 Davide Simone
Davide Simone
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Genet., 22 September 2021
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.703242
This article is part of the Research TopicGenetic Studies on Spondyloarthritis: from Disease Predictors to Therapeutic TargetsView all 10 articles
In Spondyloarthropathies (SpA), a common group of immune-mediated diseases characterised by excessive inflammation of musculo-skeletal structures and extra-articular organs, T helper 17 (Th17) cells are widely considered the main drivers of the disease. Th17 are able to modulate their genes according to the immune environment: upon differentiation, they can adopt either housekeeping, anti-bacterial gene modules or inflammatory, pathogenic functions, and only the latter would mediate immune diseases, such as SpA. Experimental work aimed at characterising Th17 heterogeneity is largely performed on murine cells, for which the in vitro conditions conferring pathogenic potential have been identified and replicated. Interestingly, Th17 recognising different microorganisms are able to acquire specific cytokine signatures. An emerging area of research associates this heterogeneity to the preferential metabolic needs of the cell. In summary, the tissue environment could be determinant for the acquisition of pathogenetic features; this is particularly important at barrier sites, such as the intestine, considered one of the key target organs in SpA, and likely a site of immunological changes that initiate the disease. In this review, we briefly summarise genetic, environmental and metabolic factors that could explain how homeostatic, anti-microbial Th17 could turn into disease-causing cells in Spondyloarthritis.
T helper lymphocytes, characterised by the expression of CD4 on their surface, are the central cell subset of adaptive immunity. They are able to recognise protein antigens belonging to microorganisms thanks to a unique receptor expressed on their surface [T-cell receptor (TCR)] and shape an organised response against them. When a self-antigen is erroneously recognised, or when the activation threshold is altered, CD4+ T cells can cause pathological responses, characterised by uncontrolled inflammation. Together with the recognition of the antigen and the TCR engagement signal, integrated stimuli dictate the transcriptional changes that guide the differentiation of the naïve T cell towards a specialised function. These stimuli include cytokines, soluble mediators or bacterial products in the microenvironment. Specific intracellular pathways, including Stat (signal transducer and activator of transcription) proteins, regulate this process, which eventually leads to the induction of a dominant transcription factor (TF). The lineage-specific (‘master’) TF controls the transcriptional programme of the cell, including specific cytokine production and the expression of chemokine receptors that mediate trafficking to the organs: this network helps each T-cell subset to exert specific functions in response to antigens, and in the tissues. The central TFs are as: T-box protein expressed in T cells (T-bet) in Th1, GATA-binding protein 3 in Th2, retinoic acid-related orphan receptor gamma-t (ROR-ɣt) in T helper 17 (Th17) and Forkhead box P3 in Tregs (Zheng and Flavell, 1997; Szabo et al., 2000; Fontenot et al., 2003; Harrington et al., 2005).
Dysregulated mechanisms in various steps of T-cell commitment, maturation and response to challenges can contribute to pathogenic responses, such as immune-mediated conditions for Th1 and Th17 cells, and allergic responses for Th2 cells. Among immune-mediated diseases driven by Th17 cells are Spondyloarthropathies (SpA), a group of inflammatory arthritides, including Ankylosing Spondylitis (AS) and Psoriatic Arthritis (PsA), characterised by inflammation and structural damage of several musculo-skeletal structures and organs. In these diseases, genetic and immunological alterations suggest a dysregulated ‘type 17’ response (Simone et al., 2018), that is the arm of cellular immunity characterised by the production of IL-17 and orchestrated by Th17. The focus of this short review will be on Th17 cells, and the genetic and environmental mechanisms that likely determine their pathogenic behaviour thought to cause SpA.
The cytokine environment provides an important contribution to the T naïve fate decision. Although being a disputed matter for years, owing to between-species differences and experimental settings, it is accepted that the process of differentiation towards Th17 requires the presence of IL-6, IL-1β and variable concentrations of TGF-β (Bettelli et al., 2006; Veldhoen et al., 2006; Zhou et al., 2007). The IL-6 receptor, in particular, activates the JAK1/STAT3 pathway to induce the lineage defining TF ROR-ɣt (Yang et al., 2007). The STAT3/ROR-ɣt axis is the cornerstone of Th17 differentiation, but more TFs come into play in the process, such as RORa, Ahr, IRF4 and BATF (Brüstle et al., 2007; Schraml et al., 2009), with the last two possibly induced even before ROR-ɣt, characterising a sort of ‘pre-Th17’ status (Ciofani et al., 2012). These markers set the initial chromatin accessibility that allows a transcriptional programme, further defined by ROR-ɣt and decisive for the expression of effector genes (Il17a and Il17f). As multiple players are involved in the differentiation at different stages, the previously accepted paradigm of T helper differentiation as a linear, irreversible process is not actual. As demonstrated by knock down experiments, inactivation of these regulatory nodes leaves space to transcriptional instability and plasticity: in this scenario, the cytokine milieu could favour, or inhibit, molecular determinants at various stages of the differentiation phase, or even divert the programme towards other T helper lineage. TGF-β is a paradigmatic case: initially identified as essential for murine Th17 differentiation (Mangan et al., 2006), but not for human Th17 cells (Acosta-Rodriguez et al., 2007), and subsequently reinstated as necessary for both human and murine differentiation (Manel et al., 2008; Volpe et al., 2008). In practice, according to a parallel interpretation, two different cytokine cocktails lead to two different Th17 ‘flavours’: TGF-β, together with IL-6, induces ‘non-pathogenic’ Th17 cells characterised by the co-expression of IL-10 (McGeachy et al., 2007); on the other hand, IL-6 and IL-23 (and no TGF-β) lead to differentiation into ‘pathogenic’ Th17 cells (Ghoreschi et al., 2010; Figure 1). Both subsets would express ROR-ɣt, but ‘pathogenic’ Th17 cells, more plastic and polymorphic, have a tendency to transition towards Th1, and consequently the production of IFN-ɣ. This distinction, popularised by studies that use in vitro differentiation assays of murine Th17, likely does not find strict correspondence in vivo. In experimental autoimmune encephalomyelitis (EAE), Th17 cells grown in presence of TGF-β are, in fact, also pathogenic (Kebir et al., 2007). It is also difficult to conceive conditions completely devoid of TGF-β, very common in plasma and tissues in vivo, and often present (in active or inactive form) in the serum enriching in vitro culture media. What might be crucial is the concentration, and the gradient, of TGF-β: high concentrations of TGF-β induce Treg-associated genes while restraining T-bet and other Th1 genes and possibly inhibit Th17 pathogenic responses. The developmental overlap between Th1, Th17 and Treg might in fact be caused by complex cytokine dynamics. At the opposite side of the spectrum, IL-23 has long been considered the main ingredient for pathogenic differentiation and the initiator of the pro-inflammatory module.
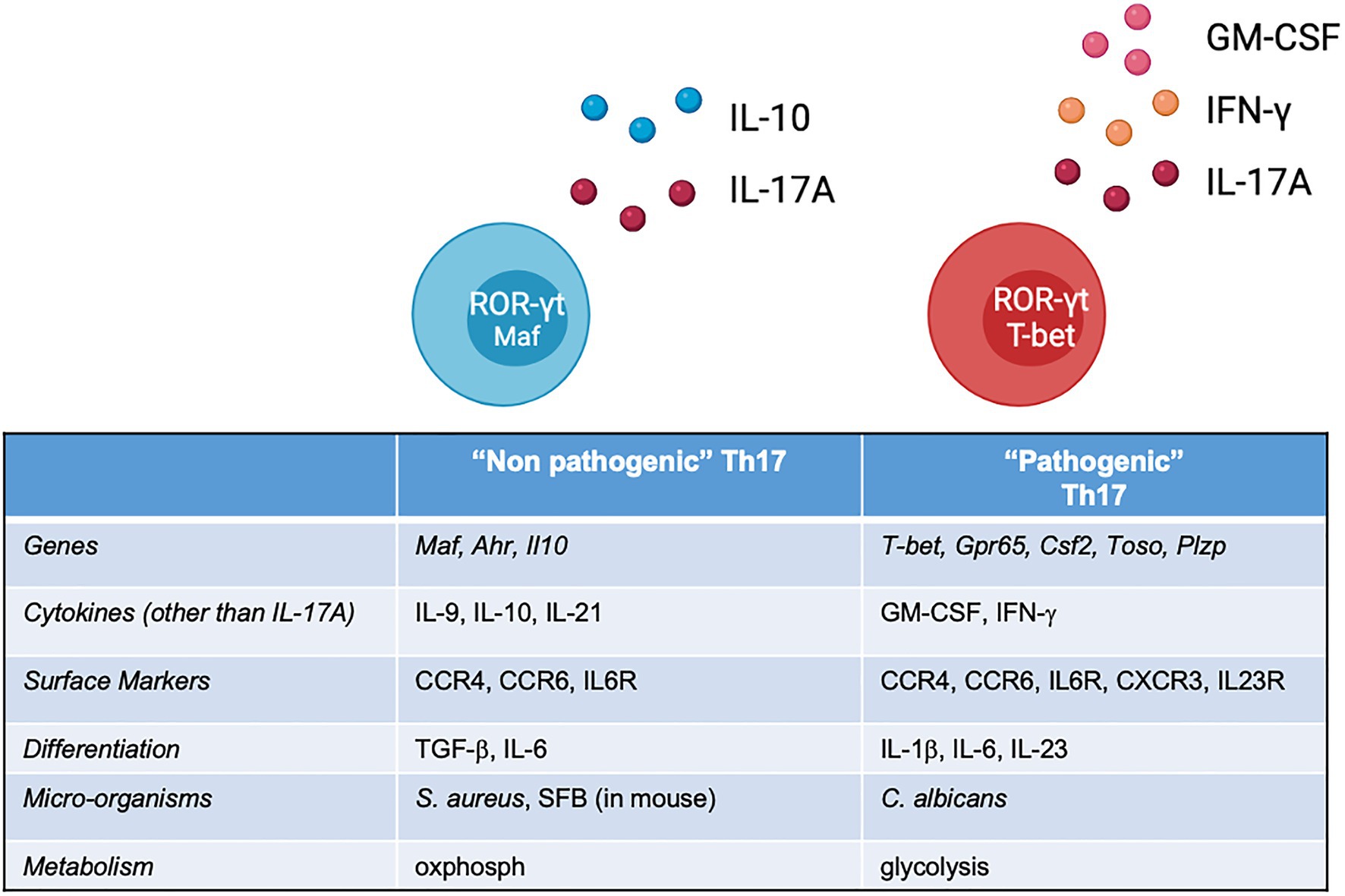
Figure 1. List of features of ‘non-pathogenic’ and ‘pathogenic’ Th17 cells identified across several studies, most of which performed on murine cells. In vivo, this classification should not be considered bimodal, but rather a continuum of states, shaped by the local environment.
There is considerable evidence that indicates a central role of Th17 cells in the pathogenesis of SpA. The proportion of Th17 is higher in the blood of AS patients (Shen et al., 2009) and so are the serum concentrations of IL-17A, as reported in a number of studies by different groups (reviewed in Taams et al., 2018). In addition to experimental evidences, current clinical practice offers the empirical demonstration of the importance of ‘type 17’ lymphocytes in the pathogenesis of SpA: secukinumab (an anti-IL-17A monoclonal antibody) is currently widely used in AS and PsA, while a number of novel agents against IL-17 superfamily cytokines are in the final experimental stages or about to be licenced for clinical use (van der Heijde et al., 2018, 2020).
An important series of evidences originates from genetic studies: Genome-Wide Association Studies (GWAS; International Genetics of Ankylosing Spondylitis Consortium (IGAS) et al., 2013) have convincingly established the association between AS and genetic variants in loci involved in the Th17 pathway, including TYK2, IL6R, IL1R1, IL1R2 and IL23R, whose products are important for the induction and survival of the Th17 response. In particular, the existence of a genetic variant of the IL-23 receptor gene, possibly causing the altered signalling predisposing to AS, emerged early from one of the first association studies (Wellcome Trust Case Control Consortium et al., 2007). Although one AS-associated SNP (rs11209026, also seen in psoriasis and IBD) causes a missense variation that alters IL-23R signalling, impairing Th17 responses (Di Meglio et al., 2011, 2013), the effect of the other gene variants is unclear. Recently, epigenetic data on chromatin remodelling and transcription factor (TF) binding helped to characterise a second independently AS-associated SNP located in the IL23R-IL12RB2 intergenic region. The observed higher number of IFN-g-secreting cells in the risk allele carriers (Roberts et al., 2016) could in fact explain the functional association with the disease. Although this work could not confirm this alteration is present on Th17 specifically, it provides an example of how allelic variations affect the genetic/epigenetic regulation of inflammatory pathways, thus potentially induce pathogenic functions in Th17 cells. Other genes, whose variants associate with AS, have been carefully studied in the search of an alteration of Th17 responses that can cause AS. STAT3 and TYK2, for example are mediators of Th17 immunity. Their protein products play a central role in IL-6 and IL-23 signalling: they are activated by phosphorylation and indirectly act as transcriptional activators of Th17 differentiation, via a chain of other intermediates. Several SNPs within STAT3 locus have been associated to AS and CD (Danoy et al., 2010), but their impact on Th17 function in these conditions has never been clarified, while different causing SNPs of STAT3 showed to impact Th17 differentiation in other autoimmune diseases (Tripathi et al., 2017). Another important GWAS hit, involving a gene involved in Th17 responses, is TYK2. The AS-associated SNPs (rs12720356) at the TYK2 locus were found to be associated with increased Th1 frequency and AS disease progression: this, together with the evidence that Tyk2 inhibition is an effective strategy in the experimental model of AS (Gracey et al., 2020), makes the search for functional variants of genes that orchestrate the Th17 response a biologically relevant enterprise, that is also very promising from a translational point of view.
IL-23, a heterodimeric cytokine, is closely related to IL-12, with whom it shares one of the two subunits. Consistent with its kinship to IL-12, it is able to induce both IL-17 and IFN-γ (Aggarwal et al., 2003) in CD4+ cells. More precisely, it is dispensable for Th17 development, but it enhances an accessory transcriptional module, thought to be pathogenic (Volpe et al., 2008). Indeed, IL-23 persistence induces Th17 to acquire a Th1-like phenotype (Lee et al., 2009), and lack of IL-23R on T cells prevents experimental colitis (Ahern et al., 2010). IL-23, together with IL-1, has been associated with Th17 in autoimmune models (Cua et al., 2003; Langrish et al., 2005; McGeachy et al., 2009). In particular, IL-23 is the key ingredient of pathogenicity in EAE because it induces high levels of T-bet, IL-23R and GM-CSF in the Th17 cell (Lee et al., 2012). Importantly, IL-23R is not constitutively expressed on the naïve T cell, but it appears during the Th17 differentiation secondarily to IL-6 signalling (Chung et al., 2009; Ghoreschi et al., 2010). Through this and other feed forward mechanisms (Kishi et al., 2016; Meyer Zu Horste et al., 2016), the Th17 inflammatory programme is stabilised.
The aforementioned association of genetic variants of the IL23R gene region with AS (International Genetics of Ankylosing Spondylitis Consortium (IGAS) et al., 2013) led to hypothesise that IL-23 was the main driver for pathogenic responses in AS as well. Early experimental data in humans did not confirm this: IL-23 blockade proved ineffective on the axial symptoms in AS (Baeten et al., 2018). Few hypotheses have been proposed to explain this: IL-23 might still be a key player in the initiation phase, losing importance when the disease is already established. An alternative explanation postulates the presence of other IL-23-independent drivers of Th17 pathogenicity. In reality, immune cytokine ‘axes’ (such as IL-23/17) are almost never linear: IL-23 and IL-17 do participate in the same branch of the immune response but at the same time, have distinct biology and affect different cell subsets in different tissues. Regardless of its induction, we now know that the effector functions of the Th17 pathogenic module consist in IL-17A plus additional inflammatory cytokines, such as IFN-ɣ and GM-CSF, the latter an emerging Th17-associated player of autoimmunity (El-Behi et al., 2011). Recent translational approaches in SpA are focusing on novel cytokine targets, such as GM-CSF (Al-Mossawi et al., 2017; Wade et al., 2019), or upstream pro-inflammatory intracellular signals, such as JAK/STAT (Hammitzsch et al., 2018), a strategy that would simultaneously regulate cell differentiation and inhibit multiple inflammatory cytokines.
Other genetic events leading to unrestrained, disease-causing differentiation are still not clear: ex vivo single-cell sequencing led to identify novel pathogenic genes (Gpr65, Toso and Plzp) promoting Th17-mediated CNS inflammation in EAE mice (Gaublomme et al., 2015; Wang et al., 2015), but none of which, as of today, have been validated in humans, with the partial exception of GPR65, an important TF in human GM-CSF + Th17 (Al-Mossawi et al., 2017). The gene, a GWAS hit associated with AS, encodes for a G-protein coupled receptor with an extracellular proton sensing domain, and strongly characterises GM-CSF-producing Th17, where its activation likely concurs to the production of said cytokine in the acidic environment, expanding the inflammatory potential of the Th17 cell.
Not only cytokines, but also different microorganisms have the ability to prime distinct Th17 responses (McGeachy and McSorley, 2012). A study in humans showed, through in vitro experiments, that Th17 cells primed with different pathogens are able to acquire specific cytokine signatures (Zielinski et al., 2012). Th17 are inherently plastic having evolved to tailor their response to different pathogens at different anatomical sites: one well-characterised example is provided by a work that described how gut-resident Th17 cells in mice, primed by segmented filamentous bacteria (SFB), exhibit little plasticity and are not involved in tissue inflammation, while Th17 induced by C. rodentium have pathogenic potential, and a preferential glycolytic metabolism (Omenetti et al., 2019). Our interpretation is that the combination of host features (e.g., presence of predisposing genetic variants and HLA haplotype) associated with either commensal or pathogenic microbiome would dictate the characteristics of Th17 responses to bacteria and fungi. A recent paper hypothesised that airway Th17 cells in airway inflammation, e.g., during acute allergic bronchopulmonary aspergillosis, could all be cross-reactive to commensal Candida (Bacher et al., 2019): it is conceivable that a similar phenomenon of cross-reactivity could be observed in Th17 from SpA patients in response to gut or skin microbes. At the moment, it is not clear whether Th17 cells driving the manifestations of SpA are primed in the barrier organs, such as gut or skin (Gracey et al., 2019). Certainly, specific microbe-induced experimental models, or even gnotobiotic animals, could help replicating different modes of Th17 induction in various disease models and highlight both pathogenic mechanism and natural regulatory responses, such as in the case of cMaf + ROR-ɣt+Tregs, a cell subset specialised in restraining pathogenic bacteria-induced Th17s (Xu et al., 2018).
As anticipated, emerging features of T-cell biology are their inherent instability (loss of expression of transcriptional signature) and plasticity (acquisition of TFs or cytokines typical of other lineages). They are often being seen upon in vitro restimulation (Hirota et al., 2011), but also in vivo: Th17 cells can acquire Th1-features and transdifferentiate into the so-called Th17.1 cells, a subset that is particularly evident in the inflammatory environment, such as the synovial fluid in the course of arthritis (Nistala et al., 2010). Conversely, Th17 can lose pathogenic features, or even transdifferentiate into Treg-like cells: under pro-inflammatory conditions in the gut, intestinal Th17 cells can differentiate into IL-10-producing Tr1-like cells, important in the resolution of inflammation (Gagliani et al., 2015). This aspect of Th17 biology can provide therapeutic opportunities, through the induction of an anti-inflammatory fate in former pathogenic Th17 cells.
In the steady state, the natural role of Th17 cells is to maintain tissue homeostasis, by maintaining barrier integrity (Lee et al., 2015) and anti-microbial functions, in particular vs. pathogenic microbes, such as, in humans, Candida and Salmonella (McGeachy and McSorley, 2012). In mice, colonisation with the commensal SFB (Ivanov et al., 2009) is sufficient to provide Th17-mediated resistance to the intestinal pathogen Citrobacter rodentium.
These gut-specific Th17 cells with a homeostatic role can be defined as ‘non-pathogenic Th17 cells’ being largely responsive to microbial or food-derived antigens and not involved in inflammatory mediated diseases. However, additional environmental cues, such as colonisation by specific microbial species, genetic predispositions and toxins, alter the equilibrium by enforcing effector profiles of autoimmune Th17 cells that can cause pathogenic responses that could then drive disease in the musculo-skeletal structures. The possible driving role of intestinal immunity in the pathogenesis of AS is indirectly confirmed by the susceptibility genes for Crohn’s disease (CD), a form of inflammatory bowel disease akin to the colitis observed in SpA, whose list includes a number of Th17-associated genes (CCR6, JAK2, TYK2, STAT3 and IL23R; Anderson et al., 2011).
Finally, it is worth reminding that the IL-17 family includes six cytokines. The role of some of them is poorly understood (McGeachy et al., 2019) but they seem central in intestinal immunity: IL-17D has a homeostatic role in the gut (it maintains ILC3 function; Huang et al., 2021), and conversely, IL-17F could promote an inflammation-promoting microbiota, by interfering with the colonisation of Treg-inducing bacteria (Tang et al., 2018), making it a potentially attractive target for pathologic, non-homeostatic, responses. IL-23 is also upregulated in the gut of AS patients (Ciccia et al., 2009), probably secondary molecular events, such as autophagy (Ciccia et al., 2014), and also relevant in CD (Eken et al., 2014).
Experimental data suggesting heterogeneity within the Th17 compartment come largely from mouse studies, where a number of conditioning elements and some molecular networks conferring pathogenic potential have been identified. Whether the majority of these is also relevant in human Th17 is not yet clear. What is increasingly apparent is that Th17 are able to adopt both housekeeping, anti-bacterial gene modules and inflammatory, pathogenic functions, and only the latter would cause immune diseases, such as SpA. The tissue environment is possibly decisive, particularly at barrier sites, where cytokine concentrations or the presence of metabolites can dictate the cell fate. To understand the biology of tissue Th17, careful immune characterisation in steady state and in tissue context will be required. GWAS have discovered associations between genomic loci associated with Th17 function, and the occurrence of AS, providing suggestive hints into pathogenesis (Knight, 2013), but comprehending the functional meaning of the variants is particularly challenging when the genetic association is agnostic of the cell subset and tissue of origin. Since small genetic defects could originate at every phase of cell differentiation or in specific tissues, we believe that combining functional genomics with system immunology will be the next biggest challenge for translational science of immune-mediated diseases.
All authors listed have made substantial, direct and intellectual contribution to the work and approved it for publication.
Publication fees were supported by the Foundation for Research in Rheumatology (FOREUM) under award number 1016807. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a past co-authorship with one of the authors FC.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acosta-Rodriguez, E. V., Napolitani, G., Lanzavecchia, A., and Sallusto, F. (2007). Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat. Immunol. 8, 942–949. doi: 10.1038/ni1496
Aggarwal, S., Ghilardi, N., Xie, M.-H., de Sauvage, F. J., and Gurney, A. L. (2003). Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J. Biol. Chem. 278, 1910–1914. doi: 10.1074/jbc.M207577200
Ahern, P. P., Schiering, C., Buonocore, S., McGeachy, M. J., Cua, D. J., Maloy, K. J., et al. (2010). Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity 33, 279–288. doi: 10.1016/j.immuni.2010.08.010
Al-Mossawi, M. H., Chen, L., Fang, H., Ridley, A., Wit, J., Yager, N., et al. (2017). Unique transcriptome signatures and GM-CSF expression in lymphocytes from patients with spondyloarthritis. Nat. Commun. 8:1510. doi: 10.1038/s41467-017-01771-2
Anderson, C. A., Boucher, G., Lees, C. W., Franke, A., D’Amato, M., Taylor, K. D., et al. (2011). Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 43, 246–252. doi: 10.1038/ng.764
Bacher, P., Hohnstein, T., Beerbaum, E., Röcker, M., Blango, M. G., Kaufmann, S., et al. (2019). Human anti-fungal Th17 immunity and pathology rely on cross-reactivity against Candida albicans. Cell 176, 1340.e15–1355.e15. doi: 10.1016/j.cell.2019.01.041
Baeten, D., Østergaard, M., Wei, J. C.-C., Sieper, J., Järvinen, P., Tam, L.-S., et al. (2018). Risankizumab, an IL-23 inhibitor, for ankylosing spondylitis: results of a randomised, double-blind, placebo-controlled, proof-of-concept, dose-finding phase 2 study. Ann. Rheum. Dis. 77, 1295–1302. doi: 10.1136/annrheumdis-2018-213328
Bettelli, E., Carrier, Y., Gao, W., Korn, T., Strom, T. B., Oukka, M., et al. (2006). Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–238. doi: 10.1038/nature04753
Brüstle, A., Heink, S., Huber, M., Rosenplänter, C., Stadelmann, C., Yu, P., et al. (2007). The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat. Immunol. 8, 958–966. doi: 10.1038/ni1500
Chung, Y., Chang, S. H., Martinez, G. J., Yang, X. O., Nurieva, R., Kang, H. S., et al. (2009). Critical regulation of early Th17 cell differentiation by Interleukin-1 signaling. Immunity 30, 576–587. doi: 10.1016/j.immuni.2009.02.007
Ciccia, F., Accardo-Palumbo, A., Rizzo, A., Guggino, G., Raimondo, S., Giardina, A., et al. (2014). Evidence that autophagy, but not the unfolded protein response, regulates the expression of IL-23 in the gut of patients with ankylosing spondylitis and subclinical gut inflammation. Ann. Rheum. Dis. 73, 1566–1574. doi: 10.1136/annrheumdis-2012-202925
Ciccia, F., Bombardieri, M., Principato, A., Giardina, A., Tripodo, C., Porcasi, R., et al. (2009). Overexpression of interleukin-23, but not interleukin-17, as an immunologic signature of subclinical intestinal inflammation in ankylosing spondylitis. Arthritis Rheum. 60, 955–965. doi: 10.1002/art.24389
Ciofani, M., Madar, A., Galan, C., Sellars, M., Mace, K., Pauli, F., et al. (2012). A validated regulatory network for Th17 cell specification. Cell 151, 289–303. doi: 10.1016/j.cell.2012.09.016
Cua, D. J., Sherlock, J., Chen, Y., Murphy, C. A., Joyce, B., Seymour, B., et al. (2003). Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421, 744–748. doi: 10.1038/nature01355
Danoy, P., Pryce, K., Hadler, J., Bradbury, L. A., Farrar, C., Pointon, J., et al. (2010). Association of Variants at 1q32 and STAT3 with ankylosing spondylitis suggests genetic overlap with Crohn’s disease. PLoS Genet. 6:e1001195. doi: 10.1371/journal.pgen.1001195
Di Meglio, P., Di Cesare, A., Laggner, U., Chu, C.-C., Napolitano, L., Villanova, F., et al. (2011). The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced Th17 effector response in humans. PLoS One 6:e17160. doi: 10.1371/journal.pone.0017160
Di Meglio, P., Villanova, F., Napolitano, L., Tosi, I., Terranova Barberio, M., Mak, R. K., et al. (2013). The IL23R A/Gln381 allele promotes IL-23 unresponsiveness in human memory T-helper 17 cells and impairs Th17 responses in psoriasis patients. J. Invest. Dermatol. 133, 2381–2389. doi: 10.1038/jid.2013.170
Eken, A., Singh, A. K., and Oukka, M. (2014). Interleukin 23 in Crohn’s disease. Inflamm. Bowel Dis. 20, 587–595. doi: 10.1097/01.MIB.0000442014.52661.20
El-Behi, M., Ciric, B., Dai, H., Yan, Y., Cullimore, M., Safavi, F., et al. (2011). The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol. 12, 568–575. doi: 10.1038/ni.2031
Fontenot, J. D., Gavin, M. A., and Rudensky, A. Y. (2003). Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 4, 330–336. doi: 10.1038/ni904
Gagliani, N., Amezcua Vesely, M. C., Iseppon, A., Brockmann, L., Xu, H., Palm, N. W., et al. (2015). Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 523, 221–225. doi: 10.1038/nature14452
Gaublomme, J. T., Yosef, N., Lee, Y., Gertner, R. S., Yang, L. V., Wu, C., et al. (2015). Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell 163, 1400–1412. doi: 10.1016/j.cell.2015.11.009
Ghoreschi, K., Laurence, A., Yang, X.-P., Tato, C. M., McGeachy, M. J., Konkel, J. E., et al. (2010). Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature 467, 967–971. doi: 10.1038/nature09447
Gracey, E., Dumas, E., Yerushalmi, M., Qaiyum, Z., Inman, R. D., and Elewaut, D. (2019). The ties that bind: skin, gut and spondyloarthritis. Curr. Opin. Rheumatol. 31, 62–69. doi: 10.1097/BOR.0000000000000569
Gracey, E., Hromadová, D., Lim, M., Qaiyum, Z., Zeng, M., Yao, Y., et al. (2020). TYK2 inhibition reduces type 3 immunity and modifies disease progression in murine spondyloarthritis. J. Clin. Invest. 130, 1863–1878. doi: 10.1172/JCI126567
Hammitzsch, A., Chen, L., Wit, J.de, Al-Mossawi, M. H., Ridley, A., Sekine, T., et al. (2018). Inhibiting ex-vivo Th17 responses in ankylosing spondylitis by targeting Janus kinases. Sci. Rep. 8:15645. doi: doi: 10.1038/s41598-018-34026-1
Harrington, L. E., Hatton, R. D., Mangan, P. R., Turner, H., Murphy, T. L., Murphy, K. M., et al. (2005). Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 6, 1123–1132. doi: 10.1038/ni1254
Hirota, K., Duarte, J. H., Veldhoen, M., Hornsby, E., Li, Y., Cua, D. J., et al. (2011). Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol. 12, 255–263. doi: 10.1038/ni.1993
Huang, J., Lee, H., Zhao, X., Han, J., Su, Y., Sun, Q., et al. (2021). Interleukin-17D regulates group 3 innate lymphoid cell function through its receptor CD93. Immunity 54, 673.e4–686.e4. doi: 10.1016/j.immuni.2021.03.018
International Genetics of Ankylosing Spondylitis Consortium (IGAS)Cortes, A., Hadler, J., Pointon, J. P., Robinson, P. C., Karaderi, T., et al. (2013). Identification of multiple risk variants for ankylosing spondylitis through high-density genotyping of immune-related loci. Nat. Genet. 45, 730–738. doi: 10.1038/ng.2667
Ivanov, I. I., Atarashi, K., Manel, N., Brodie, E. L., Shima, T., Karaoz, U., et al. (2009). Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498. doi: 10.1016/j.cell.2009.09.033
Kebir, H., Kreymborg, K., Ifergan, I., Dodelet-Devillers, A., Cayrol, R., Bernard, M., et al. (2007). Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat. Med. 13, 1173–1175. doi: 10.1038/nm1651
Kishi, Y., Kondo, T., Xiao, S., Yosef, N., Gaublomme, J., Wu, C., et al. (2016). Protein C receptor (PROCR) is a negative regulator of Th17 pathogenicity. J. Exp. Med. 213, 2489–2501. doi: 10.1084/jem.20151118
Knight, J. C. (2013). Genomic modulators of the immune response. Trends Genet. 29, 74–83. doi: 10.1016/j.tig.2012.10.006
Langrish, C. L., Chen, Y., Blumenschein, W. M., Mattson, J., Basham, B., Sedgwick, J. D., et al. (2005). IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 201, 233–240. doi: 10.1084/jem.20041257
Lee, Y., Awasthi, A., Yosef, N., Quintana, F. J., Xiao, S., Peters, A., et al. (2012). Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 13, 991–999. doi: 10.1038/ni.2416
Lee, J. S., Tato, C. M., Joyce-Shaikh, B., Gulen, M. F., Gulan, F., Cayatte, C., et al. (2015). Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity 43, 727–738. doi: 10.1016/j.immuni.2015.09.003
Lee, Y. K., Turner, H., Maynard, C. L., Oliver, J. R., Chen, D., Elson, C. O., et al. (2009). Late developmental plasticity in the T helper 17 lineage. Immunity 30, 92–107. doi: 10.1016/j.immuni.2008.11.005
Manel, N., Unutmaz, D., and Littman, D. R. (2008). The differentiation of human T H -17 cells requires transforming growth factor-β and induction of the nuclear receptor RORγt. Nat. Immunol. 9, 641–649. doi: 10.1038/ni.1610
Mangan, P. R., Harrington, L. E., O’Quinn, D. B., Helms, W. S., Bullard, D. C., Elson, C. O., et al. (2006). Transforming growth factor-β induces development of the T H 17 lineage. Nature 441, 231–234. doi: 10.1038/nature04754
McGeachy, M. J., Bak-Jensen, K. S., Chen, Y., Tato, C. M., Blumenschein, W., McClanahan, T., et al. (2007). TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain TH-17 cell–mediated pathology. Nat. Immunol. 8, 1390–1397. doi: 10.1038/ni1539
McGeachy, M. J., Chen, Y., Tato, C. M., Laurence, A., Joyce-Shaikh, B., Blumenschein, W. M., et al. (2009). The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17–producing effector T helper cells in vivo. Nat. Immunol. 10, 314–324. doi: 10.1038/ni.1698
McGeachy, M. J., Cua, D. J., and Gaffen, S. L. (2019). The IL-17 family of cytokines in health and disease. Immunity 50, 892–906. doi: 10.1016/j.immuni.2019.03.021
McGeachy, M. J., and McSorley, S. J. (2012). Microbial-induced Th17: superhero or supervillain? J. Immunol. 189, 3285–3291. doi: 10.4049/jimmunol.1201834
Meyer Zu Horste, G., Wu, C., Wang, C., Cong, L., Pawlak, M., Lee, Y., et al. (2016). RBPJ controls development of pathogenic Th17 cells by regulating IL-23 receptor expression. Cell Rep. 16, 392–404. doi: 10.1016/j.celrep.2016.05.088
Nistala, K., Adams, S., Cambrook, H., Ursu, S., Olivito, B., de Jager, W., et al. (2010). Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc. Natl. Acad. Sci. 107, 14751–14756. doi: 10.1073/pnas.1003852107
Omenetti, S., Bussi, C., Metidji, A., Iseppon, A., Lee, S., Tolaini, M., et al. (2019). The intestine harbors functionally distinct homeostatic tissue-resident and inflammatory Th17 cells. Immunity 51, 77.e6–89.e6. doi: 10.1016/j.immuni.2019.05.004
Roberts, A. R., Vecellio, M., Chen, L., Ridley, A., Cortes, A., Knight, J. C., et al. (2016). An ankylosing spondylitis-associated genetic variant in the IL23R-IL12RB2 intergenic region modulates enhancer activity and is associated with increased Th1-cell differentiation. Ann. Rheum. Dis. 75, 2150–2156. doi: 10.1136/annrheumdis-2015-208640
Schraml, B. U., Hildner, K., Ise, W., Lee, W.-L., Smith, W. A.-E., Solomon, B., et al. (2009). The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature 460, 405–409. doi: 10.1038/nature08114
Shen, H., Goodall, J. C., and Hill Gaston, J. S. (2009). Frequency and phenotype of peripheral blood Th17 cells in ankylosing spondylitis and rheumatoid arthritis. Arthritis Rheum. 60, 1647–1656. doi: 10.1002/art.24568
Simone, D., Al Mossawi, M. H., and Bowness, P. (2018). Progress in our understanding of the pathogenesis of ankylosing spondylitis. Rheumatology 57, vi4–vi9. doi: 10.1093/rheumatology/key001
Szabo, S. J., Kim, S. T., Costa, G. L., Zhang, X., Fathman, C. G., and Glimcher, L. H. (2000). A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669. doi: 10.1016/s0092-8674(00)80702-3
Taams, L. S., Steel, K. J. A., Srenathan, U., Burns, L. A., and Kirkham, B. W. (2018). IL-17 in the immunopathogenesis of spondyloarthritis. Nat. Rev. Rheumatol. 14, 453–466. doi: 10.1038/s41584-018-0044-2
Tang, C., Kakuta, S., Shimizu, K., Kadoki, M., Kamiya, T., Shimazu, T., et al. (2018). Suppression of IL-17F, but not of IL-17A, provides protection against colitis by inducing Treg cells through modification of the intestinal microbiota. Nat. Immunol. 19, 755–765. doi: 10.1038/s41590-018-0134-y
Tripathi, S. K., Chen, Z., Larjo, A., Kanduri, K., Nousiainen, K., Äijo, T., et al. (2017). Genome-wide analysis of STAT3-mediated transcription during early human Th17 cell differentiation. Cell Rep. 19, 1888–1901. doi: 10.1016/j.celrep.2017.05.013
Heijde, D.van der, Gensler, L. S., Deodhar, A., Baraliakos, X., Poddubnyy, D., Kivitz, A., et al. (2020). Dual neutralisation of interleukin-17A and interleukin-17F with bimekizumab in patients with active ankylosing spondylitis: results from a 48-week phase IIb, randomised, double-blind, placebo-controlled, dose-ranging study. Ann. Rheum. Dis. 79, 595–604. doi: 10.1136/annrheumdis-2020-216980
Heijde, D.van der, Wei, J. C.-C., Dougados, M., Mease, P., Deodhar, A., Maksymowych, W. P., et al. (2018). Ixekizumab, an interleukin-17A antagonist in the treatment of ankylosing spondylitis or radiographic axial spondyloarthritis in patients previously untreated with biological disease-modifying anti-rheumatic drugs (COAST-V): 16 week results of a phase 3 randomised, double-blind, active-controlled and placebo-controlled trial. Lancet 392, 2441–2451. doi: 10.1016/S0140-6736(18)31946-9
Veldhoen, M., Hocking, R. J., Atkins, C. J., Locksley, R. M., and Stockinger, B. (2006). TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24, 179–189. doi: 10.1016/j.immuni.2006.01.001
Volpe, E., Servant, N., Zollinger, R., Bogiatzi, S. I., Hupé, P., Barillot, E., et al. (2008). A critical function for transforming growth factor-β, interleukin 23 and proinflammatory cytokines in driving and modulating human T H -17 responses. Nat. Immunol. 9, 650–657. doi: 10.1038/ni.1613
Wade, S. M., Canavan, M., McGarry, T., Low, C., Wade, S. C., Mullan, R. H., et al. (2019). Association of synovial tissue polyfunctional T-cells with DAPSA in psoriatic arthritis. Ann. Rheum. Dis. 78, 350–354. doi: 10.1136/annrheumdis-2018-214138
Wang, C., Yosef, N., Gaublomme, J., Wu, C., Lee, Y., Clish, C. B., et al. (2015). CD5L/AIM regulates lipid biosynthesis and restrains Th17 cell pathogenicity. Cell 163, 1413–1427. doi: 10.1016/j.cell.2015.10.068
Wellcome Trust Case Control ConsortiumAustralo-Anglo-American Spondylitis Consortium (TASC)Burton, P. R., Clayton, D. G., Cardon, L. R., Craddock, N., et al. (2007). Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 39, 1329–1337. doi: 10.1038/ng.2007.17
Xu, M., Pokrovskii, M., Ding, Y., Yi, R., Au, C., Harrison, O. J., et al. (2018). c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 554, 373–377. doi: 10.1038/nature25500
Yang, X. O., Panopoulos, A. D., Nurieva, R., Chang, S. H., Wang, D., Watowich, S. S., et al. (2007). STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 282, 9358–9363. doi: 10.1074/jbc.C600321200
Zheng, W., and Flavell, R. A. (1997). The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 89, 587–596. doi: 10.1016/s0092-8674(00)80240-8
Zhou, L., Ivanov, I. I., Spolski, R., Min, R., Shenderov, K., Egawa, T., et al. (2007). IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 8, 967–974. doi: 10.1038/ni1488
Keywords: T helper 17 cell, Spondyloarthropathies, ankylosing spondylitis, genetic risk, genome-wide association studies, interleukin 17, pathogenicity
Citation: Simone D, Stingo A and Ciccia F (2021) Genetic and Environmental Determinants of T Helper 17 Pathogenicity in Spondyloarthropathies. Front. Genet. 12:703242. doi: 10.3389/fgene.2021.703242
Edited by:
Matteo Vecellio, University of Oxford, United KingdomReviewed by:
Félicie Costantino, Université de Versailles Saint-Quentin-en-Yvelines, FranceCopyright © 2021 Simone, Stingo and Ciccia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Davide Simone, davide.simone@unicampania.it
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.