 Sebastian H. N. Munk
Sebastian H. N. Munk Vasileios Voutsinos
Vasileios Voutsinos Vibe H. Oestergaard
Vibe H. Oestergaard
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Genet., 20 July 2021
Sec. Human and Medical Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.695172
This article is part of the Research TopicChromosomal Fragile Sites, Genome Instability and Human DiseasesView all 10 articles
Common chromosomal fragile sites (CFSs) are genomic regions prone to form breaks and gaps on metaphase chromosomes during conditions of replication stress. Moreover, CFSs are hotspots for deletions and amplifications in cancer genomes. Fragility at CFSs is caused by transcription of extremely large genes, which contributes to replication problems. These extremely large genes do not encode large proteins, but the extreme sizes of the genes originate from vast introns. Intriguingly, the intron sizes of extremely large genes are conserved between mammals and birds. Here, we have used reverse genetics to address the function and significance of the largest intron in the extremely large gene PRKN, which is highly fragile in our model system. Specifically, we have introduced an 80-kilobase deletion in intron 7 of PRKN. We find that gene expression of PRKN is largely unaffected by this intronic deletion. Strikingly, the intronic deletion, which leads to a 12% reduction of the overall size of the PRKN gene body, results in an almost twofold reduction of the PRKN fragility. Our results stress that while the large intron clearly contributes to the fragility of PRKN, it does not play an important role for PRKN expression. Taken together, our findings further add to the mystery concerning conservation of the seemingly non-functional but troublesome large introns in PRKN.
CFSs are specific regions of the genome that often fail to replicate before mitosis, which results in chromosome breakage and high mutation rates (Debatisse et al., 2012). Several pan-cancer genome analyses have also revealed that CFSs are hotspots for structural variants in cancer genomes (Beroukhim et al., 2010; Bignell et al., 2010) with large deletions at the center of CFSs and insertions at the borders of CFSs (Li et al., 2020). In addition, DNA double-strand breaks are remarkably recurrent at CFSs in neuronal progenitor cells (Schwer et al., 2016; Wei et al., 2016).
It has become apparent that breakage and mutations at CFSs are due to replication problems caused by transcription of extremely large genes located at CFSs (Le Tallec et al., 2013; Wilson et al., 2015; Pentzold et al., 2018). To understand how transcription of large genes perturb replication, it is important to keep in mind that eukaryotic replication is initiated bidirectionally from origin of replication complexes (ORCs) scattered across the genome. The distance between these complexes thus determines the minimum distance that two opposing replication forks have to travel to complete replication of the region. Most origins of replication are not used during a normal cell cycle, but during replication stress, excess origins of replication are engaged to ensure complete replication of the genome. The process of transcription repositions the ORCs and thereby clears active intragenic regions of replication origins (Gros et al., 2015; Macheret and Halazonetis, 2018). Hence, transcription of extremely large genes clears vast genomic regions of ORCs and in that way suppresses the firing of backup replication origins in these regions, thus impeding genome replication in an indirect manner. Moreover, clashes between transcription and replication machineries may directly challenge replication of CFSs (Helmrich et al., 2011; Oestergaard and Lisby, 2016; Hamperl et al., 2017). Finally, AT-dinucleotide rich regions capable of forming secondary structures can further perturb replication of certain regions within CFSs (Kaushal and Freudenreich, 2019).
One of the most fragile regions of the human genome is called FRA6E. Here, transcription of the 1.4 Mb PRKN underlies its fragility (Glover et al., 2017). Intriguingly, the mature PRKN mRNA is only 4 kb despite the fact that the RNA polymerase has to synthesize 1.4 Mb of pre-mRNA. This is because PRKN as well as other extremely large genes mainly consist of introns (Voutsinos et al., 2018). Despite their unstable nature and scarce coding information, we recently showed that the size of PRKN and other extremely large genes at CFSs are conserved in vertebrates, suggesting that the large introns of these genes possess currently unknown biological functions (Pentzold et al., 2018).
The PRKN gene product, parkin, is an E3 ubiquitin-protein ligase that plays a key role in removal of damaged mitochondria through mitophagy (Frank et al., 2012). This process prevents excessive production of reactive oxygen species from dysfunctional mitochondria. Inherited mutations in PRKN are the most common cause of autosomal recessive juvenile form of Parkinson’s disease, thus emphasizing the neuroprotective importance of PRKN (Klein and Westenberger, 2012). Numerous studies suggest that impaired mitophagy is involved in Parkinson’s disease etiology (Frank et al., 2012; Guo, 2012). Additionally, loss or down-regulation of PRKN has been associated with various types of cancer (Gupta et al., 2017), and its loss has been shown to result in a switch to aerobic glycolysis, known as the Warburg effect, which is a characteristic of many cancer types (Zhang et al., 2011).
To investigate the functional significance of extremely large introns, we deleted 80 kb of intron 7 in PRKN in our model system, the avian cell line DT40. We previously showed that PRKN is transcribed and fragile in this cell line (Pentzold et al., 2018). Here, we find that the deletion does not affect PRKN expression levels but leads to a drastic reduction in PRKN fragility.
All constructs generated in this study are listed in Supplementary Table 1 and all primers plus other DNA oligos used in this study are listed in Supplementary Table 2. The PRKN homology arms for C-terminal fluorescent tagging were amplified with the primer pairs VV5/VV6 and VV7/VV8 for the 5′ arm or the 3′ arm, respectively. The Venus-YFP (2YFP) was amplified using VV47 and VV49. All primers were designed to facilitate directional cloning and they were synthesized by TAG Copenhagen. The amplified products were cloned into pCR2.1-TOPO (Invitrogen) and confirmed by Sanger sequencing (performed by Eurofins Genomics).
The fragments for the PRKN 2YFP-tagging construct were then assembled in pBluescript (SK+). Specifically, the 5′ homology arm was inserted as a KpnI SalI fragment, the 3′ homology arm was inserted as a BamHI NotI fragment, and a resistance cassette (BSR or PURO) was inserted as a BamHI fragment. Finally, the 2YFP fragment was inserted as a XhoI SalI fragment into the SalI site. Correct orientation was confirmed by restriction digest. The resulting constructs were named pVV6 and pVV15 encoding puromycin (PURO) or blasticidin (BSR) resistance, respectively.
To construct the repair template for PRKN intron-7 deletion, genomic regions flanking gRNA Target Site 1 (TS1) and gRNA TS2 were first amplified by PCR templated by genomic DNA (gDNA) from DT40 cells to obtain homology arms.
To create the 5′ homology arm extending 2 kb 5′ of gRNA TS1, PCR was conducted on gDNA with the primers 5′ fwd and 5′ rev adapted with ApaI and BamHI restriction sites, respectively. Similarly, PCR was conducted on gDNA with primers 3′ fwd and 3′ rev adapted with BamHI and XbaI restriction sites, respectively, to create the 3′ homology arm extending 3′ of gRNA TS2. The homology arm fragments were subcloned into TOPO TA vectors (Invitrogen; according to manufacturer’s protocol) and sequenced (Eurofins Genomics).
Then, the 5′ homology arm was subcloned from the TOPO TA vector into pBluescript (SK+) as an ApaI-BamHI fragment. Subsequently, the 3′ homology arm was subcloned from the TOPO TA vector into the 5′homology arm-pBluescript as a XbaI-BamHI fragment. Finally, the BSR cassette fragment was cloned in as a BamHI fragment. The final construct was sequenced to confirm correct assembly (Eurofins Genomics).
The backbone for the Cas9/gRNA constructs was pX458 (Addgene). The expression of specific target gRNAs was obtained by annealing the oligonucleotides listed in Supplementary Table 2 and integrating them into pX458 at the BbsI site. Correct integration was confirmed by sequencing (Eurofins Genomics). The constructs were named pX458 PRKN TS1 and pX458 PRKN TS2.
All DT40 cell lines used in this study are listed in Supplementary Table 3. DT40 cells were cultured in RPMI 1640 medium GlutaMAX (Thermo Fisher Scientific) supplemented with 2% chicken serum (Sigma-Aldrich), 8% fetal bovine serum (Thermo Fisher Scientific), 50 U/ml penicillin, 50 μg/ml streptomycin, and 50 μM β-mercaptoethanol at 39°C with 5% CO2.
Transfections for targeted integration were performed by electroporation with Gene Pulser XcellTM (BioRad) with the settings 25 μF and 0.6 kV. Approximately 35 μg of linearized plasmid DNA was used for transfection with the 2YFP targeting construct. For deletion of PRKN intron 7, 20 million cells were transfected with 50 μg linearized repair template and 30 μg of each of the two Cas9/gRNA expression vectors (110 μg DNA in total). Transfection with Cas9/gRNA was transient.
For transient expression of the Cre recombinase, 3.5 million cells were transfected with 15 μg plasmid DNA using the nucleofector system developed by Amaxa Biosystems GmbH (Franklin and Sale, 2006).
For quantification of fluorescently tagged protein levels, the Xcyto® 10 image cytometer (ChemoMetec A/S) was used. Cells were stained by Vybrant Ruby Stain (5 μM, V10309, Thermo Fisher Scientific) to exclude dead cells based on their DNA content. Only cells meeting the following criteria were included in the analysis: not in aggregate, circularity > 0.6, and with DNA content of viable cells.
Total RNA was isolated using the GeneJET RNA Purification Kit (Thermo Fisher Scientific). For RNA samples used for analysis of PACRG mRNA levels, 1 μg RNA was pre-treated with DNase I (Fermentas) to remove gDNA according to manufacturer’s instructions in the GeneJET RNA Purification Kit (Thermo Fisher Scientific). cDNA was made using RevertAid Premium Reverse Transcriptase (Thermo Fisher Scientific) with random hexamers and oligo(dT) primers.
Each qPCR reaction was performed in triplicates. All the primer pairs used for qPCR are shown in Supplementary Table 2. All the primer pair efficiencies were close to 1 (100%) and within the acceptable range according to the Minimum Information for Publication of Quantitative Real-Time PCR Experiments guidelines (Bustin et al., 2009). qPCR was performed with Maxima SYBR Green/ROX qPCR Master Mix (Thermo Scientific) according to manufacturer’s instructions for three-step RT-qPCR cycling protocol on CFX96 Real-Time PCR Detection System (BioRad). Fold changes were calculated using the 2–ΔΔCt method (Livak and Schmittgen, 2001). In all cases GAPDH was used as reference gene.
Metaphase spreads were prepared as previously described (Smith et al., 1990; Pentzold et al., 2018). Briefly, cells were treated with DMSO or aphidicolin (APH) (0.3 μM) (Sigma-Aldrich) over 16 h followed by a 3-h treatment of 0.1 μg/ml colcemid (Life Technologies). Next, cells were swelled in 8 ml hypotonic buffer [20% FBS (vol/vol), 15 mM KCl] for 15 min. Subsequently, 1 ml fixation buffer (25% acetone, 75% methanol) was gradually added. Cells were harvested by centrifugation and then resuspended in 10 ml fixation buffer. Cells were stored at −20°C O/N. Finally, the cells were splatted onto the slides to spread the metaphase chromosomes (Pentzold et al., 2018).
FISH was carried out as previously described with minor modifications (El Achkar et al., 2005; Pentzold et al., 2018). Briefly, the probe used for PRKN detection was made with the BAC CH261-119N16 from the CHORI library, and the probes for intron 7 detection were made by amplifying ≈10 kb fractions of intron 7 with the primers listed in Supplementary Table 2. Probes were labeled either by biotin or by digoxygenin by using BioPrime DNA Labeling system (Invitrogen). Metaphase spreads had been treated with RNase H (Thermo Fisher Scientific) before they were incubated with probes.
Metaphase spreads were incubated with blocking reagent and Streptavidin-Cy3 (Alexa 594) (1:200), Biotinylated rabbit anti-streptavidin (Rockland) (1:266) for biotin-labeled probe detection and mouse anti-digoxygenin FITC (Interchim) (1:50) and goat anti-mouse (Alexa 488) for digoxygenin-labeled probe detection.
Slides with metaphase spreads were mounted with coverslips using 15 μl of mounting medium containing DAPI (4% n-propyl gallate, 80% glycerol, 1 μg/ml DAPI). Metaphase chromosomes were visualized on a widefield microscope (AxioImager Z1; Carl Zeiss) equipped with a 100× objective lens (Plan Apochromat, NA 1.4; Carl Zeiss), a cooled CCD camera (Orca-ER; Hamamatsu Photonics), differential interference contrast (DIC), and an illumination source (HXP120C; Carl Zeiss).
To investigate the role of large introns in genes coinciding with CFSs, we chose to study the PRKN gene, which is highly fragile in our model cell line (Pentzold et al., 2018). To enable live-cell detection of parkin protein levels, we first generated a DT40 cell line with PRKN endogenously tagged with a Venus-YFP (2YFP) tandem tag in a background where the non-fragile gene TOPBP1 was endogenously tagged with TFP on one of its three alleles. The resulting cell line thus has the genotype PRKNWT/2YFP TOPBP1WT/WT/TFP and is referred to as “P2Y-TT”. Following tagging of PRKN, the allele remained fragile in response to replication stress and the tagged gene product was expressed at full length (Supplementary Figures 1A–C). This cell line, P2Y-TT, was used as background for all further genetic manipulations unless otherwise stated.
PRKN, which is located on the long arm of gallus chromosome 3, contains 11 introns of varying sizes (Figure 1A). PRKN is not enriched for repetitive sequences and replicates in the middle of the S phase (Shang et al., 2013; Pentzold et al., 2018). The total AT-dinucleotide percentage of PRKN is 8.5 (Figure 1A, lower panel). We generated cell lines deleted for most of intron 7, which is the largest intron in PRKN and has a representative AT-dinucleotide frequency (Figure 1A). Deletion of this intron was achieved by combining a selectable targeting construct, with homology to each side of the region targeted for deletion, with CRISPR/Cas9-mediated cleavage at two target sites flanking the desired 80-kb deletion as outlined in Figure 1B. Successful targeting yields clones with 80 kb of PRKN intron 7 replaced with a blasticidin resistance (BSR) cassette. Flanking loxP sites enabled subsequent removal of the BSR cassette. Initial PCR screening suggested that some clones potentially had the 80 kb region replaced by the cassette (Supplementary Figures 2A–E). Subsequently, we tested whether the clones still contained a wild-type PRKN allele with PCR analyses of the two guide RNA target sites (Figure 1C). Most clones retained a wild-type allele, but three clones appeared to have lost both wild-type alleles (Figures 1D,E). Then, one potential homozygote and six potential heterozygotes for the 80-kb deletion were transiently transfected with the Cre recombinase to mediate removal of the BSR cassette followed by isolation of single clones. The resulting clones were analyzed with PCR across the region targeted for deletion, and an amplicon of the expected size confirmed successful deletion of 80 kb in PRKN intron 7 in a subset of the clones (Figure 1F). The successful homozygote (clone 27) and heterozygotes (clone 11, 18, 29, and 36) are referred to as PRKNΔ in7/Δ in7 and PRKNWT/Δ in7, respectively.
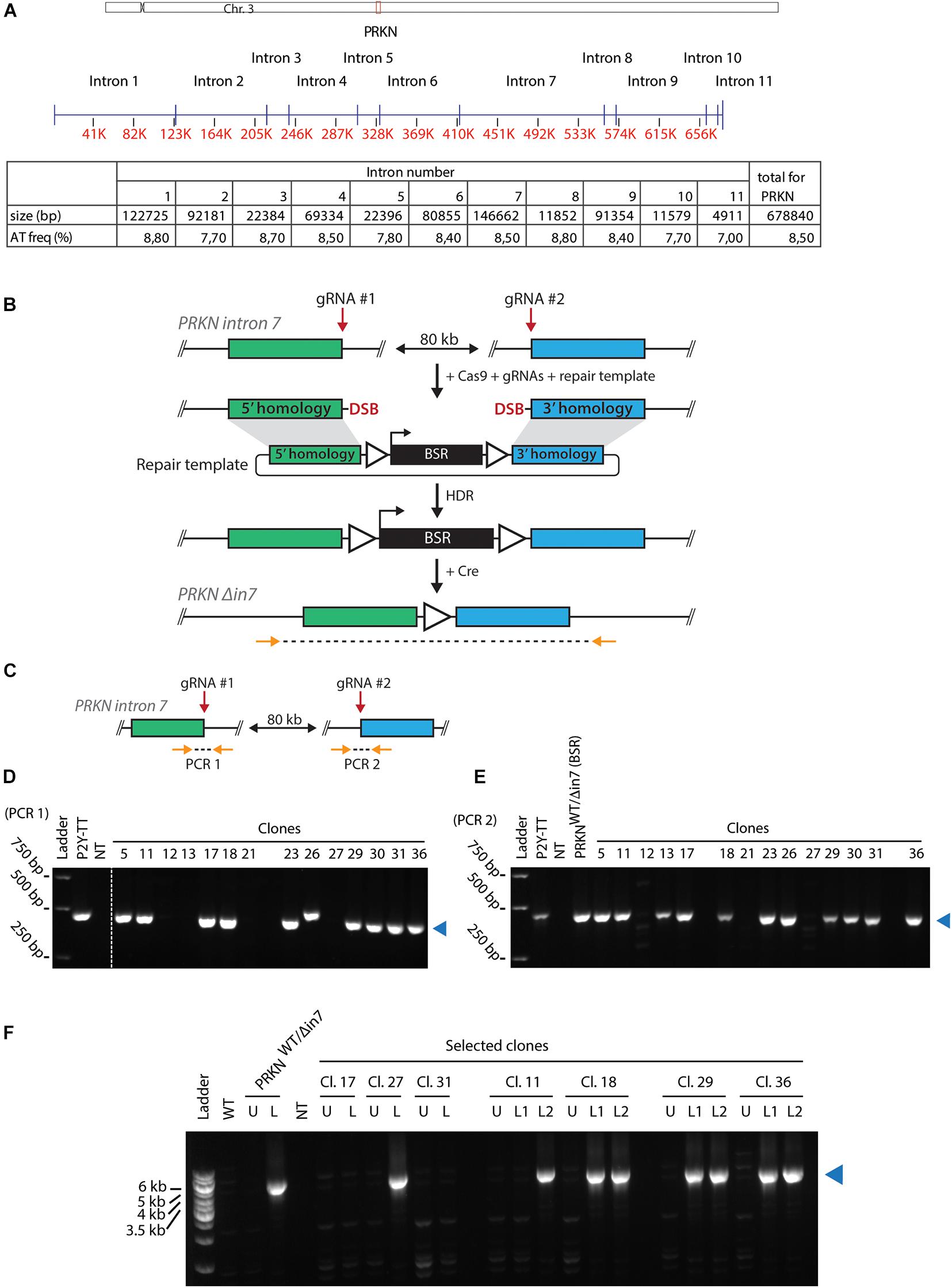
Figure 1. Generating cell lines with large intronic deletion in the fragile site gene PRKN. (A) Upper panel, schematic representation of chicken chromosome 3 drawn to scale. Positions of the centromere (x) and PRKN (red box) are indicated. Middle panel, schematic representation of PRKN drawn to scale. Intron numbers are indicated. Exons are represented as vertical pins on a horizontal line. Ruler indicates position along the gene (kb). Lower panel, table showing sizes of introns given in basepairs (bp) as well as percentage of AT-dinucleotide frequency (AT freq). (B) Outline of the strategy for targeted deletion of 80 kb in PRKN intron 7. To delete 80 kb in PRKN intron 7, cells were transfected with Cas9 and guide RNA (gRNA) expression vectors along with a repair template. The Cas9 nuclease was directed to induce double-strand breaks (DSB) at two target sites (gRNA #1 and gRNA #2) 80 kb apart in PRKN intron 7. The DSBs can be repaired by homology-directed repair (HDR) using the repair template containing a selectable blasticidin resistance (BSR) cassette flanked by loxP sites (triangles) and homology regions (green and blue rectangles). After Cre-mediated removal of the floxed BSR cassette, PCR was used to amplify across the 80-kb deletion with primers (horizontal arrows) binding at the indicated positions outside of the homology regions. (C) Outline of the PCR strategy used to amplify across each of the two gRNA target sites (gRNA #1 and gRNA #2) denoted PCR 1 and PCR 2, respectively. Primers are shown as orange arrows. (D,E) PCR amplification across gRNA #1 (D) and gRNA #2 (E). Analysis of PCR products from the indicated clones and the parental P2Y-TT cell line (positive control). In E, an additional positive control (PRKNWT/Δin7(BSR)) was included. A no-template control (NT) was included in both analyses. A blue triangle on the right of each gel indicates the product of the predicted size. (F) PCR amplification across the 80-kb deletion (as shown in B). Analysis of PCR products from WT cells and selected clones before (U) or after (L) removal of the BSR cassette. If two clonal populations were tested after loxing, this is indicated with L1 and L2. A positive control with the indicated genotype was also included in the analysis. NT denotes the no-template control. The product of the predicted size is indicated by a blue triangle on the right of the gel.
Interestingly, in a previous attempt to generate cell lines with the 80-kb deletion, we isolated two clones with the deletion that both turned out to be trisomic for chromosome 3, which is the chromosome that contains the PRKN gene (not shown). Thus, we examined the karyotype of the clones derived from this transfection for aneuploidy. Here, we found that 2 out of 14 clones were trisomic for chromosome 3 (Supplementary Figure 2F). This suggests that there is a high risk of chromosome mis-segregation associated with targeting of the PRKN gene.
Taken together, the fact that we were able to isolate a homozygous PRKN intron 7-deleted DT40 cell line demonstrates that this part of the genome does not contain functional elements essential for cell viability.
Although intron 7 is not essential for cell viability, it may contain regulatory elements that influence PRKN expression. We thus asked if deletion of the intron has an effect on parkin levels in the cell. First, we used fluorescence image cytometry to evaluate how PRKN-2YFP expression was affected by biallelic intron 7 deletion (Figure 2A). The levels of parkin-2YFP were similar in the intron 7-deleted clone and the parental cell line. Because cells with this genotype must contain the deletion in the 2YFP-tagged PRKN allele this indicates that the 80-kb deletion in PRKN intron 7 does not alter PRKN expression.
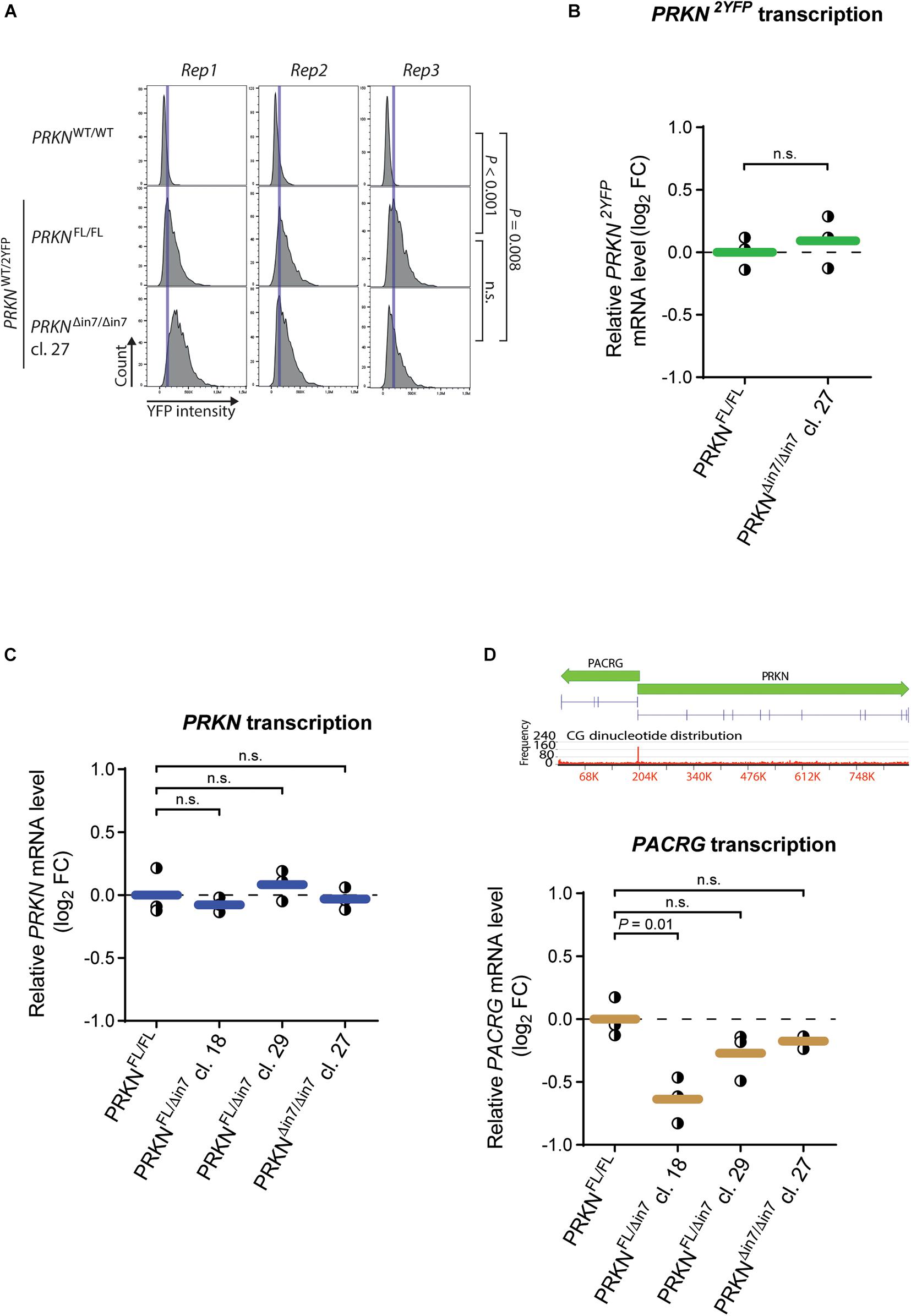
Figure 2. 80-kb deletion of intron 7 does not influence prkn expression levels. (A) Histograms of parkin-2YFP fluorescence levels measured by fluorescence image cytometry in WT cells (PRKNWT/WT) and the P2Y-TT cell line with full-length PRKN (PRKNFL/FL) or intron 7-deleted PRKN (PRKNΔin7/Δin7). Rep1-3 denote three individual experiments. Blue vertical lines indicate the histogram peak for P2Y-TT cells in each replicate. Between 900 and 3,800 cells were analyzed per cell line per replicate. P-values were calculated using the mean YFP intensities from all replicates with Student’s t-test (n.s. = not significant). (B,C) RT-qPCR analysis of PRKN-2YFP (B) and PRKN (C) mRNA levels in P2Y-TT cells with full-length PRKN (PRKNFL/FL) or intron 7 deletion in one (PRKNFL/Δin7) or both alleles (PRKNΔin7/Δin7). Y-axis shows log2 fold change (log2 FC) in mRNA levels relative to PRKNFL/FL. Dots represent individual experiments (n = 3), and horizontal lines indicate the mean. Dashed line denotes log2 FC = 0. P-values were calculated with Student’s t-test (n.s. = not significant). (D) Upper panel, Schematic representation of PACRG and PRKN including their shared promoter. Gene sizes and orientations are indicated with green arrows. Exons are represented as vertical pins on a horizontal line. Ruler indicates position along the genes (kb). Bar chart showing CG dinucleotide frequency is shown below the genes (2,930-bp windows). Lower panel, RT-qPCR analysis of PACRG mRNA levels as in (B,C).
To evaluate the effect of intron 7 deletion on PRKN transcript levels, we performed reverse transcription quantitative PCR (RT-qPCR) with two different primer sets: One set binding specifically to transcripts from the 2YFP-tagged allele (PRKN2YFP) and one set binding to transcripts from both the 2YFP-tagged and untagged (PRKNWT) allele (referred to as “total PRKN transcripts”) (Supplementary Figure 3). Only the clone with homozygous intron 7 deletion was included in the analysis of PRKN2YFP transcript levels while the homozygote and two heterozygotes were included in the analysis of total PRKN transcript levels. No change in either PRKN2YFP transcript levels (Figure 2B) or total PRKN transcript levels (Figure 2C) were detected in any of the clones, supporting that the intron 7 deletion does not alter PRKN expression.
We further investigated whether the intron 7 deletion induced changes in the promoter activity of PRKN. Specifically, we exploited that PRKN shares its promoter (marked by high GC content in Figure 2D, upper panel) with the gene PACRG (parkin coregulated) (West et al., 2003), which is transcribed in the opposite direction of PRKN. Thus, we would expect changes in the promoter activity to affect both genes, and we therefore extended our RT-qPCR investigations to include PACRG transcription (Figure 2D). While a significant decrease in PACRG transcript levels were detected in PRKNWT/Δ in 7 clone 18 compared to the parental cell line, no significant differences were detected in any of the other clones including the homozygote for intron 7 deletion, suggesting that the difference seen in one of the clones is due to clonal variation.
Altogether, this indicates that the 80-kb deletion in PRKN intron 7 does not markedly alter PRKN or PACRG expression.
To test the hypothesis that the large introns in PRKN are underlying its fragility, we performed FISH on metaphase spreads from the two PRKNWT/Δ in 7 clones after inducing replication stress by treatment with the DNA polymerase inhibitor aphidicolin (APH). These clones enabled us to use the full-length wild-type allele of PRKN as an internal control. For FISH, we used a probe that binds PRKN outside of intron 7, which detects both full-length and intron 7-deleted PRKN, as well as a probe that binds the deleted region of intron 7 and therefore only detects the full-length PRKN (Figures 3A,B). While the full-length PRKN locus in the PRKNWT/Δ in 7 clones was as fragile as the full-length PRKN in the parental cell line, the intron 7-deleted PRKN allele was significantly less fragile in both PRKNWT/Δ in 7 clones (Figure 3C). Notably, the deletion, which is equivalent to approximately 12% of the length of PRKN, resulted in an approximately 50% reduction of the fragility of the gene. Thus, the 80 kb region in intron 7 clearly contributes to PRKN fragility even though this region does not have a clear role in PRKN expression or cell viability.
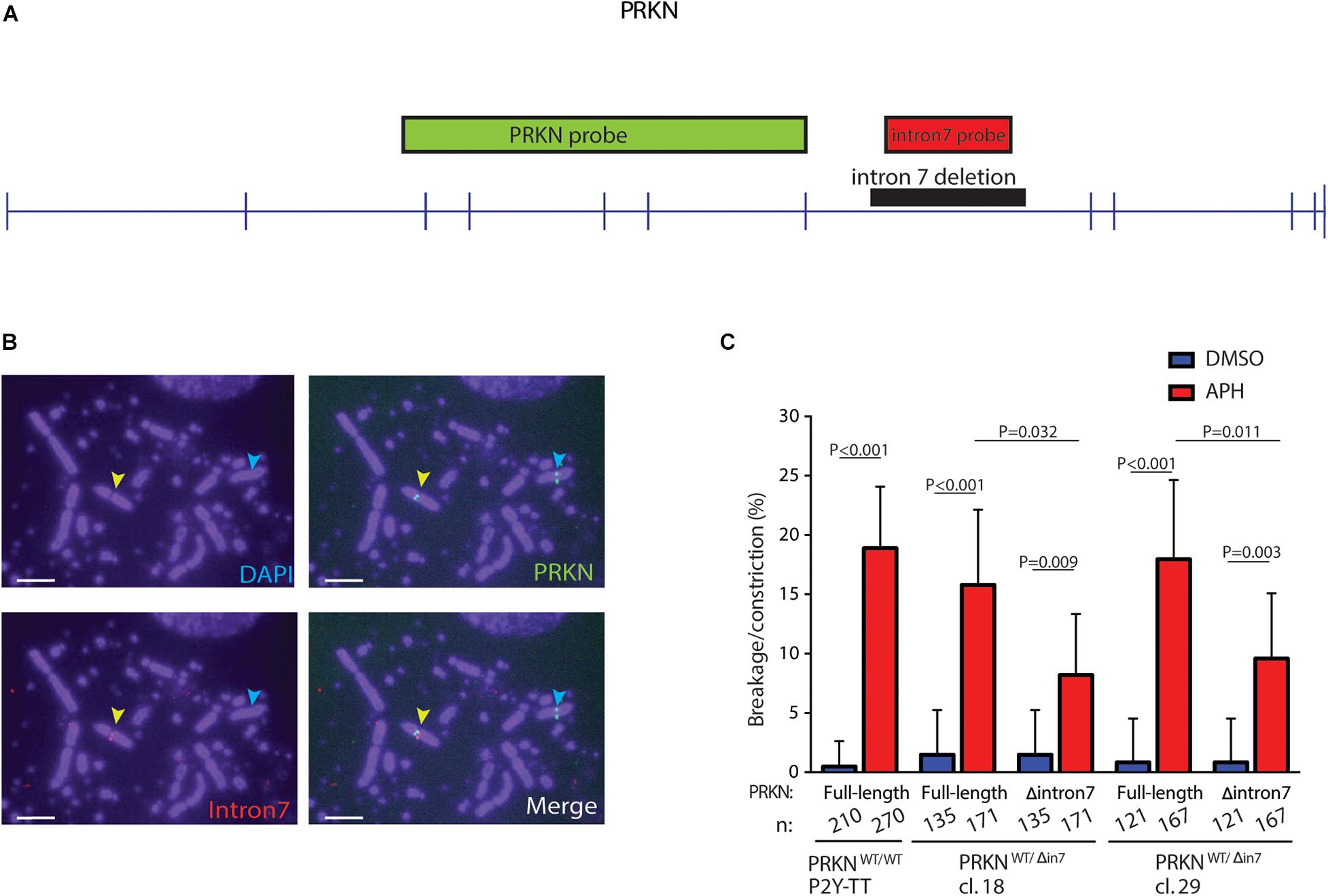
Figure 3. 80 kb deletion in intron 7 significantly affects prkn fragility. (A) Scaled schematic representation of PRKN. Positions of the FISH probes for PRKN are indicated with green and red boxes. The deleted region of intron 7 is indicated by a black box. Exons are represented as vertical pins on a horizontal line. (B) Representative images of metaphase spreads with FISH probes against PRKN (green) and PRKN intron 7 (red) on DAPI-stained metaphase spreads. Cells were treated with 0.3 μM of APH for 16 h before harvesting. Yellow arrows point to a break/constriction at the full-length PRKN allele and the cyan arrows point to the intron 7-deleted PRKN allele only recognized by the PRKN FISH probe. Scale bars are 5 μm. (C) Quantification of breakage/constriction at PRKN in P2Y-TT cells and two independently derived clones of PRKNWT/Δ in7. Cells were treated with DMSO or 0.3 μM APH for 16 h. n denotes the total number of the indicated PRKN allele that was quantified for each clone. The data were analyzed by Fisher’s exact test. Error bars indicate 95% confidence intervals.
Extremely large genes arose in an early vertebrate ancestor due to intron expansions (Voutsinos et al., 2018). Furthermore, the size of extremely large genes seems to be conserved during evolution even though they pose a threat to genome integrity (Pentzold et al., 2018; Voutsinos et al., 2018). In this paper, we have investigated the cellular role of the largest intron in the PRKN gene, which is located in a highly fragile CFS (Wilson et al., 2015; Okamoto et al., 2018; Pentzold et al., 2018; Voutsinos et al., 2018). This is to our knowledge the first controlled experiment addressing the function of an extremely large intron. We were able to generate a cell line homozygous for an 80 kb deletion in PRKN intron 7, clearly demonstrating that the deleted region is not essential for cell viability. Moreover, we find that this intron 7 truncation does not have any significant effect on PRKN gene expression. Yet, the 80-kb intronic deletion leads to an almost 50% reduction of PRKN fragility although only shortening the gene length by 12%, which does not appear to be a consequence of altered transcriptional activity. Thus, these 80 kb of intronic sequence with no apparent function are significantly contributing to PRKN fragility, most likely reflecting that extreme gene size is a trigger for chromosomal fragility, which suggests a disproportional significance of gene length on chromosomal fragility. The reason for the reduction of fragility upon intron deletion might be that conflicts between transcription and replication are avoided due to the shorter traveling time for the RNA polymerase. However, given the importance of replication timing for fragility, the reduced fragility may result from change in replication timing, which we expect to occur because the transcription unit is shortened and thereby the distance between replication origins at each side of the gene will be reduced. However, further studies are needed to experimentally determine the effect of intron deletion on replication timing. We note that the size of the gene or elements within the intron may play a functional role in certain tissues. It may even be possible that genomic instability at CFSs play a physiological role for instance in neurons where it might serve to generate genetic diversity (Schwer et al., 2016; Wei et al., 2016; Voutsinos et al., 2018). Alternatively, the replication difficulties induced by long introns may provoke epigenetic diversification as shown for replication problems induced by G4 quadruplex forming DNA sequences (Schiavone et al., 2014).
Here, we find that intronic truncation does not lead to changes in gene expression, thus adding to the mystery regarding the conservation of large introns. Therefore, further studies are needed to unravel the functional significance of large introns in genes at CFSs that clearly cause problems for dividing cells.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethical review and approval was not required for the animal study because the DT40 parental bursal lymphoblast cell line is commercially available from ATCC.
SM performed the 80-kb deletion of PRKN intron 7 and most of the experiments that relate to this. VV constructed the cell lines expressing tagged parkin and generated the intron 7 FISH probe, and also assisted with supervision and data analysis. VO conceived, coordinated, and supervised the project. VO, SM, and VV wrote the manuscript. All authors contributed to the article and approved the submitted version.
This project was supported by the Villum Foundation (00011407) and the Novo Nordisk Foundation (NNF18OC0052089) to VO. SM was supported by a Novo Scholarship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer VB declared a shared affiliation with one of the authors SM to the handling editor at the time of review.
We are grateful to Michael Lisby for sharing reagents. We would also like to thank Chemometec A/S for providing the Xcyto image cytometer and training for the instrument.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.695172/full#supplementary-material
Beroukhim, R., Mermel, C. H., Porter, D., Wei, G., Raychaudhuri, S., Donovan, J., et al. (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905. doi: 10.1038/nature08822
Bignell, G. R., Greenman, C. D., Davies, H., Butler, A. P., Edkins, S., Andrews, J. M., et al. (2010). Signatures of mutation and selection in the cancer genome. Nature 463, 893–898. doi: 10.1038/nature08768
Bustin, S. A., Benes, V., Garson, J. A., Hellemans, J., Huggett, J., Kubista, M., et al. (2009). The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55:611. doi: 10.1373/clinchem.2008.112797
Debatisse, M., Le Tallec, B., Letessier, A., Dutrillaux, B., and Brison, O. (2012). Common fragile sites: mechanisms of instability revisited. Trends Genet. 28, 22–32. doi: 10.1016/j.tig.2011.10.003
El Achkar, E., Gerbault-Seureau, M., Muleris, M., Dutrillaux, B., and Debatisse, M. (2005). Premature condensation induces breaks at the interface of early and late replicating chromosome bands bearing common fragile sites. Proc. Natl. Acad. Sci. U. S. A. 102, 18069–18074. doi: 10.1073/pnas.0506497102
Frank, M., Duvezin-Caubet, S., Koob, S., Occhipinti, A., Jagasia, R., Petcherski, A., et al. (2012). Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. Biophys. Acta 1823, 2297–2310. doi: 10.1016/j.bbamcr.2012.08.007
Franklin, R., and Sale, J. E. (2006). Transient transfection of DT40. Subcell. Biochem. 40, 379–382. doi: 10.1007/978-1-4020-4896-8_29
Glover, T. W., Wilson, T. E., and Arlt, M. F. (2017). Fragile sites in cancer: more than meets the eye. Nat. Rev. Cancer 17, 489–501. doi: 10.1038/nrc.2017.52
Gros, J., Kumar, C., Lynch, G., Yadav, T., Whitehouse, I., and Remus, D. (2015). Post-licensing Specification of Eukaryotic Replication Origins by Facilitated Mcm2-7 Sliding along DNA. Mol. Cell 60, 797–807. doi: 10.1016/j.molcel.2015.10.022
Guo, M. (2012). Drosophila as a model to study mitochondrial dysfunction in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2:a009944. doi: 10.1101/cshperspect.a009944
Gupta, A., Anjomani-Virmouni, S., Koundouros, N., and Poulogiannis, G. (2017). PARK2 loss promotes cancer progression via redox-mediated inactivation of PTEN. Mol. Cell Oncol. 4:e1329692. doi: 10.1080/23723556.2017.1329692
Hamperl, S., Bocek, M. J., Saldivar, J. C., Swigut, T., and Cimprich, K. A. (2017). Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 170, 774–786e719. doi: 10.1016/j.cell.2017.07.043
Helmrich, A., Ballarino, M., and Tora, L. (2011). Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol. Cell 44, 966–977. doi: 10.1016/j.molcel.2011.10.013
Kaushal, S., and Freudenreich, C. H. (2019). The role of fork stalling and DNA structures in causing chromosome fragility. Genes Chromosomes Cancer 58, 270–283. doi: 10.1002/gcc.22721
Klein, C., and Westenberger, A. (2012). Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2:a008888. doi: 10.1101/cshperspect.a008888
Le Tallec, B., Millot, G. A., Blin, M. E., Brison, O., Dutrillaux, B., and Debatisse, M. (2013). Common fragile site profiling in epithelial and erythroid cells reveals that most recurrent cancer deletions lie in fragile sites hosting large genes. Cell Rep. 4, 420–428. doi: 10.1016/j.celrep.2013.07.003
Li, Y., Roberts, N. D., Wala, J. A., Shapira, O., Schumacher, S. E., Kumar, K., et al. (2020). Patterns of somatic structural variation in human cancer genomes. Nature 578, 112–121. doi: 10.1038/s41586-019-1913-9
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Macheret, M., and Halazonetis, T. D. (2018). Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 555, 112–116. doi: 10.1038/nature25507
Oestergaard, V. H., and Lisby, M. (2016). Transcription-replication conflicts at chromosomal fragile sites—consequences in M phase and beyond. Chromosoma 126, 213–222. doi: 10.1007/s00412-016-0617-2
Okamoto, Y., Iwasaki, W. M., Kugou, K., Takahashi, K. K., Oda, A., Sato, K., et al. (2018). Replication stress induces accumulation of FANCD2 at central region of large fragile genes. Nucleic Acids Res. 46, 2932–2944. doi: 10.1093/nar/gky058
Pentzold, C., Shah, S. A., Hansen, N. R., Le Tallec, B., Seguin-Orlando, A., Debatisse, M., et al. (2018). FANCD2 binding identifies conserved fragile sites at large transcribed genes in avian cells. Nucleic Acids Res. 46, 1280–1294. doi: 10.1093/nar/gkx1260
Schiavone, D., Guilbaud, G., Murat, P., Papadopoulou, C., Sarkies, P., Prioleau, M. N., et al. (2014). Determinants of G quadruplex-induced epigenetic instability in REV1-deficient cells. EMBO J. 33, 2507–2520. doi: 10.15252/embj.201488398
Schwer, B., Wei, P. C., Chang, A. N., Kao, J., Du, Z., Meyers, R. M., et al. (2016). Transcription-associated processes cause DNA double-strand breaks and translocations in neural stem/progenitor cells. Proc. Natl. Acad. Sci. U. S. A. 113, 2258–2263. doi: 10.1073/pnas.1525564113
Shang, W. H., Hori, T., Martins, N. M., Toyoda, A., Misu, S., Monma, N., et al. (2013). Chromosome engineering allows the efficient isolation of vertebrate neocentromeres. Dev. Cell 24, 635–648. doi: 10.1016/j.devcel.2013.02.009
Smith, K. A., Gorman, P. A., Stark, M. B., Groves, R. P., and Stark, G. R. (1990). Distinctive chromosomal structures are formed very early in the amplification of CAD genes in Syrian hamster cells. Cell 63, 1219–1227. doi: 10.1016/0092-8674(90)90417-d
Voutsinos, V., Munk, S. H. N., and Oestergaard, V. H. (2018). Common Chromosomal Fragile Sites-Conserved Failure Stories. Genes 9:580. doi: 10.3390/genes9120580
Wei, P. C., Chang, A. N., Kao, J., Du, Z., Meyers, R. M., Alt, F. W., et al. (2016). Long Neural Genes Harbor Recurrent DNA Break Clusters in Neural Stem/Progenitor Cells. Cell 164, 644–655. doi: 10.1016/j.cell.2015.12.039
West, A. B., Lockhart, P. J., O’Farell, C., and Farrer, M. J. (2003). Identification of a novel gene linked to parkin via a bi-directional promoter. J. Mol. Biol. 326, 11–19. doi: 10.1016/s0022-2836(02)01376-1
Wilson, T. E., Arlt, M. F., Park, S. H., Rajendran, S., Paulsen, M., Ljungman, M., et al. (2015). Large transcription units unify copy number variants and common fragile sites arising under replication stress. Genome Res. 25, 189–200. doi: 10.1101/gr.177121.114
Keywords: common chromosomal fragile sites, large genes, PRKN, parkin, genomic instability, genome editing
Citation: Munk SHN, Voutsinos V and Oestergaard VH (2021) Large Intronic Deletion of the Fragile Site Gene PRKN Dramatically Lowers Its Fragility Without Impacting Gene Expression. Front. Genet. 12:695172. doi: 10.3389/fgene.2021.695172
Received: 14 April 2021; Accepted: 28 June 2021;
Published: 20 July 2021.
Edited by:
Stefano Gnan, Institut Curie, FranceReviewed by:
Lora Boteva, University of Edinburgh, United KingdomCopyright © 2021 Munk, Voutsinos and Oestergaard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vibe H. Oestergaard, VmliZUBiaW8ua3UuZGs=
†These authors have contributed equally to this work
‡Present address: Sebastian H. N. Munk, DNA Replication and Cancer Group, Genome Integrity Unit, Danish Cancer Society Research Center, Copenhagen, Denmark
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.