 Wenqing Xia1,2†Zhumei Ni1†Zheng Zhang1
Wenqing Xia1,2†Zhumei Ni1†Zheng Zhang1 Hongfei Sang1,2Huifang Liu3
Hongfei Sang1,2Huifang Liu3 Zhenzhen Chen1,4Lin Jiang1,2
Zhenzhen Chen1,4Lin Jiang1,2 Congguo Yin1,2
Congguo Yin1,2 Jinyu Huang1,5
Jinyu Huang1,5 Lingfei Li1,2*Xiaoguang Lei6*
Lingfei Li1,2*Xiaoguang Lei6*- 1The Fourth School of Clinical Medicine, Zhejiang Chinese Medical University, Hangzhou, China
- 2Department of Neurology, Hangzhou First People's Hospital, Hangzhou, China
- 3Division of Neurology, Department of Medicine, University of Hong Kong, Hong Kong, SAR China
- 4Department of Hematology, Affiliated Hangzhou First People's Hospital, Zhejiang University, Hangzhou, China
- 5Department of Cardiology, Affiliated Hangzhou First People's Hospital, Zhejiang University School of Medicine, Hangzhou, China
- 6Department of Neurology, First Affiliated Hospital of Kunming Medical University, Kunming Medical University, Kunming, China
Introduction: Congenital muscular dystrophy (CMD) is a group of early-onset disorders with clinical and genetic heterogeneity. Patients always present with muscle weakness typically from birth to early infancy, delay or arrest of gross motor development, and joint and/or spinal rigidity. There are various genes related to the development of CMD. Among them, mutations in integrin alpha 7 (ITGA7) is a rare subtype. The identification of disease-causing genes facilitates the diagnosis and treatment of CMD.
Methods: We screened ITGA7 mutations in four people by whole exome sequencing and targeted sequencing from a consanguineous family. We then carried out electromyography and neuroelectrophysiological examinations to clarify a clinical picture of the patient diagnosed with CMD.
Results: We report a Chinese boy diagnosed with CMD who carries a homozygous variant (c.1088dupG, p.H364Sfs*15) of the ITGA7 gene. According to the genotype analysis of his family members, this is an autosomal recessive inheritance.
Conclusions: Our case further shows that ITGA7 mutation is related to CMD. Genetic counseling and multidisciplinary management of CMD play an important role in helping patients and their family. Further elucidation of the significant clinical and genetic heterogeneity, therapeutic targets, and the clinical care for patients remains our challenge for the future.
Introduction
Congenital muscular dystrophy (CMD) is a group of hereditary muscular dystrophy beginning at birth or early life. The clinical manifestations are postnatal hypotonia and motor development, joint contracture, and increased serum creatine kinase (CK) (Bonne et al., 2018). Initially, CMD has been diagnosed based on clinical features and histopathology. Nowadays, CMDs are classified into different subtypes according to the pathogenetic genes (Hayashi et al., 1998). According to the 2019 version of the GeneTable, there are 34 genes related to CMD phenotypes (Bonne et al., 2018). Among them, laminin subunit alpha 2–related (merosin deficient), collagen VI–related, and α-dystroglycan–related CMDs are the most common types. The underlying mechanisms are predominantly related to disruption of components of the muscle extracellular matrix and its interaction with the sarcolemmal membrane. CMD caused by mutation in integrin α7 (ITGA7) is a rare subtype. Only a few patients diagnosed with CMD were found to have ITGA7 mutation (Hayashi et al., 1998; Esposito et al., 2013; Yu et al., 2017; Karaca et al., 2018). Here, we report a rare case of a boy from a consanguineous family diagnosed with CMD caused by ITGA7 mutation.
Case Description
The proband is the second child in the family, aged 10 years old. The boy presented with muscle weakness at the age of 3 years old and the symptoms were slowly progressive. He had difficulties in squatting in daily life and sit-ups with knee flexed in excise. He walked on tiptoes. Physical examination showed proximal muscular atrophy of both limbs (Figure 1A). The patient walked in a gait indicating proximal muscular atrophy. Lab investigation showed significantly increased serum creatine kinase (CK) activity (286 U/L, normal = 24–195 U/L). We also found increased red blood cell count (6.22 × 1012/L, normal range: 3.5–5.5 × 1012/L), decreased mean cell volume (MCV, 65.60 fl, normal range: 82–95 fl) and mean cell hemoglobin (MVH, 21.20 pg, normal range: 27–31 pg), and increased platelets (PLT). The serum electrolytes, calcium, lactate, and thyroid function test results were normal. EMG showed multiple lesions and most muscles showed neurogenic lesion, especially in both quadriceps femoris. There was muscular lesion in part of the tibialis anterior muscle. There was no abnormality in the cardiovascular or respiratory system. Echocardiography was normal. Muscle biopsy was not performed due to parental preference. Whole exome sequencing showed that the patient has a homozygous variant (c.1088dupG, p.H364Sfs*15) of the ITGA7 gene.
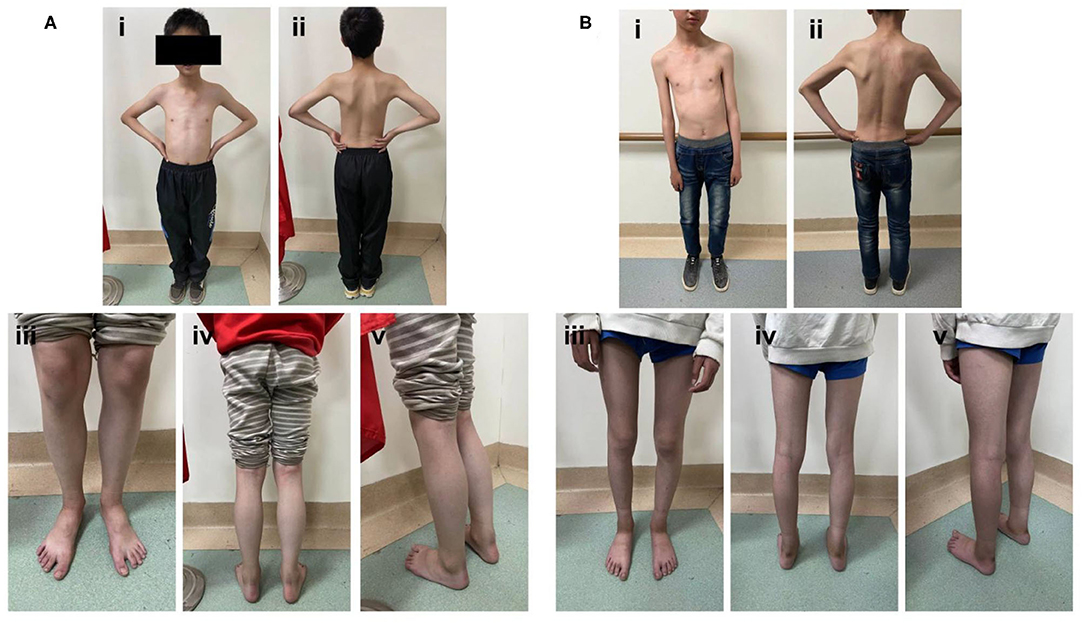
Figure 1. Photographs of the proband at 10 years old. (A, B) The proband presented with obvious limb muscular atrophy and waddling gait. A neurological examination showed proximal muscle weakness.
The big brother of the proband presented with recurrent pain of the lower limbs, mild muscle wasting, and weakness of the upper limbs. He was thin and unable to unscrew a bottle cap. He had no difficulty in walking and running. Physical examination found obvious scoliosis and atrophy of the limb muscle (Figure 1B). Lab investigation showed increased red blood cell count (6.42 × 1012/L, normal range: 4.2–5.5 × 1012/L), decreased MCV and MVH, and increased PLT. Slightly increased angiotensin-converting enzyme (ACE, 89.5 U/L, normal range: 5.0–89.0 U/L) marked increased alkaline phosphatase (ALP, 324.1 IU/L, normal range: 45.0–125.0 IU/L), normal CK (102.0 IU/L, normal range: 50.0–310.0 IU/L), and slightly increased creatine phosphokinase myocardial band (CK-MB, 24.1 U/L, normal range: <24.0 U/L). NCV showed that the brother has left quadriceps muscular disruption. Others (MRI of thoracic vertebra, lower limbs) are normal. The symptoms were relieved after vitamin B12 treatment. Two kids are being followed up at a pediatric tertiary care hospital with physiotherapy and rehabilitative care. Their parents had no symptoms and Sanger sequencing confirmed that they have heterozygous mutation in ITGA7 (Figures 2, 3).
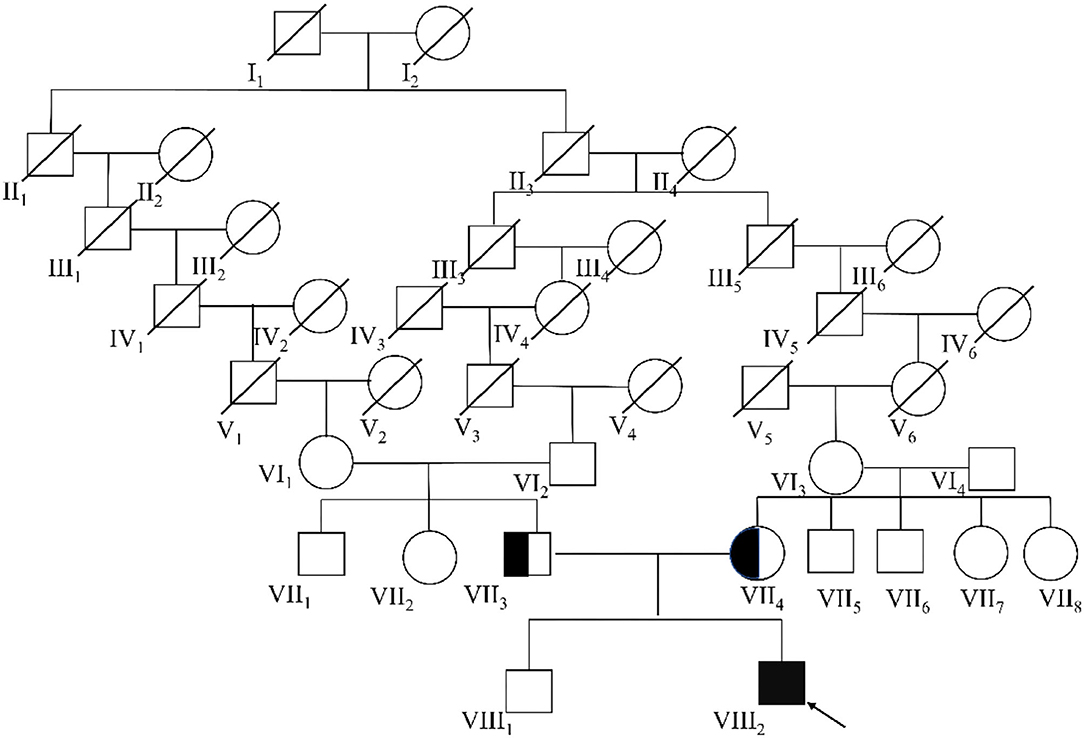
Figure 2. The complete eight-generational Chinese pedigree. The proband affected by ITGA7 is indicated with an arrow and a dark box.
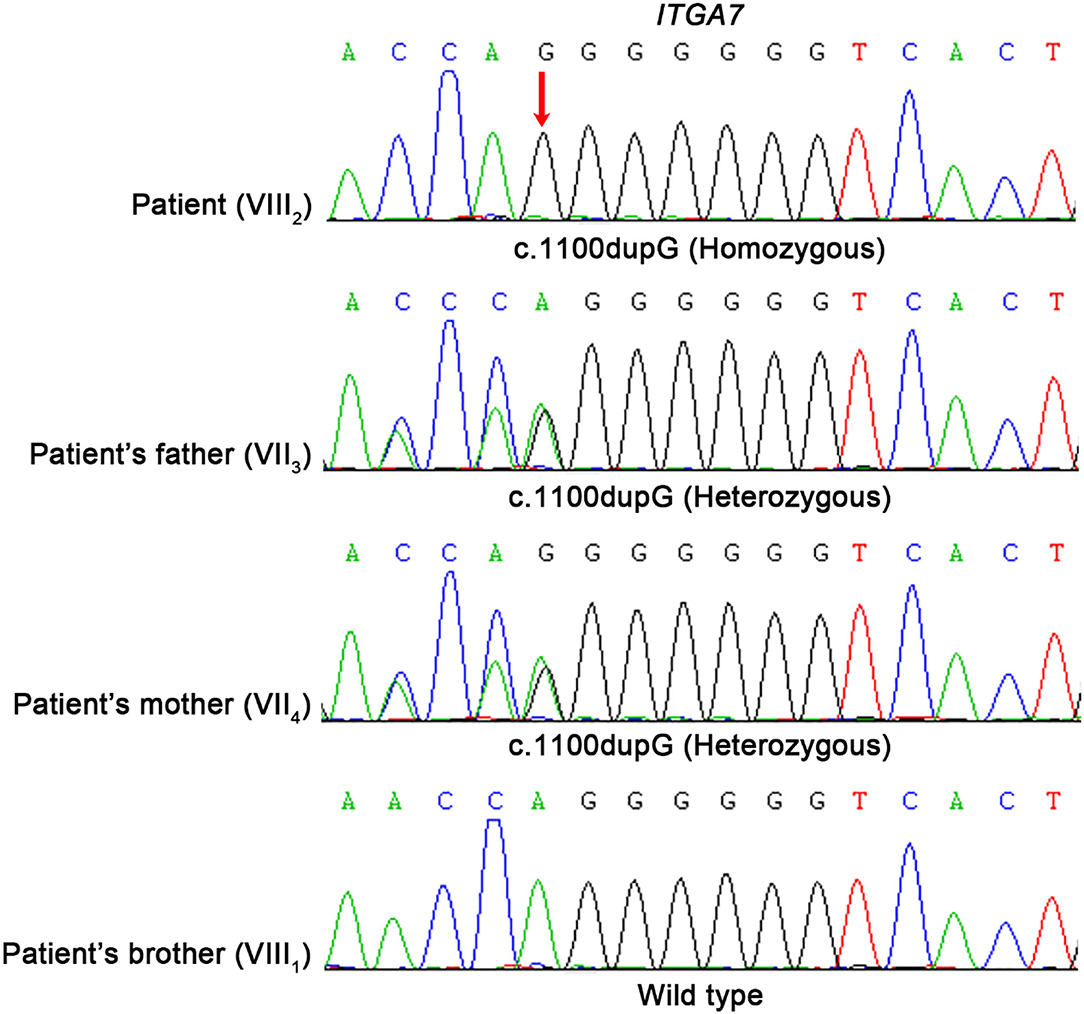
Figure 3. Molecular analysis of ITGA7 gene mutation. Mutation analysis of the ITGA7 gene. Red arrowhead marks the sites of base alterations. The DNA sequences of the family subjects are shown; the pedigree number is indicated on the left.
Methods
Clinical Assessment
The clinical assessment of the patient comprised a neurological behavior examination and evaluation of his nerve conduction velocity is detailed in Supplementary Tables 1, 2.
Whole Exome Sequencing
Peripheral blood (5 ml) of each child and their parents was collected. DNA was extracted using whole blood magnetic bead purification kit. Full exon genes were captured by IDTXGen Exome Research Panel v1.0 and sequenced on Illumina Novaseq 6000 platform. Afterwards, the sequencing data were evaluated according to Illumina Sequence Control Software (SCS) and analyzed as follows. The original sequencing data were deconstructed, removing the joint sequences, and then filtered and aligned to the NCBI database of human reference genome (hg19) using the BWA software (http://bio-bwa.sourceforge.net/). The single-nucleotide mutation [single-nucleotide variation (SNV)] and absence of insertion mutation (inserts and deletions, INDEL) were called using GATK software (https://software.broadinstitute.org/gatk/) and annotated by ANNOVAR software (http://annovar.openbioinformatics.org/en/latest/). All candidate variants were filtered first against 1,000 genomes project database, for a minor allele frequency (MAF) ≥ 1%, and ExAC hom AC ≥ 3. Obtained variants were further selected according to co-segregation, genetic model, and a MAF <1% in three databases (1,000 genomes project_EAS, ExAC, and gnomAD_EAS). After analyzing the sequencing results as above, a variant of ITGA7 was found in the child, which had not been reported in the HGMDPRO database.
Sanger Sequencing
The candidate causal genes discovered via WES were then confirmed by Sanger sequencing and co-segregation analyses among the family were also conducted. The primers were designed using Primer Premier 5.0 (Premier Biosoft, USA) and PCR was carried out to amplify the fragments covering the mutated sites on LifeECO Thermal Cycler TC-96/G/H(b)C (Bioer Technology Co. Ltd., China). The PCR products were further purified with agarose gel electrophoresis and then sequenced by ABI 3730XL DNA Sequencer (Applied Biosystems, Thermo Fisher Scientific, USA). Sanger sequencing results were analyzed by Chromas Lite v2.01 (Technelysium Pty Ltd., Tewantin, QLD, Australia).
Discussion and Conclusion
To date, only a few ITGA7 variants have been reported, and it is not clear whether all variants are pathogenic mutations (Table 1). In 1998, Hayashi et al. first described three patients with CMD, two of whom carried the ITGA7 mutation and one did not. The first patient had one 21-bp insertion mutation on one allele and a 98-bp deletion on the other allele of the ITGA7 gene, which were both splice mutations. The second patient had the same 98-bp deletion and had a 1-bp frame-shift deletion in the other allele, which is a compound heterozygote. The third patient showed a marked deficiency in the ITGA7 mRNA, but no mutations in the coding region were described (Hayashi et al., 1998). The three patients reported were affected with congenital myopathy with variable clinical phenotype, and all showed a complete absence of integrin α7 in their muscle biopsies due to primary integrin α7 nonsense/splicing mutations or to a downregulation of integrin α7 mRNA (Hayashi et al., 1998). In 2013, Esposito et al. reported a female with homozygous missense mutations in two genes, the myosin heavy chain 7B and the ITGA7, who presented with complicated congenital myopathy with left ventricular non-compact cardiomyopathy (Esposito et al., 2013). Later in 2017, Yu et al. described a female with proximal muscle weakness, who had c.2701A>G and c.1828G>A in ITGA7 (Yu et al., 2017). In 2018, Karaca et al. found an individual with a deleterious variant in ITGA7, conferring a molecular diagnosis of CMD. This proband has microcephaly, agenesis of the corpus callosum, cerebellar hypoplasia, seizures, horseshoe kidney, scoliosis, hemivertebrae, asymmetric extremities, and hypopigmented skin macules (Karaca et al., 2018). Although the case described above had various symptoms and different grades of severity, the consistent clinical features were muscle weakness and increased CK level, which were also found in our proband. Among the published studies, patients reported by Esposito's team had combined mutations (Esposito et al., 2013). This condition was not found in our patient.
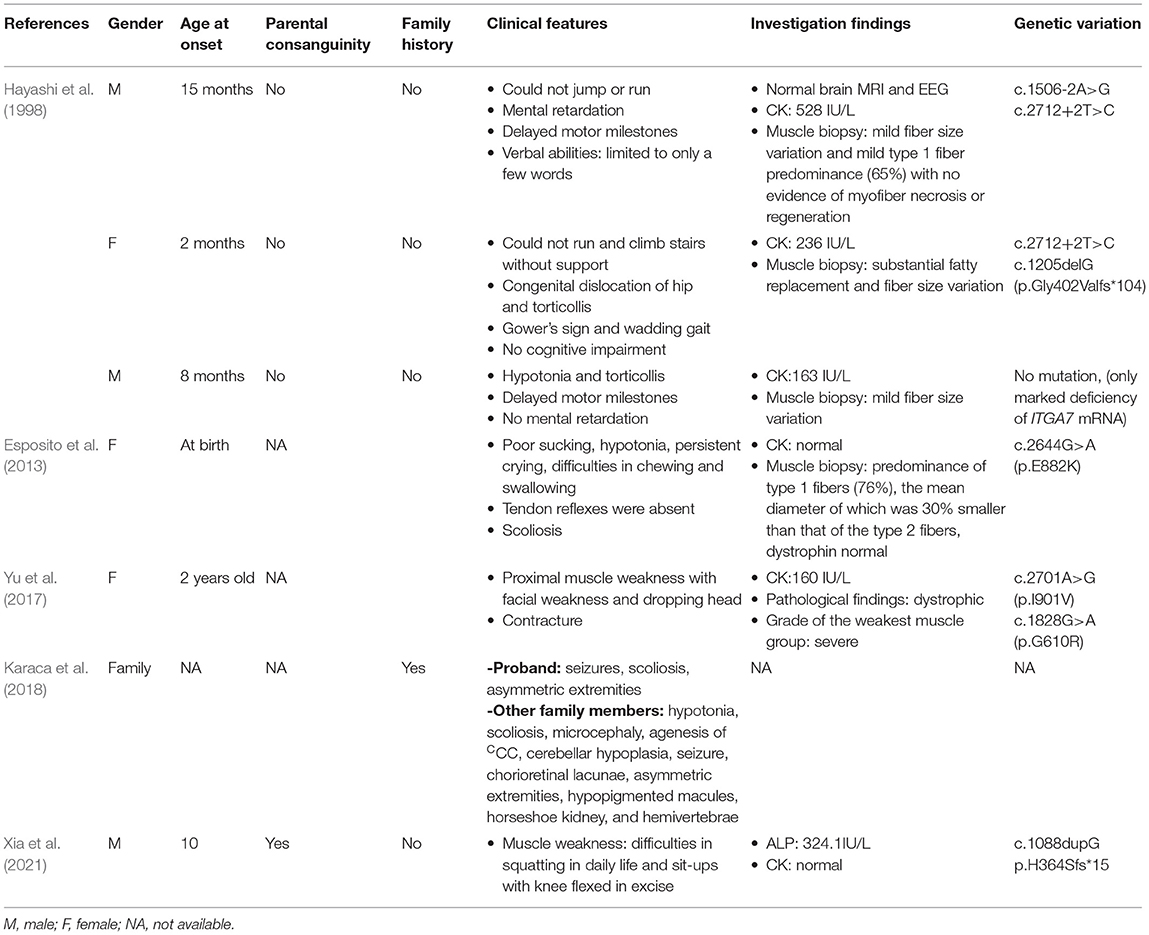
Table 1. Characteristics of CMD induced by ITGA7 mutation (NM_002206.3).
Integrins are a group of heterodimeric integral membrane glycoproteins with diverse combinations of α and β subunits. They mediate cell-to-cell and cell-to-matrix interactions, thus playing roles in cell migration, morphologic development, differentiation, and metastasis (Di Maggio et al., 2017). ITGA7 (GenBank: NG_012343.1) is on chromosome 12. This gene has 28 exons, among which 26 code for protein. ITGA7 has been reported to be associated with various types of cancers (Guan et al., 2020), stem cell differentiation (Zhang et al., 2019), and skeletal muscle development (Rooney et al., 2012; McClure et al., 2016). Mice lacking integrin α7 have demonstrated impaired muscle healing after cardiotoxin injury (Rooney et al., 2012). Previous studies indicate that the integrin α7 subunit is upregulated during myoblast differentiation. Myoblasts with silenced integrin α7 were found to regulate myogenic differentiation and demonstrate defective fusion (McClure et al., 2016). Loss of integrin α7 exacerbates a newly discovered muscle phenotype in mice lacking major adhesion complexes in skeletal muscle (Marshall et al., 2012). ITGA7-expressing muscle-resident glial cells can be activated by loss of neuromuscular junction integrity (Proietti et al., 2021). ITGA7-expressing muscle-resident glial cells are activated by loss of neuromuscular junction integrity, indicating α7 in late differentiation (Proietti et al., 2021). The homozygous mutation found in our study led to a shift in the reading frame, resulting in truncating proteins with 15 miscoded amino acids (Supplementary Figure 1). The expression of biologically functional proteins was absent, causing CMD in our patient as a result. However, we did not obtain muscle biopsy, so we cannot prove that the patient lacks the ITGA7 protein.
Despite the recent advances in our understanding of the molecular basis of neuromuscular disorders, the underlying molecular defect can be identified only in a subset of the cases. CMD present with a wide spectrum of severity and onset. Progression and other features are variable depending on the subtype and severity of the specific genetic mutation.
Data Availability Statement
The datasets for this article are not publicly available due to concerns regarding participant/patient anonymity. Requests to access the datasets should be directed to the corresponding author.
Ethics Statement
The studies involving human participants were reviewed and approved by Review Board of the Affiliated Hangzhou First People's Hospital, Zhejiang University School of Medicine. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
WX, ZN, and LL designed the study. ZZ, HS, and ZC performed the genetic analysis and bioinformatics evaluations. LL and XL drafted the manuscript. LJ, CY, and JH conducted the clinical evaluations. All authors analyzed the data and approved the final manuscript.
Funding
Yunnan provincial medical reserve talents (H-2017032), Zhejiang Traditional Chinese Medicine Science and Technology Plan (2021ZQ074), and Zhejiang Provincial Medical and health Science and Technology Plan (2021KY849).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to the patient and his family. We are grateful to Running Gene Inc. (Beijing, China) for their technical assistance. Dr. LL thanks Dr. Xiaoli Si for her professional knowledge and warm support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.706823/full#supplementary-material
Supplementary Figure 1. Schema of integrin alpha-7 with the mutation found in our patient. This homozygous mutation leads to a shift in the reading frame, resulting in truncating proteins with 15 miscoded amino acids.
Supplementary Table 1. Nerve conduction studies of the proband. L, left; R, right; Lat, latency; Amp, amplitude; Dis, distance; CV, conduction velocity.
Supplementary Table 2. Nerve conduction studies of the brother of the proband. L, left; R, right; Lat, latency; Amp, amplitude; Dis, distance; CV, conduction velocity.
References
Bonne, G., Rivier, F., and Hamroun, D. (2018). The 2019 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul. Disord. 28, 1031–1063. doi: 10.1016/j.nmd.2018.09.006
Di Maggio, N., Martella, E., Frismantiene, A., Resink, T. J., Schreiner, S., Lucarelli, E., et al. (2017). Extracellular matrix and alpha5beta1 integrin signaling control the maintenance of bone formation capacity by human adipose-derived stromal cells. Sci. Rep. 7:44398. doi: 10.1038/srep44398
Esposito, T., Sampaolo, S., Limongelli, G., Varone, A., Formicola, D., Diodato, D., et al. (2013). Digenic mutational inheritance of the integrin alpha 7 and the myosin heavy chain 7B genes causes congenital myopathy with left ventricular non-compact cardiomyopathy. Orphanet. J. Rare Dis. 8:91. doi: 10.1186/1750-1172-8-91
Guan, Y., Bhandari, A., Xia, E., Kong, L., Zhang, X., and Wang, O. (2020). Downregulating integrin subunit alpha 7 (ITGA7) promotes proliferation, invasion, and migration of papillary thyroid carcinoma cells through regulating epithelial-to-mesenchymal transition. Acta Biochim. Biophys. Sin. (Shanghai) 52, 116–124. doi: 10.1093/abbs/gmz144
Hayashi, Y. K., Chou, F. L., Engvall, E., Ogawa, M., Matsuda, C., Hirabayashi, S., et al. (1998). Mutations in the integrin alpha7 gene cause congenital myopathy. Nat. Genet. 19, 94–97. doi: 10.1038/ng0598-94
Karaca, E., Posey, J. E., Coban Akdemir, Z., Pehlivan, D., Harel, T., Jhangiani, S. N., et al. (2018). Phenotypic expansion illuminates multilocus pathogenic variation. Genet. Med. 20, 1528–1537. doi: 10.1038/gim.2018.33
Marshall, J. L., Chou, E., Oh, J., Kwok, A., Burkin, D. J., and Crosbie-Watson, R. H. (2012). Dystrophin and utrophin expression require sarcospan: loss of alpha7 integrin exacerbates a newly discovered muscle phenotype in sarcospan-null mice. Hum. Mol. Genet. 21, 4378–4393. doi: 10.1093/hmg/dds271
McClure, M. J., Clark, N. M., Hyzy, S. L., Chalfant, C. E., Olivares-Navarrete, R., Boyan, B. D., et al. (2016). Role of integrin alpha7beta1 signaling in myoblast differentiation on aligned polydioxanone scaffolds. Acta Biomater. 39, 44–54. doi: 10.1016/j.actbio.2016.04.046
Proietti, D., Giordani, L., De Bardi, M., D'Ercole, C., Lozanoska-Ochser, B., Amadio, S., et al. (2021). Activation of skeletal muscle-resident glial cells upon nerve injury. JCI Insight. 6:e143469. doi: 10.1172/jci.insight.143469
Rooney, J. E., Knapp, J. R., Hodges, B. L., Wuebbles, R. D., and Burkin, D. J. (2012). Laminin-111 protein therapy reduces muscle pathology and improves viability of a mouse model of merosin-deficient congenital muscular dystrophy. Am. J. Pathol. 180, 1593–1602. doi: 10.1016/j.ajpath.2011.12.019
Xia, W.Q., Ni, Z., Zhang, Z., Sang, H., Liu, H., Chen, Z., et al. (2021). Case report: a boy from a consanguineous family diagnosed with congenital muscular dystrophy caused by integrin alpha 7 (ITGA7) mutation. Front. Genet. 12:706823. doi: 10.3389/fgene.2021.706823
Yu, M., Zheng, Y., Jin, S., Gang, Q., Wang, Q., Yu, P., et al. (2017). Mutational spectrum of Chinese LGMD patients by targeted next-generation sequencing. PLoS ONE. 12:e0175343. doi: 10.1371/journal.pone.0175343
Keywords: congenital muscular dystrophy, a consanguineous family, rare genetic mutation, genetic consultation, integrin alpha 7
Citation: Xia W, Ni Z, Zhang Z, Sang H, Liu H, Chen Z, Jiang L, Yin C, Huang J, Li L and Lei X (2021) Case Report: A Boy From a Consanguineous Family Diagnosed With Congenital Muscular Dystrophy Caused by Integrin Alpha 7 (ITGA7) Mutation. Front. Genet. 12:706823. doi: 10.3389/fgene.2021.706823
Received: 08 May 2021; Accepted: 30 July 2021;
Published: 06 September 2021.
Edited by:
Rita Selvatici, University of Ferrara, ItalyReviewed by:
Jun Mitsui, The University of Tokyo, JapanDean J. Burkin, University of Nevada, Reno, United States
Copyright © 2021 Xia, Ni, Zhang, Sang, Liu, Chen, Jiang, Yin, Huang, Li and Lei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lingfei Li, bGVlbGluZ2ZlaUAxMjYuY29t; Xiaoguang Lei, bGVpeGlhb2d1YW5nQHpqdS5lZHUuY24=
†These authors have contributed equally to this work