 Linlin Xu1†
Linlin Xu1† Dina Zhu1†Yanxia Zhang1Guanxia Liang1Min Liang1Xiaofeng Wei1
Dina Zhu1†Yanxia Zhang1Guanxia Liang1Min Liang1Xiaofeng Wei1 Xiaoqing Feng2Xuedong Wu2Xuan Shang1*
Xiaoqing Feng2Xuedong Wu2Xuan Shang1*- 1Department of Medical Genetics, School of Basic Medical Sciences, Southern Medical University, Guangzhou, China
- 2Department of Pediatrics, Nanfang Hospital, Southern Medical University, Guangzhou, China
Background: Anemia is one of the most common diseases affecting children worldwide. Hereditary forms of anemia due to gene mutations are difficult to diagnose because they only rely on clinical manifestations. In regions with high prevalence of thalassemia such as southern China, pediatric patients with a hereditary hemolytic anemia (HHA) phenotype are often diagnosed with β-thalassemia. However, HHA can be caused by other gene defects. Here, a case previously diagnosed with thalassemia in a local hospital was sent to our laboratory for further genetic diagnosis. Preliminary molecular testing did not identify any mutations in globin genes.
Methods: All blood samples were collected after informed consent had been obtain from the proband’s parents. Both clinical and genetic analyses were conducted for the patient and her family members, including clinical data collection and sequencing of the KLF1 gene. Relevant literature was reviewed, including genetically confirmed cases with well-documented clinical summaries.
Results: Based on the detailed clinical data for this case, we diagnosed the patient with severe HHA. Sanger sequencing confirmed that there was a mutation on each KLF1 allele in the proband, which is missense mutation c.892G > C (p.Ala298Pro) inherited from father and frameshift mutation c.525_526insCGGCGCC (p.Gly176Argfs∗179) from the mother, respectively. A summary of the KLF1 mutation spectrum and a clarification of genotype–phenotype correlation were performed through a combined analysis of the case and literature studies.
Conclusion: This study corrected the misdiagnosis and identified the etiology in a Chinese patient with HHA. Identification of the disease-causing gene is important for the treatment and care of the patient and prevention of another affected childbirth in her family. In addition, this study provided insight to better distinguish HHA patients with β-thalassemia mutations from those with KLF1 mutations.
Introduction
Anemia is the most common blood disease worldwide (Kassebaum et al., 2014), which is defined by a hemoglobin (Hb) level below the normal value, leading to decreased oxygen-carrying capacity. Severe anemia impairs growth, cardiac function, and cognitive development in infants. As such, anemia is a leading cause of morbidity and mortality in children in tropical countries (Reyburn, 2010; Khan, 2018).
Anemia is often multifactorial, including genetic and environment factors. Although nutritional-deficiency anemia, due to iron deficiency, is typically assumed to be the most frequent cause in children (Allali et al., 2017), hereditary forms of anemia caused by mutations are not uncommon in the clinic. Moreover, patients with hereditary anemia are often concerned by pediatricians because they are difficult to diagnose and cannot be cured completely with conventional treatment. For such patients, especially those who depend on regular transfusions, a better understanding of etiology and more accurate diagnostic methods are crucial for subsequent treatment and care.
Hereditary hemolytic anemia (HHA) is a group of diseases that result from the destruction of red blood cells (RBCs), and hemolytic anemia shows varied clinical and molecular heterogeneity (Iolascon et al., 2009; Kim et al., 2017). The typical clinical presentation includes pallor, anemia, jaundice, and often enlargement of the liver and spleen. Laboratory results that help confirm HHA include elevated reticulocyte count, increased lactate dehydrogenase, elevated unconjugated bilirubin, and decreased haptoglobin. In southern China, most pediatric HHA cases are caused by β-thalassemia, which is highly prevalent in this region (Shang et al., 2017). β-thalassemia is caused by mutations in the β-globin gene, which results in impaired Hb synthesis (Shang and Xu, 2017). Hematological phenotype analysis, combined with molecular testing for the β-globin gene, provides an avenue for accurate diagnosis. However, HHA can be caused by mutations in other genes. For example, a case of HHA in a Chinese patient was originally misdiagnosed as thalassemia and ultimately diagnosed as hereditary spherocytosis because of mutations in the ankyrin 1 (ANK1) gene (Zhu et al., 2020). More notably, compound heterozygous mutations in the Kruppel-like factor 1 (KLF1) gene, an important erythroid transcriptional factor involved in erythrocyte development, can lead to chronic non-spherocytic hemolytic anemia (CNSHA). The phenotypic traits of CNSHA are very similar to those of β-thalassemia (Viprakasit et al., 2014; Huang et al., 2015; Perkins et al., 2016). Because KLF1 mutations are found significantly more frequently in thalassemia endemic regions such as southern China (Liu et al., 2014), it is important for physicians in these regions to distinguish between HHA caused by β-thalassemia from HHA caused by KLF1 mutations.
Here, we report the case of a Chinese patient with transfusion-dependent hemolytic anemia. The patient was originally misdiagnosed with β-thalassemia by local physicians and was finally diagnosed with CNSHA caused by compound KLF1 mutations. We also present a summary of relevant literature on the mutation spectrum of the KLF1 gene. Moreover, to help clinicians better recognize HHA caused by KLF1 compound heterozygotes, we compared hematological parameters in patients with β-thalassemia and KLF1 mutations.
Materials and Methods
Subject
The proband was a 3-year-old girl from Yunfu City in Guangdong Province, southern China. She and her parents were experiencing problems with clinical diagnosis and were referred to a free clinic held by a hospital for “World Thalassemia Day—May 8.” The patient had previously experienced hyperbilirubinemia and severe anemia, and she had received eight blood transfusions before age 3 years. A local hospital initially assumed that she had β-thalassemia because she had microcytic hypochromic anemia and high HbF levels (>5%). Furthermore, she was a native of Guangdong province, which is a region with high prevalence of thalassemia. However, following molecular testing for her and her parents, no known thalassemia mutation was detected. Therefore, the patient was referred to our laboratory for further investigation. Blood samples were collected from the patient and her parents after obtaining informed consent. The pedigree of this family is shown in Figure 1A).
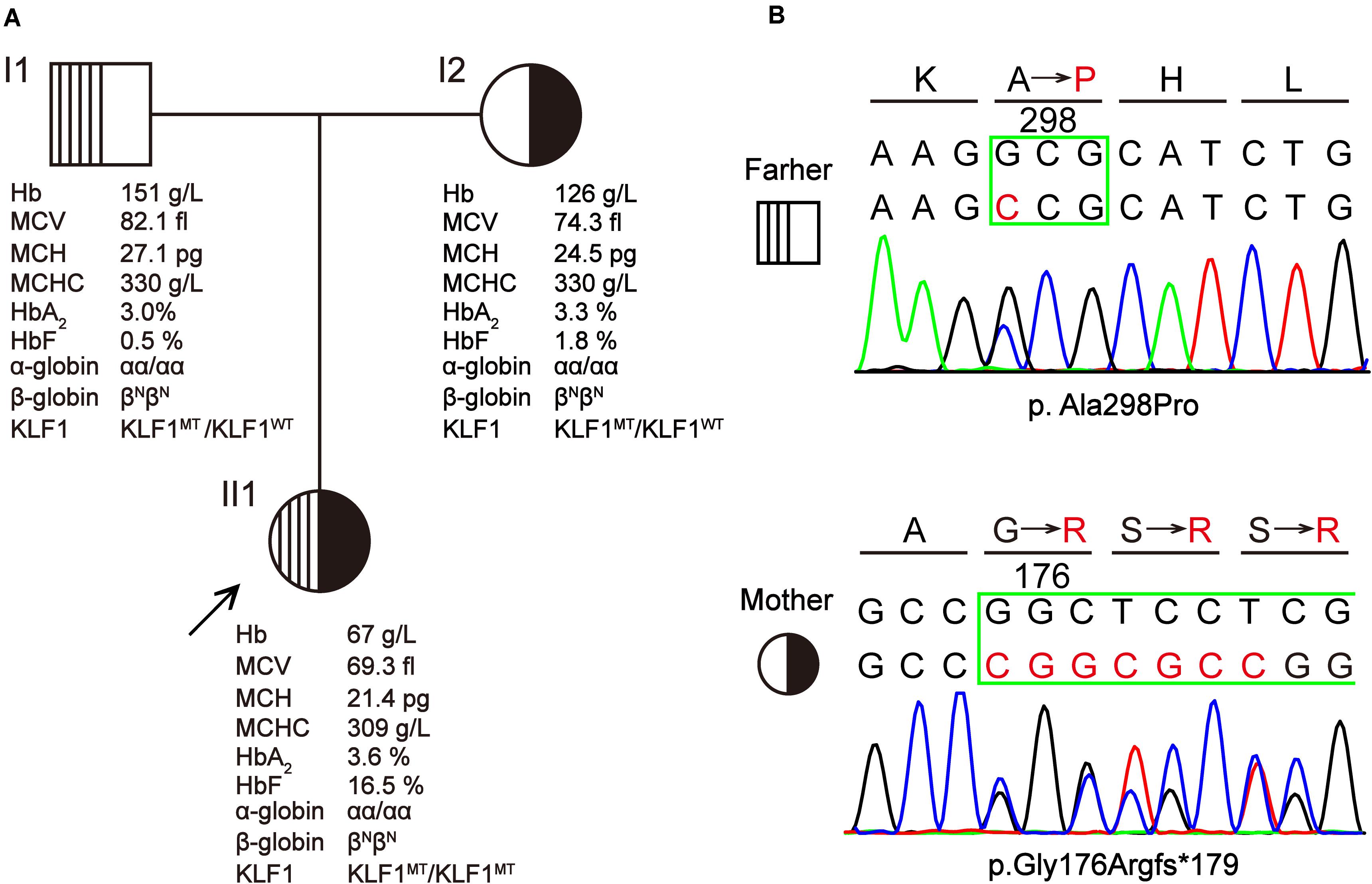
Figure 1. A Chinese family with KLF1 compound heterozygote mutation causing transfusion-dependent hemolytic anemia. (A) The pedigree, hematological parameters, and genotype data of family members. (B) Sanger sequencing of the KLF1 gene shows the p.Ala298Pro and p.Gly176Argfs*179 mutations. Amino acids are indicated above each chromatogram, with base pair and amino acid changes highlighted in red.
Phenotype Analysis
Fresh peripheral blood samples were collected. The patient had not received a blood transfusion in the 2 months prior to sample collection. The hematological data were analyzed using an automated blood cell counter (Sysmex KX-21; Sysmex Corp., Kobe, Japan). The levels of HbA, HbA2, and HbF were analyzed using the Bio-Rad Variant II HPLC system (HPLC, VARIANTTM, Bio-Rad Laboratories, Hercules, CA, United States). Red cell osmotic fragility test, glucose-6-phosphate dehydrogenase (G6PD) activity, and other immune hemolysis test were performed according to standard operation procedure. Routine biochemical and hemolytic parameters were measured using standard techniques.
Molecular Analysis
Sanger sequencing was used to detect point mutations in the α-globin, β-globin, and KLF1 genes, as previously described (Zhu et al., 2019). Polymerase chain reaction (PCR) fragment were sequenced with BigDyeTM Terminators V3.1 Ready Reaction Kit using 3130 Genetic Analyzer (Applied Biosystems). Reference sequences were downloaded from NCBI database1 and used for sequence alignment and analysis. The multiplex ligation-dependent probe amplification assay (MLPA; SALSA P140-C1 HBA or P140-B2 HBB; MRC-Holland, Amsterdam, the Netherlands) was performed to exclude large rearrangements of the α-/β-globin gene clusters.
Literature Review
An electronic literature search of the PubMed database was performed for current and past findings of the molecular genetics of KLF1 from the years 1990 to 2020. The following keywords were used to search: “Kruppel-like factor 1,” “KLF1,” “EKLF,” “hemolytic anemia,” “mutation,” and “variant.” All related publications were reviewed to identify other relevant articles that were not identified in the initial research. The publications were limited to English-language articles. Cases with detailed clinical presentation and KLF1 genotype information were selected.
Statistical Analysis
Statistical differences between groups were determined by one-way analysis of variance, and further comparison across multiple groups was tested by Tukey method. Mean and standard error of the mean were used to describe clinical data in each group. Statistical significance was determined by a P-value of less than 0.05. All statistical analyses were performed using the statistical software GraphPad Prism 6.
Results
Hematological Evaluation and Molecular Analysis of the Patient
Hematological data for this family are summarized in Figure 1A. The father displayed a normal phenotype, except for borderline mean corpuscular Hb (MCH) and HbF level. The mother displayed microcytic and hypochromic features, as well as a slightly elevated HbF level. Patient II1 presented with severe microcytic hypochromic anemia, borderline HbA2 (3.6%) and significantly elevated HbF (16.5%); she has required regular transfusions for survival. This patient was born at full term (38 weeks + 2 days’ gestation) with a birth weight of 2.740 kg. The day after birth, she developed severe jaundice and neonatal hyperbilirubinemia (total and indirect bilirubin were 133.4 and 109.9 μmol/L, respectively), for which she received phototherapy and was referred to a superior hospital. She was followed up as a high-risk baby, at which point she was found to be anemic with hepatosplenomegaly (4 and 3 cm, respectively); she received her first blood transfusion within 1 month after birth. Peripheral blood smear analysis showed increased poikilocytosis and anisocytosis, with hypochromic microcytosis, similar to what is observed with thalassemia. The patient has required regular blood transfusion every 3 months since 4 months of age to keep Hb > 90 g/L, although the interval of blood transfusions was reduced during times when the patient experienced fever or infection. Routine evaluations excluded the possibility of immune hemolysis and RBC membrane disorder. At a local hospital, she was initially diagnosed with β-thalassemia by age 1 year based on her abnormal Hb profiles and the hematological features of microcytic hypochromic anemia. The routine test for common β-thalassemia mutations in Chinese patients by reverse dot blot and gap-PCR methods showed a negative result. Therefore, the patient was thought to have hereditary anemia due to an unidentified thalassemia allele (possible de novo mutation), and she was referred to our laboratory for further study.
We have provided an in-depth review of the patient’s hematological parameters in (Table 1). Complete blood cell analysis showed white blood cell, and platelet values were within normal levels. However, RBC parameters suggested moderate anemia with hypochromic microcytosis. Elevated reticulocyte count and red cell osmotic fragility indicated the existence of hemolysis. Immune hemolysis and erythrocyte enzymatic disease (G6PD deficiency) were excluded by related hemolysis tests. Biochemical tests suggested mild damage to liver function with increased levels of alanine aminotransferase, aspartate aminotransferase, and alkaline phosphatase. Elevated bilirubin and decreased globulins further confirmed hemolysis and jaundice. Iron deficiency and megaloblastic anemia were excluded by the normal values of ferritin, folic acid, and vitamin B12, respectively. Therefore, the patient was ultimately diagnosed with non-spherocytic hemolytic anemia.
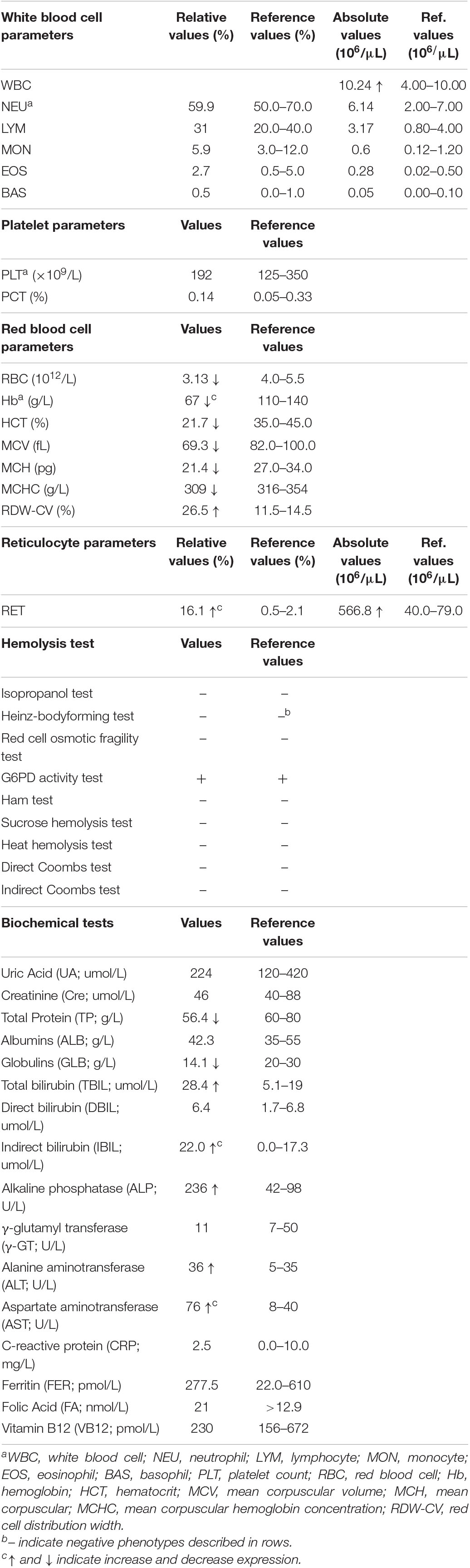
Table 1. Current, in-depth hematological parameters of subject II1.
Based on hematological data from the parents (Figure 1A), it is likely that the patient inherited a recessive disease. However, mutations in the α-, β-, or γ-globin genes that could cause thalassemic phenotypes similar to those seen in the patient were excluded by direct sequencing of above genes and MLPA analysis of α- or β-globin cluster. This indicated that there might be another gene responsible for the observed hemolytic anemia phenotypes. Considering the high HbF levels in the patient, KLF1 was chosen to be the first candidate gene (Borg et al., 2010). Sequencing of the KLF1 gene identified a compound heterozygote of missense variant c.892G > C (p.Ala298Pro) and frameshift variant c.525_526insCGGCGCC (p.Gly176Argfs∗179) in the patient, which she inherited from her father and mother, respectively (Figure 1B). Both variants have been previously reported as pathogenic mutations that affect the biological function of KLF1 (Viprakasit et al., 2014; Huang et al., 2015).
Literature Review of KLF1 Mutation Spectrum and Genotype–Phenotype Correlation in Individuals With KLF1 Mutations
KLF1 (OMIM# 600599) is located at chromosome 19p13.2 and contains three exons that encode 362 amino acids. There are two major functional domains in the KLF1 protein product: two PRA (proline-rich amino) domains located at the N-terminus function as a transcriptional activator and three zinc finger (ZF) domains located on the C-terminus act as a DNA-binding domain (Quadrini and Bieker, 2002).
After searching the literature for KLF1 mutations, 113 mutations were identified (Supplementary Table 1). These mutations can be divided into three groups according to their position on KLF1 gene. Seven mutations are located on the promoter region, and therefore may affect transcription of the KLF1 gene. Additionally, 104 mutations (72 missense mutations, 13 nonsense mutations, and 19 frameshift mutations) are located on the exon region and cause alterations to the primary structure of the KLF1 protein. Although the ZF domains comprise only 20% of KLF1, almost 40% of the functionally effective mutations are found within and between these domains. Two intronic mutations affect splicing. In addition, the 104 mutations in the coding sequence (CDS) region can be divided into four subclasses, based primarily on differing effects on KLF1 protein function (Figure 2A). Class 1 includes 33 mutations with no effects or minor functional effects, most of which are located outside the ZF domains. Class 2 comprises 40 mutations, most of which are missense mutations in ZF domains that interfere with normal function of KLF1. Class 3 includes 30 nonsense or frameshift mutations, which lead to a truncated KLF1 peptide that lacks all or part of the ZF domains. Class 4 contains only one missense mutation (p.Glu325Lys) (Arnaud et al., 2010), which is a special dominant variant.
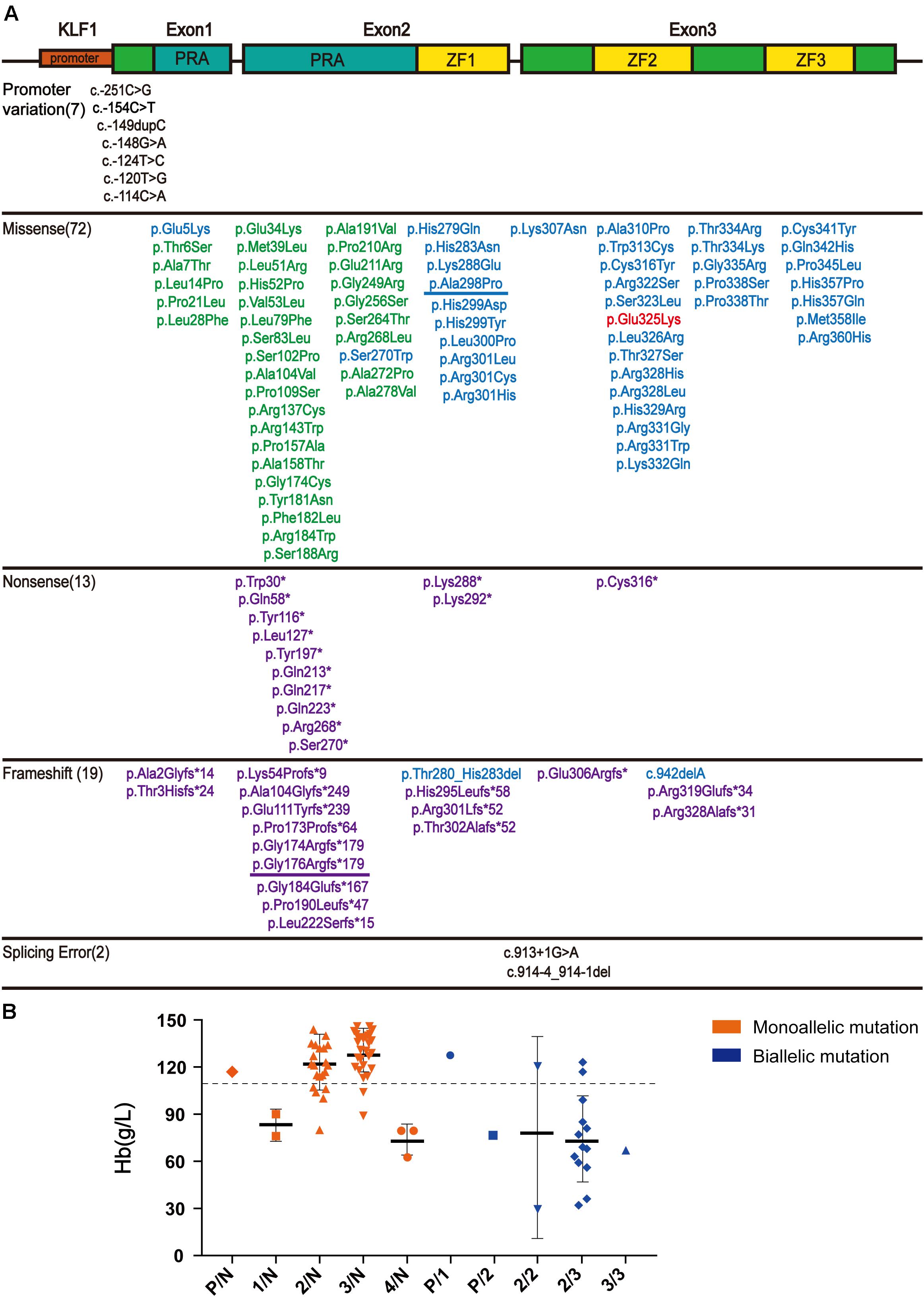
Figure 2. The literature review of KLF1 mutations and phenotype. (A) Schematic diagram of the KLF1 gene (NM_006563.5 and NP_006554.1) structure and variants, as reported in the literature. The gene structure includes the promoter (small red box) and two introns and three exons (big box). The proline-rich amino domain (PRA) located at the N-terminus is responsible for transcriptional activation (blue box). The zinc finger (ZF) domains (yellow box) located at C-terminus enable KLF1 to bind specific sites on DNA. Mutations are color-coded: promoter mutations and splicing mutations, black; class 1, green; class 2, blue; class 3, purple; class 4, red. The two mutations described in this article are underlined. See Supplementary Table 1 for details and references. (B) Hb level in reported cases carrying KLF1 mutations. All KLF1 alleles were classified into six types: N, normal allele; P, promoter mutations; 1–4, classes 1–4 mutations. The dotted line shows the anemia boundary line. See Supplementary Table 2 for details and references.
According to published literature, 80 cases with detailed hematological phenotype data and KLF1 genotype were collected (Supplementary Table 2). We have presented the genotype–phenotype relationship in Figure 2B. Most cases carrying monoallelic KLF1 mutations do not display anemia. On the contrary, most cases carrying biallelic mutations do exhibit an anemia phenotype, as their Hb level is often less than 110 g/L.
Discussion
HHA includes a diverse group of genetically and phenotypically heterogeneous diseases. The major causes of HHA are defects in Hb, the RBC membrane, and RBC enzymes, which are commonly referred to as thalassemia, hereditary spherocytosis, and G6PD deficiency, respectively. However, these common disorders do not represent all HHA patients, and precise diagnosis of HHA remains a challenge for clinical doctors. As clinical presentation often overlaps in cases with different etiologies, it is difficult to make a reliable diagnosis based only on phenotype analysis. Identification of disease-causing mutations at the DNA level can increase the accuracy of HHA diagnosis. Our study provided a new cause of HHA in pediatric patients, KLF1 compound mutations. The patient discussed here had jaundice, hepatomegaly, and microcytic hypochromic anemia with remarkably elevated HbF levels.
KLF1 is known as a hematopoietic-specific transcription factor that regulates erythroid lineage commitment, globin switching, and the terminal maturation of RBCs (Miller and Bieker, 1993; Wijgerde et al., 1996; Hariharan et al., 2019). It is no wonder that KLF1 mutations can lead to shortened survival of RBCs, resulting in inherited hemolytic anemias. Currently, there are reports of 19 compound heterozygotes of KLF1 mutations (including our case) with detailed hematological phenotypes (Table 2). Four cases (Table 2, numbers 1–4) showed normal Hb levels with high HbF and were described as hereditary persistence of fetal Hb (HPFH) (Satta et al., 2011; Radmilovic et al., 2013; Rani et al., 2018), a kind of benign hemoglobinopathy. Fifteen cases (Table 2, numbers 5–19) showed mild to severe anemia (Viprakasit et al., 2014; Huang et al., 2015; Magor et al., 2015; Lee et al., 2016; Jiang et al., 2019; Belgemen-Ozer and Gorukmez, 2020). Although some cases carry pathogenic mutations in globin genes that further affect the hematological phenotype, these cases suggest that compound heterozygotes have a high probability of showing a transfusion-dependent anemia phenotype in childhood.
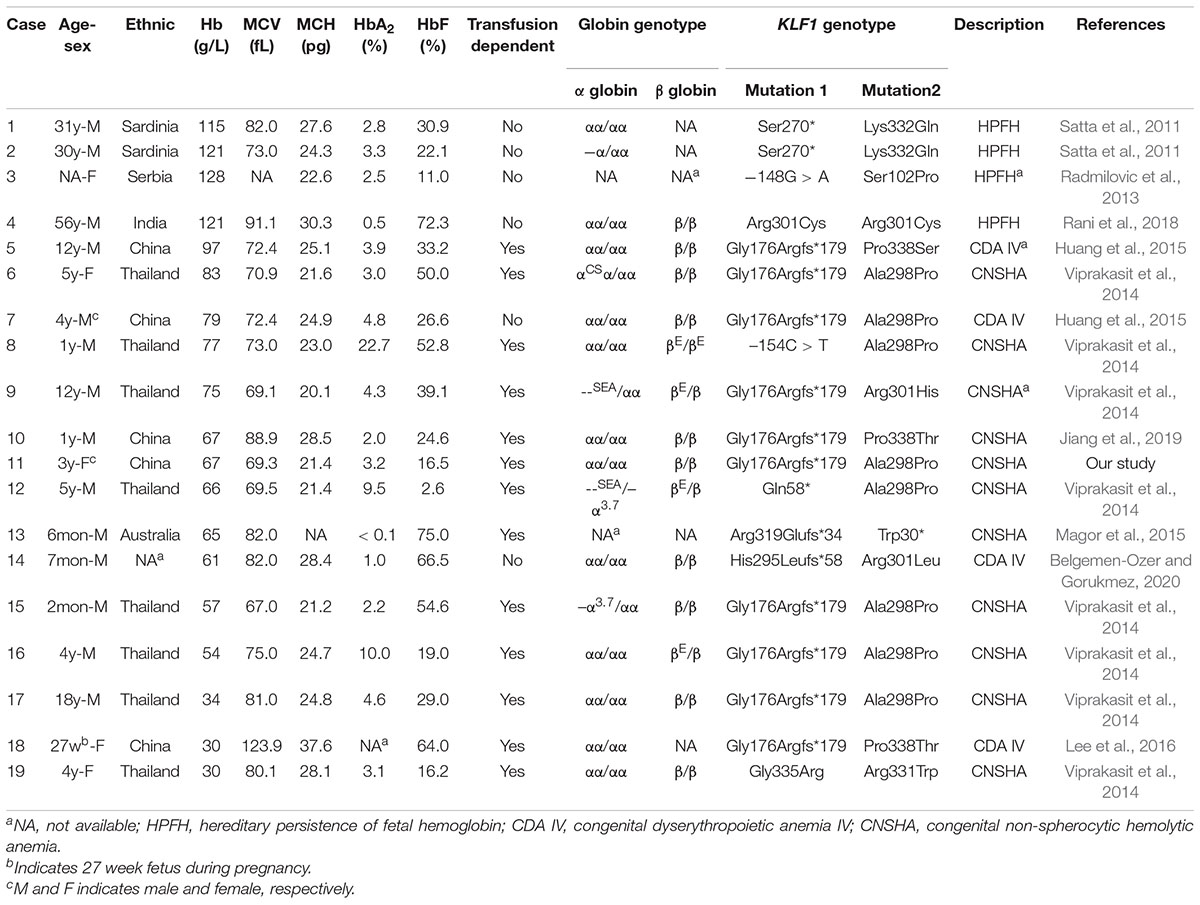
Table 2. Literature reports of clinical and laboratory characteristics of compound heterozygotes for KLF1 mutations.
Southern China is a region with a high incidence of β-thalassemia, and local pediatricians receive considerable training in thalassemia through long-term public health education and clinical practices. However, this has raised another problem in that patients in this region with a hemolytic anemia phenotype and recessive inheritance were more likely to be diagnosed with β-thalassemia. According to a previous publication (Liu et al., 2014), southern China is also a region with a high prevalence of KLF1 mutations. Therefore, patients with KLF1 compound mutations should account for a certain proportion of all hemolytic anemia patients. To avoid misdiagnosis, it is necessary to effectively distinguish between anemia caused by KLF1 mutations and anemia caused by β-thalassemia mutations. According to previous reports (Huang et al., 2015), most patients with HHA caused by compound mutations of KLF1 were from Chinese or southeast Asian populations, and they tended to be diagnosed with β-thalassemia intermedia (β-TI) at the local hospital. Here, 44 TI patients from southern China (Chen et al., 2010) along with 30 TI patients from Indonesia (Rujito et al., 2016) (reported previously) were selected for comparison with the 15 previously referenced cases that were caused by KLF1 compound mutations (KLF1-AM; cases 5–19 in Table 2). A summary of typical clinical features is shown in Figure 3A. Some important symptoms were overlapping; for example, both groups presented with hypochromic microcytic anemia, jaundice, splenomegaly, severe iron overload, and elevated HbF levels. The specific symptoms [zinc protoporphyrin, In(Lu) phenotype, and CD44 deficiency] are not routine examination items in clinical practices. Therefore, it is not surprising that misdiagnosis occurs. Because whole-blood cell analysis and Hb electrophoresis are required items for anemia diagnosis and thalassemia screening, the key RBC parameters and the constitution of Hb were compared in patients with TI or KLF1-AM (Figures 3B–F). Hb values and HbF levels were significantly lower in KLF1-AM patients. Additionally, significant differences in mean corpuscular volume (MCV) and MCH values were evident between Chinese TI patients and KLF1-AM patients, suggesting that these two values may be useful in distinguishing KLF1-AM from β-TI in southern China. Patients with an HHA phenotype but borderline-low MCV and MCH values (rather than markedly decreased MCV and MCH values) are more likely to have KLF1 mutations.
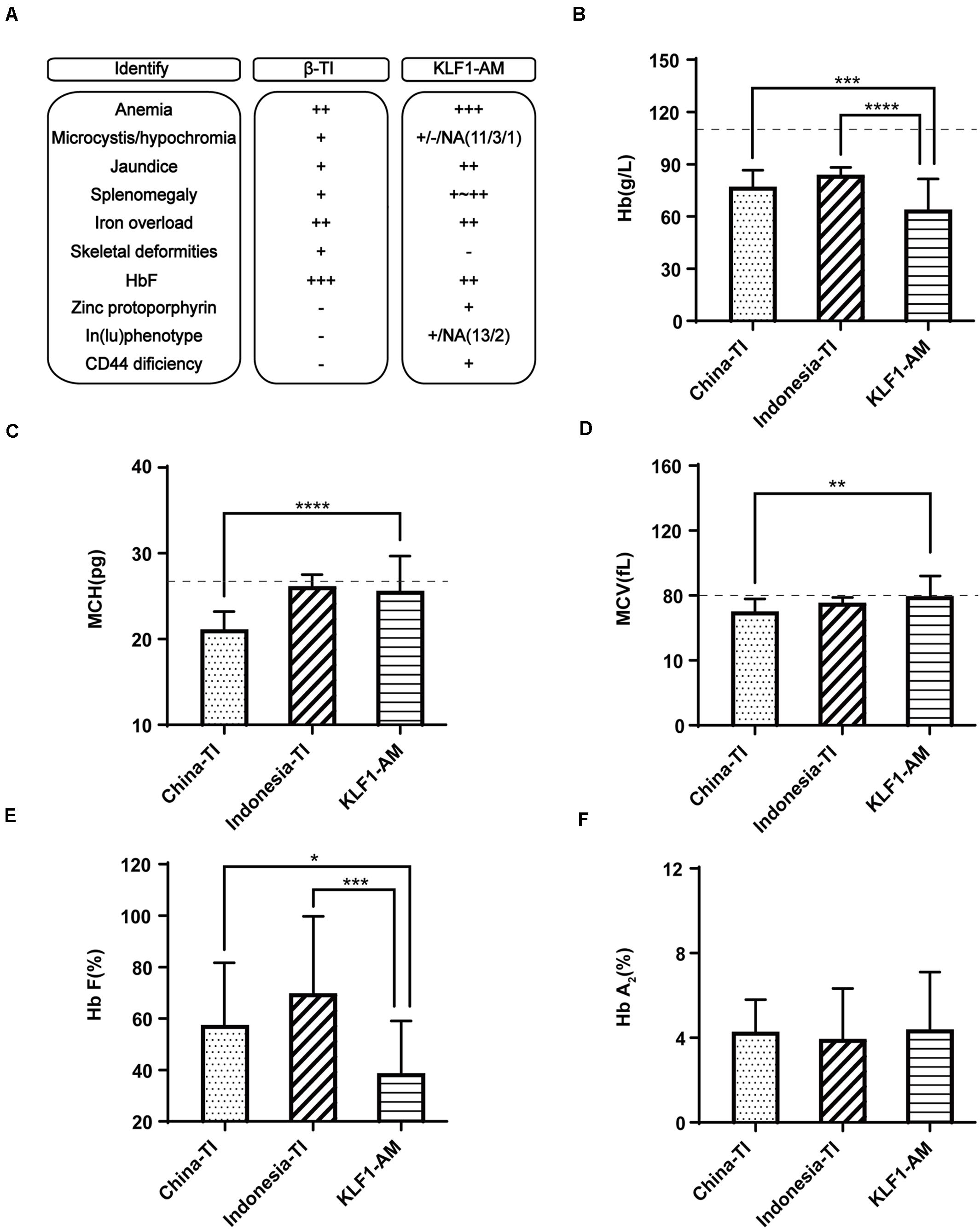
Figure 3. Differential diagnosis of KLF1 anemia and β-thalassemia intermedia. (A) Summary of clinical characteristic features in Chinese patients with β-thalassemia intermedia (China-TI), Indonesian patients with β-thalassemia intermedia (Indonesia-TI) and anemia caused by compound heterozygotes of KLF1 mutations (KLF1-AM). NA indicates not available; + and −, positive and negative phenotypes described in the columns; ++ or +++, degree of symptom. (B–F) Comparisons of Hb, MCV, MCH, HbF, and HbA2 levels between China-TI, Indonesia-TI, and KLF1-AM. Data are presented as mean ± standard deviation (SD). Reference range values are shown as a dotted line: MCV, 80 fL; MCH, 27 pg; China-TI, β-thalassemia intermedia patients in China; Indonesia-TI, β-thalassemia intermedia patients in Indonesia; KLF1-AM, cases caused by KLF1 compound mutations. Asterisks represent statistically significant differences: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
In conclusion, our study corrected the misdiagnosis and identified the true cause of disease in an HHA patient. The mutation spectrum of KLF1 and the genotype–phenotype correlation of KLF1 were reviewed. Furthermore, we propose that pediatricians in southern China should be cautious in making a diagnosis in children presenting with HHA. For difficult cases, further molecular testing should be performed, and current clinical and molecular reports should be reviewed to ensure that the final diagnosis is accurate.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Ethical Committee of Nanfang Hospital, Southern Medical University. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
LX performed the experiments, analyzed the data, and wrote the manuscript. DZ, YZ, and GL performed the experiments. ML and XFW provided technical support for this work. XF and XDW were responsible for accumulating data and referring patients for the laboratory investigations. XS designed the study and drafted the manuscript. All authors reviewed, edited, and approved the version to be submitted.
Funding
This research was supported by the National Natural Science Foundation of China (81870148), the Guangdong Basic and Applied Basic Research Foundation (2019A1515011545), and the Southern Medical University Innovation Training Program for Undergraduate Students (201912121027 and S202012121059).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.691461/full#supplementary-material
Footnotes
References
Allali, S., Brousse, V., Sacri, A., Chalumeau, M., and de Montalembert, M. (2017). Anemia in children: prevalence, causes, diagnostic work-up, and long-term consequences. Expert Rev. Hematol. 10, 1023–1028. doi: 10.1080/17474086.2017.1354696
Arnaud, L., Saison, C., Helias, V., Lucien, N., Steschenko, D., Giarratana, M., et al. (2010). A dominant mutation in the gene encoding the erythroid transcription factor KLF1 Causes a congenital dyserythropoietic anemia. Am. J. Hum. Genet. 87, 721–727. doi: 10.1016/j.ajhg.2010.10.010
Belgemen-Ozer, T., and Gorukmez, O. (2020). A very rare congenital dyserythropoietic anemia variant-type IV in a patient with a novel mutation in the KLF1 gene: a case report and review of the literature. J. Pediatric Hematol.Oncol. 42, e536–e540. doi: 10.1097/MPH.0000000000001727
Borg, J., Papadopoulos, P., Georgitsi, M., Gutiérrez, L., Grech, G., Fanis, P., et al. (2010). Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat. Genet. 42, 801–805. doi: 10.1038/ng.630
Chen, W., Zhang, X., Shang, X., Cai, R., Li, L., Zhou, T., et al. (2010). The molecular basis of beta-thalassemia intermedia in southern China: genotypic heterogeneity and phenotypic diversity. BMC Med. Genet. 11:31. doi: 10.1186/1471-2350-11-31
Hariharan, P., Colah, R., Ghosh, K., and Nadkarni, A. (2019). Differential role of Kruppel like factor 1 (KLF1) gene in red blood cell disorders. Genomics 111, 1771–1776. doi: 10.1016/j.ygeno.2018.11.032
Huang, J., Zhang, X., Liu, D., Wei, X., Shang, X., Xiong, F., et al. (2015). Compound heterozygosity for KLF1 mutations is associated with microcytic hypochromic anemia and increased fetal hemoglobin. Eur. J. Hum. Genet. 23, 1341–1348. doi: 10.1038/ejhg.2014.291
Iolascon, A., De Falco, L., and Beaumont, C. (2009). Molecular basis of inherited microcytic anemia due to defects in iron acquisition or heme synthesis. Haematologica 94, 395–408. doi: 10.3324/haematol.13619
Jiang, H., Jiang, F., Li, J., Tang, F., and Li, D. (2019). Congenital nonspherocytic hemolytic anemia caused by krüppel-like factor 1 gene variants: another case report. Hemoglobin 43, 292–295. doi: 10.1080/03630269.2019.1680384
Kassebaum, N. J., Jasrasaria, R., Naghavi, M., Wulf, S. K., Johns, N., Lozano, R., et al. (2014). A systematic analysis of global anemia burden from 1990 to 2010. Blood 123, 615–624. doi: 10.1182/blood-2013-06-508325
Kim, Y., Park, J., and Kim, M. (2017). Diagnostic approaches for inherited hemolytic anemia in the genetic era. Blood Res. 52, 84–94. doi: 10.5045/br.2017.52.2.84
Lee, H. H. L., Mak, A. S. L., Kou, K. O., Poon, C. F., Wong, W. S., Chiu, K. H., et al. (2016). An unusual hydrops fetalis associated with compound heterozygosity for krüppel-like factor 1 mutations. Hemoglobin 40, 431–434. doi: 10.1080/03630269.2016.1267017
Liu, D., Zhang, X., Yu, L., Cai, R., Ma, X., Zheng, C., et al. (2014). KLF1 mutations are relatively more common in a thalassemia endemic region and ameliorate the severity of β-thalassemia. Blood 124, 803–811. doi: 10.1182/blood-2014-03-561779
Magor, G. W., Tallack, M. R., Gillinder, K. R., Bell, C. C., McCallum, N., Williams, B., et al. (2015). KLF1-null neonates display hydrops fetalis and a deranged erythroid transcriptome. Blood 125, 2405–2417. doi: 10.1182/blood-2014-08-590968
Miller, I. J., and Bieker, J. J. (1993). A novel, erythroid cell-specific murine transcription factor that binds to the CACCC element and is related to the Kruppel family of nuclear proteins. Mol. Cell Biol. 13, 2776–2786. doi: 10.1128/mcb.13.5.2776
Perkins, A., Xu, X., Higgs, D. R., Patrinos, G. P., Arnaud, L., Bieker, J. J., et al. (2016). Kruppeling erythropoiesis: an unexpected broad spectrum of human red blood cell disorders due to KLF1 variants. Blood 127, 1856–1862. doi: 10.1182/blood-2016-01-694331
Quadrini, K. J., and Bieker, J. J. (2002). Krüppel-like zinc fingers bind to nuclear import proteins and are required for efficient nuclear localization of erythroid krüppel-like factor. J. Biol. Chem. 277, 32243–32252. doi: 10.1074/jbc.M205677200
Radmilovic, M., Zukic, B., Petrovic, M. S., Bartsakoulia, M., Stankovic, B., Kotur, N., et al. (2013). Functional analysis of a novel KLF1 gene promoter variation associated with hereditary persistence of fetal hemoglobin. Ann. Hematol. 92, 53–58. doi: 10.1007/s00277-012-1625-9
Rani, N., Jamwal, M., Kaur, J., Sharma, P., Malhotra, P., Maitra, A., et al. (2018). Homozygous KLF1 mutation c.901C>T (p.Arg301Cys) resulting in mild thalassemia intermedia in an Indian: a next-generation sequencing diagnosis. Blood Cells Mol. Dis. 72, 19–21. doi: 10.1016/j.bcmd.2018.06.003
Reyburn, H. (2010). New WHO guidelines for the treatment of malaria. BMJ 340:c2637. doi: 10.1136/bmj.c2637
Rujito, L., Basalamah, M., Siswandari, W., Setyono, J., Wulandari, G., Mulatsih, S., et al. (2016). Modifying effect of XmnI, BCL11A, and HBS1L-MYB on clinical appearances: a study on β-thalassemia and hemoglobin E/β-thalassemia patients in Indonesia. Hematol. Oncol. Stem Cell Ther. 9, 55–63. doi: 10.1016/j.hemonc.2016.02.003
Satta, S., Perseu, L., Moi, P., Asunis, I., Cabriolu, A., Maccioni, L., et al. (2011). Compound heterozygosity for KLF1 mutations associated with remarkable increase of fetal hemoglobin and red cell protoporphyrin. Haematologica 96, 767–770. doi: 10.3324/haematol.2010.037333
Shang, X., and Xu, X. (2017). Update in the genetics of thalassemia: what clinicians need to know. Best Pract. Res. Clin. Obstet. Gynaecol. 39, 3–15. doi: 10.1016/j.bpobgyn.2016.10.012
Shang, X., Peng, Z., Ye, Y., Asan, Zhang, X., Chen, Y., et al. (2017). Rapid targeted next-generation sequencing platform for molecular screening and clinical genotyping in subjects with hemoglobinopathies. Ebiomedicine 23, 150–159. doi: 10.1016/j.ebiom.2017.08.015
Viprakasit, V., Ekwattanakit, S., Riolueang, S., Chalaow, N., Fisher, C., Lower, K., et al. (2014). Mutations in Kruppel-like factor 1 cause transfusion-dependent hemolytic anemia and persistence of embryonic globin gene expression. Blood 123, 1586–1595. doi: 10.1182/blood-2013-09-526087
Wijgerde, M., Gribnau, J., Trimborn, T., Nuez, B., Philipsen, S., Grosveld, F., et al. (1996). The role of EKLF in human beta-globin gene competition. Genes Dev. 10, 2894–2902. doi: 10.1101/gad.10.22.2894
Zhu, F., Liang, M., Xu, L., Peng, Z., Cai, D., Wei, X., et al. (2020). A tetranucleotide deletion in the ANK1 gene causes hereditary spherocytosis; a case of misdiagnosis. Gene 726:144226. doi: 10.1016/j.gene.2019.144226
Keywords: KLF1, compound heterozygote, hereditary hemolytic anemia, children, chronic non-spherocytic hemolytic anemia, β-thalassemia
Citation: Xu L, Zhu D, Zhang Y, Liang G, Liang M, Wei X, Feng X, Wu X and Shang X (2021) Compound Heterozygosity for KLF1 Mutations Causing Hemolytic Anemia in Children: A Case Report and Literature Review. Front. Genet. 12:691461. doi: 10.3389/fgene.2021.691461
Received: 06 April 2021; Accepted: 18 May 2021;
Published: 25 June 2021.
Edited by:
Merlin G. Butler, University of Kansas Medical Center, United StatesReviewed by:
Theresa V. Strong, Foundation for Prader-Willi Research, United StatesFulya Taylan, Karolinska Institutet (KI), Sweden
Copyright © 2021 Xu, Zhu, Zhang, Liang, Liang, Wei, Feng, Wu and Shang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuan Shang, c2hhbmdyYWJiaXRAMTYzLmNvbQ==
†These authors have contributed equally to this work and share first authorship