 Huishu E.
Huishu E. Lili Liang
Lili Liang Huiwen Zhang
Huiwen Zhang Wenjuan Qiu
Wenjuan Qiu Jun Ye
Jun Ye Feng Xu
Feng Xu Zhuwen Gong
Zhuwen Gong Lianshu Han
Lianshu Han
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 07 July 2021
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.702374
Purpose: To characterize the phenotypic and genotypic variations associated with Glutaric aciduria type 1 (GA1) in Chinese patients.
Methods: We analyzed the clinical, neuroradiological, biochemical, and genetic information from 101 GA1 patients in mainland China.
Results: 20 patients were diagnosed by newborn screening and the remaining 81 cases were identified following clinical intervention. Macrocephaly was the most common presentation, followed by movement disorders and seizures. A total of 59 patients were evaluated by brain MRI and 58 patients presented with abnormalities, with widening of the sylvian fissures being the most common symptom. The concentration of glutarylcarnitine in the blood, glutarylcarnitine/capryloylcarnitine ratio, and urine levels of glutaric acid were increased in GA1 patients and were shown to decrease following intervention. A total of 88 patient samples were available for genotyping and 74 variants within the GCDH gene, including 23 novel variants, were identified. The most common variant was c.1244-2A > C (18.4%) and there were no significant differences in the biochemical or clinical phenotypes described for patients with the four most common variants: c.1244-2A > C, c.1064G > A, c.533G > A, and c.1147C > T. Patients identified by newborn screening had better outcomes than clinical patients.
Conclusion: Our findings expand the spectrum of phenotypes and genotypes for GA1 in Chinese populations and suggest that an expanded newborn screening program using tandem mass spectrometry may facilitate the early diagnosis and treatment of this disease, improving clinical outcomes for patients in China.
Glutaric aciduria type 1 (GA1, OMIM #231670) is a rare autosomal recessive disorder caused by the variants in glutaryl-CoA dehydrogenase (GCDH) gene, leading to a marked decrease in glutaryl- GCDH activity, which will resulting in the accumulation of glutaric acid (GA), 3-hydroxyglutaric acid (3-OH-GA), and glutarylcarnitine (C5DC) in various tissues, especially the brain (Kolker et al., 2006). The GCDH gene is mapped to chromosome 19p13.2 and spans approximately 7 kb of genomic DNA. The worldwide incidence of GA1 is estimated to be 1/110,000 (Boy et al., 2017b); however, prevalence rates are higher in certain genetically homogeneous communities such as the Old Order Amish of Lancaster County, Pennsylvania (1/300–1/400), and the aboriginal Ojibway-Cree Indians of Northern Canada (1/300) where common variants are often found in homozygous states (Haworth et al., 1991; Morton et al., 1991). The nationwide prevalence of GA1 is between 1/171,411 and 1/52,078 in China, accounting for 7.6% of the organic acidemia (Han et al., 2015; Lin et al., 2019; Yang et al., 2020).
Glutaric aciduria type 1 is further divided into two biochemical subgroups based on urinary GA concentrations: low excretors (GA in urine < 100 mmol/mol Cr) and high excretors (GA in urine > 100 mmol/mol Cr) (Busquets et al., 2000). Both subtypes show similar clinical progression with an increased risk for striatal injury if untreated (Christensen et al., 2004; Kolker et al., 2006). There are three phenotypic presentations for GA1, asymptomatic, motor disability, and insidious onset (Strauss et al., 2007). In general, neonates present as asymptomatic, but some present with macrocephaly and frontotemporal atrophy. Affected individuals may present with brain hypoplasia and basal ganglia lesions, which in most cases, are triggered by an acute infection, fever and/or vomiting (Strauss et al., 2003; Bijarnia et al., 2008). Chronic progressive cerebral deterioration and acute encephalopathic crises accompanied by seizures, coma, and basal ganglia degeneration occur in two-thirds of untreated patients and are always followed by motor and neurological manifestations, including dyskinesia, dystonia, and hypotonia (Guerreiro et al., 2020). GA1 therapy consists of a low lysine, tryptophan diet, L-carnitine supplementation, and prompt management of intercurrent illnesses (Kolker et al., 2007). If not promptly and properly treated, severe morbidity and mortality can be caused by the irreversible neurological damage (Lindner et al., 2006). Asymptomatic neonates can be identified through expanded newborn screening (NBS) using tandem mass spectrometry (MS/MS), which has become a disease-changing intervention for GA1 (Boy et al., 2018).
In this study, we evaluated the clinical, biochemical, neuroradiological, and genetic features and outcomes of 101 Chinese patients with GA1 in an effort to expand our understanding of the variant spectrum and corresponding clinical manifestations of these patients.
A total of 101 patients (60 males and 41 females) with GA1 who were followed up in the Shanghai Xinhua Hospital between December 2007 and July 2019 were enrolled in this study. Patients were between 9 days and 11.2 years of age, and the demographic, clinical, and laboratory data were collected from patient charts. Written informed consent was obtained from the parents of study participants and the study protocol was approved by the Ethics Committee of the Xinhua Hospital Affiliated with Shanghai Jiao Tong University School of Medicine (No. XHEC-D-2020-153).
Tandem mass spectrometry was used to evaluate amino acid and acylcarnitine concentrations in dried blood spots using a tandem mass spectrometer (API 4000, American Bio-Systems Inc.) (Han et al., 2015). Gas chromatography and mass spectrometry (GC-MS) was conducted on a GCMS-QP 2010 (Shimadzu Corporation, Kyoto, Japan) and was used to detect the concentration of organic acids, including glutaric acid in the urine, using a previously established protocol (Luo et al., 2003).
Genomic DNA was isolated from the peripheral blood of patients and their parents using a TIANamp Blood DNA Kit (Tiangen Biotech Co. Ltd., Beijing, China), following the manufacturer’s instructions. All 12 coding exons and flanking regions of the GCDH gene were amplified using polymerase chain reaction (PCR) as previously described (E et al., 2017), and these PCR products were sequenced on an ABI3700 sequencer (Applied Biosystems, Foster City, California, United States) after purification. The pathogenicity of novel variants was evaluated using the American College of Medical Genetics and Genomics (ACMG) standards and guidelines (Richards et al., 2015). The pathogenicity of the missense variants was predicted by Mutation Taster1, PolyPhen-22, and SIFT3.
Glutaric aciduria type 1 diagnosis was made based on the presence of C5DC in the blood and the C5DC/capryloylcarnitine (C8) ratio detected by MS/MS. In addition, urine GA levels detected by GC–MS as previously reported (E et al., 2017), together with the clinical features and conventional laboratory tests were used for diagnostic purposes. 88 patients’ diagnoses were confirmed by GCDH gene analysis. Patients diagnosed from the NBS program could be asymptomatic.
Metabolic maintenance treatment designed in accordance with the guideline recommendations (Boy et al., 2017b) consisted of the following therapies: (1) A lysine-free, tryptophan-reduced amino acid supplement for all patients up to 6 years of age; (2) After 6 years, a low lysine content natural protein diet avoiding lysine-rich foods was recommended; (3) Lifelong oral carnitine supplementation. Emergency treatment requires specialized nutritional products and medication.
Clinical follow-up parameters were used to assess basic motor function and language development. Based on prognosis, the patients were divided into two main groups–normal and abnormal. The normal group showed no significant disability in daily life while the abnormal group presented with either deficits and/or delayed achievement of motor and/or speech milestones, including the need for support when walking and being unable to communicate or pronounce words effectively.
Statistical analysis was performed using Prism software (GraphPad, version 7.0). Continuous variables were not normally distributed; therefore, they were reported as the median and interquartile range. Mann–Whitney U and Kruskal–Wallis H tests were used to compare continuous variables between groups if the variables were non-parametric. Chi-squared tests were used to compare the categorical variables between groups.
This study recruited 101 individuals with a confirmed GA1 diagnosis from 22 provinces across China. The top three provinces were Jiangsu, Anhui, and Shandong, accounting for 43.6% of all the GA1 patients, followed by Fujian province accounting for 13.13% of all the GA1 patients. A total of 20 (19.8%) affected individuals (12 males and 8 females) were identified using NBS screening. Of the remaining 81 (80.2%) clinical patients, 78 (96.3%) were symptomatic with a median age at onset of 6.6 months (range 9 days–53.7 months) and a median age at diagnosis of 14.5 months (range 1.2–99 months). A total of 32 of the clinical patients (39.5%) had their first crises before the age of 24 months.
The clinical characteristics of all 101 patients are summarized in Table 1. Macrocephaly was the most common symptom, with this being the sole symptom in five patients, followed by movement disorders and seizures. Phenotypically, 20 patients (19.8%) were asymptomatic, 46 patients (45.5%) had at least one acute encephalopathic crisis presenting as seizures or vomiting, while 35 individuals (34.7%) presented as insidious onset and developed neurological disease and striatal injury in the absence of encephalopathic crises. One of these patients was a 3-year-old with recurrent headaches.
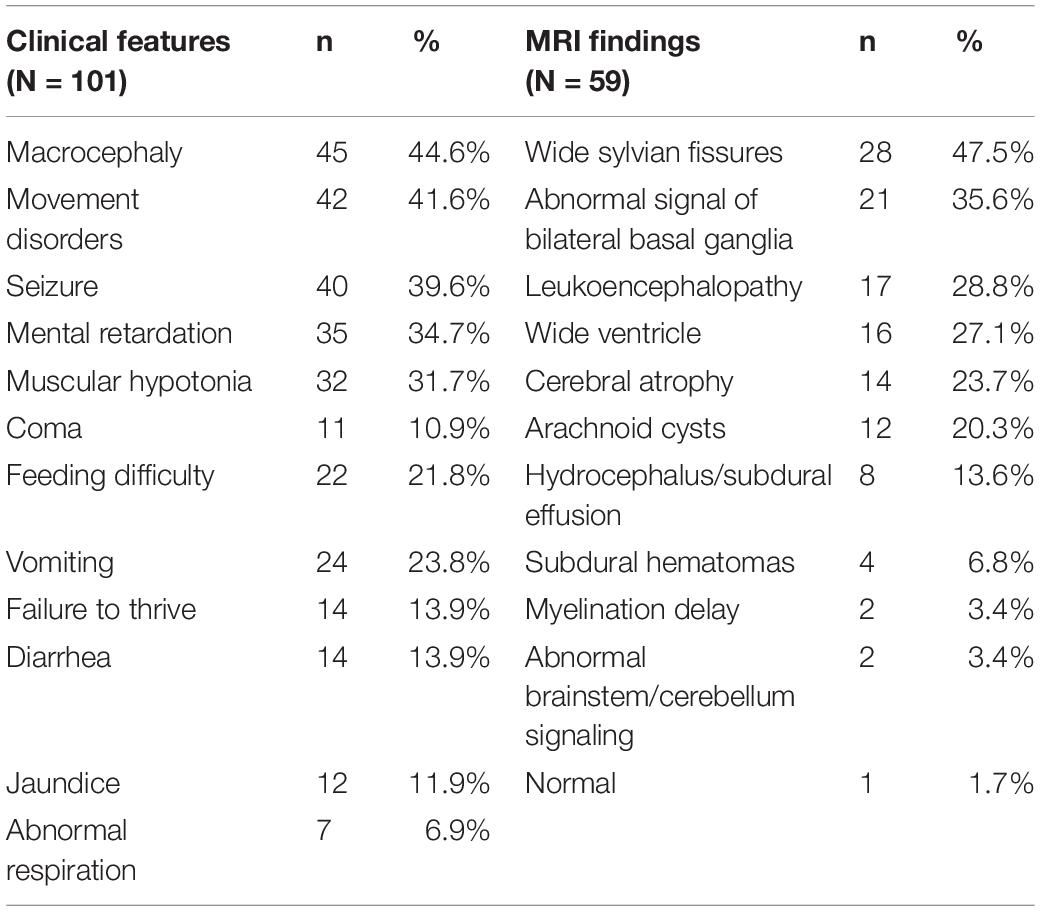
Table 1. Clinical features and MRI findings of patients with Glutaric aciduria type 1 (GA1).
A total of 59 patients were evaluated by MRI brain scans (Table1) with a median test age of 1 year (range 1 month–7.43 years), 55 of whom (93.2%) were clinical patients and four (6.8%) were asymptomatic (diagnosed by NBS). A total of 58 patients presented with abnormal images. The results demonstrated that widened sylvian fissures of the frontotemporal lobes was the most common symptom of this disease, followed by abnormal signals from the basal ganglia and leukoencephalopathy. Eight patients received a second MRI scan within 1 year, and while seven showed no changes or progress, one patient presented with a slightly smaller arachnoid cyst compared to that in their previous scan.
The serum C5DC, C5DC/C8 ratio, and urinary GA values for each group of patients before and after medical intervention are summarized in Table 2. All 101 patients received at least one MS/MS test, and 89 patients were available for GC-MS analysis, with 78 patients identified as high excretors. There were significant differences in the C5DC concentration, C5DC/C8 ratio, and urinary GA levels in each of the patient groups following treatment. In addition, when these variables were compared between the NBS and clinical groups, significant differences were noted in all of them with the exception of the C5DC value in NBS patients. On comparing the NBS and clinical groups, only a significant difference in the C5DC values after treatment could be identified. The C5DC levels of three patients were normal at their initial evaluations and their GA1 diagnosis were based on an increased C5DC/C8 ratio, which was then further confirmed by a GCDH gene test.
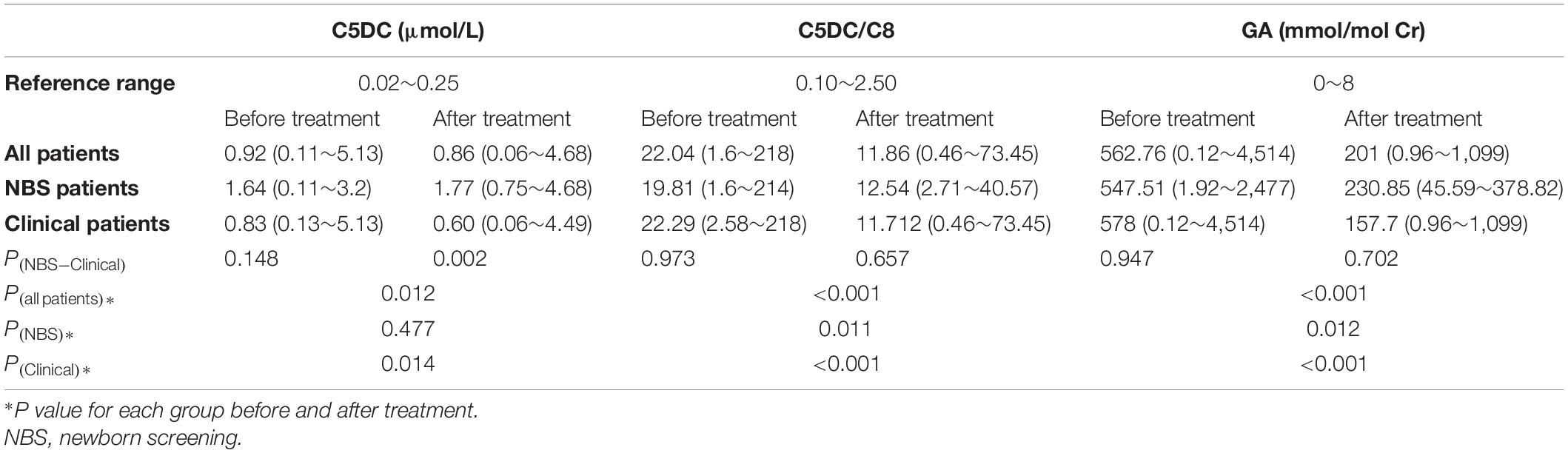
Table 2. Levels of C5DC in the blood, C5DC/C8 ratios and urinary GA in each of the three groups.
The details of the nine patients with unmatched biochemical and clinical manifestations are listed in Table 3. Patient Nos. 1 and 2 were siblings and patient Nos. 3 and 4 were siblings, while the rest of the patients were unrelated. All the relevant biochemical indices were significantly elevated in patient Nos. 1–4, but their clinical characterizations were reasonably mild. In contrast, patient Nos. 5–9 showed only moderate increase in their biochemical indices but had much more severe clinical presentations than patient Nos. 1–4.
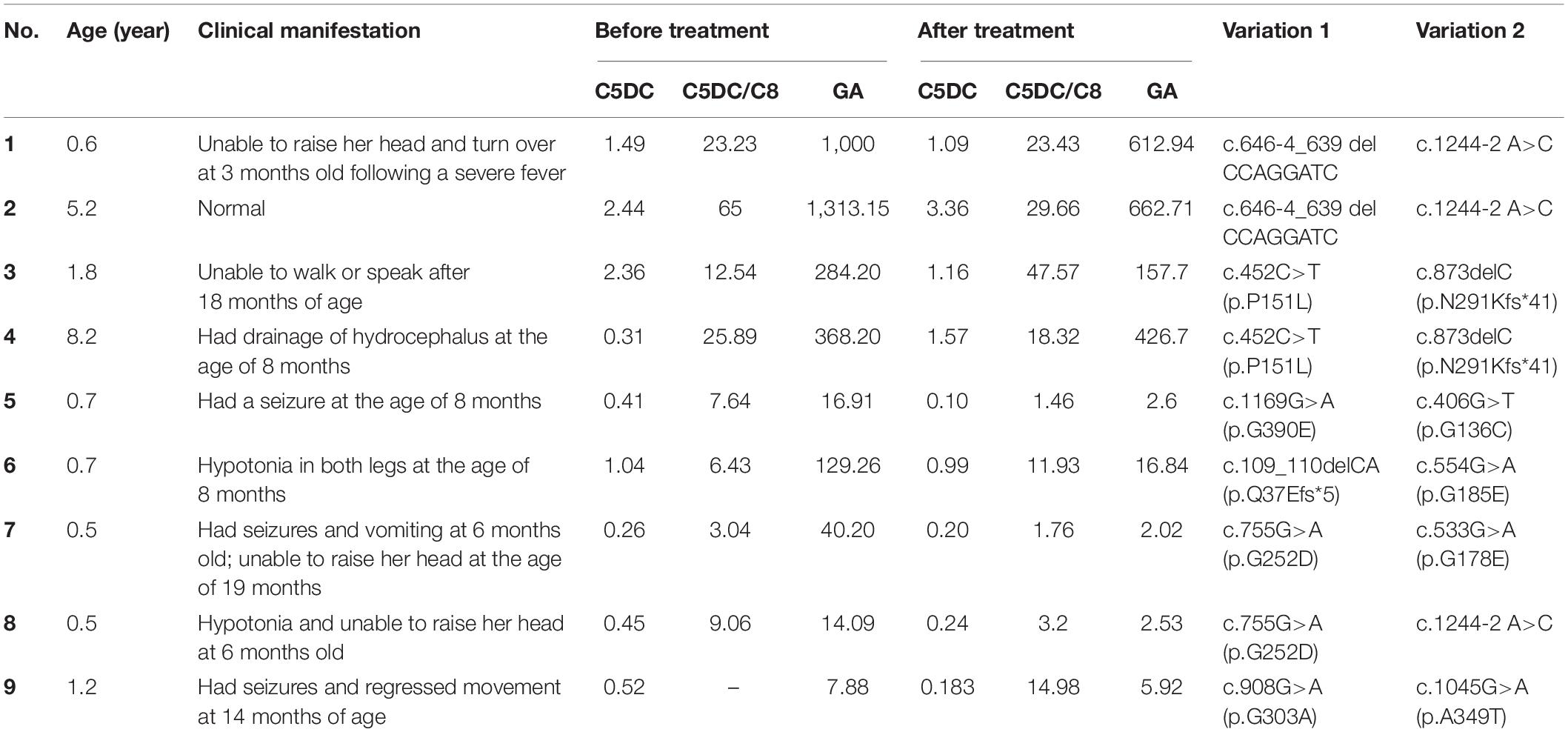
Table 3. Phenotype and biochemical results for nine patients with unmatched manifestations.
A total of 88 patient samples (87.1%) were sequenced for GCDH. As a result, we found that 87 patients were compound heterozygotes or homozygotes, while only one patient had a single variant. A total of 74 different variants were identified (Figure 1), including 61 missense variants (82.4%), six frameshifts (8.1%), five nonsense (6.8%), and two splicing (2.7%) mutations. The most frequent variant was c.1244-2A > C (18.3%), followed by c.1064G > A (p. Arg355His) (7.4%), c.533G > A (p. Gly178Glu) (3.5%), and c.1147C > T (p. Arg383Cys) (2.5%). A total of 17 patients carried homozygous variants, and nine of these carried the c.1244-2A > C variant. In addition, 23 novel variants were identified, including 18 missense variants, four frameshifts, and one nonsense variant. Among these, 17 of the novel missense variants were predicted to be pathogenic using Mutation Taster SIFT and polyphen2. Variant c.493C > A (p. Leu165Met) was predicted to be a neutral mutation when evaluated by SIFT, but was shown to be pathogenic using the other two algorithms. The genotype-phenotype correlation analysis focused on the four most common variants: c.1244-2A > C, c.1064G > A (p. Arg355His), c.533G > A (p. Gly178Glu), and c.1147C > T (p. Arg383Cys), and 47 patients carrying these variants were analyzed (Table 4). Kruskal–Wallis test showed that all four variants exhibited no significant difference for age of onset/diagnosis, clinical manifestations, biochemical results, or outcomes. An acute encephalopathic crisis was identified in one patient with c.1147C > T (p. Arg383Cys) (20%), one with c.533G > A (p. Gly178Glu) (14.3%), one with c.1064G > A (p. Arg355His) (25%), and four with c.1244-2A > C (17.4%). All the patients were classified as high excretors, except one, who carried a c.1244-2A > C variation. Taken together, these results suggest that there were no significant differences in biochemical or clinical phenotypes for these four genotypes.
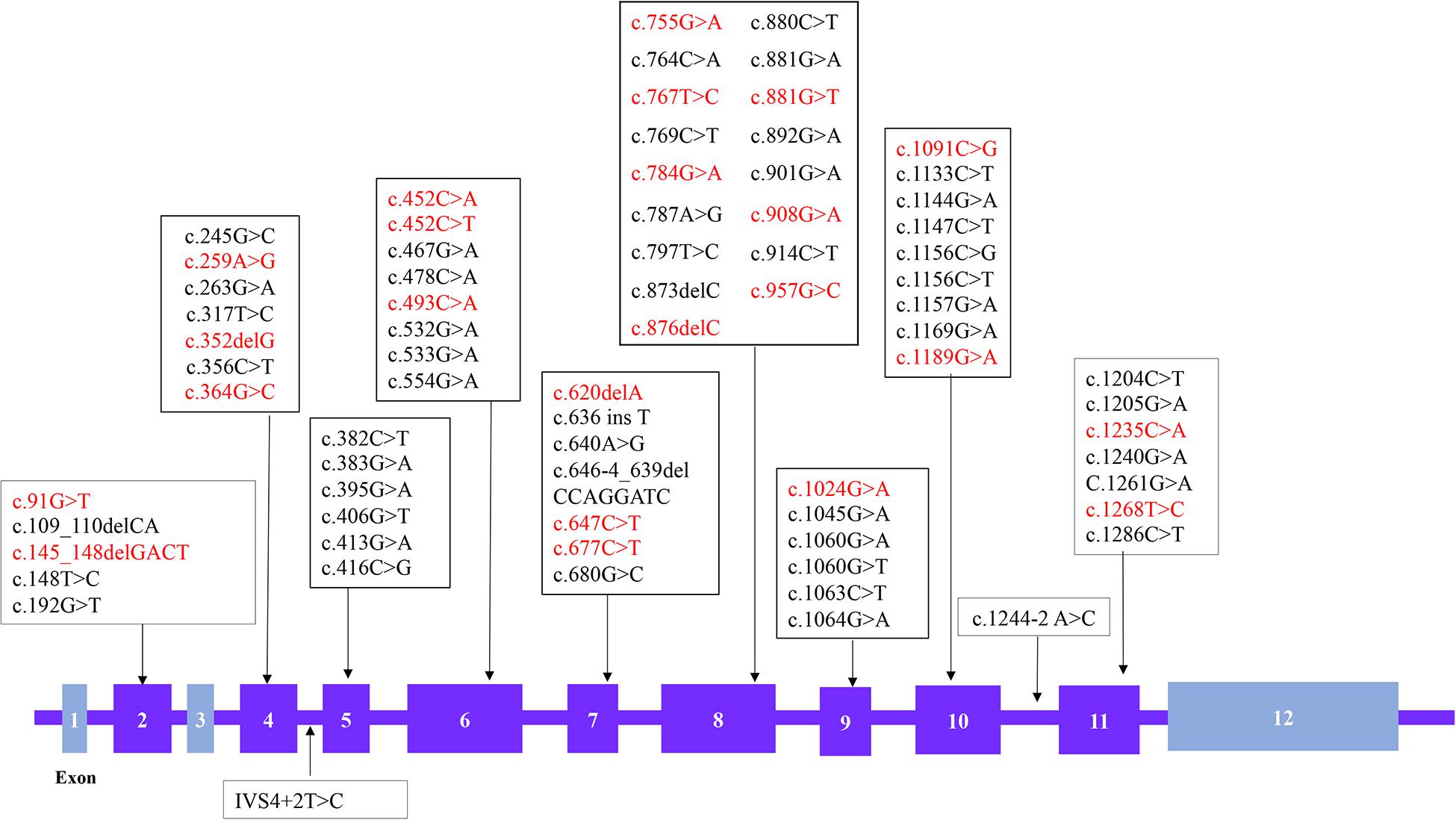
Figure 1. Distribution of the GCDH variations identified in 88 Glutaric aciduria type 1 (GA1) patients. The boxes represent the exons and novel variations are shown in red (The chromosome position is chr19:12,891,160-12,899,999 and the number of transcription version is NM-000159.3).
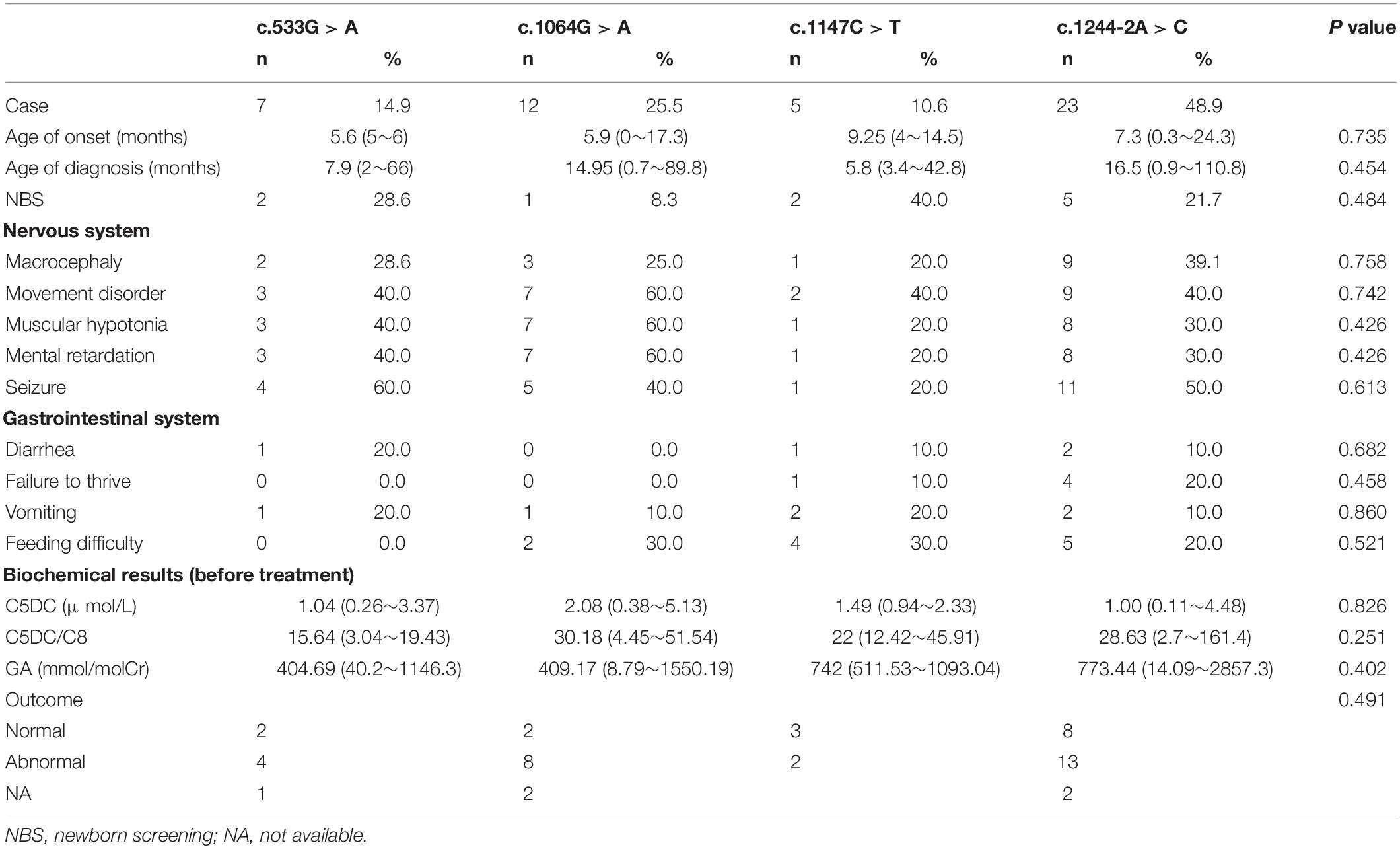
Table 4. Correlation between genotype and phenotype in GA1 patients: comparison of the four most common variants from 47 patients.
All patients received appropriate therapy from the time of diagnosis. The follow-up study was performed on 96 patients (Figure 2), as the other five patients discontinued the trial. Of the 19 NBS patients, 18 were asymptomatic at diagnosis and continued to develop normally, only one patient showed a slight delay in motor milestones without any acute encephalopathic crises and was unable to walk at 3 years of age. A total of 77 patients were in the clinical group, while three asymptomatic patients were diagnosed due to the presence of a GA1 sibling. Outcomes in the symptomatic patients varied, 58 presented with motor and/or speech dysfunctions, one patient died at 18 months of age and the remaining 15 developed normally. This data shows that NBS and clinical patient outcomes are significantly different (P < 0.001).
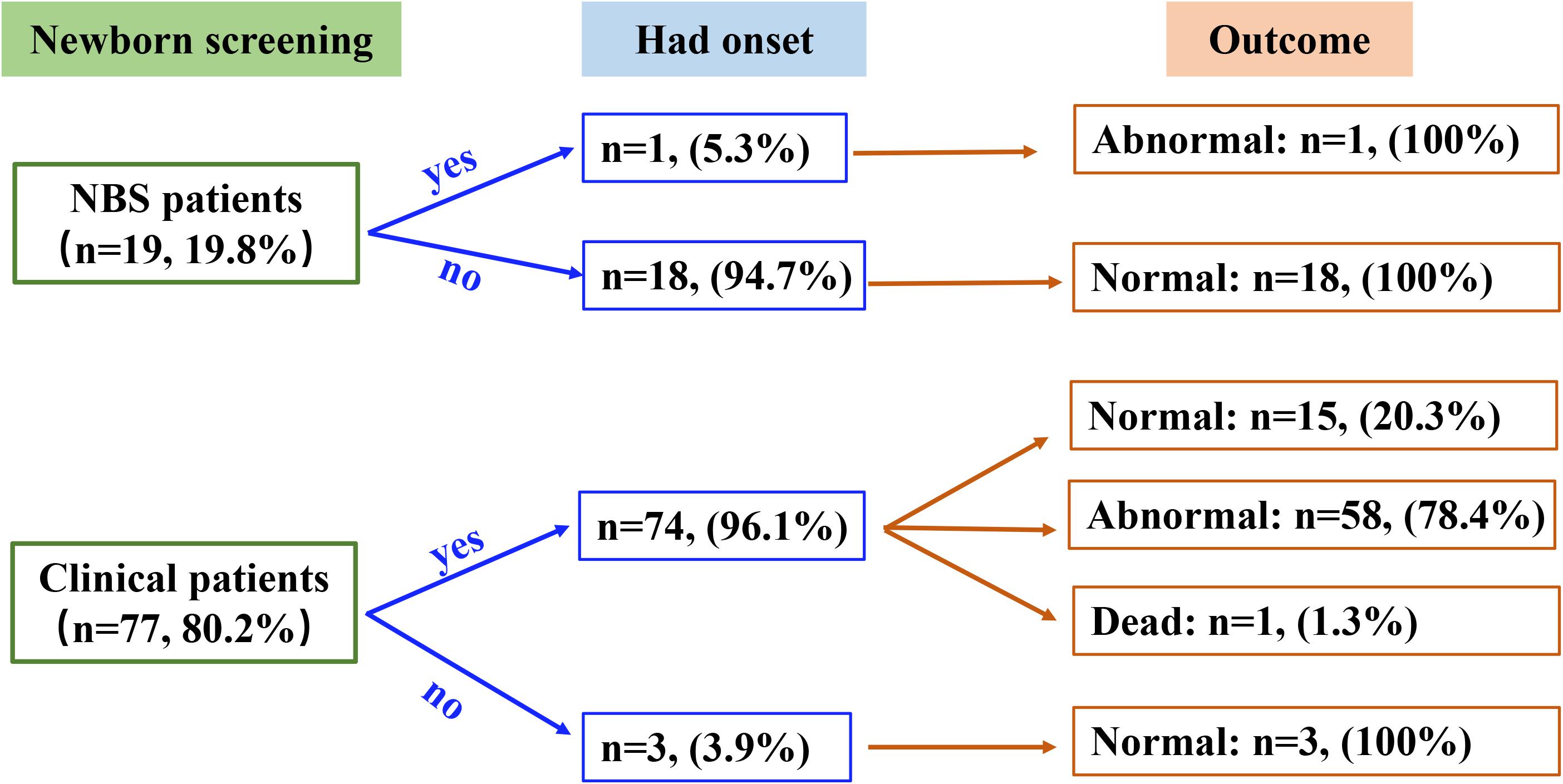
Figure 2. Summary of the newborn screening status, disease onset, and clinical outcome data for the participants in this study.
Of the 78 high excretor patients, 30 (38.5%) developed normally, 43 (55.1%) became disabled, and one died (four patients were lost to follow-up). Similar results were found in the 11 low excretor patients–four (36.4%) developed normally, while seven (63.6%) had movement and/or speech impediments (P > 0.05).
Our study was designed to systematically investigate the clinical, biochemical, and genetic characteristics of 101 GA1 patients from mainland China ultimately identifying 23 novel variants. Unlike Turkish and Indian patients with GA1 (Radha Rama Devi et al., 2016; Gurbuz et al., 2020), none of our patients were from consanguineous families. Approximately 50% of the study participants were from Jiangsu, Shandong, Anhui, and Fujian Province, with Jiangsu being the most common. Studies have shown that GA1 prevalence is higher in the Fujian province (Lin et al., 2019). However, our data, derived from Shanghai, might be influenced by several geographical factors, including the fact that Jiangsu is much closer to Shanghai making it more convenient for these patients.
Typically, GA1 presents in infancy after an acute metabolic encephalopathy that results in striatal necrosis, such as dystonia, mostly between the ages of 3 and 36 months, while some children only present with macrocephaly during their neonatal development (Strauss et al., 2003; Boy et al., 2017b). The onset of dystonia may be caused by catabolism and infectious diseases (Kolker et al., 2006; Harting et al., 2009). Our study population was made up of patients with acute onset (45.5%) and insidious onset (34.7%). The most common presentation for our acute onset patients were seizures, vomiting, and/or diarrhea. This is in agreement with previous studies, which suggest that seizures could be the initial clinical presentation of GA1 and that these patients experience a higher frequency of epilepsy (Young-Lin et al., 2013; Kolker et al., 2015). This was confirmed by our data that demonstrated that 40 patients experienced single or recurrent seizures and that in 28 of these patients, seizures were the first symptom. Macrocephaly is the most common characteristic of GA1 and was found in nearly half of the patients in this cohort; however, this value is lower than that reported in other studies (Bjugstad et al., 2000; Renaud, 2012). In addition, movement disorders and mental retardation were common in this cohort. Although the severity of the acquired movement disorders and motor disabilities remained stable with increasing age, dystonia tended to become fixed and was associated with the development of parkinsonism with increasing age (Tuncel et al., 2018). In addition to acute and insidious onset patients, there have been several reports of patients with late onset symptoms (Pierson et al., 2015; Boy et al., 2017a). This late-onset type has been used to describe patients diagnosed after 6 years of age, who present with a variety of non-specific neurological symptoms, including headaches, nausea, and muscular weakness (Tuncel et al., 2018; Gelener et al., 2020). Our data set included three late onset patients (aged from 6.4 to 11.2 years), all of whom were diagnosed because of their GA1 siblings and showed normal development. Two of these patients underwent an MRI scan and both resented with white matter changes. Since neurological signs are also common in late onset GA1 patients, brain MRI evaluations can help with diagnosis.
Half of the macrocephaly patients received MRI evaluations with the majority of the reports finding striatal changes, subdural collection, and widening of both sylvian fissures (Mohammad et al., 2015). We found that sylvian fissure widening, also known as Batwing sign, was the most common presentation. However, a large proportion of our patients underwent brain MRIs under 1 year of age. During this period, the widening of the cerebral operculum is related to underdevelopment rather than atrophy or the absence of the cerebral gyri comprising the operculum (Twomey et al., 2003; Strauss et al., 2007). Therefore, re-examining the brain MRIs of these patients after 1–2 years should be required. The second most common MRI finding was abnormal signaling in the bilateral basal ganglia, and injuries within this brain region are the main determinant of morbidity. More than 50% of the patients in this study who presented with bilateral basal ganglia abnormalities experienced seizures, and nearly 40% of these patients developed movement disorders. The third most common MRI characteristic was leukoencephalopathy, which is not a specific manifestation in GA1; however, it is an important symptom in most cerebral organic acid disorders (Sauer et al., 2011; Kölker et al., 2013). The neuropathological implications of the presence of white matter abnormalities seem to focus on spongiform myelinopathy, which is probably caused by the toxin accumulation associated with GA1 resulting in desmyelination or demyelination (Gerstner et al., 2005; Zinnanti et al., 2006). In addition, subdural hematomas, which might be caused by stretched cortical veins and venous hypertension (Hou et al., 2007; Strauss et al., 2010), were identified in three patients. Notably, the frequency of arachnoid cysts was higher in our data, and when this non-specific neurological sign is found in conjunction with frontoparietal brain atrophy and widened sylvian fissures, it may reflect abnormal brain growth during intrauterine development. There are some limitations to these results because most of the MRI results came from different hospitals across the country suggesting that there may be some differences in the interpretation of the scans.
Glutaric acid 1 is a treatable neurometabolic disorder when diagnosed in early neonates (Pusti et al., 2014). Our study showed that the outcomes were much better in patients diagnosed by NBS, who were able to receive intensive management interventions. In contrast, clinical patients showed a higher frequency of complications and reduced life expectancy. It has been demonstrated that the onset of encephalopathic crises is the predictable outcome of GCDH deficiency (Strauss et al., 2003; Kyllerman et al., 2004). Although only one patient died in this cohort, the disability rates were frustrating with more than 80% of the clinical patients presenting with motor and/or speech disabilities. While in NBS patients, these disability rates were significantly decreased. Thus our study confirmed the findings of several others and demonstrate that NBS is a beneficial, disease-changing intervention for GA1 and that early intervention is critical for preventing irreversible central nervous system (CNS) damage associated with GA1 (Viau et al., 2012; Lee et al., 2013; Boy et al., 2018). To our knowledge, GA1 has been included in the NBS plans in many of the provinces across mainland China, Taiwan province, and Hongkong, but increased vigilance is needed, since some low excretors can be missed by NBS. Therefore, further testing and gene analysis is necessary.
A definite diagnosis of GA1 relies on biochemical and genetic analyses. In this study, nearly 90% of the patients were high excretors and their clinical outcomes were similar to the low excretors, which was consist with previous studies (Christensen et al., 2004). Of note, GA1 diagnosis was usually based on C5DC, GA, and 3-OH-GA (Boy et al., 2017b; Shaik et al., 2019) concentrations; however, our previous study demonstrated that the C5DC/C8 ratio is also important in the diagnosis of GA1 and might be more sensitive than C5DC (E et al., 2017). As GC–MS is not included in NBS, some NBS patients were diagnosed on the basis of their MS/MS results and re-examined using an additional MS/MS and GC–MS analyses. Thus, when the first round of MS/MS results demonstrate abnormal or slightly increased C5DC, a positive diagnosis of GA1 should be considered especially where the C5DC/C8 ratio suggests it as a possibility. In addition, all patients should undergo a GCDH gene analysis.
A total of 88 patients underwent GCDH gene sequencing, and we identified 74 unique variants, most of which were classified as missense variants. Pathogenic variants responsible for GA1 vary between ethnic groups. The c.1204C > T (p.Arg402Trp) is the most common in the European population (Christensen et al., 2004), and it has been reported that c.1244-2A > C was a recurrent variant in the Chinese population both in mainland China and Taiwan province (Tang et al., 2000). This was further confirmed by our study where the c.1244-2A > C accounted for 18.3% of variant alleles and resulted in a splicing error. The variants identified in this study were widely distributed throughout the gene, with the largest number in exon 8. Besides c.493C > A (p.Leu165Met), all 17 other novel missense variants were predicted to have some pathogenic potential when evaluated by Mutation Taster, SIFT, and Polyphen2. p.Leu165Met was a compound heterozygous variant found in a patient who carried another novel variant, c.91G > T (p.Glu31Ter). This patient was a high excretor who experienced their first crisis before 12 months of age and who went on to develop mental retardation. It has been postulated that p.Arg402Trp and p.Ala293Thr were most frequently identified in high excretors, while p.Val400Met and p.Arg227Pro are only found in low excretors (Busquets et al., 2000; Mühlhausen et al., 2003; Funk et al., 2005; Gallagher et al., 2005). Although p.Arg402Trp accounts for 40% of alleles in patients of German origin (Radha Rama Devi et al., 2016), it was less common in our study. p.Ala293Thr and p.Val400Met were not detected in our study, but we did find p.Arg402Trp and p.Arg227Pro in both our high excretor and low excretor patients. However, we could not determine a clear correlation between these variants and biochemical phenotype. In addition, we identified several unmatched phenotype-biochemical relationships that should be evaluated in the future: c.646-4_639 del CCAGGATC and c.452C > T, which may be related to high excretors who present with a milder phenotype, while c.109_110delCA, c.755G > A, and c.908G > A may be related to low excretors who present with more severe phenotypes. To explore these genotype-phenotype relationships, we analyzed the four most common variants, with each identified in at least three cases. The distribution of the clinical and biochemical phenotypes was similar in all groups and no differences in outcome were noted amongst these variants. Researchers have tried for years to find a link between the genotype and phenotype of these patients in an effort to predict their prognosis with limited success (Sanju et al., 2021; Sitta et al., 2021). Recently, an Egyptian study suggested that six variants: p.Trp50Arg, p.Glu64Asp, p.Ser119Leu, p.Arg128Gln, p.Ser139Leu, and p.Arg402Trp, might present with a genotype-phenotype correlation, indicating a poor outcome (Mosaeilhy et al., 2017). Notably, five of these variants were detected in our study, and most patients carrying these variants did become disabled. However, we cannot draw a similar conclusion as our sample size remains relatively small. More patients should be recruited so that we can explore the relationships between phenotype and genotype.
In conclusion, a cohort of 101 Chinese patients with GA1 were analyzed and genotyped. Our findings demonstrated that macrocephaly was the most common presentation of GA1, and C5DC/C8 ratios should be used in the diagnosis of GA1. We could not draw any clear genotype-phenotype correlations due to the high number of variants and the relatively limited number of patients. However, our study has expanded the spectrum of genotypes for Chinese patients with GA1. Our data supports the expansion of the NBS and genotyping programs for GA1 in an effort to promote early diagnosis and improved clinical outcomes.
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Ethics Committee of the Xinhua Hospital Affiliated with Shanghai Jiao Tong University School of Medicine (No. XHEC-D-2020-153). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
LH initiated and designed the study. XG contributed to the conception of the study. HZ, WQ, and JY contributed to the acquisition and analysis of data. FX and ZG contributed to the testing of MS/MS and GC/MS. HE and LL contributed to drafting the text, preparing the figures and tables, they contributed equally to the work. All authors contributed to the article and approved the submitted version.
This work was supported by the National Key R&D Program of China (No: 2016YFC0901505), Shanghai Municipal Health Commission (No. 202040448), and the National Natural Science Foundation of China (No. 81600701).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We gratefully acknowledge all the patients, healthy children and medical staff involved in this study.
Bijarnia, S., Wiley, V., Carpenter, K., Christodoulou, J., Ellaway, C. J., and Wilcken, B. (2008). Glutaric aciduria type I: outcome following detection by newborn screening. J. Inherit. Metab. Dis. 31, 503–507. doi: 10.1007/s10545-008-0912-z
Bjugstad, K. B., Goodman, S. I., and Freed, C. R. (2000). Age at symptom onset predicts severity of motor impairment and clinical outcome of glutaric acidemia type 1. J. Pediatr. 137, 681–686. doi: 10.1067/mpd.2000.108954
Boy, N., Heringer, J., Brackmann, R., Bodamer, O., Seitz, A., Kolker, S., et al. (2017a). Extrastriatal changes in patients with late-onset glutaric aciduria type I highlight the risk of long-term neurotoxicity. Orphanet J. Rare Dis. 12:77. doi: 10.1186/s13023-017-0612-6
Boy, N., Muhlhausen, C., Maier, E. M., Heringer, J., Assmann, B., Burgard, P., et al. (2017b). Proposed recommendations for diagnosing and managing individuals with glutaric aciduria type I: second revision. J. Inherit. Metab. Dis. 40, 75–101. doi: 10.1007/s10545-016-9999-9
Boy, N., Mengler, K., Thimm, E., Schiergens, K. A., Marquardt, T., Weinhold, N., et al. (2018). Newborn screening: a disease-changing intervention for glutaric aciduria type 1. Ann. Neurol. 83, 970–979. doi: 10.1002/ana.25233
Busquets, C., Merinero, B., Christensen, E., Gelpí, J. L., Campistol, J., Pineda, M., et al. (2000). Glutaryl-CoA dehydrogenase deficiency in Spain: evidence of two groups of patients, genetically, and biochemically distinct. Pediatr. Res. 48, 315–322. doi: 10.1203/00006450-200009000-00009
Christensen, E., Ribes, A., Merinero, B., and Zschocke, J. (2004). Correlation of genotype and phenotype in glutaryl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 27, 861–868. doi: 10.1023/B:BOLI.0000045770.93429.3c
E, H. S., Han, L. S., Ye, J., Qiu, W. J., Zhang, H. W., and Liang, L. L. (2017). Analysis of clinical features and the results of mass spectrometry with glutaric acidemia type I. Chin. J. Endocrinol. Metab. 033, 730–734.
Funk, C. B., Prasad, A. N., Frosk, P., Sauer, S., Kölker, S., Greenberg, C. R., et al. (2005). Neuropathological, biochemical and molecular findings in a glutaric acidemia type 1 cohort. Brain 128, 711–722. doi: 10.1093/brain/awh401
Gallagher, R. C., Cowan, T. M., Goodman, S. I., and Enns, G. M. (2005). Glutaryl-CoA dehydrogenase deficiency and newborn screening: retrospective analysis of a low excretor provides further evidence that some cases may be missed. Mol. Genet. Metab. 86, 417–420. doi: 10.1016/j.ymgme.2005.08.005
Gelener, P., Severino, M., Diker, S., Terali, K., Tuncel, G., Tuzlali, H., et al. (2020). Adult-onset glutaric aciduria type I: rare presentation of a treatable disorder. Neurogenetics 21, 179–186. doi: 10.1007/s10048-020-00610-9
Gerstner, B., Gratopp, A., Marcinkowski, M., Sifringer, M., Obladen, M., and Buhrer, C. (2005). Glutaric acid and its metabolites cause apoptosis in immature oligodendrocytes: a novel mechanism of white matter degeneration in glutaryl-CoA dehydrogenase deficiency. Pediatr. Res. 57, 771–776. doi: 10.1203/01.Pdr.0000157727.21503.8d
Guerreiro, G., Diaz Jaques, C. E., Wajner, M., and Vargas, C. R. (2020). Elevated levels of BDNF and cathepsin-d as possible peripheral markers of neurodegeneration in plasma of patients with glutaric acidemia type I. Int. J. Dev. Neurosci. 80, 42–49. doi: 10.1002/jdn.10006
Gurbuz, B. B., Yilmaz, D. Y., Coskun, T., Tokatli, A., Dursun, A., and Sivri, H. S. (2020). Glutaric aciduria type 1: genetic and phenotypic spectrum in 53 patients. Eur. J. Med. Genet. 63:104032. doi: 10.1016/j.ejmg.2020.104032
Han, L., Han, F., Ye, J., Qiu, W., Zhang, H., Gao, X., et al. (2015). Spectrum analysis of common inherited metabolic diseases in Chinese patients screened and diagnosed by tandem mass spectrometry. J. Clin. Lab. Anal. 29, 162–168. doi: 10.1002/jcla.21745
Harting, I., Neumaier-Probst, E., Seitz, A., Maier, E. M., Assmann, B., Baric, I., et al. (2009). Dynamic changes of striatal and extrastriatal abnormalities in glutaric aciduria type I. Brain 132, 1764–1782. doi: 10.1093/brain/awp112
Haworth, J. C., Dilling, L. A., and Seargeant, L. E. (1991). Increased prevalence of hereditary metabolic diseases among native Indians in Manitoba and northwestern Ontario. CMAJ 145, 123–129.
Hou, L. C., Veeravagu, A., Hsu, A. R., Enns, G. M., and Huhn, S. L. (2007). Glutaric acidemia type I: a neurosurgical perspective. Report of two cases. J. Neurosurg. 107, 167–172. doi: 10.3171/ped-07/08/167
Kölker, S., Burgard, P., Sauer, S. W., and Okun, J. G. (2013). Current concepts in organic acidurias: understanding intra- and extracerebral disease manifestation. J. Inherit. Metab. Dis. 36, 635–644. doi: 10.1007/s10545-013-9600-8
Kolker, S., Christensen, E., Leonard, J. V., Greenberg, C. R., Burlina, A. B., Burlina, A. P., et al. (2007). Guideline for the diagnosis and management of glutaryl-CoA dehydrogenase deficiency (glutaric aciduria type I). J. Inherit. Metab. Dis. 30, 5–22. doi: 10.1007/s10545-006-0451-4
Kolker, S., Garbade, S. F., Greenberg, C. R., Leonard, J. V., Saudubray, J. M., Ribes, A., et al. (2006). Natural history, outcome, and treatment efficacy in children and adults with glutaryl-CoA dehydrogenase deficiency. Pediatr. Res. 59, 840–847. doi: 10.1203/01.pdr.0000219387.79887.86
Kolker, S., Garcia-Cazorla, A., Valayannopoulos, V., Lund, A. M., Burlina, A. B., Sykut-Cegielska, J., et al. (2015). The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 1: the initial presentation. J. Inherit. Metab. Dis. 38, 1041–1057. doi: 10.1007/s10545-015-9839-3
Kyllerman, M., Skjeldal, O., Christensen, E., Hagberg, G., Holme, E., Lonnquist, T., et al. (2004). Long-term follow-up, neurological outcome and survival rate in 28 Nordic patients with glutaric aciduria type 1. Eur. J. Paediatr. Neurol. 8, 121–129. doi: 10.1016/j.ejpn.2003.12.007
Lee, C. S., Chien, Y. H., Peng, S. F., Cheng, P. W., Chang, L. M., Huang, A. C., et al. (2013). Promising outcomes in glutaric aciduria type I patients detected by newborn screening. Metab. Brain Dis. 28, 61–67. doi: 10.1007/s11011-012-9349-z
Lin, Y., Zheng, Q., Zheng, T., Zheng, Z., Lin, W., and Fu, Q. (2019). Expanded newborn screening for inherited metabolic disorders and genetic characteristics in a southern Chinese population. Clin. Chim. Acta 494, 106–111. doi: 10.1016/j.cca.2019.03.1622
Lindner, M., Ho, S., Fang-Hoffmann, J., Hoffmann, G. F., and Kolker, S. (2006). Neonatal screening for glutaric aciduria type I: strategies to proceed. J. Inherit. Metab. Dis. 29, 378–382. doi: 10.1007/s10545-006-0284-1
Luo, X. P., Wang, M. T., Wei, H., Liang, Y., Wang, H. W., Lin, H. H., et al. (2003). Application of gas chromatography-mass spectrometry analysis on urine filter paper in the high-risk screening and diagnosis of inherited metabolic diseases. Zhonghua Er Ke Za Zhi 41, 245–248.
Mohammad, S. A., Abdelkhalek, H. S., Ahmed, K. A., and Zaki, O. K. (2015). Glutaric aciduria type 1: neuroimaging features with clinical correlation. Pediatr. Radiol. 45, 1696–1705. doi: 10.1007/s00247-015-3395-8
Morton, D. H., Bennett, M. J., Seargeant, L. E., Nichter, C. A., and Kelley, R. I. (1991). Glutaric aciduria type I: a common cause of episodic encephalopathy and spastic paralysis in the Amish of Lancaster County, Pennsylvania. Am. J. Med. Genet. 41, 89–95. doi: 10.1002/ajmg.1320410122
Mosaeilhy, A., Mohamed, M. M., C, G. P. D., El Abd, H. S. A., Gamal, R., Zaki, O. K., et al. (2017). Genotype-phenotype correlation in 18 Egyptian patients with glutaric acidemia type I. Metab. Brain Dis. 32, 1417–1426. doi: 10.1007/s11011-017-0006-4
Mühlhausen, C., Christensen, E., Schwartz, M., Muschol, N., Ullrich, K., and Lukacs, Z. (2003). Severe phenotype despite high residual glutaryl-CoA dehydrogenase activity: a novel mutation in a Turkish patient with glutaric aciduria type I. J. Inherit. Metab. Dis. 26, 713–714. doi: 10.1023/b:boli.0000005604.90621.e2
Pierson, T. M., Nezhad, M., Tremblay, M. A., Lewis, R., Wong, D., Salamon, N., et al. (2015). Adult-onset glutaric aciduria type I presenting with white matter abnormalities and subependymal nodules. Neurogenetics 16, 325–328. doi: 10.1007/s10048-015-0456-y
Pusti, S., Das, N., Nayek, K., and Biswas, S. (2014). A treatable neurometabolic disorder: glutaric aciduria type 1. Case Rep. Pediatr. 2014:256356. doi: 10.1155/2014/256356
Radha Rama Devi, A., Ramesh, V. A., Nagarajaram, H. A., Satish, S. P., Jayanthi, U., and Lingappa, L. (2016). Spectrum of mutations in Glutaryl-CoA dehydrogenase gene in glutaric aciduria type I–Study from South India. Brain Dev. 38, 54–60. doi: 10.1016/j.braindev.2015.05.013
Renaud, D. L. (2012). Leukoencephalopathies associated with macrocephaly. Semin. Neurol. 32, 34–41. doi: 10.1055/s-0032-1306384
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Sanju, S., Tullu, M. S., Seshadri, N., and Agrawal, M. (2021). Glutaric Aciduria Type 1: a case report and review of literature. J. Pediatr. Intensive Care 10, 65–70. doi: 10.1055/s-0040-1709704
Sauer, S. W., Opp, S., Hoffmann, G. F., Koeller, D. M., Okun, J. G., and Kölker, S. (2011). Therapeutic modulation of cerebral L-lysine metabolism in a mouse model for glutaric aciduria type I. Brain 134, 157–170. doi: 10.1093/brain/awq269
Shaik, M., Kruthika-Vinod, T. P., Kamate, M., and Vedamurthy, A. B. (2019). Is Expanded Newborn Screening Adequate to Detect Indian Biochemical Low Excretor Phenotype Patients of Glutaric Aciduria Type I? Indian J. Pediatr. 86, 995–1001. doi: 10.1007/s12098-019-03017-z
Sitta, A., Guerreiro, G., de Moura Coelho, D., da Rocha, V. V., Dos Reis, B. G., Sousa, C., et al. (2021). Clinical, biochemical and molecular findings of 24 Brazilian patients with glutaric acidemia type 1: 4 novel mutations in the GCDH gene. Metab. Brain Dis. 36, 205–212. doi: 10.1007/s11011-020-00632-0
Strauss, K. A., Donnelly, P., and Wintermark, M. (2010). Cerebral haemodynamics in patients with glutaryl-coenzyme A dehydrogenase deficiency. Brain 133, 76–92. doi: 10.1093/brain/awp297
Strauss, K. A., Lazovic, J., Wintermark, M., and Morton, D. H. (2007). Multimodal imaging of striatal degeneration in Amish patients with glutaryl-CoA dehydrogenase deficiency. Brain 130, 1905–1920. doi: 10.1093/brain/awm058
Strauss, K. A., Puffenberger, E. G., Robinson, D. L., and Morton, D. H. (2003). Type I glutaric aciduria, part 1: natural history of 77 patients. Am. J. Med. Genet. C Semin. Med. Genet. 121C, 38–52. doi: 10.1002/ajmg.c.20007
Tang, N. L., Hui, J., Law, L. K., Lam, Y. Y., Chan, K. Y., Yeung, W. L., et al. (2000). Recurrent and novel mutations of GCDH gene in Chinese glutaric acidemia type I families. Hum. Mutat. 16:446. doi: 10.1002/1098-1004(200011)16:5<446::Aid-humu14<3.0.Co;2-y
Tuncel, A. T., Boy, N., Morath, M. A., Horster, F., Mutze, U., and Kolker, S. (2018). Organic acidurias in adults: late complications and management. J. Inherit. Metab. Dis. 41, 765–776. doi: 10.1007/s10545-017-0135-2
Twomey, E. L., Naughten, E. R., Donoghue, V. B., and Ryan, S. (2003). Neuroimaging findings in glutaric aciduria type 1. Pediatr. Radiol. 33, 823–830. doi: 10.1007/s00247-003-0956-z
Viau, K., Ernst, S. L., Vanzo, R. J., Botto, L. D., Pasquali, M., and Longo, N. (2012). Glutaric acidemia type 1: outcomes before and after expanded newborn screening. Mol. Genet. Metab. 106, 430–438. doi: 10.1016/j.ymgme.2012.05.024
Yang, C., Zhou, C., Xu, P., Jin, X., Liu, W., Wang, W., et al. (2020). Newborn screening and diagnosis of inborn errors of metabolism: a 5-year study in an eastern Chinese population. Clin. Chim. Acta 502, 133–138. doi: 10.1016/j.cca.2019.12.022
Young-Lin, N., Shalev, S., Glenn, O. A., Gardner, M., Lee, C., Wynshaw-Boris, A., et al. (2013). Teaching neuroimages: infant with glutaric aciduria type 1 presenting with infantile spasms and hypsarrhythmia. Neurology 81, e182–3. doi: 10.1212/01.wnl.0000437291.75075.53
Keywords: glutaric aciduria type 1, glutaryl-CoA dehydrogenase, glutarylcarnitine, capryloylcarnitine, GCDH gene
Citation: E H, Liang L, Zhang H, Qiu W, Ye J, Xu F, Gong Z, Gu X and Han L (2021) Evaluation of the Clinical, Biochemical, Neurological, and Genetic Presentations of Glutaric Aciduria Type 1 in Patients From China. Front. Genet. 12:702374. doi: 10.3389/fgene.2021.702374
Received: 29 April 2021; Accepted: 14 June 2021;
Published: 07 July 2021.
Edited by:
Theodora Katsila, National Hellenic Research Foundation, GreeceReviewed by:
Emanuele Micaglio, IRCCS Policlinico San Donato, ItalyCopyright © 2021 E, Liang, Zhang, Qiu, Ye, Xu, Gong, Gu and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lianshu Han, aGFubGlhbnNodUB4aW5odWFtZWQuY29tLmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.