other BM/ECM components, such as the β3LN domain binding of the NC1 domain of type VII collagen, nidogen binding to the γ1 LE repeats (Chen et al., 1997; Rousselle et al., 1997; Rousselle and Beck, 2013).
The crystal structures of α5, β1, γ1 LN domains have been solved, and these combined with conservation of residues between chains allows inferences as to which regions are involved in domain folding and those involved αβγ ternary node formation (Figure 1C). The crystals revealed a similar overall structure of an antiparallel β sandwich with 8 β sheets forming a jelly roll motif held in conformation by cysteines C200 and C220 (Figure 1D; Hussain et al., 2011; Carafoli et al., 2012). In the α5LN domain, two conserved motifs Patch 1 and Patch 2 are of particular relevance (Hussain et al., 2011). Patch 1 within the conserved β1-β2-β7-β4-β5 “back face,” consists of E178, P189, R265, and R267. These residues are blocked by a glycan attached to N148 (Hussain et al., 2011), suggesting that Patch 1 plays a structural, non-polymerizing role (Carafoli et al., 2012). Patch 2 is located across the β6-β3-β8 “front face,” residues W132 and N168, and the β5-β6 loop, residues P229, L230, and E231. Patch 2 is not glycosylated nor conserved with β- or γ-chains but is important for polymerization as mutation of PLENGE residues in the β5-β6 loop all result in inhibition of polymerization (Hussain et al., 2011; Carafoli et al., 2012). The β-sandwiches of β1LN and γ1LN domains have similar structure with the main differences in peripheral regions (Carafoli et al., 2012). β1LN contains two particular regions of functional importance: the βa-βb hairpin and the β7-α4 loop. The βa-βb hairpin sits at the top of the domain with S80 a key residue for β-γLN interactions (Purvis and Hohenester, 2012). One notable difference in γ1LN domain is a calcium binding site located within a short α-helix and flanked by highly conserved D106 and T114 (Carafoli et al., 2012). Testing inferences about residue function is a laborious task but elegant in vitro analysis using LN domain- fusion proteins have been performed and improve interpretation of clinical findings, discussed in context below (McKee et al., 2018).
Laminin N-terminal domains, in addition to polymerization, are implicated in cell adhesion, neurite outgrowth, perlecan, heparin and heparan sulfate binding (Nielsen and Yamada, 2001; Nomizu et al., 2001; Garbe et al., 2002; Kunneken et al., 2004). The LN domain of LMα1, α2, and α5 can interact with integrins α1β1, α2β1, and α3β1, and presumed between LMα3b and integrin α3β1 based on antibody inhibition (Pfaff et al., 1994; Colognato et al., 1997; Ettner et al., 1998; Kariya et al., 2004; Hozumi et al., 2012). These interactions are lower affinity than LG domain interactions and likely are involved in localization to allow polymerization rather than signal propagation.
The importance of LN domains to tissue function becomes apparent when the variety of LN domains mutations which lead to human disease are considered. Each affected LM results in a distinct set of syndromic disorders reflecting isoform-specific distribution and functions (Table 1).
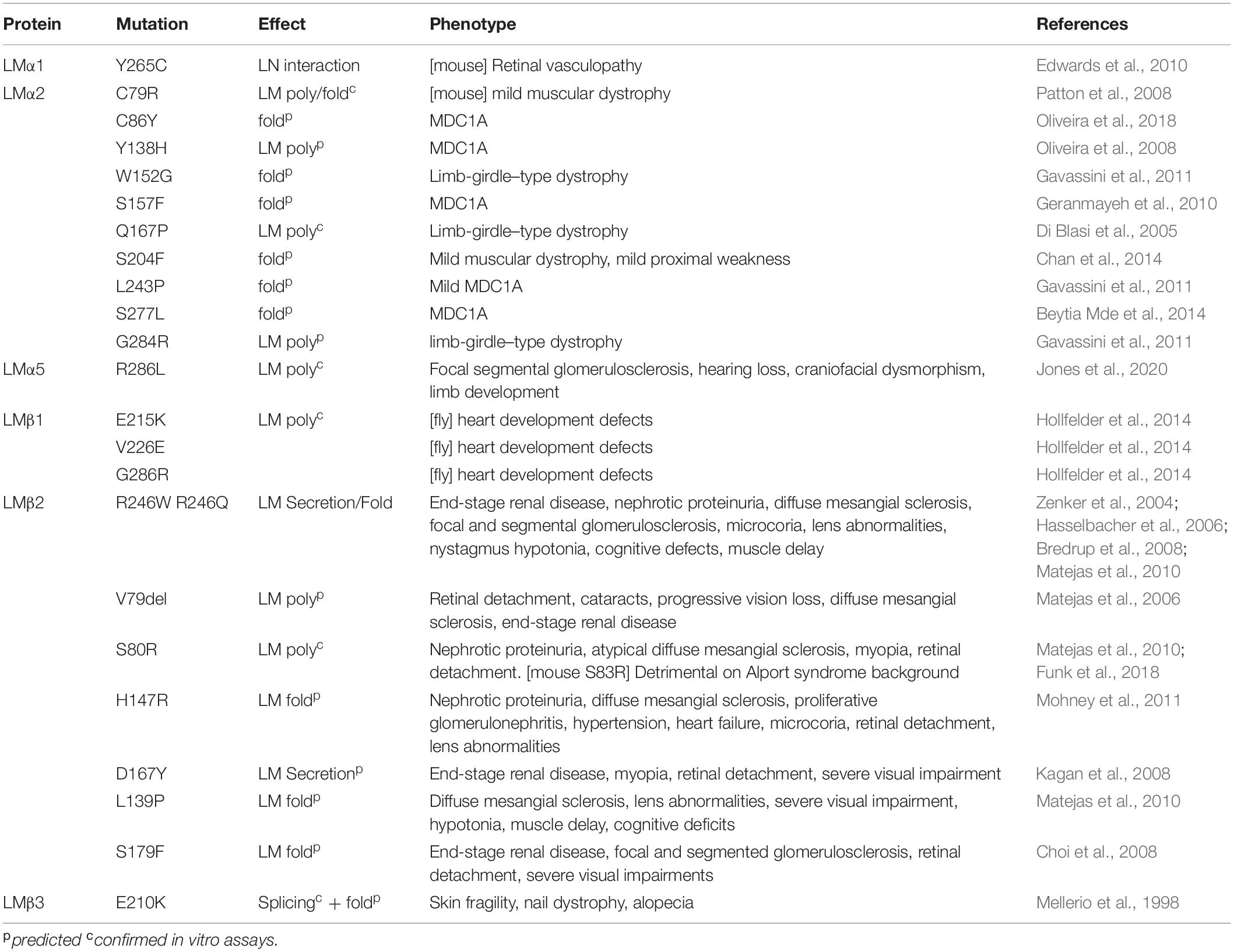
Table 1. Pathogenic LN domain mutations.
Pierson syndrome first described in 1963, is a severe congenital nephrotic syndrome with eye abnormalities (Pierson et al., 1963), caused by mutations in LAMB2 (LMβ2). LMβ2 is highly expressed in the glomerular basement membrane, multiple ocular structures (lens, retina, and cornea), and neuromuscular synapses (Hunter et al., 1989; Noakes et al., 1995; Libby et al., 2000; Bystrom et al., 2006). In addition to the defining features of congenital nephrotic syndrome that rapidly progresses to end-stage renal failure, and microcoria, many patients develop complications of motor delay, speech difficulties, intellectual disability, and seizures (Bowen et al., 1964). Indeed, the spectrum of LAMB2-related phenotypes is vast. The severest forms of the disease are associated with knockout mutations, whereas the majority of missense and indel mutations cluster to the LN domain and result in milder forms of the disease (Table 1).
One of the earliest studied LMβ2 LN mutations, R246W, is characterized by severe end-stage renal disease and microcoria (McKee et al., 2018). Similarly, R246Q, resulted in severe glomerular abnormalities and impaired secretion (Chen et al., 2011). Conservation of this arginine led to predictions that mutations impair LM polymerization, and in vitro this mutant polymerize substantially less effectively than the wild type protein (Zenker et al., 2004). However, R246W also reduced abundance of LM in BMs, likely due to disturbed protein processing (Matejas et al., 2010). Together these data indicate that this arginine has a role in protein folding. A second highly studied missense mutation, S80R, affects the highly conserved βa-βb and directly prevents LN–LN domain interactions with polymerization disrupted in vitro (Matejas et al., 2010; Carafoli et al., 2012; Purvis and Hohenester, 2012). Again, the importance of this region was further highlighted by an adjacent V79del patient (Matejas et al., 2006), presenting with milder symptoms of atypical diffuse mesangial sclerosis, retinal detachment, and myopia.
Other β2LN mutations with variable phenotypes include H147R (Mohney et al., 2011), L139P (Matejas et al., 2010), D167Y (Kagan et al., 2008), and S179F (Choi et al., 2008). Similar to R26Q, H147R caused a partial reduction in polymerization ability in vitro. L139P and D167Y mutations are near each other and are predicted to affect LN domain folding, and together suggest this region to be particularly sensitive to changes. L139P interferes with the hydrophobic core, is associated with a particularly severe phenotype. In contrast, the D167, and H147 result in milder phenotypes.
Mutations affecting α LN domains affect the stabilization step of ternary node assembly. The best example is merosin-deficient congenital muscular dystrophy type 1A (MDC1A), caused by mutations to LAMA2 (LMα2) (Helbling-Leclerc et al., 1995). This affects LM211 and LM221, the most abundant LMs in skeletal muscles (Ehrig et al., 1990), peripheral nerves, astrocytes and pericytes in the brain (Voit et al., 1995).
In LMα2 knockout conditions, MDC1A presents with disabilities of the proximal and distal limb muscles, with patients unable to walk more than a few steps unaided (Philpot et al., 1995; Jones et al., 2001). Weakness in facial muscles result in reduced sucking and swallowing capabilities, life-threatening problems can arise from failure of the respiratory muscles (Mendell et al., 2006), and cases with intellectual disability and epilepsy have been reported (Philpot et al., 1995; Muntoni and Voit, 2004; Mendell et al., 2006). In knockout situations, LM411 replaces LM211 in muscle basement membranes (Ringelmann et al., 1999). LMα4 lacks an LN domain and is unable to polymerize, resulting in a weakened BM. LMα4 and LMα2 also differ in their receptor binding interaction repertoire and affinities (Talts et al., 2000), for example, LMα2 binds integrin α7β1 whereas LMα4 cannot, and LMα4 has weaker affinity for α-dystroglycan (Nishiuchi et al., 2006). Comparison between missense mutations and knockout mutations allows differentiation between polymerization and receptor-mediated effects, although these inferences are complicated by not every affected tissue expressing LM411.
Many mutations have been reported throughout LAMA2’s 65 exons in MDC1A and are cataloged in LAMA2 gene variant database1 (Oliveira et al., 2018). Again, the LN domain contains a cluster of missense and in frame deletions (Patton et al., 2008; Oliveira et al., 2018). For example, a point mutation in the highly conserved CxxC motif, C79R, led to a milder form of MDC1A, which affects the myelination of Schwann cells in spinal roots and the stability of the skeletal muscles (Patton et al., 2008). This amyelination was not attributed to a change in abundance or mislocalization, and in vitro assays confirmed a dramatic effect on LM polymerization (McKee et al., 2018). Other pathogenic missense variants include Q167P, Y138H, G284R on the surface of α2 LN domain and C86Y, W152G, S157F, S277L, S204F, L243P in the interior (Yurchenco et al., 2018; Table 1). The S204F mutation lies at one extreme of the phenotypic spectrum, whereby the patient was misdiagnosed with a peripheral neuropathy, presenting with mild proximal weakness. Muscle biopsy revealed depletion of LMα2 in intramuscular nerve, subtly depleted LMα2 expression in muscle BMs and diffusely upregulated LMα5 expression (Chan et al., 2014). To the other extreme, Q167P maps to near the polymerization face, and consistent with this, causes a 60% drop in in vitro polymerization capability. This leads to ambulatory muscular dystrophy (McKee et al., 2018). More severe still, G284R causes proximal weakness, with a loss of functional gait with age accompanied by frequent falls, and epilepsy. The mutation effect is yet to be confirmed but predicted to inhibit LM polymerization (Gavassini et al., 2011).
LMα5 is almost ubiquitous to all adult BMs. Unsurprisingly, knockout mice die before birth with a failure in neural tube closure, and no human knockouts have been reported (Miner et al., 1998). However, a patient with R286L in LMα5 LN has been identified. They presented with a complex syndromic disease characterized by defects in kidney, craniofacial and limb development (Jones et al., 2020). The affected residue lies adjacent to the conserved PLENGE sequence required for LM polymerization (Hussain et al., 2011), and R286L abrogated in vitro polymerization potential (Jones et al., 2020). We cannot compare the LN mutation against knockout; however, a patient with V3140M, in the LG3 domain has been reported (Sampaolo et al., 2017). Both the LG3 mutation and R286L lead to complex syndromic disorders with similarity in tissues affected but with notable differences. Specifically, in the skin V3140M caused alopecia, lack of eyebrows and body hair, features not present in the R286L patient. V3140M patients also had retinal rod degeneration whereas the R286L had hearing loss but no sight abnormalities. Kidney defects were common to both with R286L presenting with atypical focal segmental glomerulosclerosis progressing to end stage kidney disease compared with floating kidney syndrome in V3140M. Finally, R286L presented with numerous dysmorphic issues include craniofacial dysmorphism, syndactyly, and pyloric web.
LMβ3 is expressed in most epithelial tissues where it forms part of LM3a32 and LM3b32 (Matsui et al., 1995; Ferrigno et al., 1997). The resulting heterotrimers have either one or two LN domains and are unable to polymerize independently (Yurchenco and Cheng, 1994; Cheng et al., 1997). One would assume that LN domain mutations are tolerated for this LM chain. However, patients were identified where the pathogenic mutation caused E210K, which gives rise to a phenotype of trauma-induced blisters, nail dystrophy and alopecia (mild junctional epidermolysis bullosa) (McGrath et al., 1995; Mellerio et al., 1998; Posteraro et al., 2004). In comparison, homozygous knockout of LMβ3 leads to much more extensive skin blistering complications and early lethality (Pulkkinen et al., 1994; Kuster et al., 1997; Ryan et al., 1999; Meng et al., 2003).
Interpretation of the E210K mutation is complicated; the affected base pair is at a splice junction and in a knock-in mouse model led to skipping of the out-of-frame, and no detectable LMβ3 in the skin. However, in humans, miss-splicing has been reported for some, but not all patients, which can be rescued by second-site mutations (Pasmooij et al., 2007). Numerous alternative splice products are produced, including some full-length transcripts. Modeling of the E210K mutation indicates it is unlikely to be required for laminin polymerization but also is not predicted to affect protein folding or secretion (Mittwollen et al., 2020). The most common in-frame deletion is predicted to remove several of the central β-strands and disrupt the fold. Overall, the evidence from these patients does not point toward a LM polymerization effect but does suggest a role for the LMβ3 LN domain in protein function. Direct evidence for the importance of the LMβ3 LN domain has been obtained from keratinocytes expressing either full-length or LN domain-deleted LMβ3 and grafted as skin equivalents onto immunodeficient mice (Sakai et al., 2010). Here, the LN deleted versions displayed subepidermal blistering, erosions, and prominent granulation tissue, not associated with reduced LM332 deposition pointing LN domain roles beyond polymerization.
LMα1 is extremely important for developmental processes, with knockout mice embryonic lethal. However, Y256C mice are viable with retinal defects of vitreal fibroplasia, vascular tortuosity and hypervascularization, and abnormalities to the retinal inner limiting membrane (Edwards et al., 2010). No reduction in LMα1 was noted, and a two-hybrid screen identified the mutation affects LN–LN interaction. Random mutagenesis in Drosophila has identified three LN domain mutations in LMβ1 that led to heart developmental defects, E215K, V226E, and G286R (Hollfelder et al., 2014). Of these, E215K was tested in in vitro assays and reduced polymerization (McKee et al., 2018).
The netrins family of proteins are structurally and ancestrally related to LMs (Tessier-Lavigne and Goodman, 1996; Fahey and Degnan, 2012). Each netrin comprises a LN domain and stretch of LE repeats followed by a unique C-terminal region (Kappler et al., 2000; Yurchenco and Wadsworth, 2004). The LN domains of most netrins have diverged that they do not influence LM network assembly. However, for netrin-4 the situation is dramatically different where the β-type LN domain of netrin-4 can potently disrupt LM networks (Schneiders et al., 2007; Reuten et al., 2016, 2021). The physiological implications of this ability are beginning to be appreciated; recent work has demonstrated that netrin-4 levels are a key determinant of basement membrane stiffness with knock-on effects to cell behavior and tumor metastasis (Reuten et al., 2016, 2021).
Whereas netrins have evolved as independent genes, alternative splicing from LM genes or proteolytic processing of LM proteins leads to generation of LN domain containing fragments (Kariya et al., 2004; Hamill et al., 2009; Horejs et al., 2014). These fragments contain “perfect” LN domains that are likely to compete for binding sites (with reduced potency compared with netrin-4). One LAMA3-derived alternative splice isoform, Laminin N terminus α31 (LaNt α31) has widespread expression in human tissues (Troughton et al., 2020b), is upregulated during wounding and corneal limbal stem cell activation (Barrera et al., 2018) and emerging data indicate that it can modulate LM organization in vitro (Troughton et al., 2020a). In vivo overexpression is embryonic lethal during development with tissue defects that resemble LM network disruption phenotypes (Sugden et al., 2020).
From an evolutionary perspective, netrin-4, LaNt α31 and proteolytically released LN fragments represent multiple mechanisms to fine-tune LM network assembly. Although human diseases directly associated with loss-of-function mutations have not been identified, a SNP in the netrin-4 gene (causing Y205H) has been associated with late onset Alzheimer’s disease (Saad et al., 2015), and dysregulation of expression appear to contribute to tumor pathogenesis and point toward an additional important aspect of BM biology (Schneiders et al., 2007; Reuten et al., 2016, 2021; Troughton et al., 2020c).
Although the standard gene and protein therapy toolbox are available to treat LN domain disorders, the large size of LM genes and associated challenges of producing and delivering recombinant therapy-grade LM protein presents challenges. However, promising results have been obtained recently from delivering the 800 kDa LM521 to the blood stream of LAMB2-null mice which rescues some aspects of Pierson syndrome. The delivered LM521 accumulated in the glomerular basement membrane in the correct orientation and led to reduced expression of the podocyte injury markers, and delayed the onset of proteinuria. However, the exogenous LM521 did not migrate to the podocytes nor fully restore the glomerular filtration barrier. Smaller, or hybrid proteins, may be a solution to overcome these challenges (Lin et al., 2018). For some LM disorders, upregulating expression of a compensatory LM may be a viable option. While there are differences, LMα1 and LMα2 are very similar both structurally and functionally, therefore in LMα2-deficient MDC1A, increasing LMα1 could compensate for the lack of functional LMα2. LMα1 expression is usually downregulated following development; however, encouraging progress has been made here using guide RNA to target the LMα1 promoter with inactive Cas9 coupled to VP160 transcription activation domain. In mouse models, electroporation of the gRNA-containing plasmids into the tibialis anterior of 4-week old animals led to increased expression of LM111 with appropriate localization 2-weeks post-electroporation (Perrin et al., 2017). This data provides an encouraging base for development that may be exploitable for other conditions using a similar approach.
A particularly innovative solution exploiting the knowledge gained from studying LM polymerization and counteracting the inherent LM size problems is using protein chimeras to act as linkers (McKee et al., 2009, 2017; Reinhard et al., 2017). Three such “Frankenstein” chimeric proteins have been created, a fusion of a functional LN domain to the LM binding region of nidogen, a miniature form of agrin (mini-agrin) containing only the LM-binding regions and α-dystroglycan binding regions, and a fusion between LM-binding domains of agrin and the dystroglycan binding domain of perlecan. As LM411 is upregulated in MDC1A but cannot compensate for LM211 dysfunction, the nidogen/LN domain chimeric protein can be used to bind the γ1 chain of LM411 via the nidogen region and provide the missing αLN domain needed to allow LM411 polymerization (Reinhard et al., 2017). The mini-agrin/perlecan chimeras can be used in concert with the nidogen chimera to compensate for α-dystroglycan binding (Talts et al., 2000; Moll et al., 2001). Where patients harbor LN mutations, only the nidogen fusion would be required, whereas for knockout both the LN/nidogen and mini-agrin would be necessary. Promising results have been observed with these chimeras in mouse models. Moreover, switching the LN domain from an αLN to βLN, this approach is likely to also be effective for Pierson syndrome patients.
Comparison between knockout and missense mutation associated phenotypes in LM genes has provided valuable information to identify which LMs are essential for individual tissues, but also which domains are involved. Rather than a binary outcome caused by ability or inability to polymerize, we see system-wide differences highlighting the multifaceted roles of LN domains. The variety of pathologies arising from mutations within a stretch of ∼250 amino acids illustrate the importance of LN domains to tissue function.
LS, CS, and KH wrote and edited the manuscript. All authors read and approved the final version of the manuscript for publication.
This work was supported by the Biotechnology and Biological Sciences Research Council (Grant No. BB/P025773/1) and The University of Liverpool Crossley Barnes Bequest fund.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aumailley, M., Bruckner-Tuderman, L., Carter, W. G., Deutzmann, R., Edgar, D., Ekblom, P., et al. (2005). A simplified laminin nomenclature. Matrix Biol. 24, 326–332. doi: 10.1016/j.matbio.2005.05.006
Barrera, V., Troughton, L. D., Iorio, V., Liu, S., Oyewole, O., Sheridan, C. M., et al. (2018). Differential distribution of Laminin N-Terminus alpha31 across the ocular surface: implications for corneal wound repair. Invest. Ophthalmol. Vis. Sci. 59, 4082–4093. doi: 10.1167/iovs.18-24037
Beytia Mde, L., Dekomien, G., Hoffjan, S., Haug, V., Anastasopoulos, C., and Kirschner, J. (2014). High creatine kinase levels and white matter changes: clinical and genetic spectrum of congenital muscular dystrophies with laminin alpha-2 deficiency. Mol. Cell Probes 28, 118–122. doi: 10.1016/j.mcp.2013.11.002
Bowen, P., Lee, C. S., Zellweger, H., and Lindenberg, R. (1964). A familial syndrome of multiple congenital defects. Bull. Johns Hopkins Hosp. 114, 402–414.
Bredrup, C., Matejas, V., Barrow, M., Blahova, K., Bockenhauer, D., Fowler, D. J., et al. (2008). Ophthalmological aspects of Pierson syndrome. Am. J. Ophthalmol. 146, 602–611.
Bystrom, B., Virtanen, I., Rousselle, P., Gullberg, D., and Pedrosa-Domellof, F. (2006). Distribution of laminins in the developing human eye. Invest. Ophthalmol. Vis. Sci. 47, 777–785. doi: 10.1167/iovs.05-0367
Carafoli, F., Hussain, S. A., and Hohenester, E. (2012). Crystal structures of the network-forming short-arm tips of the laminin beta1 and gamma1 chains. PLoS One 7:e42473. doi: 10.1371/journal.pone.0042473
Champliaud, M. F., Lunstrum, G. P., Rousselle, P., Nishiyama, T., Keene, D. R., and Burgeson, R. E. (1996). Human amnion contains a novel laminin variant, laminin 7, which like laminin 6, covalently associates with laminin 5 to promote stable epithelial-stromal attachment. J. Cell Biol. 132, 1189–1198. doi: 10.1083/jcb.132.6.1189
Chan, S. H., Foley, A. R., Phadke, R., Mathew, A. A., Pitt, M., Sewry, C., et al. (2014). Limb girdle muscular dystrophy due to LAMA2 mutations: diagnostic difficulties due to associated peripheral neuropathy. Neuromuscul. Disord. 24, 677–683. doi: 10.1016/j.nmd.2014.05.008
Chen, M., Marinkovich, M. P., Veis, A., Cai, X., Rao, C. N., O’Toole, E. A., et al. (1997). Interactions of the amino-terminal noncollagenous (NC1) domain of type VII collagen with extracellular matrix components. A potential role in epidermal-dermal adherence in human skin. J. Biol. Chem. 272, 14516–14522. doi: 10.1074/jbc.272.23.14516
Chen, Y. M., Kikkawa, Y., and Miner, J. H. (2011). A missense LAMB2 mutation causes congenital nephrotic syndrome by impairing laminin secretion. J. Am. Soc. Nephrol. 22, 849–858. doi: 10.1681/asn.2010060632
Cheng, Y. S., Champliaud, M. F., Burgeson, R. E., Marinkovich, M. P., and Yurchenco, P. D. (1997). Self-assembly of laminin isoforms. J. Biol. Chem. 272, 31525–31532. doi: 10.1074/jbc.272.50.31525
Choi, H. J., Lee, B. H., Kang, J. H., Jeong, H. J., Moon, K. C., Ha, I. S., et al. (2008). Variable phenotype of Pierson syndrome. Pediatr. Nephrol. 23, 995–1000. doi: 10.1007/s00467-008-0748-7
Colognato, H., MacCarrick, M., O’Rear, J. J., and Yurchenco, P. D. (1997). The laminin alpha2-chain short arm mediates cell adhesion through both the alpha1beta1 and alpha2beta1 integrins. J. Biol. Chem. 272, 29330–29336. doi: 10.1074/jbc.272.46.29330
Di Blasi, C., Piga, D., Brioschi, P., Moroni, I., Pini, A., Ruggieri, A., et al. (2005). LAMA2 gene analysis in congenital muscular dystrophy: new mutations, prenatal diagnosis, and founder effect. Arch. Neurol. 62, 1582–1586.
Domogatskaya, A., Rodin, S., and Tryggvason, K. (2012). Functional diversity of laminins. Annu. Rev. Cell Dev. Biol. 28, 523–553. doi: 10.1146/annurev-cellbio-101011-155750
Edwards, M. M., Mammadova-Bach, E., Alpy, F., Klein, A., Hicks, W. L., Roux, M., et al. (2010). Mutations in Lama1 disrupt retinal vascular development and inner limiting membrane formation. J. Biol. Chem. 285, 7697–7711. doi: 10.1074/jbc.m109.069575
Ehrig, K., Leivo, I., Argraves, W. S., Ruoslahti, E., and Engvall, E. (1990). Merosin, a tissue-specific basement membrane protein, is a laminin-like protein. Proc. Natl. Acad. Sci. U.S.A. 87, 3264–3268. doi: 10.1073/pnas.87.9.3264
Engel, J., Hunter, I., Schulthess, T., Beck, K., Dixon, T. W., and Parry, D. A. (1991). Assembly of laminin isoforms by triple- and double-stranded coiled-coil structures. Biochem. Soc. Trans. 19, 839–843. doi: 10.1042/bst0190839
Ettner, N., Gohring, W., Sasaki, T., Mann, K., and Timpl, R. (1998). The N-terminal globular domain of the laminin alpha1 chain binds to alpha1beta1 and alpha2beta1 integrins and to the heparan sulfate-containing domains of perlecan. FEBS Lett. 430, 217–221. doi: 10.1016/s0014-5793(98)00601-2
Fahey, B., and Degnan, B. M. (2012). Origin and evolution of laminin gene family diversity. Mol. Bio.l Evol. 29, 1823–1836. doi: 10.1093/molbev/mss060
Ferrigno, O., Virolle, T., Galliano, M. F., Chauvin, N., Ortonne, J. P., Meneguzzi, G., et al. (1997). Murine laminin alpha3A and alpha3B isoform chains are generated by usage of two promoters and alternative splicing. J. Biol. Chem. 272, 20502–20507. doi: 10.1074/jbc.272.33.20502
Funk, S. D., Bayer, R. H., Malone, A. F., McKee, K. K., Yurchenco, P. D., and Miner, J. H. (2018). Pathogenicity of a human laminin beta2 mutation revealed in models of alport syndrome. J. Am. Soc. Nephrol. 29, 949–960. doi: 10.1681/asn.2017090997
Garbe, J. H., Gohring, W., Mann, K., Timpl, R., and Sasaki, T. (2002). Complete sequence, recombinant analysis and binding to laminins and sulphated ligands of the N-terminal domains of laminin alpha3B and alpha5 chains. Biochem. J. 362(Pt 1), 213–221. doi: 10.1042/bj3620213
Gavassini, B. F., Carboni, N., Nielsen, J. E., Danielsen, E. R., Thomsen, C., Svenstrup, K., et al. (2011). Clinical and molecular characterization of limb-girdle muscular dystrophy due to LAMA2 mutations. Muscle Nerve 44, 703–709. doi: 10.1002/mus.22132
Geranmayeh, F., Clement, E., Feng, L. H., Sewry, C., Pagan, J., Mein, R., et al. (2010). Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul. Disord. 20, 241–250. doi: 10.1016/j.nmd.2010.02.001
Hamill, K. J., Langbein, L., Jones, J. C., and McLean, W. H. (2009). Identification of a novel family of laminin N-terminal alternate splice isoforms: structural and functional characterization. J. Biol. Chem. 284, 35588–35596. doi: 10.1074/jbc.m109.052811
Hasselbacher, K., Wiggins, R. C., Matejas, V., Hinkes, B. G., Mucha, B., Hoskins, B. E., et al. (2006). Recessive missense mutations in LAMB2 expand the clinical spectrum of LAMB2-associated disorders. Kidney Int. 70, 1008–1012. doi: 10.1038/sj.ki.5001679
Helbling-Leclerc, A., Zhang, X., Topaloglu, H., Cruaud, C., Tesson, F., Weissenbach, J., et al. (1995). Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat. Genet. 11, 216–218. doi: 10.1038/ng1095-216
Hohenester, E., and Yurchenco, P. D. (2013). Laminins in basement membrane assembly. Cell Adh. Migr. 7, 56–63. doi: 10.4161/cam.21831
Hollfelder, D., Frasch, M., and Reim, I. (2014). Distinct functions of the laminin beta LN domain and collagen IV during cardiac extracellular matrix formation and stabilization of alary muscle attachments revealed by EMS mutagenesis in Drosophila. BMC Dev. Biol. 14:26. doi: 10.1186/1471-213X-14-26
Horejs, C. M., Serio, A., Purvis, A., Gormley, A. J., Bertazzo, S., Poliniewicz, A., et al. (2014). Biologically-active laminin-111 fragment that modulates the epithelial-to-mesenchymal transition in embryonic stem cells. Proc. Natl. Acad. Sci. U.S.A. 111, 5908–5913. doi: 10.1073/pnas.1403139111
Hozumi, K., Ishikawa, M., Hayashi, T., Yamada, Y., Katagiri, F., Kikkawa, Y., et al. (2012). Identification of cell adhesive sequences in the N-terminal region of the laminin alpha2 chain. J. Biol. Chem. 287, 25111–25122. doi: 10.1074/jbc.m112.348151
Hunter, D. D., Shah, V., Merlie, J. P., and Sanes, J. R. (1989). A laminin-like adhesive protein concentrated in the synaptic cleft of the neuromuscular junction. Nature 338, 229–234. doi: 10.1038/338229a0
Hussain, S. A., Carafoli, F., and Hohenester, E. (2011). Determinants of laminin polymerization revealed by the structure of the alpha5 chain amino-terminal region. EMBO Rep. 12, 276–282. doi: 10.1038/embor.2011.3
Jones, K. J., Morgan, G., Johnston, H., Tobias, V., Ouvrier, R. A., Wilkinson, I., et al. (2001). The expanding phenotype of laminin alpha2 chain (merosin) abnormalities: case series and review. J. Med. Genet. 38, 649–657. doi: 10.1136/jmg.38.10.649
Jones, L. K., Lam, R., McKee, K. K., Aleksandrova, M., Dowling, J., Alexander, S. I., et al. (2020). A mutation affecting laminin alpha 5 polymerisation gives rise to a syndromic developmental disorder. Development 147:dev189183.
Kagan, M., Cohen, A. H., Matejas, V., Vlangos, C., and Zenker, M. (2008). A milder variant of Pierson syndrome. Pediatr. Nephrol. 23, 323–327. doi: 10.1007/s00467-007-0624-x
Kalb, E., and Engel, J. (1991). Binding and calcium-induced aggregation of laminin onto lipid bilayers. J. Biol. Chem. 266, 19047–19052. doi: 10.1016/s0021-9258(18)55170-x
Kappler, J., Franken, S., Junghans, U., Hoffmann, R., Linke, T., Muller, H. W., et al. (2000). Glycosaminoglycan-binding properties and secondary structure of the C-terminus of netrin-1. Biochem. Biophys. Res. Commun. 271, 287–291. doi: 10.1006/bbrc.2000.2583
Kariya, Y., Yasuda, C., Nakashima, Y., Ishida, K., Tsubota, Y., and Miyazaki, K. (2004). Characterization of laminin 5B and NH2-terminal proteolytic fragment of its alpha3B chain: promotion of cellular adhesion, migration, and proliferation. J. Biol. Chem. 279, 24774–24784. doi: 10.1074/jbc.m400670200
Kunneken, K., Pohlentz, G., Schmidt-Hederich, A., Odenthal, U., Smyth, N., Peter-Katalinic, J., et al. (2004). Recombinant human laminin-5 domains. Effects of heterotrimerization, proteolytic processing, and N-glycosylation on alpha3beta1 integrin binding. J. Biol. Chem. 279, 5184–5193.
Kuster, J. E., Guarnieri, M. H., Ault, J. G., Flaherty, L., and Swiatek, P. J. (1997). IAP insertion in the murine LamB3 gene results in junctional epidermolysis bullosa. Mamm. Genome 8, 673–681. doi: 10.1007/s003359900535
Li, S., Harrison, D., Carbonetto, S., Fassler, R., Smyth, N., Edgar, D., et al. (2002). Matrix assembly, regulation, and survival functions of laminin and its receptors in embryonic stem cell differentiation. J. Cell Biol. 157, 1279–1290. doi: 10.1083/jcb.200203073
Li, S., Liquari, P., McKee, K. K., Harrison, D., Patel, R., Lee, S., et al. (2005). Laminin-sulfatide binding initiates basement membrane assembly and enables receptor signaling in Schwann cells and fibroblasts. J. Cell Biol. 169, 179–189. doi: 10.1083/jcb.200501098
Libby, R. T., Champliaud, M. F., Claudepierre, T., Xu, Y., Gibbons, E. P., Koch, M., et al. (2000). Laminin expression in adult and developing retinae: evidence of two novel CNS laminins. J. Neurosci. 20, 6517–6528. doi: 10.1523/jneurosci.20-17-06517.2000
Lin, M. H., Miller, J. B., Kikkawa, Y., Suleiman, H. Y., Tryggvason, K., Hodges, B. L., et al. (2018). Laminin-521 protein therapy for glomerular basement membrane and podocyte abnormalities in a model of pierson syndrome. J. Am. Soc. Nephrol. 29, 1426–1436. doi: 10.1681/asn.2017060690
Matejas, V., Al-Gazali, L., Amirlak, I., and Zenker, M. (2006). A syndrome comprising childhood-onset glomerular kidney disease and ocular abnormalities with progressive loss of vision is caused by mutated LAMB2. Nephrol. Dial. Transplant 21, 3283–3286. doi: 10.1093/ndt/gfl463
Matejas, V., Hinkes, B., Alkandari, F., Al-Gazali, L., Annexstad, E., Aytac, M. B., et al. (2010). Mutations in the human laminin beta2 (LAMB2) gene and the associated phenotypic spectrum. Hum. Mutat. 31, 992–1002. doi: 10.1002/humu.21304
Matsui, C., Wang, C. K., Nelson, C. F., Bauer, E. A., and Hoeffler, W. K. (1995). The assembly of laminin-5 subunits. J. Biol. Chem. 270, 23496–23503. doi: 10.1074/jbc.270.40.23496
McGrath, J. A., Pulkkinen, L., Christiano, A. M., Leigh, I. M., Eady, R. A., and Uitto, J. (1995). Altered laminin 5 expression due to mutations in the gene encoding the beta 3 chain (LAMB3) in generalized atrophic benign epidermolysis bullosa. J. Invest. Dermatol. 104, 467–474. doi: 10.1111/1523-1747.ep12605904
McKee, K. K., Aleksandrova, M., and Yurchenco, P. D. (2018). Chimeric protein identification of dystrophic, Pierson and other laminin polymerization residues. Matrix Biol. 67, 32–46. doi: 10.1016/j.matbio.2018.01.012
McKee, K. K., Capizzi, S., and Yurchenco, P. D. (2009). Scaffold-forming and adhesive contributions of synthetic laminin-binding proteins to basement membrane assembly. J. Biol. Chem. 284, 8984–8994. doi: 10.1074/jbc.m809719200
McKee, K. K., Crosson, S. C., Meinen, S., Reinhard, J. R., Ruegg, M. A., and Yurchenco, P. D. (2017). Chimeric protein repair of laminin polymerization ameliorates muscular dystrophy phenotype. J. Clin. Invest. 127, 1075–1089. doi: 10.1172/jci90854
McKee, K. K., Harrison, D., Capizzi, S., and Yurchenco, P. D. (2007). Role of laminin terminal globular domains in basement membrane assembly. J. Biol. Chem. 282, 21437–21447. doi: 10.1074/jbc.m702963200
Mellerio, J. E., Eady, R. A., Atherton, D. J., Lake, B. D., and McGrath, J. A. (1998). E210K mutation in the gene encoding the beta3 chain of laminin-5 (LAMB3) is predictive of a phenotype of generalized atrophic benign epidermolysis bullosa. Br. J. Dermatol. 139, 325–331. doi: 10.1046/j.1365-2133.1998.02377.x
Mendell, J. R., Boue, D. R., and Martin, P. T. (2006). The congenital muscular dystrophies: recent advances and molecular insights. Pediatr. Dev. Pathol. 9, 427–443. doi: 10.2350/06-07-0127.1
Meng, X., Klement, J. F., Leperi, D. A., Birk, D. E., Sasaki, T., Timpl, R., et al. (2003). Targeted inactivation of murine laminin gamma2-chain gene recapitulates human junctional epidermolysis bullosa. J. Invest. Dermatol. 121, 720–731. doi: 10.1046/j.1523-1747.2003.12515.x
Miner, J. H., Cunningham, J., and Sanes, J. R. (1998). Roles for laminin in embryogenesis: exencephaly, syndactyly, and placentopathy in mice lacking the laminin alpha5 chain. J. Cell Biol. 143, 1713–1723. doi: 10.1083/jcb.143.6.1713
Mittwollen, R., Wohlfart, S., Park, J., Grosch, E., Has, C., Hohenester, E., et al. (2020). Aberrant splicing as potential modifier of the phenotype of junctional epidermolysis bullosa. J. Eur. Acad. Dermatol Venereol. 34, 2127–2134. doi: 10.1111/jdv.16332
Mohney, B. G., Pulido, J. S., Lindor, N. M., Hogan, M. C., Consugar, M. B., Peters, J., et al. (2011). A novel mutation of LAMB2 in a multigenerational mennonite family reveals a new phenotypic variant of Pierson syndrome. Ophthalmology 118, 1137–1144. doi: 10.1016/j.ophtha.2010.10.009
Moll, J., Barzaghi, P., Lin, S., Bezakova, G., Lochmuller, H., Engvall, E., et al. (2001). An agrin minigene rescues dystrophic symptoms in a mouse model for congenital muscular dystrophy. Nature 413, 302–307. doi: 10.1038/35095054
Muntoni, F., and Voit, T. (2004). The congenital muscular dystrophies in 2004: a century of exciting progress. Neuromuscul. Disord. 14, 635–649. doi: 10.1016/j.nmd.2004.06.009
Nielsen, P. K., and Yamada, Y. (2001). Identification of cell-binding sites on the Laminin alpha 5 N-terminal domain by site-directed mutagenesis. J. Biol. Chem. 276, 10906–10912. doi: 10.1074/jbc.m008743200
Nishiuchi, R., Takagi, J., Hayashi, M., Ido, H., Yagi, Y., Sanzen, N., et al. (2006). Ligand-binding specificities of laminin-binding integrins: a comprehensive survey of laminin-integrin interactions using recombinant alpha3beta1, alpha6beta1, alpha7beta1 and alpha6beta4 integrins. Matrix Biol. 25, 189–197. doi: 10.1016/j.matbio.2005.12.001
Nissinen, M., Vuolteenaho, R., Boot-Handford, R., Kallunki, P., and Tryggvason, K. (1991). Primary structure of the human laminin A chain. Limited expression in human tissues. Biochem. J. 276(Pt 2), 369–379. doi: 10.1042/bj2760369
Noakes, P. G., Gautam, M., Mudd, J., Sanes, J. R., and Merlie, J. P. (1995). Aberrant differentiation of neuromuscular junctions in mice lacking s-laminin/laminin beta 2. Nature 374, 258–262. doi: 10.1038/374258a0
Nomizu, M., Yokoyama, F., Suzuki, N., Okazaki, I., Nishi, N., Ponce, M. L., et al. (2001). Identification of homologous biologically active sites on the N-terminal domain of laminin alpha chains. Biochemistry 40, 15310–15317. doi: 10.1021/bi011552c
Oliveira, J., Gruber, A., Cardoso, M., Taipa, R., Fineza, I., Goncalves, A., et al. (2018). LAMA2 gene mutation update: toward a more comprehensive picture of the laminin-alpha2 variome and its related phenotypes. Hum. Mutat. 39, 1314–1337. doi: 10.1002/humu.23599
Oliveira, J., Santos, R., Soares-Silva, I., Jorge, P., Vieira, E., Oliveira, M. E., et al. (2008). LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients. Clin. Genet. 74, 502–512. doi: 10.1111/j.1399-0004.2008.01068.x
Pasmooij, A. M., Pas, H. H., Bolling, M. C., and Jonkman, M. F. (2007). Revertant mosaicism in junctional epidermolysis bullosa due to multiple correcting second-site mutations in LAMB3. J. Clin. Invest. 117, 1240–1248. doi: 10.1172/jci30465
Patton, B. L., Wang, B., Tarumi, Y. S., Seburn, K. L., and Burgess, R. W. (2008). A single point mutation in the LN domain of LAMA2 causes muscular dystrophy and peripheral amyelination. J. Cell Sci. 121(Pt 10), 1593–1604. doi: 10.1242/jcs.015354
Paulsson, M. (1992). Basement membrane proteins: structure, assembly, and cellular interactions. Crit. Rev. Biochem. Mol. Biol. 27, 93–127.
Paulsson, M., Deutzmann, R., Timpl, R., Dalzoppo, D., Odermatt, E., and Engel, J. (1985). Evidence for coiled-coil alpha-helical regions in the long arm of laminin. EMBO J. 4, 309–316. doi: 10.1002/j.1460-2075.1985.tb03630.x
Perrin, A., Rousseau, J., and Tremblay, J. P. (2017). Increased Expression of Laminin Subunit Alpha 1 Chain by dCas9-VP160. Mol. Ther. Nucleic Acids 6, 68–79. doi: 10.1016/j.omtn.2016.11.004
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Pfaff, M., Gohring, W., Brown, J. C., and Timpl, R. (1994). Binding of purified collagen receptors (alpha 1 beta 1, alpha 2 beta 1) and RGD-dependent integrins to laminins and laminin fragments. Eur. J. Biochem. 225, 975–984. doi: 10.1111/j.1432-1033.1994.0975b.x
Philpot, J., Sewry, C., Pennock, J., and Dubowitz, V. (1995). Clinical phenotype in congenital muscular dystrophy: correlation with expression of merosin in skeletal muscle. Neuromuscul. Disord. 5, 301–305. doi: 10.1016/0960-8966(94)00069-l
Pierson, M., Cordier, J., Hervouuet, F., and Rauber, G. (1963). [an unusual congenital and familial congenital malformative combination involving the eye and kidney]. J. Genet. Hum. 12, 184–213.
Posteraro, P., De Luca, N., Meneguzzi, G., El Hachem, M., Angelo, C., Gobello, T., et al. (2004). Laminin-5 mutational analysis in an Italian cohort of patients with junctional epidermolysis bullosa. J. Invest. Dermatol. 123, 639–648. doi: 10.1111/j.0022-202x.2004.23302.x
Pulkkinen, L., Christiano, A. M., Airenne, T., Haakana, H., Tryggvason, K., and Uitto, J. (1994). Mutations in the gamma 2 chain gene (LAMC2) of kalinin/laminin 5 in the junctional forms of epidermolysis bullosa. Nat. Genet. 6, 293–297. doi: 10.1038/ng0394-293
Purvis, A., and Hohenester, E. (2012). Laminin network formation studied by reconstitution of ternary nodes in solution. J. Biol. Chem. 287, 44270–44277. doi: 10.1074/jbc.m112.418426
Reinhard, J. R., Lin, S., McKee, K. K., Meinen, S., Crosson, S. C., Sury, M., et al. (2017). Linker proteins restore basement membrane and correct LAMA2-related muscular dystrophy in mice. Sci. Transl. Med. 9:eaal4649. doi: 10.1126/scitranslmed.aal4649
Reuten, R., Patel, T. R., McDougall, M., Rama, N., Nikodemus, D., Gibert, B., et al. (2016). Structural decoding of netrin-4 reveals a regulatory function towards mature basement membranes. Nat. Commun. 7:13515.
Reuten, R., Zendehroud, S., Nicolau, M., Fleischhauer, L., Laitala, A., Kiderlen, S., et al. (2021). Basement membrane stiffness determines metastases formation. Nat. Mater. 20, 892–903. doi: 10.1038/s41563-020-00894-0
Ringelmann, B., Roder, C., Hallmann, R., Maley, M., Davies, M., Grounds, M., et al. (1999). Expression of laminin alpha1, alpha2, alpha4, and alpha5 chains, fibronectin, and tenascin-C in skeletal muscle of dystrophic 129ReJ dy/dy mice. Exp. Cell Res. 246, 165–182. doi: 10.1006/excr.1998.4244
Rousselle, P., and Beck, K. (2013). Laminin 332 processing impacts cellular behavior. Cell Adh. Migr. 7, 122–134. doi: 10.4161/cam.23132
Rousselle, P., Keene, D. R., Ruggiero, F., Champliaud, M. F., Rest, M., and Burgeson, R. E. (1997). Laminin 5 binds the NC-1 domain of type VII collagen. J. Cell Biol. 138, 719–728. doi: 10.1083/jcb.138.3.719
Ryan, M. C., Lee, K., Miyashita, Y., and Carter, W. G. (1999). Targeted disruption of the LAMA3 gene in mice reveals abnormalities in survival and late stage differentiation of epithelial cells. J. Cell Biol. 145, 1309–1323. doi: 10.1083/jcb.145.6.1309
Ryan, M. C., Tizard, R., VanDevanter, D. R., and Carter, W. G. (1994). Cloning of the LamA3 gene encoding the alpha 3 chain of the adhesive ligand epiligrin. Expression in wound repair. J. Biol. Chem. 269, 22779–22787. doi: 10.1016/s0021-9258(17)31713-1
Saad, M., Brkanac, Z., and Wijsman, E. M. (2015). Family-based genome scan for age at onset of late-onset Alzheimer’s disease in whole exome sequencing data. Genes Brain Behav. 14, 607–617. doi: 10.1111/gbb.12250
Sakai, N., Waterman, E. A., Nguyen, N. T., Keene, D. R., and Marinkovich, M. P. (2010). Observations of skin grafts derived from keratinocytes expressing selectively engineered mutant laminin-332 molecules. J. Invest. Dermatol. 130, 2147–2150. doi: 10.1038/jid.2010.85
Sampaolo, S., Napolitano, F., Tirozzi, A., Reccia, M. G., Lombardi, L., Farina, O., et al. (2017). Identification of the first dominant mutation of LAMA5 gene causing a complex multisystem syndrome due to dysfunction of the extracellular matrix. J. Med. Genet. 54, 710–720. doi: 10.1136/jmedgenet-2017-104555
Schneiders, F. I., Maertens, B., Bose, K., Li, Y., Brunken, W. J., Paulsson, M., et al. (2007). Binding of netrin-4 to laminin short arms regulates basement membrane assembly. J. Biol. Chem. 282, 23750–23758. doi: 10.1074/jbc.m703137200
Smyth, N., Vatansever, H. S., Meyer, M., Frie, C., Paulsson, M., and Edgar, D. (1998). The targeted deletion of the LAMC1 gene. Ann. N. Y. Acad. Sci. 857, 283–286. doi: 10.1111/j.1749-6632.1998.tb10133.x
Sugden, C. J., Iorio, V., Troughton, L. D., Liu, K., Bou-Gharios, G., and Hamill, K. J. (2020). Laminin N-terminus α31 expression during development is lethal and causes widespread tissue-specific defects in a transgenic mouse model. bioRxiv [Preprint]. doi: 10.1101/2020.07.26.221663
Talts, J. F., Sasaki, T., Miosge, N., Gohring, W., Mann, K., Mayne, R., et al. (2000). Structural and functional analysis of the recombinant G domain of the laminin alpha4 chain and its proteolytic processing in tissues. J. Biol. Chem. 275, 35192–35199. doi: 10.1074/jbc.m003261200
Tessier-Lavigne, M., and Goodman, C. S. (1996). The molecular biology of axon guidance. Science 274, 1123–1133. doi: 10.1126/science.274.5290.1123
Timpl, R., Tisi, D., Talts, J. F., Andac, Z., Sasaki, T., and Hohenester, E. (2000). Structure and function of laminin LG modules. Matrix Biol. 19, 309–317. doi: 10.1016/s0945-053x(00)00072-x
Troughton, L. D., Iorio, V., Shaw, L., Sugden, C. J., Yamamoto, K., and Hamill, K. J. (2020a). Laminin N-terminus α31 regulates keratinocyte adhesion and migration through modifying the organisation and proteolytic processing of laminin 332. bioRxiv [Preprint]. doi: 10.1101/617597
Troughton, L. D., Reuten, R., Sugden, C. J., and Hamill, K. J. (2020b). Laminin N-terminus alpha31 protein distribution in adult human tissues. PLoS One 15:e0239889. doi: 10.1371/journal.pone.0239889
Troughton, L. D., Zech, T., and Hamill, K. J. (2020c). Laminin N-terminus α31 is upregulated in invasive ductal breast cancer and changes the mode of tumour invasion. bioRxiv [Preprint]. doi: 10.1101/2020.05.28.120964
Utani, A., Nomizu, M., Timpl, R., Roller, P. P., and Yamada, Y. (1994). Laminin chain assembly. Specific sequences at the C terminus of the long arm are required for the formation of specific double- and triple-stranded coiled-coil structures. J. Biol. Chem. 269, 19167–19175. doi: 10.1016/s0021-9258(17)32290-1
Voit, T., Sewry, C. A., Meyer, K., Hermann, R., Straub, V., Muntoni, F., et al. (1995). Preserved merosin M-chain (or laminin-alpha 2) expression in skeletal muscle distinguishes Walker-Warburg syndrome from Fukuyama muscular dystrophy and merosin-deficient congenital muscular dystrophy. Neuropediatrics 26, 148–155. doi: 10.1055/s-2007-979745
Yurchenco, P. D., and Cheng, Y. S. (1994). Laminin self-assembly: a three-arm interaction hypothesis for the formation of a network in basement membranes. Contrib. Nephrol. 107, 47–56. doi: 10.1159/000422960
Yurchenco, P. D., McKee, K. K., Reinhard, J. R., and Ruegg, M. A. (2018). Laminin-deficient muscular dystrophy: molecular pathogenesis and structural repair strategies. Matrix Biol. 7, 174–187. doi: 10.1016/j.matbio.2017.11.009
Yurchenco, P. D., and Wadsworth, W. G. (2004). Assembly and tissue functions of early embryonic laminins and netrins. Curr. Opin. Cell Biol. 16, 572–579. doi: 10.1016/j.ceb.2004.07.013
Zenker, M., Aigner, T., Wendler, O., Tralau, T., Muntefering, H., Fenski, R., et al. (2004). Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum. Mol. Genet. 13, 2625–2632. doi: 10.1093/hmg/ddh284
Keywords: laminin, netrin, Pierson syndrome, MDC1A, basement membrane, junctional epidermolysis bullosa
Citation: Shaw L, Sugden CJ and Hamill KJ (2021) Laminin Polymerization and Inherited Disease: Lessons From Genetics. Front. Genet. 12:707087. doi: 10.3389/fgene.2021.707087
Received: 08 May 2021; Accepted: 13 July 2021;
Published: 12 August 2021.
Edited by:
Tom Van Agtmael, University of Glasgow, United KingdomReviewed by:
Patricia Rousselle, Centre National de la Recherche Scientifique (CNRS), FranceCopyright © 2021 Shaw, Sugden and Hamill. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kevin J. Hamill, a2hhbWlsbEBsaXZlcnBvb2wuYWMudWs=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.