 Qian Li†
Qian Li† Zhu-Qing Shao
Zhu-Qing Shao- School of Life Sciences, Nanjing University, Nanjing, China
Barley is one of the top 10 crop plants in the world. During its whole lifespan, barley is frequently infected by various pathogens. In this study, we performed genome-wide analysis of the largest group of plant disease resistance (R) genes, the nucleotide binding site–leucine-rich repeat receptor (NLR) gene, in an updated barley genome. A total of 468 NLR genes were identified from the improved barley genome, including one RNL subclass and 467 CNL subclass genes. Proteins of 43 barley CNL genes were shown to contain 25 different integrated domains, including WRKY and BED. The NLR gene number identified in this study is much larger than previously reported results in earlier versions of barley genomes, and only slightly fewer than that in the diploid wheat Triticum urartu. Barley Chromosome 7 contains the largest number of 112 NLR genes, which equals to seven times of the number of NLR genes on Chromosome 4. The majority of NLR genes (68%) are located in multigene clusters. Phylogenetic analysis revealed that at least 18 ancestral CNL lineages were presented in the common ancestor of barley, T. urartu and Arabidopsis thaliana. Among them fifteen lineages expanded to 533 sub-lineages prior to the divergence of barley and T. urartu. The barley genome inherited 356 of these sub-lineages and duplicated to the 467 CNL genes detected in this study. Overall, our study provides an updated profile of barley NLR genes, which should serve as a fundamental resource for functional gene mining and molecular breeding of barley.
Introduction
Plants are consistently challenged by various pathogens during its whole lifespan. A two-layered immune system has been developed along the plant long-term evolution to defense infectious pathogens from environments (Wang et al., 2020; Zhang J. et al., 2020). The first layer immune system can recognize pathogen-associated molecular patterns (PAMPs) through plant cell surface-localized receptors, which induce PAMP-triggered immunity (PTI) (Wang et al., 2020; Zhang J. et al., 2020). Some pathogens can release effector proteins into plant cells to dampen signal transduction of PTI (Wang et al., 2020; Zhang J. et al., 2020). In response, the second layer immune system is required to detect those effectors, through proteins encoded by intracellular disease resistance genes (R genes), which induce effector-triggered immunity (ETI) (Wang et al., 2020; Zhang J. et al., 2020). Several types of R genes have been identified in the past twenty years. Among them, the nucleotide binding site (NBS)-leucine-rich repeat (LRR) receptor (NBS-LRR, also termed as NLR) gene family comprise the majority of R genes identified to date (Kourelis and van der Hoorn, 2018).
NLR genes are specifically discovered in the plant lineage, and their origin could be traced back to the common ancestor of all green plants (Shao et al., 2019). Phylogenetic analysis suggested that NLR genes had diverged into different subclasses prior to the divergence of green plants (Shao et al., 2019). Distinct N-terminal protein domains, including Toll/Interleukin-1 receptor (TIR) domain, Coiled-coil (CC) domain and Resistance to powdery mildew8 (RPW8) domain, have been found from different NLR subclasses. Accordingly, the three NLR subclasses were named as TIR-NLR (TNL), CC-NLR (CNL), and RPW8-NLR (RNL), respectively (Meyers et al., 2003; Shao et al., 2016). Genome-wide analysis revealed that angiosperm genomes contain abundant and variable number of NLR genes. For example, the NLR gene number in Poaceae species ranges from 145 in Zea mays to 2298 in Triticum aestivum (Liu et al., 2021). NLR subclasses composition is also different among angiosperm species. Generally, all monocots and most sequenced magnoliids lack the TNL subclasses, whereas the majority of dicot species genomes have all three NLR subclasses (Liu et al., 2021).
Defining the NLR gene composition in a species is not only helpful for exploring the evolutionary pattern of NLR gene family, but also important for mining and utilization of functional NLR genes. Genome-wide NLR gene analysis have greatly promoted functional NLR gene cloning in several crops. For example, dozens of NLR genes against rice blast have been identified from rice and other Poaceae species by genome-wide identification and comparative genomic analysis (Yang et al., 2013; Wang et al., 2019). Recently, analysis of multiple wheat genomes contributed to the successful cloning of Sm1, a R gene resistant to the orange wheat blossom midge (OWBM, Sitodiplosis mosellana Géhin) (Walkowiak et al., 2020).
Cultivated barley, Hordeum vulgare L. ssp. vulgare, is one of the top ten crop plants in the world1. The product is not only used for animal feeding and malt production, but also serves as a major food staple in many contraries and regions of the world. In 2018, the world-wide production of barley ranks the fourth among all cereal crops (FAOSTAT, 2018). However, like other cereals, barley is also frequently infected by a variety of pathogens. Dozens of different diseases caused by fungi, bacteria, viruses and nematodes have been reported in barley, which result in significant yield reduction and poor grain quality (Murray and Brennan, 2009). However, only a few R genes have been identified from barley, including the Rph1, Rph15 and some MLA alleles (Seeholzer et al., 2010; Chen et al., 2021).
Two previous studies performed genome-wide analyses of NLR genes, using earlier versions of the barley genome assemblies by short-read sequence strategy, and only 100 or so NLR genes were identified (Andersen et al., 2016; Habachi-Houimli et al., 2018). These numbers are much smaller than those in diploid wheat genomes, which have more than 500 NLR genes (Liu et al., 2021). A recent study reported a newly assembly of barley genome of the cultivar Morex by long-read sequencing technology (Mascher et al., 2021). Investigation of the gene composition in the MLA locus revealed that three tandem CNL genes at this locus were missed in the earlier assemblies but present in the new assembly (Mascher et al., 2021). The quality of genome assembly and annotation is critical to genome-wide analysis to gene families, especially to R genes with resembled repeats and duplicates. The above comparison indicated that the NLR genes in barley genome might be greatly underestimated. In this study, we performed a genome-wide NLR gene analysis based on the newly released barley genome, which should provide full and more comprehensive information of NLR genes in this important crop.
Materials and Methods
Data Used in This Study
The protein coding DNA sequences, amino acid sequences and gff3 annotation files of the reference genome sequence assembly of barley cv. Morex V3. were downloaded from the electronic data archive library (e!DAL)2 (Mascher et al., 2021). The Arabidopsis thaliana NLR genes were retrieved from our previous study (Zhang et al., 2016). The T. urartu NLR genes were downloaded from the angiosperm NLR atlas ANNA3 (Liu et al., 2021).
Identification and Classification of Barley NLR Genes
NLR gene identification in the barley genome was performed using BLAST and hidden Markov models search (HMMsearch) methods as described previously (Shao et al., 2014). Briefly, the amino acid sequence of the NBS (also named as NB-ARC) domain was downloaded from the Pfam database (accession number: PF00931) and used as a query to search for NLR proteins using the BLASTp program of the NCBI BLAST software, with expectation value (E-value) setting to 1.0. Simultaneously, the HMM profile of the NBS domain was used as a query to perform a HMMsearch against protein sequences of barley with an E-value setting of 1.0. Then, the results from the two methods were merged together. A round of HMMscan was performed for all the obtained hits against the Pfam-A database (E-value set to 0.0001) to confirm the presence of the NBS domain. Genes do not encode a conserved NBS domain were removed from the datasets. The non-redundant candidate sequences were subjected to the online NCBI Conserved Domains Database (CDD) to identify the CC, RPW8, LRR and other integrated domains. MEME analysis (Bailey et al., 2009) was performed to discover conserved motifs in the NBS domain of the identified NLR genes. The number of displayed motifs was set to 20 with all other parameters default settings as described by Nepal and Benson (2015).
Chromosomal Distribution of Barley NLR Genes
Chromosomal distribution of barley NLR genes was analyzed as described previously (Ameline-Torregrosa et al., 2008). The barley gff3 annotation file was parsed to extract the genomic locations of identified NLR genes. A sliding window analysis was performed with a window size of 250 kb. If two successive annotated NLR genes were located within 250 kb on a chromosome, they were considered as clustered.
Phylogenetic Analysis
Sequence alignment and phylogenetic analysis were performed as described by Shao et al. (2014); Zhang Y. M. et al. (2020). Briefly, amino acid sequences of the conserved NBS domain encoded by barley NLR genes were retrieved and aligned using ClustalW with default options, and then manually corrected in MEGA 7.0 (Kumar et al., 2016). Too short or extremely divergent sequences were excluded from the analysis. Phylogenetic analysis was carried out by IQ-TREE using the maximum likelihood method (Nguyen et al., 2015) after selecting the best-fit model by ModelFinder (Kalyaanamoorthy et al., 2017). Branch support values were estimated using SH-aLRT and UFBoot2 tests (Minh et al., 2013). The phylogeny was reconciled as previously described (Shao et al., 2014) to reconstruction the ancestral state of the NLR genes.
Synteny and Gene Duplication Analysis
Pair-wise all-against-all BLAST was performed for the barley protein sequences. The obtained results and the gff3 annotation file were then subjected to MCScanX for determination of the gene duplication type (Wang et al., 2012). Microsynteny relationships were analyzed and displayed using Tbtools (Chen et al., 2020).
Results
Barley Genome Contains Over 400 NLR Genes
By surveying the annotated protein coding genes of the improved barley genome, a total of 468 NLR genes were identified (Supplementary Table 1), accounting for approximately 0.7% of the more than 62,648 annotated protein coding genes. The number of NLR genes identified from the improved barley genome is three to fourfold larger than those reported in the previous studies (Andersen et al., 2016; Habachi-Houimli et al., 2018). To assign the identified NLR genes into different subclasses, a BLASTp analysis was performed for all obtained NLR genes against the well-defined A. thaliana NLR proteins (Zhang et al., 2016). The results showed that the 468 barley NLR genes comprise one RNL and 467 CNL genes. TNL genes were not detected in the barley genome, which is consistent with the notion that TNL genes are lost in the common ancestor of monocots (Collier et al., 2011; Shao et al., 2016; Liu et al., 2021).
Domain structure analysis revealed high structure diversity of barley NLR proteins. Proteins encoded by the 467 CNL genes could be classified into 14 groups according to their domain composition and arrangement (Figure 1A). Among them, only 119 CNL genes encode intact CNL proteins that contain both the N-terminal CC domain and the C-terminal LRR domain, in addition to the central NBS domain (Figure 1A). Seven of these intact CNL genes encode additional integrated domains (IDs) at the C-terminal, forming a CNL-ID structure; and one has both N-terminal and C-terminal IDs, forming a ID-CNL-ID structure (Figure 1A). There are 32 CNL genes encoding proteins without the N-terminal CC domain. The presence/absence of additional IDs at N-terminal and/or C-terminal further separated these genes into NL (29 genes), NL-ID (1), ID-NL (1), and N-ID-NL (1) groups (Figure 1A). There are 229 CNL genes encoding proteins without the C-terminal LRR domain, including 206 CN, 19 CN-ID, three ID-CN and one ID-CN-ID protein (Figure 1A). 87 CNL genes lost both the N-terminal CC domain and the C-terminal LRR domain (Figure 1A). Five and three of them fused IDs at N-terminal and C-terminal, respectively (Figure 1A). A total of 25 different IDs were detected from 43 barley NLR proteins, accounting for 9% of all NLR proteins. Several of them have been shown to have important function in NLR protein function, e.g., WRKY and BED family domains.
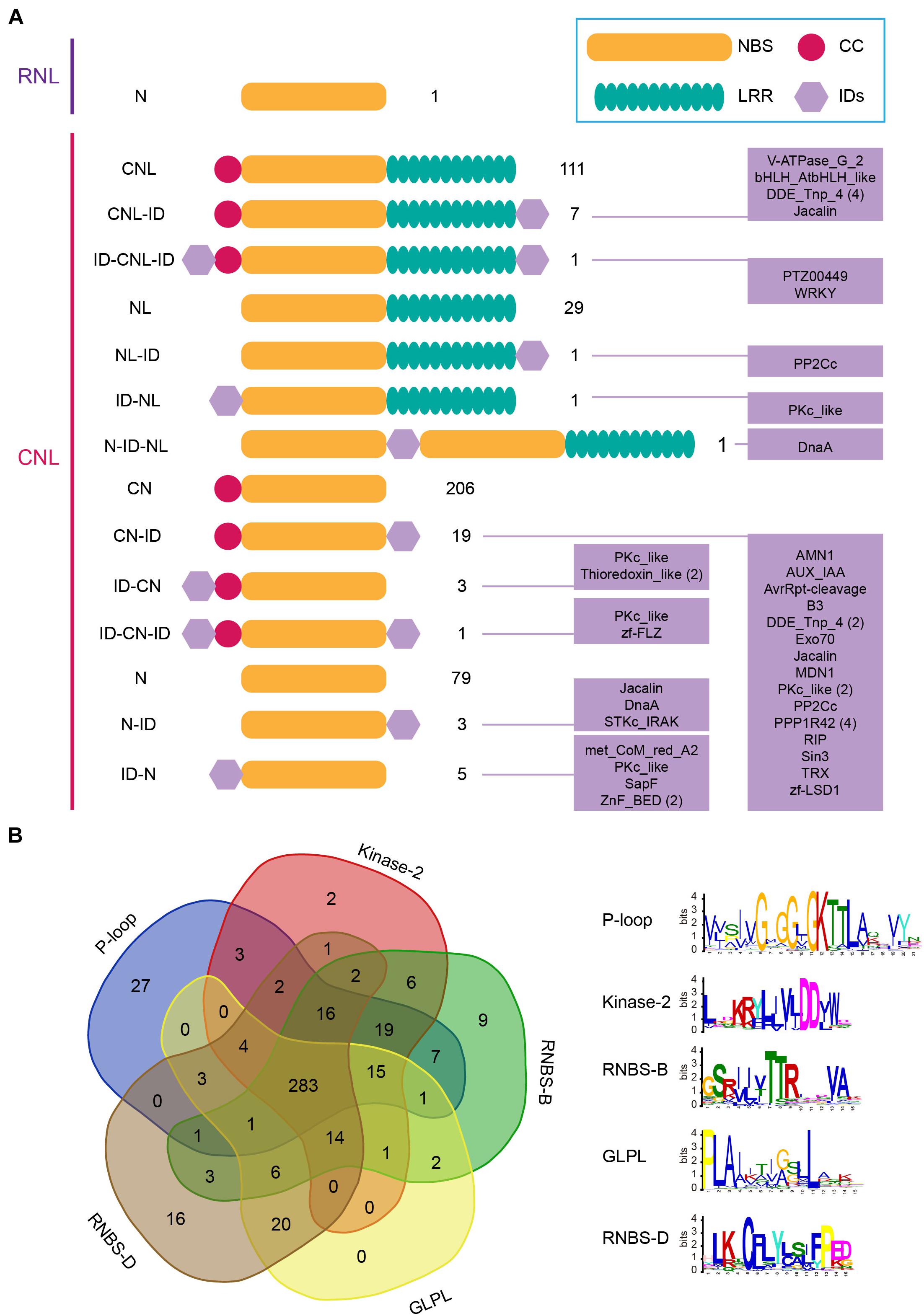
Figure 1. Identification and classification of barley NLR genes. (A) Domain compositions and arrangements of proteins encoded by 468 barley NLR genes. (B) Presence of five key motifs in the amino acid sequence of the NBS domain of 468 barley NLR genes.
We detected the presence of five key motifs in the amino acid sequence of NBS domain by MEME analysis (Bailey et al., 2009). The result showed that the five motifs P-loop, Kinase- 2, RNBS-B, GLPL, and RNBS-D are readily detected and highly conserved in barley NLR proteins as reported in other angiosperms (Shao et al., 2016). Likewise, frequent losses of motifs were detected in many barley NLR proteins. Among the 468 NLR proteins, only 283 preserve all five motifs, accounting for 60% of all NLR proteins (Figure 1B). In contrast, nearly 40% NLR proteins lost at least one key motif in the NBS domain (Figure 1B).
A Majority of Barley NLR Genes Are Presented in Cluster on Chromosomes
All 468 barley NLR genes were mapped to specific chromosomes except one. Calculation of the NLR gene numbers on the seven chromosomes suggested an uneven gene distribution among different chromosomes (Figure 2A). Chromosome 4 has only 16 NLR genes; in contrast, Chromosome 7 contains the maximal of 112 NLR genes, which equals to seven times of NLR gene number on Chromosome 4. Chromosomes 1, 2, 3, 5, and 6 each has 77, 59, 67, 66, and 69 NLR genes, respectively.
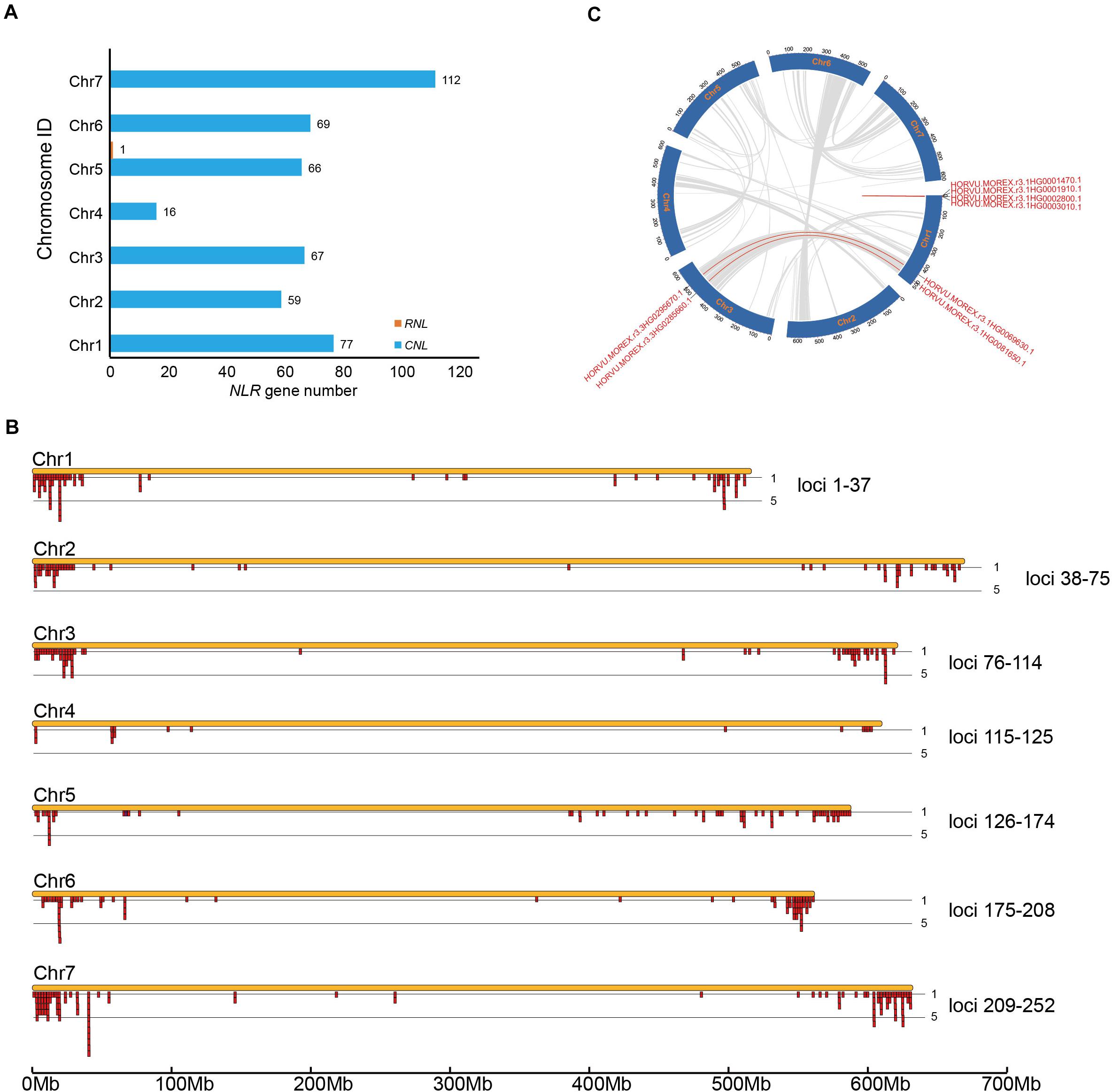
Figure 2. Chromosomal distribution of barley NLR genes. (A) NLR gene number variation among barley genomes. (B) Physical locations of NLR genes on barley chromosomes. NLR genes within an interval less than 250 kb were treated as a cluster (Ameline-Torregrosa et al., 2008). (C) Syntenic relationship of the eight segmental-duplicated NLR genes.
The distribution of NLR genes on barley chromosomes were further deciphered by retrieve their physical locations from the genomic gff3 file. Within each chromosome, most NLR genes are enriched near the telomeric region, whereas very few NLR genes are located at the centromere region. A total of 252 NLR loci were defined on the seven chromosomes, including 150 singletons and 102 multigene clusters (Figure 2B and Supplementary Table 1). The result revealed that 318 NLR genes are present in the 102 clusters, occupying 68% of the total NLR genes. This ratio is slightly lower than that in A. thaliana (Meyers et al., 2003). There are three NLR genes per cluster on average. Among the 102 defined clusters, 54 of them contains only two NLR genes, including 9, 8, 11, 1, 9, 8, and 8 such loci on Chromosome 1–7, respectively. The largest cluster is on chromosome 7, which has 11 NLR genes (Figure 2B). Over 15 clusters have more than 5 NLR genes.
NLR gene may duplicate through different mechanisms. We determined the duplication types of barley NLR genes using the MCScanX (Wang et al., 2012). The result shows that 74 NLR genes show tandem arrays, 146 are proximal duplicates (with no more than 8 interval non-NLR genes), 240 dispersed duplicates and eight are segmental duplicates (Figure 2C).
Species-Specific Preservation and Amplification of Ancestral NLR Lineages During the Speciation of Barley and Wheat
To trace the evolutionary history of barley NLR genes, phylogenetic analysis was conducted by incorporating NLR genes from a diploid wheat T. urartu genome and a dicot species A. thaliana genome. Only CNL and RNL genes of A. thaliana were included in the analysis, because the two monocot species do not contain TNL genes. The phylogenetic analysis result revealed that NLR genes from the three species form two deeply separated clades with a high support value, representing the ancient divergence of RNL and CNL subclasses (Figure 3 and Supplementary Figure 1). RNL genes from the three species further separated into two lineages, namely ADR1 and NRG1. The only RNL gene in barley (HORVU.MOREX.r3.5HG0438750) together with one T. urartu RNL gene (TRIUR3_09219) form a highly supported lineage with four A. thaliana ADR1 genes (Supplementary Figure 1). The remaining two A. thaliana RNL genes form a sister lineage to ADR1, corresponding to the NRG1 lineage. This topology is in accordance with the previous finding that the RNL-NRG1 lineage was lost in the common ancestor of monocots (Collier et al., 2011; Shao et al., 2016; Liu et al., 2021).
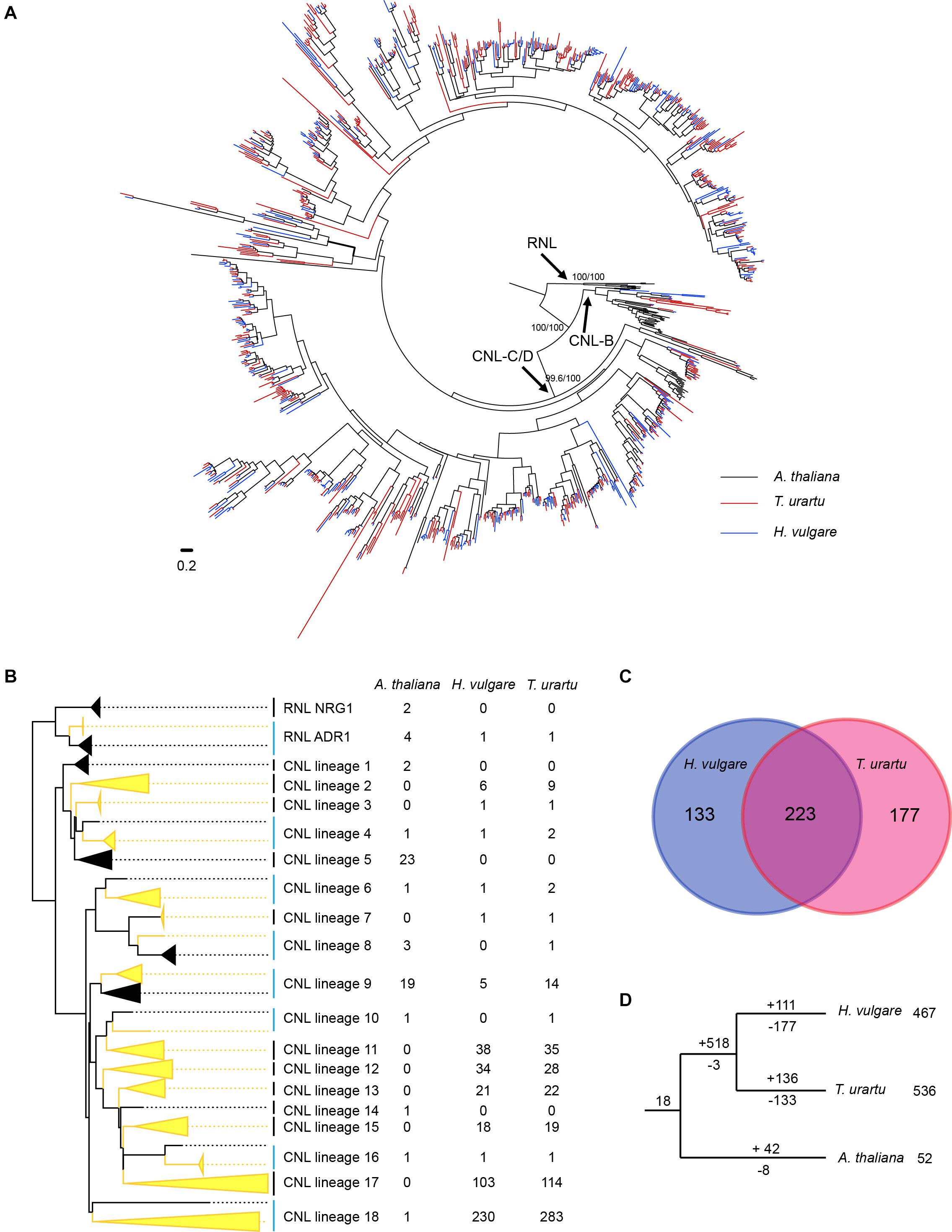
Figure 3. Phylogenetic and evolutionary analysis of RNL and CNL genes from barley, T. urartu and A. thaliana. (A) The phylogeny was constructed based on the conserved NBS domain of CNL and RNL genes from the three species. Branch support values obtained from SH-aLRT (%) and UFBoot2 (%) are labeled on basal nodes. The CNL-B, and CNL-C/D lineages are labeled according to Meyers et al. (2003). (B) Predicted ancestral lineages in the common ancestor of the three species. Gene number of each species on these lineages are indicated at the right of the phylogeny. (C) Shared and species-specific inherited of the 533 CNL sub-lineages that presented in the common ancestor of barley and T. urartu. (D) Duplication/loss events of the CNL genes during the speciation of barley, T. urartu and A. thaliana. Gene duplication/loss events are indicated by numbers with “+” or “-” on each branch, respectively.
CNL genes from the three species form two deep and well-supported clades (Figure 3A and Supplementary Figure 1). One contains the previously defined A. thaliana CNL-B clade genes, whereas the other one contains A. thaliana CNL-C and D clades genes (Meyers et al., 2003). Notably, only eight barley CNL gene and 12 T. urartu CNL genes are presented in the CNL-B clade, whereas the remaining over 400 CNL genes in each species are presented in the CNL-C/D clade. This phenomenon is quite different to that observed in A. thaliana, which has equal number of CNL genes in the two clades (Meyers et al., 2003). The results suggested that lineage-specific expansion of CNL-C/D genes occurred in the two monocot species.
Further reconciling the NLR phylogeny with species relationship revealed that at least 18 ancestral CNL lineages were presented in the progenitor of the three species before the divergence of monocots and eudicots (Figure 3B and Supplementary Figure 1). Among the 18 ancestral CNL lineages, seven (Lineage 4, 6, 8, 9, 10, 16, and 18) were inherited by A. thaliana and at least one of the Poaceae species. Among them, the lineages 4, 6, and 16 seem to have conservatively evolved in all three species, with no more than four genes per species (Figure 3B). In contrast, lineage 18 has expanded greatly to 230 and 283 genes in barley and T. urartu, respectively, whereas only maintained one copy in A. thaliana. The NLR genes in this single lineage occupies about half of all NLR genes in barley and T. urartu, providing a good example of differential expansion among different lineages. Lineage 9 experienced moderate expansion in A. thaliana and the two Poaceae species, with 5–19 NLR genes in each species.
There are three lineages only inherited by A. thaliana and eight lineages only inherited by barley and/or T. urartu, indicating that the two monocot species inherited more ancestral CNL lineages than A. thaliana. In total, the ancestor of barley and T. urartu inherited 15 of the 18 ancestral CNL lineages that emerged in the common ancestor of A. thaliana and the two Poaceae species. These ancestral CNL lineages further diverged into 533 sub-lineages before separation of barley and T. urartu (Figure 3C). Among them, 223 sub-lineages were maintained in both species after speciation, whereas 133 and 177 sub-lineages were only inherited by barley and T. urartu, respectively (Figure 3C). This means a considerable of CNL sub-lineages have been independently lost in the two species. Besides the gene loss events, species-specific gene duplication also occurred frequently. For example, some sub-lineages duplicated to up to ten copies in barley since it separated from T. urartu (Supplementary Figure 1). The species-specific gene duplication occurred more than loss of ancestral sub-lineages in T. urartu, which resulted in the fact that the NLR gene number in its current genome is larger than that in the ancestor of barley and T. urartu. However, in barley the NLR sub-lineage loss has not compensated by species-specific gene duplications, suggesting an “expansion to contraction” shift of the evolutionary pattern.
Discussion
Plant R genes play vital roles in its defense against various pathogens (Xue et al., 2020). The NLR gene family composes the largest group of plant R genes (Kourelis and van der Hoorn, 2018). With the development of DNA sequencing technology, hundreds of plant genomes have been sequenced in the past 20 years, which have greatly benefitted the evolutionary analysis and functional mining of R genes in economically important plants (Wang et al., 2019; Liu et al., 2021). Genome-wide identification and evolutionary analysis has been performed in over 300 angiosperms since the studies in rice and A. thaliana genomes 20 years ago (Bai et al., 2002; Meyers et al., 2003; Liu et al., 2021). Previous studies identified less than 200 NLR genes from barley assemblies generated from short-read sequence sequencing strategy (Andersen et al., 2016; Habachi-Houimli et al., 2018). The number was much smaller than that in wheat, a close relative of barley that separated 11.6 million years ago (Chalupska et al., 2008). The hexaploid wheat T. aestivum has over 2000 NLR genes due to recently occurred polyploidization, whereas the diploid wheat T. urartu has 537 NLR genes. However, by improving the barley genome with long-read sequence strategy, a recently study revealed that the NLR gene number in barley might have been underestimated (Mascher et al., 2021).
In this study, a total of 468 NLR genes were identified from the improved barley genome. The abundance of NLR genes in barley is only slightly smaller than that in the diploid wheat T. urartu. Since similar methodologies were used by our and previous studies (Andersen et al., 2016; Habachi-Houimli et al., 2018), the result suggested that the great difference of NLR gene numbers in barley identified in the present study and previous studies should be caused by genome assembling issues of the short-read sequence strategy. This is in accordance with the result of a recent study, which showed that the updated barley genome has more NLR genes at the MLA locus than the early version genome assembly (Mascher et al., 2021). Furthermore, the wide distribution of barley NLR genes on the phylogeny that constructed with NLR genes from T. urartu and A. thaliana suggested that NLR gene diversity in barley is also comparable with those in T. urartu. Recent studies reported that some functional NLR genes can be transformed from wheat or barley to each other for molecular breeding (Halterman et al., 2001; Zhang et al., 2019). The high abundance and diversity of NLR genes in barley reported in the present study suggested that barley could be an important resource for exploring NLR genes to serve its relatives. Tandem duplication of NLR gene can generate NLR clusters on chromosomes, which is important for maintaining NLR diversity and generating novel functional R genes (Innes et al., 2008; Shao et al., 2014). In barley, the MLA locus is also a multigene cluster with several functional alleles identified (Seeholzer et al., 2010; Mascher et al., 2021). Our data revealed that 318 of identified NLR genes in barley form 102 clusters on its seven chromosomes, accounting for 68% of all NLR genes. These NLR clusters may serve as important reservoirs for preserving and generating of barley NLR diversity. Therefore, deciphering the character of chromosomal distribution and cluster arrangement of barley NLR genes would be helpful for map-based cloning of functional R genes and molecular breeding in barley.
Plant-microbe interaction is a long-term “arms race,” which can drive rapid turnover of NLR profiles during species-speciation (Liu et al., 2021). Therefore, NLR genes often exhibit rapid losses and duplications of ancestral lineages, resulting in few conserved NLR lineages preserved across different species. For example, only seven ancestral lineages were inherited by four legume species and maintained in a conservative manner (Shao et al., 2014). The rare long-term conservatively evolved NLR genes must have been constrained by conserved functions. For example, RNL genes function as NLR signal transducers in both Arabidosis and tobacco (Castel et al., 2019; Saile et al., 2020). In this study, we identified five lineages, namely lineages 4, 6, 8, 10, and 16, that conservatively evolved in both A. thaliana and the two Poaceae species. Interestingly, the NLR genes from A. thaliana in lineage 4 and lineage 10 are RPS2 and RPM1. Proteins encoded by both genes are responsible for resistance to Pseudomonas syringae by monitoring the state changes of the host protein RIN4 (Bent et al., 1994; Mackey et al., 2002). Determining the close relationship of these NLR genes in barley to A. thaliana functional R genes and uncovering their conserved evolutionary pattern may provide clues for exploring their function in barley.
The ancestor of T. urartu and barley have expanded its NLR sub-lineage to 533 after its separation from A. thaliana about 100 million years ago. The majority of these NLR sub-lineages are descendants of CNL lineage 18. However, the 533 sub-lineages presented in the ancestor of T. urartu and barley are differently inherited by the two species. T. urartu only preserved 400 of these sub-lineages and duplicated to the 536 CNL genes in its current genome, whereas barley preserved 356 of these sub-lineages and duplicated to the 467 CNL genes in its current genome. Recently occurred polyploidization caused NLR genes in several hexaploid Triticum species expanded to more than 1000, reflecting rapidly changed NLR profiles after species-speciation by species-specific gene loss and duplication. Considering the transformable of functional NLR genes between the two species (Halterman et al., 2001; Zhang et al., 2019), the shared and species-specific NLR genes may further expand the cross-species pan-NLRome.
Conclusion
Overall, a total of 468 NLR genes were identified from the improved barley genome, including one RNL subclass and 467 CNL subclass genes. The structure diversity, chromosomal distribution and evolutionary history of barley NLR genes were comprehensively analyzed. These results extended the understanding on the abundance and diversity of NLR genes in this important crop, which may serve as a fundamental resource for the molecular breeding of barley.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
Z-QS conceived, designed the study, and revised the manuscript. QL and X-MJ obtained, analyzed the data, and wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Founding of China (32070243 to Z-QS).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We greatly appreciate the Frontiers editors and reviewers for handling our manuscript and providing critical suggestions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.694682/full#supplementary-material
Supplementary Figure 1 | The full phylogeny of NLR genes from barley, T. urartu and A. thaliana.
Supplementary Table 1 | Detailed features of NLR genes identified from barley genome.
Footnotes
- ^ https://www.croptrust.org/crop/barley/
- ^ http://doi.org/10.5447/ipk/2021/3
- ^ http://compbio.nju.edu.cn/app/ANNA/
References
Ameline-Torregrosa, C., Wang, B. B., O’Bleness, M. S., Deshpande, S., Zhu, H., Roe, B., et al. (2008). Identification and characterization of nucleotide-binding site-leucine-rich repeat genes in the model plant Medicago truncatula. Plant Physiol. 146, 5–21. doi: 10.1104/pp.107.104588
Andersen, E. J., Ali, S., Reese, R. N., Yen, Y., Neupane, S., and Nepal, M. P. (2016). Diversity and evolution of disease resistance genes in barley (Hordeum vulgare L.). Evol. Bioinformatics 12, 99–108. doi: 10.4137/Ebo.S38085
Bai, J., Pennill, L. A., Ning, J., Lee, S. W., Ramalingam, J., Webb, C. A., et al. (2002). Diversity in nucleotide binding site-leucine-rich repeat genes in cereals. Genome Res. 12, 1871–1884. doi: 10.1101/gr.454902
Bailey, T. L., Boden, M., Buske, F. A., Frith, M., Grant, C. E., Clementi, L., et al. (2009). Meme suite: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–W208. doi: 10.1093/nar/gkp335
Bent, A. F., Kunkel, B. N., Dahlbeck, D., Brown, K. L., Schmidt, R., Giraudat, J., et al. (1994). RPS2 of Arabidopsis thaliana: a leucine-rich repeat class of plant disease resistance genes. Science 265, 1856–1860. doi: 10.1126/science.8091210
Castel, B., Ngou, P. M., Cevik, V., Redkar, A., Kim, D. S., Yang, Y., et al. (2019). Diverse NLR immune receptors activate defence via the RPW8-NLR NRG1. New Phytol. 222, 966–980. doi: 10.1111/nph.15659
Chalupska, D., Lee, H. Y., Faris, J. D., Evrard, A., Chalhoub, B., Haselkorn, R., et al. (2008). Acc homoeoloci and the evolution of wheat genomes. Proc. Natl. Acad. Sci.U.S.A. 105, 9691–9696. doi: 10.1073/pnas.0803981105
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 13, 1194–1202. doi: 10.1016/j.molp.2020.06.009
Chen, C., Jost, M., Clark, B., Martin, M., Matny, O., Steffenson, B. J., et al. (2021). BED domain-containing NLR from wild barley confers resistance to leaf rust. Plant Biotechnol. J. doi: 10.1111/pbi.13542 Online ahead of print,
Collier, S. M., Hamel, L. P., and Moffett, P. (2011). Cell death mediated by the N-terminal domains of a unique and highly conserved class of NB-LRR protein. Mol. Plant Microbe Interact. 24, 918–931. doi: 10.1094/MPMI-03-11-0050
FAOSTAT. (2018). Available online at: http://www.fao.org/faostat/en/#home
Habachi-Houimli, Y., Khalfallah, Y., Mezghani-Khemakhem, M., Makni, H., Makni, M., and Bouktila, D. (2018). Genome-wide identification, characterization, and evolutionary analysis of NBS-encoding resistance genes in barley. 3 Biotech 8:453. doi: 10.1007/S13205-018-1478-6
Halterman, D., Zhou, F., Wei, F., Wise, R. P., and Schulze-Lefert, P. (2001). The MLA6 coiled-coil, NBS-LRR protein confers AvrMla6-dependent resistance specificity to Blumeria graminis f. sp. hordei in barley and wheat. Plant J. 25, 335–348. doi: 10.1046/j.1365-313x.2001.00982.x
Innes, R. W., Ameline-Torregrosa, C., Ashfield, T., Cannon, E., Cannon, S. B., Chacko, B., et al. (2008). Differential accumulation of retroelements and diversification of NB-LRR disease resistance genes in duplicated regions following polyploidy in the ancestor of soybean. Plant Physiol. 148, 1740–1759. doi: 10.1104/pp.108.127902
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Kourelis, J., and van der Hoorn, R. A. L. (2018). Defended to the nines: 25 years of resistance gene cloning identifies nine mechanisms for R protein function. Plant Cell 30, 285–299. doi: 10.1105/tpc.17.00579
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Liu, Y., Zeng, Z., Li, Q., Jiang, X. M., Jiang, Z., Tang, J. H., et al. (2021). An angiosperm NLR atlas reveals that NLR gene reduction is associated with ecological specialization and signal transduction component deletion. bioRxiv [Preprint]. doi: 10.1101/2021.02.10.430603
Mackey, D., Holt, B. F., Wiig, A., and Dangl, J. L. (2002). RIN4 interacts with Pseudomonas syringae type III effector molecules and is required for RPM1-mediated resistance in Arabidopsis. Cell 108, 743–754. doi: 10.1016/S0092-8674(02)00661-X
Mascher, M., Wicker, T., Jenkins, J., Plott, C., Lux, T., Koh, C. S., et al. (2021). Long-read sequence assembly: a technical evaluation in barley. Plant Cell koab077. doi: 10.1093/plcell/koab077 Online ahead of print,
Meyers, B. C., Kozik, A., Griego, A., Kuang, H., and Michelmore, R. W. (2003). Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant Cell 15, 809–834. doi: 10.1105/tpc.009308
Minh, B. Q., Nguyen, M. A., and von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. doi: 10.1093/molbev/mst024
Murray, G. M., and Brennan, J. P. (2009). Estimating disease losses to the Australian wheat industry. Australas. Plant Pathol. 38, 558–570. doi: 10.1071/AP09053
Nepal, M. P., and Benson, B. V. (2015). CNL disease resistance genes in soybean and their evolutionary divergence. Evol. Bioinformatics 11, 49–63. doi: 10.4137/Ebo.S21782
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Saile, S. C., Jacob, P., Castel, B., Jubic, L. M., Salas-Gonzales, I., Backer, M., et al. (2020). Two unequally redundant “helper” immune receptor families mediate Arabidopsis thaliana intracellular “sensor” immune receptor functions. PLoS Biol. 18:e3000783. doi: 10.1371/journal.pbio.3000783
Seeholzer, S., Tsuchimatsu, T., Jordan, T., Bieri, S., Pajonk, S., Yang, W. X., et al. (2010). Diversity at the Mla powdery mildew resistance locus from cultivated barley reveals sites of positive selection. Mol. Plant Microbe Interact. 23, 497–509. doi: 10.1094/Mpmi-23-4-0497
Shao, Z. Q., Xue, J. Y., Wang, Q., Wang, B., and Chen, J. Q. (2019). Revisiting the origin of plant NBS-LRR genes. Trends Plant Sci. 24, 9–12. doi: 10.1016/j.tplants.2018.10.015
Shao, Z. Q., Xue, J. Y., Wu, P., Zhang, Y. M., Wu, Y., Hang, Y. Y., et al. (2016). Large-scale analyses of angiosperm nucleotide-binding site-leucine-rich repeat genes reveal three anciently diverged classes with distinct evolutionary patterns. Plant Physiol. 170, 2095–2109. doi: 10.1104/pp.15.01487
Shao, Z. Q., Zhang, Y. M., Hang, Y. Y., Xue, J. Y., Zhou, G. C., Wu, P., et al. (2014). Long-term evolution of nucleotide-binding site-leucine-rich repeat genes: understanding gained from and beyond the legume family. Plant Physiol. 166, 217–234. doi: 10.1104/pp.114.243626
Walkowiak, S., Gao, L., Monat, C., Haberer, G., Kassa, M. T., Brinton, J., et al. (2020). Multiple wheat genomes reveal global variation in modern breeding. Nature 588, 277–283. doi: 10.1038/s41586-020-2961-x
Wang, L., Zhao, L., Zhang, X., Zhang, Q., Jia, Y., Wang, G., et al. (2019). Large-scale identification and functional analysis of NLR genes in blast resistance in the Tetep rice genome sequence. Proc. Natl. Acad. Sci. U.S.A. 116, 18479–18487. doi: 10.1073/pnas.1910229116
Wang, W., Feng, B., Zhou, J. M., and Tang, D. (2020). Plant immune signaling: advancing on two frontiers. J. Integr. Plant Biol. 62, 2–24. doi: 10.1111/jipb.12898
Wang, Y., Tang, H., Debarry, J. D., Tan, X., Li, J., Wang, X., et al. (2012). MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 40:e49. doi: 10.1093/nar/gkr1293
Xue, J. Y., Takken, F. L. W., Nepal, M. P., Maekawa, T., and Shao, Z. Q. (2020). Editorial: evolution and functional mechanisms of plant disease resistance. Front. Genet. 11:593240. doi: 10.3389/fgene.2020.593240
Yang, S., Li, J., Zhang, X., Zhang, Q., Huang, J., Chen, J. Q., et al. (2013). Rapidly evolving R genes in diverse grass species confer resistance to rice blast disease. Proc. Natl. Acad. Sci. U.S.A. 110, 18572–18577. doi: 10.1073/pnas.1318211110
Zhang, C., Huang, L., Zhang, H., Hao, Q., Lyu, B., Wang, M., et al. (2019). An ancestral NB-LRR with duplicated 3’UTRs confers stripe rust resistance in wheat and barley. Nat. Commun. 10:4023. doi: 10.1038/s41467-019-11872-9
Zhang, J., Coaker, G., Zhou, J. M., and Dong, X. N. (2020). Plant immune mechanisms: from reductionistic to holistic points of view. Mol. Plant 13, 1358–1378. doi: 10.1016/j.molp.2020.09.007
Zhang, Y. M., Chen, M., Sun, L., Wang, Y., Yin, J., Liu, J., et al. (2020). Genome-wide identification and evolutionary analysis of NBS-LRR genes from Dioscorea rotundata. Front. Genet. 11:484. doi: 10.3389/fgene.2020.00484
Keywords: barley, NLR gene, disease resistance, gene family, evolutionary analysis
Citation: Li Q, Jiang X-M and Shao Z-Q (2021) Genome-Wide Analysis of NLR Disease Resistance Genes in an Updated Reference Genome of Barley. Front. Genet. 12:694682. doi: 10.3389/fgene.2021.694682
Received: 13 April 2021; Accepted: 26 April 2021;
Published: 24 May 2021.
Edited by:
Pengtao Ma, Yantai University, ChinaCopyright © 2021 Li, Jiang and Shao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhu-Qing Shao, emh1cWluZ3NoYW9Abmp1LmVkdS5jbg==
†These authors have contributed equally to this work