
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 23 September 2021
Sec. Human and Medical Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.696624
This article is part of the Research Topic Advances in Genomic and Genetic Tools, and Their Applications for Understanding Embryonic Development and Human Diseases View all 33 articles
 Jiahao Song1†
Jiahao Song1† Qian Zhang2†Bing Lu1Zhongshan Gou1
Qian Zhang2†Bing Lu1Zhongshan Gou1 Ting Wang3Hui Tang3
Ting Wang3Hui Tang3 Jingjing Xiang3Wei Jiang1*Xuedong Deng1*
Jingjing Xiang3Wei Jiang1*Xuedong Deng1*Background: Patients with deletions involving the long arm of chromosome 1 are rare, and the main aim of this study was to refine the genotype-phenotype correlation.
Case Report: In this report, a 28-year-old pregnant woman, gravida 2 para 1, at 25+4 weeks of gestation underwent ultrasound examination in our institute. The ultrasonographic findings of the fetus were as follows: (1) fetal growth restriction; (2) cleft lip and palate; (3) bilateral renal hypoplasia; (4) lateral ventriculomegaly; (5) single umbilical artery; (6) absent stomach; (7) coronary sinus dilatation with persistent left superior vena cava, ventricular septal defect and unroofed coronary sinus syndrome. Chromosomal microarray analysis of amniotic fluid from the fetus revealed a 28.025 Mb deletion in 1q23.3q31.2, spanning from position 164,559,675 to 192,584,768 (hg19).
Conclusion: Genotype-phenotype correlation might improve prenatal diagnosis of fetuses with chromosome 1q deletion. PBX1 could be a candidate gene for fetal growth restriction, renal hypoplasia and congenital heart disease. Fetal growth restriction was accompanied by decreased renal volume in the fetus. Combined with ultrasonic examination, the application of chromosomal microarray analysis will provide accurate prenatal diagnosis.
The incidence of chromosome 1q deletion in the population has not been reported due to the limited number of reported cases. In this study, we retrospectively reviewed a cohort study of 4,119 pregnancies from September 2015 to December 2020, and only one case was detected (0.024%). Available data on these patients with the proximal and intermediate deletions on chromosome 1q, indicate that the most common clinical features include fetal growth restriction, microcephaly, intellectual disability, abnormal ears, palmprint abnormality, fifth finger clinodactyly, fingernail dysplasia, multiple hernia, short limbs and external genital malformations (Scarbrough et al., 1988). In recent years, the application of chromosomal microarray analysis (CMA) makes it possible to identify more fetuses with chromosomal abnormalities. CMA is a high-resolution technique that can detect aneuploidy, microduplication and microdeletions throughout the genome (Xiang et al., 2020). Overall, the literature clearly shows that CMA will detect clinically significant submicroscopic CNVs other than karyotypes in about 6–7% of fetuses with structural anomalies observed by ultrasound (Vora et al., 2016). Based on this, the American Congress of Obstetricians and Gynecologists (ACOG) now recommends CMA as the first tier approach for the diagnosis of fetal structural abnormalities (Levy and Wapner, 2018).
A 28-year-old pregnant woman, gravida 2 para 1, at 25+4 weeks of gestation underwent ultrasound examination in our institute. The biparietal diameter was 5.9 cm (−1.67SD), the head circumference was 20.6 cm (−3.11SD), the abdominal circumference was 18.1 cm (−2.55SD) and the length of femur was 3.7 cm (−4SD) (Figure 1). The results indicated that all the biological measurement indexes were consistent with the ultrasound findings of 22+4 weeks of pregnancy. The size of left and right kidneys was small (left: 2.1 × 1.1 × 0.9 cm, right: 1.8 × 1.0 × 1.0 cm).

Figure 1. Fetal growth curve.
The ultrasound machines used in this study were a Voluson E10 (GE Healthcare Ultrasound, Milwaukee, WI, USA) with a 1–5-MHz transabdominal 2D curvilinear transducer and a Affiniti 70 (Philips Healthcare, Bothell, WA) with a 1–5-MHz convex probe. This examination was conducted in accordance with the prenatal ultrasound examination guidelines of the Ultrasound Physicians Branch of the Chinese Physicians Association. In addition, the abnormalities of placenta, umbilical cord, amniotic fluid and other accessory structures were also observed carefully. A detailed fetal echocardiogram examination was performed according to the practice guidelines of fetal echocardiography issued by the International Society of Ultrasound in Obstetrics and Gynecology (Carvalho et al., 2013). Two dimension and color Doppler were used to evaluate the position, axis and size of the heart, the connection of atrium-ventricle-artery, and the return of systemic veins and pulmonary veins. The ultrasonographic findings of the fetus were as follows: (1) fetal growth restriction; (2) cleft lip and palate (Figure 2A); (3) bilateral renal hypoplasia; (4) lateral ventriculomegaly; (5) single umbilical artery; (6) absent stomach; (7) unroofed coronary sinus syndrome (Figure 2B); (8) coronary sinus dilatation with persistent left superior vena cava (Figure 2C); (9) membranous ventricular septal defect; (10) main pulmonary artery was dilated with inner diameter 7.6 mm, both branch pulmonary arteries arise from the right side of main pulmonary artery.

Figure 2. (A) Transabdominal scan performed at 25 weeks 4 days in coronal plane: the cleft lip appeared as an anhecogenic area at the level of the left upper lip. (B) Transverse section of fetal chest at 25 weeks 4 days: the red arrow pointed to the defect between the left atrium (LA) and the coronary sinus (CS). (C) The three-vessel and tracheal (3VT) view at the upper mediastinum showed a supernumerary vessel to the left of the pulmonary trunk and arterial duct. The red arrow pointed to the persistent left superior vena cava (PLSVC) draining into the right atrium.
After genetic counseling, amniocentesis and CMA were performed at 26 weeks of gestation. The pregnant woman signed an informed consent and was fully informed of the risks of prenatal diagnosis, the advantages and limitations of testing methods.
The genomic DNA of amniotic fluid was digested, amplified, purified, fragmented, labeled, hybridized, washed and scanned strictly according to the standard protocol of the Affymetrix CytoScan platform (Affymetrix, Santa Clara, CA, USA), and the data were analyzed with ChAS 2.0 software. CNVs ≥ 50 Kb containing at least 20 contiguous markers were called for further analysis, and public reference databases such as Clingen, OMIM, DGV, DECIPHER, ISCA, USCS and PubMed were searched for comprehensive analysis. According to the standards and guidelines released by the American College of Medical Genetics (Kearney et al., 2011), CNVs can be classified as pathogenic, likely pathogenic, likely benign, benign or variants of uncertain significance. The CMA results of the fetus (Figure 3) revealed a 28.025-Mb deletion at 1q23.3q31.2 containing 115 OMIM genes (chr1:164,559,675–192,584,768), which was classified as pathogenic. The pregnant woman ultimately chose to terminate the pregnancy at 28 weeks of gestation and underwent induced labor. The autopsy after induced labor confirmed the prenatal ultrasound findings.
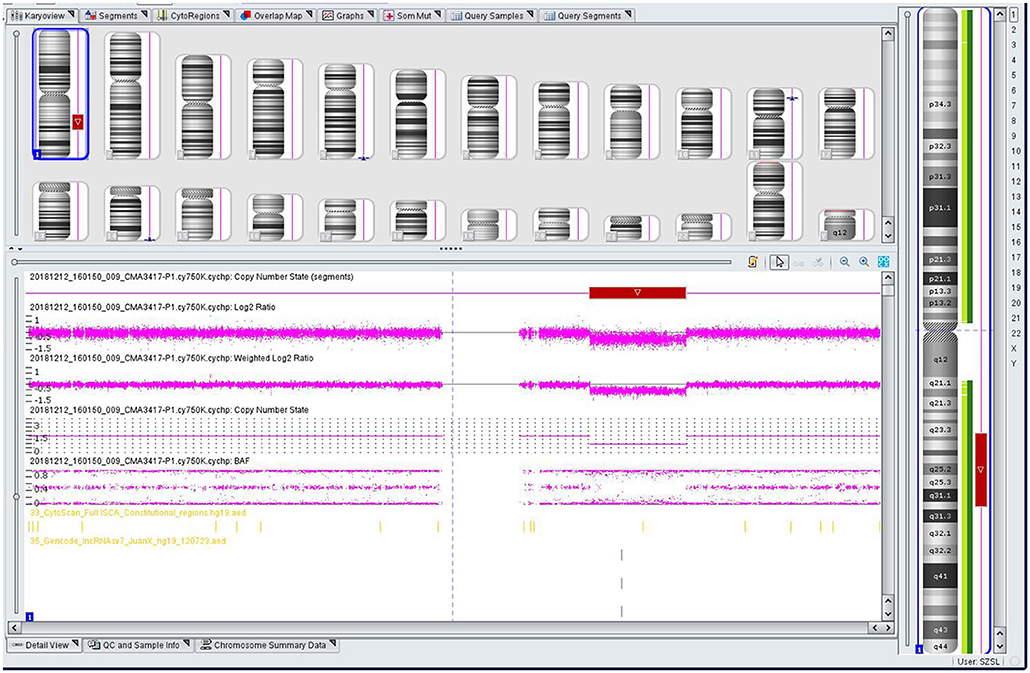
Figure 3. Single nucleotide polymorphism (SNP) array results of the fetus: the red rectangle showed the deletion region of 1q23.3q31.2.
CMA of this fetus revealed a 28.025 Mb deletion on chromosome 1q23.3q31.2 (chr1:164,559,675–192,584,768), which encompassed 115 OMIM genes. The size of deletions on chromosome 1q and the resulting phenotypes varied among patients. A previous case (Table 1) of del(1)(q23.3q31.1) showed similar phenotypes, which included fetal growth restriction, cleft-lip and cleft-palate, absent stomach, bilateral renal hypoplasia, lateral ventriculomegaly, and ventricular septal defect (Lee et al., 2018). However, unroofed coronary sinus syndrome detected by prenatal ultrasound was not reported before. Unroofed coronary sinus (UCS) refers to the communication between the coronary sinus wall and the left atrium, which is often associated with the persistent left superior vena cava (PLSVC). In fact, UCS is a special type of atrial septal defect, which is easy to be missed due to the lack of specific clinical features (Tonni and Grisolia, 2020; Khadkikar et al., 2021). The diagnosis of the lesion is highly significant for the patient's prognosis because it can result in pulmonary hypertension, brain abscess or cerebral embolism (Bonardi et al., 2012). PLSVC is the most common systemic venous abnormality, accounting for 0.5% of the general population, and 3–8% of people with congenital heart disease (Galindo et al., 2007). The fetus with PLSVC is often associated with intra-cardiac and/or extra-cardiac anomalies. Ventricular septal defect, endocardial cushion defect and tetralogy of Fallot are common intra-cardiac malformations, while single umbilical artery is a common extra-cardiac malformation (Berg et al., 2006). Intra-cardiac and/or extra-cardiac anomalies in our patient were demonstrated by ventricular septal defect and single umbilical artery. In addition, PLSVC is more common in fetuses with chromosomal abnormalities, and PLSVC fetuses with other structural malformations have a higher correlation with chromosomal diseases than fetuses with isolated PLSVC (Du et al., 2014), so CMA should be recommended.
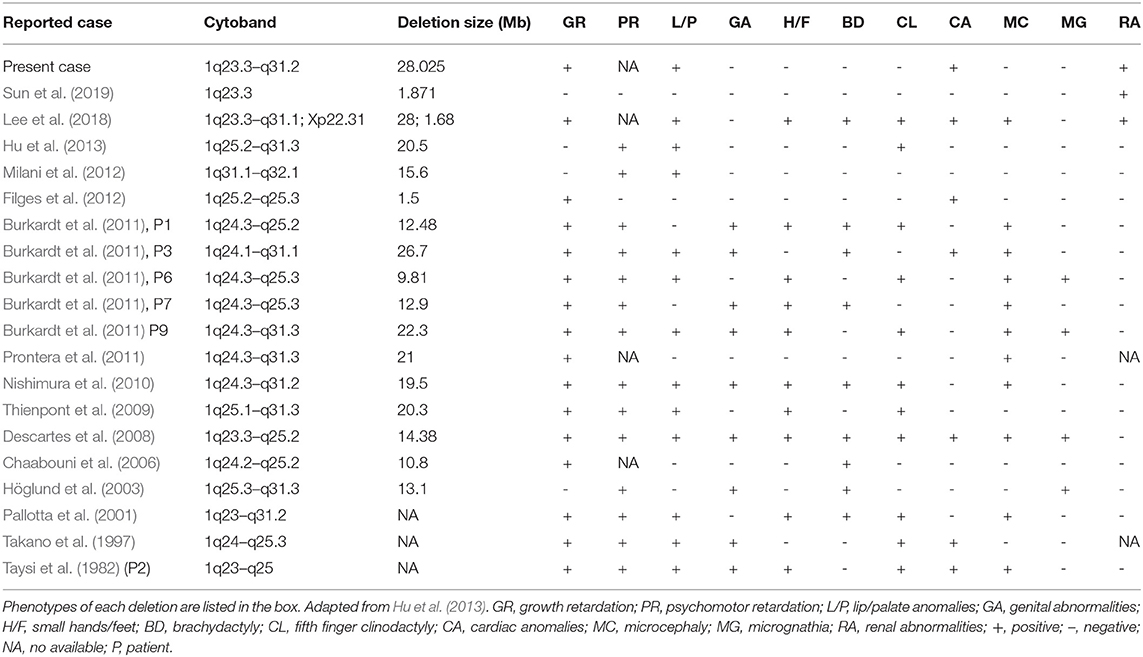
Table 1. Genotypes and phenotypes of the 20 previously identified cases of 1q23–32.1 deletions.
The abnormal expression of PBX1 have been reported to be associated with congenital heart disease (CHD) previously (Alankarage et al., 2020). Except for three cases reported by Hoglund, Milani and Hu (Höglund et al., 2003; Milani et al., 2012; Hu et al., 2013), severe pre- and/or postnatal growth retardation was found in almost all the patients with 1q23.3-q32 deletion. PBX1, LHX4 and CENPL remain to be the primary candidate genes for growth retardation.
PBX1 regulates numerous embryonic processes, including morphologic patterning, organogenesis and hematopoiesis (Ficara et al., 2008). PBX1, which is most strongly expressed in fetal kidney and brain, is an important regulator of interstitial function in renal morphogenesis and plays a key role in interstitial-epithelial signal transduction (Le Tanno et al., 2017). Patients with pathogenic PBX1 mutations/microdeletions showed multiple developmental defects similar to those in PBX1−/− mice, including external ear malformation, abnormal branchial arch derivatives, heart malformations, diaphragmatic hernia, renal hypoplasia and ambiguous genitalia (Slavotinek et al., 2017). Zhou et al. found that PBX1 was a unique functional transcription factor in dNK cells, which promoted fetal development by up-regulating the expression of GPFs. The lack of PBX1+ dNK cells led to fetal growth restriction (Zhou et al., 2020). LHX4 gene is located on 1q25 and involved in the regulation of motoneuron localization and pituitary development. Haploin sufficiency of LHX4 could lead to growth hormone deficiency in patients with a deletion of this area. Another gene, CENPL gene at 1q25.1 is important for proper kinetochore function and mitotic progression. The deletion of this gene would result in growth deficiency (Lam and Morris, 2016).
Fetal growth restriction (FGR) means that fetal growth does not reach its due genetic potential due to the influence of maternal, fetal, placental and other pathological factors, which is characterized by the fact that the fetal weight or abdominal circumference estimated by ultrasound is lower than the 10th percentile of the corresponding gestational age (Galan and Grobman, 2019). Based on the growth status of different gestational weeks, fetal weight can be estimated and dynamic monitoring can be used to understand the fetal growth trend. Detailed ultrasound scans of fetal structure are recommended for FGR.
Brenner et al. proposed that FGR caused congenital renal hypoplasia (Brenner et al., 1988). Renal hypoplasia was defined as abnormally small kidneys with normal morphology and reduced nephron number. Current clinical practice utilizes ultrasonography to measure fetal kidney size and echogenicity as a means of evaluating the severity of renal hypoplasia and dysplasia (Phua and Ho, 2016). Interestingly, the kidneys appeared uniformly echogenic on ultrasound without cysts, and were difficult to differentiate from normal kidneys. The renal artery flow on Doppler couldn't always be recorded. Decreased blood flow to fetal kidneys could be responsible for decreased renal volumes (Slabiak-Blaz et al., 2015). However, renal hypoplasia is more likely the consequence of a malformative embryologic process. PBX1 was thought to play a crucial role during embryogenesis by regulating the expression of developmental genes thus influencing proliferation or remodeling processes (Le Tanno et al., 2017). Defects in the induction and patterning of the developing kidney can cause congenital anomalies of the kidney and/or urinary tract (CAKUT), a wide range of renal-related birth defects including kidney agenesis, kidney hypoplasia or dysplasia, horseshoe kidney, cystic kidneys, or duplex kidney, as well as multiple collecting ducts or ureters abnormalities (San Agustin et al., 2016). Efforts to systematically elucidate genetic link between CAKUT and CHD have been reported, suggesting that CAKUT is highly associated with CHD (Gabriel et al., 2018). These findings may allow CHD fetuses benefit from early diagnosis and early therapeutic intervention of CAKUT. In addition, the mutations in ciliopathies also lead to kidney disease, which is called kidney ciliopathies. Unlike previous studies, we could not found any genes involved in ciliopathies or ciliogenesis in the deleted region (Table 2) (Devlin and Sayer, 2019).
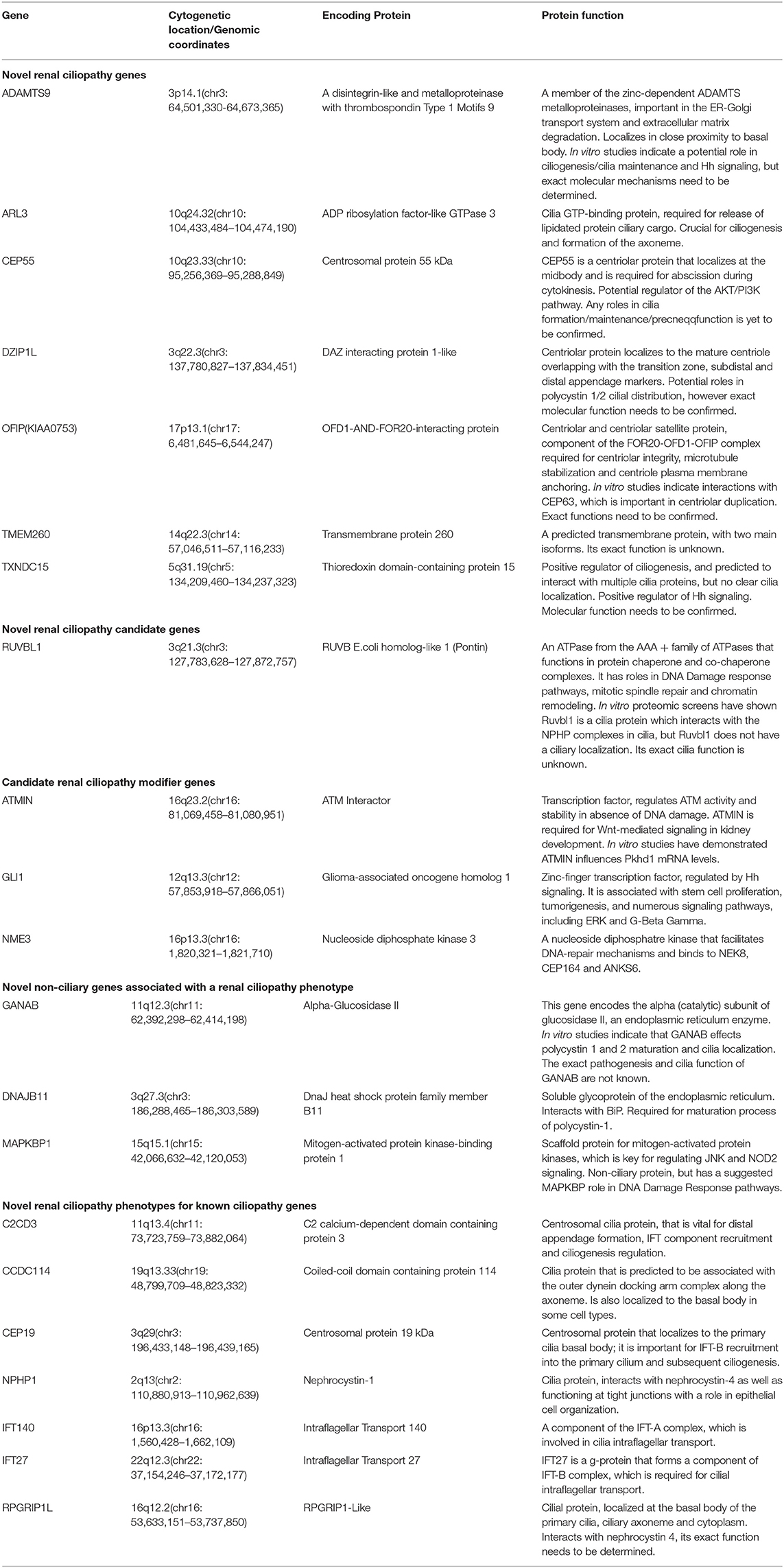
Table 2. Summary of novel renal ciliopathy genes and their function. Adapted from Devlin and Sayer (2019).
Our case is the first report of detailed prenatal ultrasound diagnosis of a fetus with del(1)(q23.3q31.2). When the fetus with FGR is complicated with abnormal ultrasound soft markers or structural abnormalities in the second trimester, interventional prenatal diagnosis and CMA are recommended. CMA is performed either by array comparative genomic hybridization or single nucleotide polymorphism array. Compared with classic cytogenetic and fluorescence in situ hybridization (FISH) techniques, the biggest advantage of using CMA for prenatal genetic diagnosis of chromosomal abnormalities is that CMA can detect much smaller imbalances (Levy and Wapner, 2018). Conventional G-banded karyotype analysis can only be able to detect the deletions or duplications over 5–10 Mb in size. The meta-analysis of the application of CMA to FGR compared with typical G-banding karyotype analysis showed that when FGR was associated with structural abnormalities, CMA could detect an additional 10% of the pathogenic CNV (Borrell et al., 2018).
In this study, we reported a detailed description of the phenotypes in a fetus with del(1)(q23.3q31.2) and proposed some phenotype-related candidate genes. However, this deletion region didn't contain previously reported ciliary genes. Identification of additional affected fetuses with similar deletion of chromosome1q23.3–q31.2 is needed to provide further insights into the pathogenesis of 1q23.3–q31.2 deletion.
The genotyping data for this article are not publicly available to assure patient confidentiality and participant privacy. Requests to access the datasets should be directed to Ting Wang, Ymlvd3RAbmptdS5lZHU=.cn.
The studies involving human participants were reviewed and approved by The Institutional Ethics Committee of The Affiliated Suzhou Hospital of Nanjing Medical University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
JS and QZ made substantial contributions to conception and design, acquisition, analysis and interpretation of the data, and involved in drafting the manuscript. BL, ZG, TW, HT, and JX made substantial contributions to the acquisition, analysis, and interpretation of the data. WJ and XD involved in critically revising the manuscript for important intellectual content and gave the final approval of the version to be published. All authors contributed to the article and approved the submitted version.
This study was supported by grants from the Gusu Health Talent Training Program (GSWS2019056).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank the patient and his family for allowing us to publish this case.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.696624/full#supplementary-material
Alankarage, D., Szot, J. O., Pachter, N., Slavotinek, A., Selleri, L., Shieh, J. T., et al. (2020). Functional characterization of a novel PBX1 de novo missense variant identified in a patient with syndromic congenital heart disease. Hum. Mol. Genet. 29, 1068–1082. doi: 10.1093/hmg/ddz231
Berg, C., Knüppel, M., Geipel, A., Kohl, T., Krapp, M., Knöpfle, G., et al. (2006). Prenatal diagnosis of persistent left superior vena cava and its associated congenital anomalies. Ultrasound Obstetr. Gynecol. 27, 274–280. doi: 10.1002/uog.2704
Bonardi, M., Valentini, A., and Camporotondo, R. (2012). Unroofed coronary sinus and persistent left superior vena cava: a case report. J. Ultrasound 15, 179–182. doi: 10.1016/j.jus.2012.06.001
Borrell, A., Grande, M., Pauta, M., Rodriguez-Revenga, L., and Figueras, F. (2018). Chromosomal microarray analysis in fetuses with growth restriction and normal karyotype: a systematic review and meta-analysis. Fetal Diagn. Ther. 44, 1–9. doi: 10.1159/000479506
Brenner, B. M., Garcia, D. L., and Anderson, S. (1988). Glomeruli and blood pressure. Less of one, more the other? Am. J. Hypertension 1(4 Pt 1), 335–347. doi: 10.1093/ajh/1.4.335
Burkardt, D. D. C., Rosenfeld, J. A., Helgeson, M. L., Angle, B., Banks, V., Smith, W. E., et al. (2011). Distinctive phenotype in 9 patients with deletion of chromosome 1q24-q25. Am. J. Med. Genet. Part A 155A, 1336–1351. doi: 10.1002/ajmg.a.34049
Carvalho, J. S., Allan, L. D., Chaoui, R., Copel, J. A., DeVore, G. R., Hecher, K., et al. (2013). ISUOG Practice Guidelines (updated): sonographic screening examination of the fetal heart. Ultrasound Obstetr. Gynecol. 41, 348–359. doi: 10.1002/uog.12403
Chaabouni, M., Martinovic, J., Sanlaville, D., Attié-Bittach, T., Caillat, S., Turleau, C., et al. (2006). Prenatal diagnosis and molecular characterization of an interstitial 1q24.2q25.2 deletion. Eur. J. Med. Genet. 49, 487–493.
Descartes, M., Hain, J. Z., Conklin, M., Franklin, J., Mikhail, F. M., Lachman, R. S., et al. (2008). Molecular characterization of a patient with an interstitial 1q deletion [del(1)(q24.1q25.3)] and distinctive skeletal abnormalities. Am. J. Med. Genet. Part A 146A, 2937–2943. doi: 10.1002/ajmg.a.32550
Devlin, L. A., and Sayer, J. A. (2019). Renal ciliopathies. Curr. Opin. Genet. Dev. 56, 49–60. doi: 10.1016/j.gde.2019.07.005
Du, L., Xie, H.-N., Zhu, Y.-X., Li, L.-J., Peng, R., and Zheng, J. (2014). Fetal persistent left superior vena cava in cases with and without chromosomal anomalies. Prenat. Diagn. 34, 797–802. doi: 10.1002/pd.4380
Ficara, F., Murphy, M. J., Lin, M., and Cleary, M. L. (2008). Pbx1 regulates self-renewal of long-term hematopoietic stem cells by maintaining their quiescence. Cell Stem Cell 2, 484–496. doi: 10.1016/j.stem.2008.03.004
Filges, I., Bischof-Renner, A., Röthlisberger, B., Potthoff, C., Glanzmann, R., Günthard, J., et al. (2012). Panhypopituitarism presenting as life-threatening heart failure caused by an inherited microdeletion in 1q25 including LHX4. Pediatrics 129, e529–e534. doi: 10.1542/peds.2010-3849
Gabriel, G. C., Pazour, G. J., and Lo, C. W. (2018). Congenital heart defects and ciliopathies associated with renal phenotypes. Front. Pediatr. 6:175. doi: 10.3389/fped.2018.00175
Galan, H., and Grobman, W. (2019). ACOG practice bulletin no. 204: fetal growth restriction. Obstetr. Gynecol. 133:3071. doi: 10.1097/AOG.0000000000003071
Galindo, A., Gutiérrez-Larraya, F., Escribano, D., Arbues, J., and Velasco, J. M. (2007). Clinical significance of persistent left superior vena cava diagnosed in fetal life. Ultrasound Obstetr. Gynecol. 30, 152–161. doi: 10.1002/uog.4045
Höglund, P., Jalkanen, R., Marttinen, E., and Alitalo, T. (2003). Interstitial 1q25.3-q31.3 deletion in a boy with mild manifestations. Am. J. Med. Genet. A 123A, 290–295. doi: 10.1002/ajmg.a.20385
Hu, P., Wang, Y., Meng, L.-L., Qin, L., Ma, D.-Y., Yi, L., et al. (2013). 1q25.2-q31.3 Deletion in a female with mental retardation, clinodactyly, minor facial anomalies but no growth retardation. Mol. Cytogenet. 6, 30. doi: 10.1186/1755-8166-6-30
Kearney, H. M., Thorland, E. C., Brown, K. K., Quintero-Rivera, F., and South, S. T. (2011). American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet. Med. 13, 680–685. doi: 10.1097/GIM.0b013e3182217a3a
Khadkikar, G., V, S. M., Patel, A., Shah, S. C., and Patel, T. M. (2021). A rare case of an unroofed coronary sinus with a persistent left superior vena cava diagnosed by two-dimensional transthoracic echocardiography. Cureus 13:e13041. doi: 10.7759/cureus.13041
Lam, F., and Morris, C. (2016). Nine year old boy with chromosome 1q23.3-q25.1 deletion. Am. J. Med. Genet. A 170, 3013–3017. doi: 10.1002/ajmg.a.37843
Le Tanno, P., Breton, J., Bidart, M., Satre, V., Harbuz, R., Ray, P. F., et al. (2017). haploinsufficiency leads to syndromic congenital anomalies of the kidney and urinary tract (CAKUT) in humans. J. Med. Genet. 54, 502–510. doi: 10.1136/jmedgenet-2016-104435
Lee, J. S., Becker, B. A., Kirby, A., and Knutsen, A. P. (2018). Chromosome 1q23.3q31.1 deletion associated with decreased newborn screening of T cell receptor rearrangement circles (TRECs). Ann. Allergy Asthma Immunol. 121, 125–126. doi: 10.1016/j.anai.2018.04.003
Levy, B., and Wapner, R. (2018). Prenatal diagnosis by chromosomal microarray analysis. Fertil. Steril. 109, 201–212. doi: 10.1016/j.fertnstert.2018.01.005
Milani, D., Bedeschi, M. F., Iascone, M., Chiarelli, G., Cerutti, M., and Menni, F. (2012). De novo deletion of 1q31.1-q32.1 in a patient with developmental delay and behavioral disorders. Cytogenet. Genome Res. 136, 167–170. doi: 10.1159/000336979
Nishimura, A., Hiraki, Y., Shimoda, H., Nishimura, G., Tadaki, H., Tsurusaki, Y., et al. (2010). De novo deletion of 1q24.3-q31.2 in a patient with severe growth retardation. Am. J. Med. Genet. Part A 152A, 1322–1325. doi: 10.1002/ajmg.a.33371
Pallotta, R., Dalprà, L., Miozzo, M., Ehresmann, T., and Fusilli, P. (2001). A patient defines the interstitial 1q deletion syndrome characterized by antithrombin III deficiency. Am. J. Med. Genet. 104, 282–286.
Phua, Y. L., and Ho, J. (2016). Renal dysplasia in the neonate. Curr. Opin. Pediatr. 28, 209–215. doi: 10.1097/MOP.0000000000000324
Prontera, P., Clerici, G., Bernardini, L., Schippa, M., Capalbo, A., Manes, I., et al. (2011). Prenatal diagnosis and molecular characterization of an interstitial 1q24.3-31.3 deletion: case report and review. Genetic counseling 22, 41–48.
San Agustin, J. T., Klena, N., Granath, K., Panigrahy, A., Stewart, E., Devine, W., et al. (2016). Genetic link between renal birth defects and congenital heart disease. Nat. Commun. 7, 11103. doi: 10.1038/ncomms11910
Scarbrough, P. R., Files, B., Carroll, A. J., Quinlan, R. W., Finley, S. C., and Finley, W. H. (1988). Interstitial deletion of chromosome 1 [del(1)(q25q32)] in an infant with prune belly sequence. Prenat. Diagn. 8, 169–174. doi: 10.1002/pd.1970080302
Slabiak-Blaz, N., Adamczak, M., Gut, N., Grajoszek, A., Nyengaard, J. R., Ritz, E., et al. (2015). Administration of cyclosporine A in pregnant rats—the effect on blood pressure and on the glomerular number in their offspring. Kidney Blood Pressure Res. 40, 413–423. doi: 10.1159/000368515
Slavotinek, A., Risolino, M., Losa, M., Cho, M. T., Monaghan, K. G., Schneidman-Duhovny, D., et al. (2017). De novo, deleterious sequence variants that alter the transcriptional activity of the homeoprotein PBX1 are associated with intellectual disability and pleiotropic developmental defects. Human Mol. Genet. 26, 4849–4860. doi: 10.1093/hmg/ddx363
Sun, M., Lou, J., Li, Q., Chen, J., Li, Y., Li, D., et al. (2019). Prenatal findings and molecular cytogenetic analyses of a de novo interstitial deletion of 1q23.3 encompassing PBX1 gene. Taiwan J. Obstet. Gynecol. 58, 292–295. doi: 10.1016/j.tjog.2019.01.022
Takano, T., Yamanouchi, Y., Mori, Y., Kudo, S., Nakayama, T., Sugiura, M., et al. (1997). Interstitial deletion of chromosome 1q [del(1)(q24q25.3)] identified by fluorescence in situ hybridization and gene dosage analysis of apolipoprotein A-II, coagulation factor V, and antithrombin III. Am. J. Med. Genet. 68, 207–210.
Taysi, K., Sekhon, G. S., and Hillman, R. E. (1982). A new syndrome of proximal deletion of the long arm of chromosome 1: 1q21-23 leads to 1q25. Am. J. Med. Genet. 13, 423–430.
Thienpont, B., Dimitriadou, E., Theodoropoulos, K., Breckpot, J., Fryssira, H., Kitsiou-Tzeli, S., et al. (2009). Refining the locus of branchio-otic syndrome 2 (BOS2) to a 5.25 Mb locus on chromosome 1q31.3q32.1. Eur. J. Med. Genet. 52, 393–397. doi: 10.1016/j.ejmg.2009.09.005
Tonni, G., and Grisolia, G. (2020). “Fetal left cardiac malformations,” in Perinatal Cardiology Part 1, eds. E. A. Júnior, N. J. M. Bravo-Valenzuela and A. B. Peixoto. (Sharjah: Bentham Science Publishers), 220–253. doi: 10.2174/9789811446801120010014
Vora, N. L., Romero, S. T., Ralston, S. J., Dugoff, L., and Kuller, J. A. (2016). Committee Opinion No.682: microarrays and next-generation sequencing technology: the use of advanced genetic diagnostic tools in obstetrics and gynecology. Obstet. Gynecol. 128, e262–e268. doi: 10.1097/AOG.0000000000001817
Xiang, J., Ding, Y., Song, X., Mao, J., Liu, M., Liu, Y., et al. (2020). Clinical utility of SNP array analysis in prenatal diagnosis: a cohort study of 5000 pregnancies. Front. Genet. 11:571219. doi: 10.3389/fgene.2020.571219
Keywords: unroofed coronary sinus syndrome, persistent left superior vena cava, chromosomal microarray analysis, kidney, ultrasound, fetal growth restriction
Citation: Song J, Zhang Q, Lu B, Gou Z, Wang T, Tang H, Xiang J, Jiang W and Deng X (2021) Case Report: Candidate Genes Associated With Prenatal Ultrasound Anomalies in a Fetus With Prenatally Detected 1q23.3q31.2 Deletion. Front. Genet. 12:696624. doi: 10.3389/fgene.2021.696624
Received: 17 April 2021; Accepted: 30 August 2021;
Published: 23 September 2021.
Edited by:
Muhammad Abu-Elmagd, King Abdulaziz University, Saudi ArabiaReviewed by:
Gabriele Tonni, Azienda Unita Sanitaria Locale di Reggio Emilia, ItalyCopyright © 2021 Song, Zhang, Lu, Gou, Wang, Tang, Xiang, Jiang and Deng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Jiang, bG9uZ2ZlbGxvd2p3QHNpbmEuY29t; Xuedong Deng, eHVlZG9uZ2RlbmdAMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.