 Xin Jia1
Xin Jia1 Yingmin Lyu
Yingmin Lyu- 1Beijing Key Laboratory of Ornamental Germplasm Innovation and Molecular Breeding, China National Engineering Research Center for Floriculture, College of Landscape Architecture, Beijing Forestry University, Beijing, China
- 2Beijing Key Laboratory of Greening Plant Breeding, Beijing Institute of Landscape Architecture, Beijing, China
Rose is one of the most fundamental ornamental crops, but its yield and quality are highly limited by drought. The key transcription factors (TFs) and co-expression networks during rose’s response to drought stress and recovery after drought stress are still limited. In this study, the transcriptomes of leaves of 2-year-old cutting seedlings of Rosa chinensis ‘Old Blush’ from three continuous droughted stages (30, 60, 90 days after full watering) and rewatering were analyzed using RNA sequencing. Weighted gene co-expression network analysis (WGCNA) was used to construct a co-expression network, which was associated with the physiological traits of drought response to discovering the hub TFs involved in drought response. More than 45 million high-quality clean reads were generated from the sample and used for comparison with the rose reference genome. A total of 46433 differentially expressed genes (DEGs) were identified. Gene Ontology (GO) term enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis indicated that drought stress caused significant changes in signal transduction, plant hormones including ABA, auxin, brassinosteroid (BR), cytokinin, ethylene (ET), jasmonic acid (JA) and salicylic acid (SA), primary and secondary metabolism, and a certain degree of recovery after rewatering. Gene co-expression analysis identified 18 modules, in which four modules showed a high degree of correlation with physiological traits. In addition, 42 TFs including members of NACs, WRKYs, MYBs, AP2/ERFs, ARFs, and bHLHs with high connectivity in navajowhite1 and blue modules were screened. This study provides the transcriptome sequencing report of R. chinensis ‘Old Blush’ during drought stress and rewatering process. The study also identifies the response of candidate TFs to drought stress, providing guidelines for improving the drought tolerance of the rose through molecular breeding in the future.
Introduction
Rose is one of the most fundamental ornamental crops. With an important economic, cultural, and symbolic value, the rose has a significant market share in the floriculture industry. Cut rose flowers account for 32% of the world’s total cut flower trade. The rose occupies nearly a quarter of the Netherlands floriculture market (Mordor Intelligence, 2020). However, their growth conditions are closely related to changes in the external environment. Most roses suffer from various degrees of drought, which result in a sizable loss of productivity and quality around the world. As a kind of abiotic stress, drought is becoming an even more serious issue due to global warming. Changes in the earth’s climate have an adverse impact on plant growth, crop production, and distribution (Zhu, 2002). It is therefore pressing to identify the response mechanism in rose under the drought stress.
To resistance drought stress, plants have evolved several adaptive mechanisms depending on the activation of the molecular networks involved in stress perception, signal transcription and transduction, expression of specific stress-related genes, and physiological reaction of tolerance (Chaves et al., 2003). Damage to plants under drought stress is manifested in the peroxidation caused by the accumulation of active oxygen in the plant (Niu et al., 2013). The role of various antioxidant protection enzymes in plants under drought stress has been extensively studied. In addition, malondialdehyde (MDA) is an important indicator of cell stability (Fang et al., 2018). Studies have shown that drought stress leads to different trends in plant antioxidant systems (Zandalinas et al., 2017; Niu et al., 2018). The activity of four antioxidant enzymes fluctuate while the MDA content continues to increase in Herbaceous peony with drought stress (Niu et al., 2018). Drought stress accumulates amounts of osmotic adjustment substances in plants such as proline and soluble sugars. In research on the drought stress of rose, it was found that as the drought stress deepened the content of proline and soluble sugar increased (Gadzinowska et al., 2019; Adamipour et al., 2020; Al-Yasi et al., 2020). It is crucial to identify the genes and biological pathways involved in drought stress tolerance and improve the understanding of the molecular mechanisms relating to drought resistance in plants (Valliyodan and Nguyen, 2006). A large number of signaling metabolic pathways and numerous genes have been determined in several model plants exposed to drought environments, such as Arabidopsis (Huang et al., 2018), rice (Liu et al., 2018), and so on. Transcription factors (TFs) control the regulating downstream stress response genes that play essential roles in various abiotic stress response processes (Joshi et al., 2016). During signal transduction, TFs directly regulate the expression of related genes by acting as a molecular switch. These TFs interact specifically with cis-elements located in the promoter region of the regulator (Franco-Zorrilla et al., 2014). As the plant drought tolerance network is extremely complex, this study started by exploring the transcriptional regulatory network of TFs for drought resistance. To date, RcLEA (Zhang et al., 2014), RcMYBPA2 (Li et al., 2019c), RhMYB96 (Jiang et al., 2018), RcNAC3 (Jiang et al., 2014), and RcNAC31 (Ding et al., 2019) genes have been reported to improve the abiotic stress resistance of the transgenic plant. However, information on the mechanism of rose response to drought stress is still limited.
As a deep sequencing technology, transcriptome technology has been successfully used to explore the gene network of non-model plant species in response to drought stress. Drought-responsive transcriptome studies have been conducted in ornamental plants such as chrysanthemum (Xu et al., 2013), tree peony (Paeonia section Moutan DC.) (Zhao et al., 2019a), and Dianthus spiculifolius (Zhou et al., 2017). Previously, only a suppression subtractive hybridization library was constructed for the dehydration stress of cut rose (Rosa hybrida cv. Samantha) petals, which contained 3513 unique expression sequence tags, and its expression profile during the dehydration cycle was analyzed (Dai et al., 2012). Although the transcriptome of R. chinensis ‘Mutabilis’ in the three stages of drought stress has been reported (Li et al., 2021a) and provided some basis, drought stress generally happens continuously, and it is more essential to discover how the rose recovers after drought stress. In other words, knowledge about the gene co-expression network involved in rose drought and recovery is very limited. WGCNA has become an important tool to identify gene co-expression related to associating with function, which has been widely used in the research of horticultural plants (Lu et al., 2019; Liu et al., 2021).
Rosa chinensis ‘Old Blush’ originated from China and has participated in modern rose hybrid breeding. R. chinensis ‘Old Blush’ is the diploid species in Rosa, which may represent a convenient genomic model for Rosa research and may be used as a model plant to study rose response to drought stress. Currently, the genome of R. chinensis ‘Old Blush’ was published, providing a valuable database for further function and genomic research in rose (Raymond et al., 2018). To obtain unique insights into the molecular mechanisms at the transcriptome level in response to drought and recovery, this study compared transcriptome profiles of R. chinensis ‘Old Blush’ at continuous drought stages (30, 60, 90 days after full watering) and rewatering, and constructed co-expression modules using WGCNA. The Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis in differentially expressed genes (DEGs) were performed on the modules with significant physiological data and the key TFs in the module were identified. The study aims to obtain an understanding of the drought responsive regulatory network at the molecular level and identify the key TFs. Furthermore, the results of this study can provide the basis for a better understanding of not only other R. hybrida cultivars but also other woody plants responding to drought stress, and odder candidate genes for further rose resistance research.
Materials and Methods
Plant Materials
Two-year-old healthy cutting seedlings of the R. chinensis ‘Old Blush’ used in the experiments were collected from the Beijing Institute of Landscape Architecture (Beijing, China). The plant material was planted in the pot with an inner size of 26 cm in diameter, 22 cm in height, and 18 cm in diameter at the bottom. It contained sterilized soil with a mixture of nutritive soil, garden soil, perlite, and vermiculite with the ratio of 5:3:1:1 (v/v). The size and height of plant materials used in all experiments were almost the same. Before drought stress, samples were cultured under the following growth conditions: 50% relative humidity, 25°C/18°C, day/night temperatures in the same artificial climate chamber in Beijing Forestry University (BJFU) (116.3°E, 40.0°N). Then, the samples were subjected to a continuously dry environment (no-watering) for 15, 30, 45, 60, 75, and 90 days. Then, after 7 days with rewatering treatment, the leaves were totally recovered. Fully watered plants were used as a control. Refer to experimental methods of drought treatment from David Gamboa-Tuz et al. (2018). At each time point, the fourth or fifth fully expanded leaves from the plants were collected, then immediately frozen in liquid nitrogen and stored at −80°C until the physiological measurement or RNA extraction. Each treatment group contained three replicates, and each sample from the three replicates was used to obtain biological replicates.
Measurement of Physiological Parameters
Before transcriptome sequencing using leaf tissue, which was collected and stored at −80°C from plants subjected to 0 to 90 days of drought stress, the relative water content of leaves, superoxide dismutase (SOD) activity, MDA, and relative soil water content (RSWC) was evaluated. RSWC was analyzed using direct drying method. The relative water content of leaves was determined by the initial fresh weight of the leaf samples, referencing from Zhou et al. (2019) with slight modifications, the samples were soaked in distilled water placed on the lab bench with dark cloth for 24 h and finally weighted, The thiobarbituric acid (TBA) reaction was used for evaluation of the MDA content as described by Heath and Packer (1968) with minor adjustments. The SOD activity was measured after the spectrophotometer by determining its ability to inhibit the photochemical reduction of nitro blue tetrazolium (NBT). Each experiment included three biological replicates. The correlation analysis for various physiological parameters used the Pearson correlation coefficient in SPSS software.
RNA Isolation, cDNA Library Preparation, and Transcriptome Sequencing
According to the manufacturer’s instructions, total RNA was isolated with an Easy Spin Plus RNA Extraction Kit (RN53, Aidlab China). RNA integrity was verified using electrophoresis on 1% agarose gel. The 2100 Bioanalyzer (Agilent Technologies, United States) and ND-2000 (NanoDrop Technologies) were employed to test the quality and purity of RNA.
The RNA-Seq library was generated using the NEB#7530 RNA Library Prep Kit (#E7530, New England Biolabs). The total mixed RNA from R. chinensis leaves in all treatments was used for cDNA library construction. The library quality was subsequently detected through the High Sensitivity DNA assay Kit (Agilent Technologies). The three biological replicates from each experiment were sequenced using an Illumina HiSeqTM 4000 platform by Genedenovo Biotechnology, Co., Ltd. (Guangzhou, China).
To investigate genes involved in the transcriptome, a cDNA sample was prepared from an equal mixture of t all RNA. The total RNA was isolated from the leaves of 2-year-old cutting seedlings R. chinensis from the control, drought-treated (30, 60, 90 days) and rehydration groups, designated as CK1, CK2, CK3, DT1-1, DT1-2, DT1-3, DT2-1, DT2-2, DT2-3, DT3-1, DT3-2, DT3-3, RW1, RW2, RW3. Three biological replicates from each group were analyzed on the Illumina HiSeq 4000 platform.
To get high quality clean reads, we were further filtered by fastp (version 0.18.0). The parameters were as follows: first removing reads containing adapters, and then removing reads containing more than 10% of unknown nucleotides (N), finally, removing low quality reads containing more than 50% low quality (Q-value ≤ 20) bases.
Mapping to the Rose Genome and Gene Expression Quantification
HISAT2.2.4 software (Kim et al., 2015) was used to carry out comparative analysis based on the rose genome (Raymond et al., 2018). The comparison results of HISAT2 comparison StringTie (Pertea et al., 2015, 2016) were used to reconstruct the transcript, and the expression of all genes in each sample was calculated.
Identification of DEGs and Genes Co-expression Network Analysis
The Fragments Per Kilobase of Exon Per Million Mapped Fragments (FPKM) method was used to analyze the expression level of each transcript. The reference-based approach was used to map the reads of each sample assembled by StringTie v1.3.1. For each transcription region, an FPKM value was calculated by StringTie software to quantify its expression abundance and variations. Differential expression analysis was performed by DESeq2 software (Love et al., 2014) between two different groups [also by edgeR (Robinson et al., 2010) between two samples]. The genes with the parameter of false discovery rate (FDR) below 0.05 and absolute fold change ≥ 2 were considered DEGs. GO enrichment analyses of DEGs (Ashburner et al., 2000) were able to recognize the main biological functions. Then, KEGG (Ogata et al., 1999) pathway enrichment analysis identified significantly enriched metabolic pathways or signal transduction pathways in DEGs.
Co-expression networks were established using WGCNA (v1.47) package in R (Langfelder and Horvath, 2008). Gene expression values were imported into WGCNA to construct co-expression modules using automatic network construction function blockwise Modules with default settings, except that the power was 10, TOMType unsigned, and minModuleSize was 50. The correlation coefficient between module eigengenes and physiological data was calculated to find out significant modules, and the hub TFs were selected from those modules (Huang et al., 2020). The networks were visualized using Cytoscape_3.3.0 (Shannon et al., 2003), GO and KEGG enrichment analyses were conducted for genes in each module. The calculated p-value was subjected to FDR and correction, taking FDR ≤ 0.05 as a threshold.
Real-Time Quantitative PCR Verification
Total RNAs were isolated from the leaves of the treatment and controlled specimens as described above. According to the manufacturer’s instructions, first-strand cDNA synthesis was performed using Prime Script II 1st strand cDNA Synthesis Kit (Takara, Shiga, Japan). Based on the manufacturer’s protocol, the qRT-PCR was performed using a Bio-Rad/CFX ConnectTM Real-Time PCR Detection System (Bio-Rad, CA, United States) with SYBR® qPCR mix (Takara, Shiga, Japan). Relative mRNA content was calculated using the 2–△△Ct method against the internal reference gene RcPP2A (Klie and Debener, 2011). The primers used in this study were designed with Primer Premier 5 and are listed in Supplementary Table 12. Three biological replicates were performed for all reactions.
Results
Changes in Phenotype and Physiology at Drought Stress in R. chinensis
The experiment applied drought stress by stop watering. Plant leaf phenotypes were recorded and physiological indexes were measured under control condition (CK) at 15, 30, 45, 60, 75, 90 days after stress and rewatering. Leaves were dark green and shiny during normal conditions (Figure 1A). After 15 days of drought treatment, the leaves has no obvious morphological changes (Figure 1B). Similarly, the RSWC and leaf relative water content were not significantly different from the control (Figures 2A,B). At 30 days the leaves were slightly wrinkled (Figure 1C). When drought stress lasted for 45 days and 60 days, the leaves were all curled (Figures 1D,E). At this time the relative water content of the soil considerably decreased, reaching a moderate degree of stress (Figure 2A). At 60 days, the relative water content of the leaves decreased significantly compared with the control, which was consistent with the phenotype (Figure 2B). The curl degree of the leaf deepened and the leaf became thinner, and the relative water content of the leaf has a material decrease during 75 days of drought (Figures 1F, 2B). Both leaf relative water content and soil relative water content reached the lowest value, and the leaves were severely curled and withered after 90 days of drought (Figures 1G, 2A,B). After rewatering, the leaves were green and thin (Figure 1H). In order to determine the exact point for transcriptome sequencing, physiological indicators were also measured. The MDA content of leaves treated with drought and rewatering was determined. With the deepening degree of drought stress, the content of MDA rises in fluctuation and reached the highest point after 90 days of the drought treatment (Figure 2D). In addition, SOD increased with the increasing of drought treatment time and achieved the maximum value after 90 days (Figure 2C). Both MDA and SOD content increased significantly during drought treatment for 30 days, 60 days, and 90 days. We conducted correlation analysis on physiological data. MDA content and SOD activity were positively correlated with different treatments and were significantly correlated at the 0.01 level (0.885, 0.979), while the relative leaf water content and soil relative water content were negatively correlated, at 0.01 significantly correlated horizontally (−0.891, −0.987). After rewatering, the MDA and SOD contents were all reduced. Based on these results, leaves were selected under normal conditions, drought treatment for 30 days, 60 days, 90 days, and rewatering treatment for transcriptome sequencing.

Figure 1. Leaf phenotypes of Rosa chinensis ‘Old Blush’ leaves during drought and rewatering stages. (A) The control plants. (B) Drought treatment for 15 days. (C) Drought treatment for 30 days. (D) Drought treatment for 45 days. (E) Drought treatment for 60 days. (F) Drought treatment for 75 days. (G) Drought treatment for 90 days. (H) Rewatering treatment.
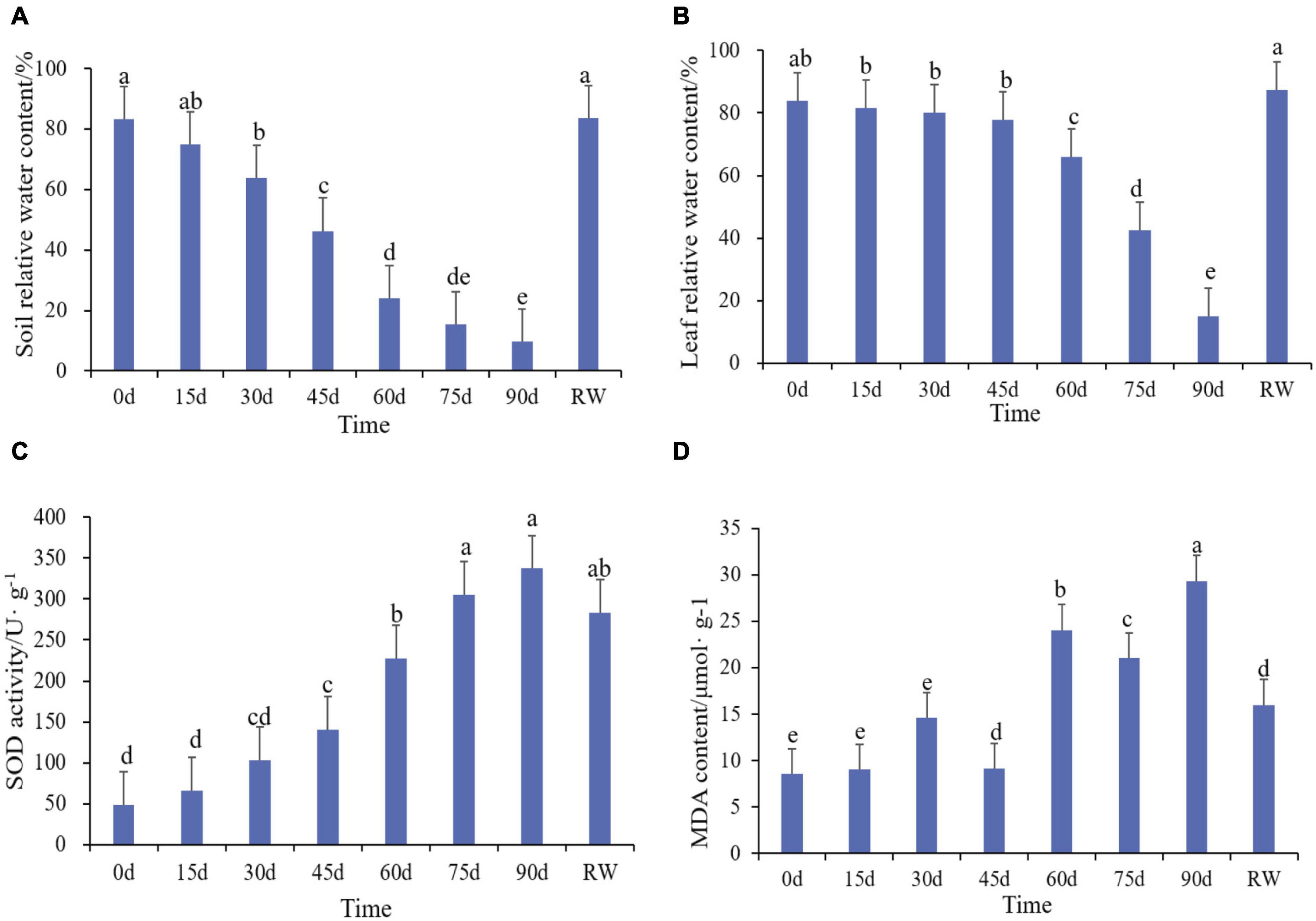
Figure 2. Physiological and biochemical changes of Rosa chinensis ‘Old Blush’ under drought and rewatering treatment. (A) Soil relative water content. (B) Leaf relative water content. (C) MDA content. (D) SOD activity.
Transcriptome Analysis of R. chinensis
The clean reads were obtained by removing adaptor sequences, ambiguous nucleotides, and low-quality reads (Q-value < 20), which ranged from 41397356 to 54756474. The Q20 and Q30 values of all 15 libraries were more than 97% and 92%. The GC content was approximately ranged from 48% to 49%. Between 92.33% and 93.89% of the sequenced reads could be aligned to the rose reference genome (Table 1). In this study, the proportion of exon sequences ranged from 95.30% to 96.38%, and the proportion of intron sequences ranged from 1.45% to 2.66% (Supplementary Figure 1).
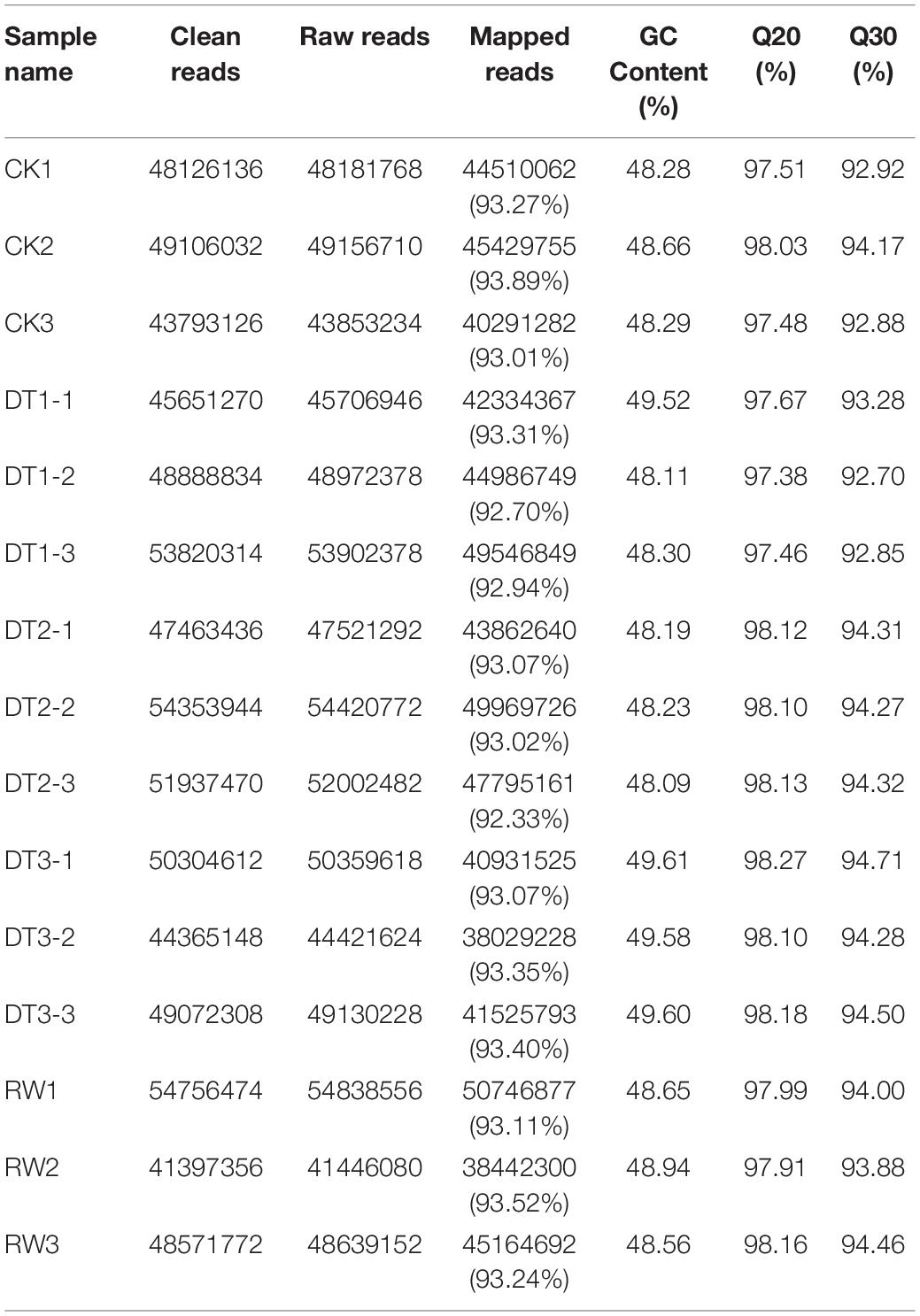
Table 1. RNA sequencing data and corresponding quality control.
Comparisons of DEGs Under the Different Drought Stress Stages
To analyze the DEGs between leaves that suffered from drought stress at four different time duration (30, 60, 90 days and rewatering) and the control, comparisons were conducted between five groups. The DEGs were filtrated according to an expression level | log2(FC)| > 1 and FDR < 0.05 in each pairwise comparison. 6380 genes were identified exclusively at DT3, while 866 genes were specific in DT1. Especially 6380 (35.14%) genes were identified in DT3, but only 1680 (9.25%) and 1804 (9.94%) genes were identified in DT1 and DT2 (Figure 3B).
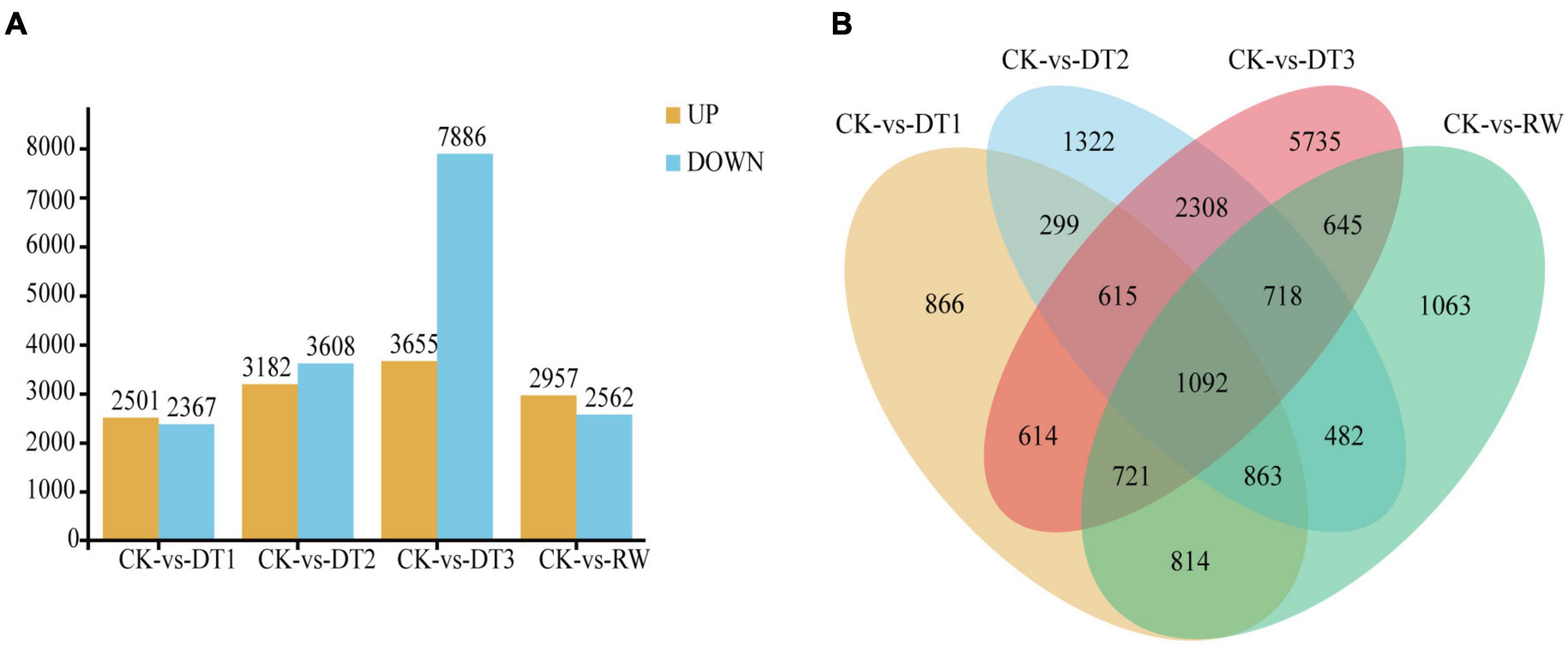
Figure 3. RNA-seq data expression profiles during drought and rewatering treatment in Rosa chinensis ‘Old Blush.’ (A) The number of genes up-regulated and down-regulated in four drought and rewatering treatment stages compared with the control. Genes up-and down-regulated are shown as yellow and blue bars. (B) Venn diagram analysis of the DEGs from four drought and rewatering treatment stages compared with control plants.
The number of DEGs is an increase from DT1 to DT3 and reached a peak at the CK vs. DT3 comparison set, indicating most of the drought regulated genes had a late response. The results showed DEG enrichment in CK vs. DT1, CK vs. DT2, and CK vs. DT3, illustrating the number of down-regulated DEGs was significantly higher than the number of upregulated DGEs. Meanwhile, a total of 11545 DEGs including 7886 downregulated and 3659 upregulated between CK and DT3 which contains the largest number of observations among all drought stress groups. Only 2503 upregulated and 2367 downregulated were observed between CK and DT1, suggesting that the differentiation of expressed genes between CK and DT3 is larger than that of CK and DT1. The result indicates that transcript abundance changed dramatically at key switched among the drought stages, which drought response genes could be induced and expressed largely. Compared with CK, there are slightly more genes upregulated than genes downregulated in RW (Figure 3A).
GO Functional Enrichment and KEGG Pathway Enrichment Analysis of DEGs
The drought resistance process in R. chinensis is complex According to GO functional enrichment analysis, as the drought stress deepened, the most enriched GO category among these DEGs was ‘metabolic process,’ followed by ‘cellular process,’ ‘biological regulation,’ ‘regulation of biological process,’ ‘response to stimulus,’ ‘catalytic activity,’ ‘binding,’ ‘transport activity,’ ‘cell part,’ ‘cell,’ ‘organelle,’ ‘membrane,’ and ‘membrane part.’ Meanwhile, based on the GO analysis of up-regulated DEGs, the GO terms of up-regulated DEGs in CK vs. DT1, CK vs. DT2, and CK vs. RW were mostly concentrated on membranes, including the cellular components category, such as ‘membrane’ (GO:0016020), ‘membrane part’ (GO:0044425), and ‘intrinsic component of membrane’ (GO:0031224) (Supplementary Table 1). CK vs. DT3 also involved more cells and metabolic processes including the biological process category (metabolic process, GO:0008152, cellular, GO:0009987, single-organism process, GO:0044699, cellular metabolic process, GO:0044237) (Supplementary Table 1). Moreover, the GO analysis of the down-regulated DEGs found that among the top 20 enriched, 13 of them belong to the category of biological processes, mainly focusing on the response to stimuli in CK vs. DT1 (Supplementary Table 2). It is worth noting that the GO analysis of DT3 vs. RW mainly focused on cells and cell membranes, indicating that the rose has been restored to a certain extent at the cell level (Supplementary Tables 1, 2).
The KEGG pathway analysis revealed that it is mainly enriched in the metabolism under the different drought degrees and rewatering treatment. Among the top 10 enriched pathways, ‘plant–pathogen interaction’ (ko04626), ‘biosynthesis of secondary metabolites’ (ko01110), ‘plant hormone signal transduction’ (ko04075), ‘MAPK signaling pathway-plant’ (ko04016), were regulated in response to DT1, indicating that drought stress was sensed and signaling transduction pathways were activated. However, more pathways related to metabolism were enriched at DT2 and DT3. The ‘photosynthesis’ (ko00195) and ‘photosynthesis-antenna proteins’ (ko00196) associated with photosynthesis were all enriched in CK vs. DT2 and CK vs. DT3 (Supplementary Table 3). Metabolite-related pathways such as ‘biosynthesis of secondary metabolites’ (ko01110), ‘alpha-Linolenic acid metabolism’ (ko00592), ‘tryptophan metabolism’ (ko00380), ‘glycosphingolipid biosynthesis-lacto and neolacto series’ (ko00601), ‘alanine, aspartate and glutamate metabolism’ (ko00250), ‘galactose metabolism’ (ko00052), and ‘glyoxylate and dicarboxylate metabolism’ (ko00630) were enriched in CK vs. DT2 and CK vs. DT3. These results indicated that the metabolism processes of R. chinensis may act as the predominant process in the middle and later stages of drought stress. This result was unsurprising, as drought causes changes in the metabolism of plants to improve their drought tolerance. Meanwhile, secondary metabolism, primary metabolism, and photosynthesis were focused in DT3 vs. RW, indicating that the metabolism and photosynthesis of the rose after rewatering recovered, and that drought stress did not cause serious damage to the rose (Supplementary Table 3). Apart from metabolism enrichment, ‘plant–pathogen interaction’ (ko04626) was also enriched in CK vs. RW, indicating that plants are vulnerable to pathogenic bacteria after recovering from drought stress (Supplementary Table 3).
Identification of Plant Hormone and Signal Transduction-Related DEGs
In this study, signal transduction in plants was influenced by drought stress which was verified by both the GO and KEGG enrichment analysis of the DEGs. More than 240 genes were predicted to encode protein kinases with varying expression levels in R. chinensis. Genes encoding receptor-like protein kinases (RLKs), in which LRR receptor kinases were the majority accounted for the largest proportion of these genes. Twenty-nine of them were up-regulated in DT1 and DT2. Among them, MIK1 (RchiOBHm_Chr6g0286591) was up-regulated 8.02-fold, respectively. More than 66 RLK genes were included in the group of serine/threonine-protein kinases during the drought stress. Furthermore, one gene related to G-type lectin S-receptor-like serine/threonine-protein kinase showed the highest degree of upregulation in DT3. In the KEGG pathway analysis of DEGs, the ‘MAPK signaling pathway’ (ko04016) was induced during drought treatment. For DEGs detected in DT1 and DT2, two MAPKKKs including RchiOBHm_Chr2g0158821 and RchiOBHm_Chr5g0002581 and four MAPKKs including, MPK9 (RchiOBHm_Chr7g0178621), MKK10 (RchiOBHm_Chr7g0211301), MEKK1 (RchiOBHm_Chr7 g0240941), MPK4 (RchiOBHm_Chr7g0196781) were up-regulated. MKK10 (RchiOBHm_Chr7g0211301) was particularly up-regulated 9.18-fold. However, three MAPKKs consist of MPK3 (RchiOBHm_Chr5g0061451), MKK4 (RchiOBHm_Chr2g0150341), and MEKK1 (RchiOBHm_Chr1g0352161) were down-regulated. Also, MPK4 (RchiOBHm_Chr7g0196781) and MPK19 (RchiOBHm_Chr5g0028971) were substantially increased in DT3 (Supplementary Table 4).
Ca2+ signal transmission is achieved through the generation, decryption, and transmission of specific calcium signals and the corresponding physiological and biochemical reactions downstream. The study identified DEGs related to calcium ion, encoding calcium-binding proteins, Ca2+-binding protein EF hand, calmodulin-like, calcium-dependent protein kinases, calcium-transporting ATPase, and calcium channel protein. One gene (RchiOBHm_Chr2g0158571) encoding the calcium channel protein sustained high expression under drought stress and was up-regulated 6.44-fold in DT1 vs. CK (Supplementary Table 5).
According to the GO and KEGG analyses of the DEGs, the GO term ‘hydrogen peroxide metabolic’ (GO:0042743) and ‘response to reactive oxygen species’ (GO:0000302) were enriched at DT1 and DT2, while peroxisome (ko04146) was induced at DT3. 69 DEGs encoded ROS production and scavenging. A total of 55 DEGs encoding enzymes related to ROS scavenging, including peroxidase (POD), ascorbate peroxidase (APX), glutathione S-transferase (GST), glutathione peroxidase (GPX), polyphenol oxidase (PPO), ferritin, glutaredoxin, thioredoxin, and peroxiredoxin. Among these antioxidant enzymes, POD45 (RchiOBHm_Chr4g0400821) encoding POD was up-regulated by 4.79-fold at DT1, another gene encoding POD PNC1 (RchiOBHm_Chr5g0072281) has no significant change from DT1 to the control, but 9.29-fold has a significant at DT2, reaching the peak of expression level at DT3 (Supplementary Table 6).
The expression of genes involved in hormone biosynthesis or signaling was substantially changed during the drought stress and rewatering stage. The changed genes included ABA, auxin, BR, cytokinin, ET, JA, and SA. Among these DEGs, most genes were involved in ABA response. Nine-cis-epoxycarotenoid dioxygenase (NCED) is a critical enzyme in ABA biosynthesis. For instance, one gene encoding NCED3 (RchiOBHm_Chr5g0014331) was up-regulated by 2.70-fold at DT1, while another gene encoding NCED6 (RchiOBHm_Chr4g0397001) was significantly down-regulated 9.40-fold at DT3. The expression of a gene (RchiOBHm_Chr5g0049951) encoding AAO is also a vital enzyme in ABA biosynthesis, which was up-regulated by 2.78-fold at DT1 and down-regulated by 7.28-fold at DT3. Moreover, some genes involved in the ABA mediated signaling pathway were also induced by drought stress. Abscisic acid receptor PYL4 (RchiOBHm_Chr1g0371101) was down-regulated. In contrast, one gene encoding PP2C (RchiOBHm_Chr5g0066401), a negative regulator of ABA, has an up-regulation (Supplementary Table 7).
A total of 24 genes involved in the auxin signaling pathway changed. Three genes encoding auxin response factors (ARF3, ARF5, ARF9) were drought induced. Particularly, ARF3 (RchiOBHm_Chr5g0009381) expression continued to decline under drought stress, which was induced 4.41-fold at DT3. At the same time, the expression level of ARF5 (RchiOBHm_Chr6g0302551) continued to increase under drought stress. Two genes encoding AUX/IAA proteins were up-regulated at DT1 and down-regulated at DT3. AUX22D (RchiOBHm_Chr4g0389611) was lowered nearly 10-fold at DT3. Furthermore, some genes participated in auxin response and signal, for example of auxin-binding protein ABP19a-like (ABP19A), auxin transporter-like protein 2 (LAX2), and auxin efflux carrier (PIN1C). The gene encoding ABP19A (RchiOBHm_Chr2g0094781) was up-regulated 7.41-fold at DT1. Five genes encoding SAUR were identified, among these, SAUR77 (RchiOBHm_Chr5g0026611) was up-regulated 9.78-fold at DT1 (Supplementary Table 7).
In this study, a large number of ethylene-responsive transcription factor (ERF) genes displayed a down-regulation trend under early drought stress. However, one gene encoding ERF023 (RchiOBHm_Chr1g0360021) had an up-regulation 3.70-fold at DT1 and was then down-regulated 5.02-fold at DT3. Another gene encoding example, ERF110 (RchiOBHm_Chr6g0274591) was up-regulated 7.34-fold at DT3. Meanwhile, ethylene receptor (ERS1), ethylene-overproduction protein 1 (ETO1), and ethylene insensitive3-like protein (EIL3) were identified (Supplementary Table 7).
Most of the genes associated with the biosynthesis or signaling of jasmonic acid (JA) were down-regulated. The gene encoding malate dehydrogenase (MDHG) was down-regulated 4.06-fold at DT3, while 12-oxophytodienoate reductase 3-like (OPR3) was up-regulated 4.13-fold at DT3. Additionally, the majority of genes were related to cytokinin, salicylic acid, and brassinosteroid pathways. One gene encoding cytokinin dehydrogenase 3 (RchiOBHm_Chr2g0093491) was down-regulated 7.40-fold at DT3 (Supplementary Table 7).
Expression of Genes Involved in Metabolism and Biosynthesis
The output of GO and KEGG enrichment analysis indicates that many DEGs were related to metabolism and biosynthesis. The ‘polysaccharide catabolic process’ (GO:0000272) and the ‘Starch and sucrose metabolism’ (ko00500) pathway were significantly induced by drought stress. Related to carbohydrate synthase and starch synthase were induced. The expression level of sucrose synthase increased, and one of the genes encoding SUS7 (RchiOBHm_Chr3g0488651) up-regulated 4.03-fold at DT3. Only a small portion of starch synthase genes were down-regulated, others appeared to be up-regulated. Additionally, drought treatment significantly induced the expression of enzyme genes involved in starch and sugar metabolism, fructose, and mannose metabolism. Among these, genes encoding fructose-1,6-bisphosphatase, fructose-bisphosphate aldolase, mannose-1-phosphate guanyltransferase alpha, pyrophosphate–fructose 6-phosphate 1-phosphotransferase subunit beta-like and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase were down-regulated at DT3 (Supplementary Table 8).
In total, 89 DEGs associated with lipid metabolism were enriched in the ‘lipid metabolic process’ (GO:0006629). Most of these were related to the biosynthesis and metabolic processes of lipoprotein, wax, keratin, lipid, and fatty acid. Moreover, most genes involved in wax biosynthesis were significantly up-regulated. For example, one gene encoding CYP86B1 (RchiOBHm_Chr4g0432631) up-regulated 9.90-fold at DT3 (Supplementary Table 9). The KEGG functional annotation of these differentially expressed genes was related to metabolism and differentially expressed genes were also related to secondary metabolisms. A total of 95 DEGs involved in secondary metabolism were enriched by KEGG function, and the results are shown in Figure 4. Drought stress affected the transcription level of genes that related to phenylpropanoid biosynthesis pathways.
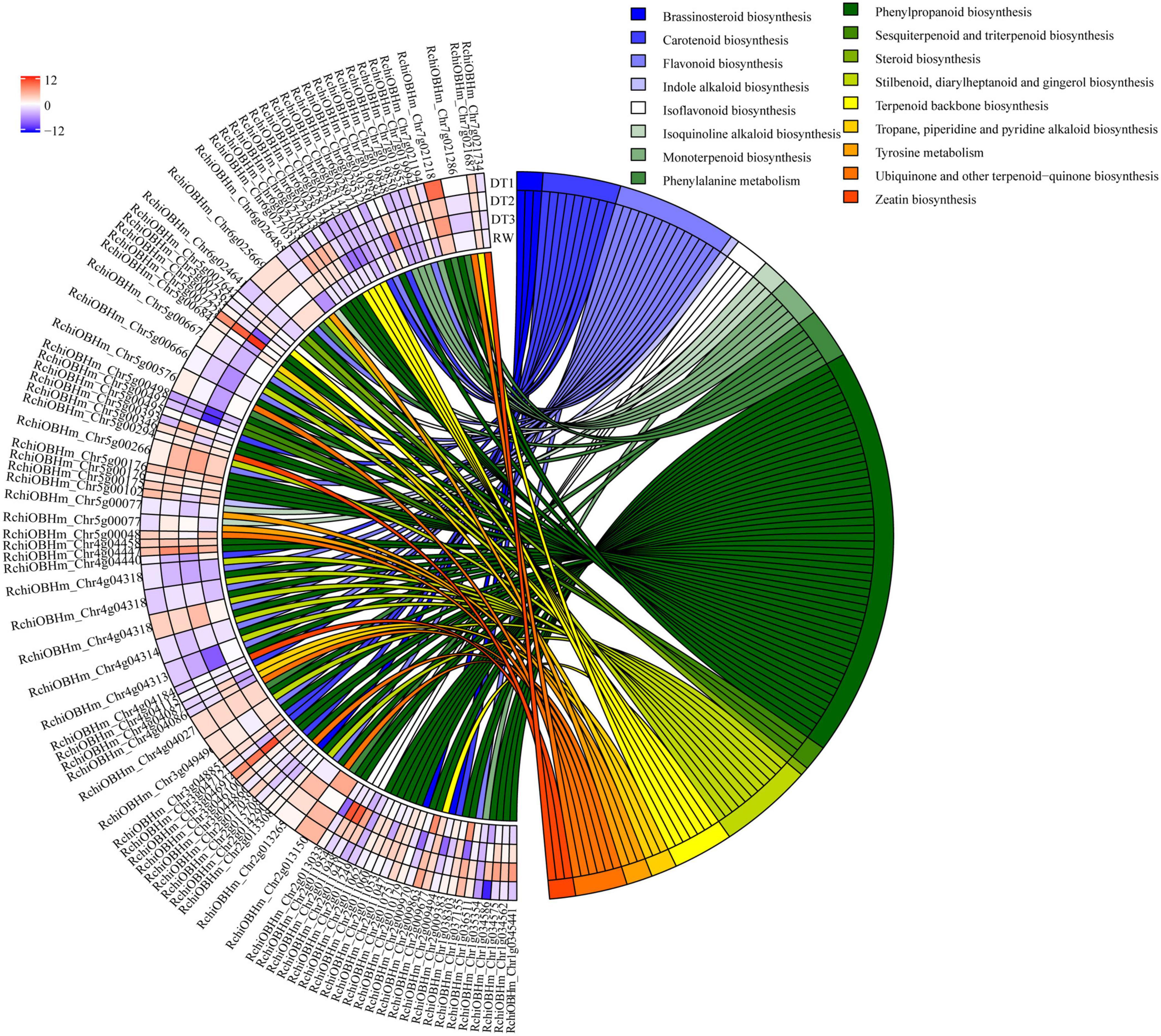
Figure 4. KEGG enrichment of differentially expressed genes involved in secondary metabolism.
Photosynthesis-Related Genes Under Drought Stress
Many DEGs enriched in the category of ‘photosynthesis’ (ko00915) were overexpressed in DEGs down-regulated by DT2 and DT3. In the process of DT2 and DT3, many genes involved in photosynthesis were significantly down-regulated. A total of 70 DEGs involved in photosynthesis were identified. In the seam, three (CAB40, LHCB5, psaA) of 16 DEGs encoding the components of photosystem I (PSI) were slightly up-regulated in DT1, and other genes were significantly down-regulated during the drought treatment period. A total of 33 genes encoding PS II were identified, including 6 up-regulated genes (psbE, three genes encoding psbC, psbD, PSB27-1), and 27 down-regulated genes. Most of these genes decreased significantly in the middle and later periods of drought, indicating that PS I and PS II were suppressed under drought stress. Meanwhile, there were 15 genes encoding redox chains, all of which were significantly down-regulated in the middle and late periods of drought stress. Additionally, the expression of six gene encoding components of chloroplasts significantly decreased (Supplementary Table 10).
Transcription Factors Responding to Drought Stress
Transcription factors specifically binding to cis-acting regulatory elements in the promoter of target genes are crucial regulatory proteins, which can modulate numbers of genes up or down-regulation (Dhriti and Ashverya, 2015). In this study, WRKY (58), NAC (43), MYB (52), bZIP (19), bHLH (55) families contained large numbers of transcription factors. The expression level of the WRKYs family exhibited large changes under different treatments. The expression of some genes reached a maximum in the middle and later periods of drought. Genes encoding WRKY75 and WRKY71 all reached the highest expression level at DT3 with an up-regulation of more than 5-fold. WRKY57 (RchiOBHm_Chr5g0083891) up-regulated regarding expression responses at different drought stress, especially increased 7.93-fold at DT3. However, more than half of the WRKY families have decreased expression levels at different drought levels. Twenty-four genes encoding WRKYs were down-regulated at DT1 by 1.02- to 3.71-fold (Supplementary Table 11).
More than half of the genes encoding NACs were up-regulated to varying degrees under drought stress. One gene encoding NAC100 (RchiOBHm_Chr2g0167461) continued to increase in the middle and later periods of drought stress by 7.20-fold. A total of 11 genes upregulated during DT1. Some genes increased and then decreased under drought stress, but finally increased after rewatering. For example, NAC073 (RchiOBHm_Chr0c19g0500091) was upregulated 5.78-fold at DT1, decreasing slightly at DT2, then down-regulated 2.64-fold at DT3, but had an increase of 6.96-fold after rewatering. NAC037 (RchiOBHm_Chr2g0099671) had a similar expression trend, but the up-regulation decreased a small amount. On the contrary, one gene encoding NAC072 (RchiOBHm_Chr5g0034761) was down-regulated at DT1 and continued to increase afterward (Supplementary Table 11).
Under different drought treatments, more than three-quarters of the MYB genes’ expression levels showed different degrees of up-regulation. Among them, a relatively large number of up-regulated genes appeared during the DT1 period. For instance, MYB46 (RchiOBHm_Chr1g0315931) and MYB61 (RchiOBHm_Chr1g0315931) showed the highest up-regulation of expression with 7.09 and 7.15-fold. Moreover, some genes up-regulated significantly at DT3. MYB75 (RchiOBHm_Chr2g0116041) displayed the most amount of up-regulation among all up-regulated genes at DT3, with an increase of 7.65-fold. Some genes showed a tendency to increase and then decrease during drought treatment, for example, gene encoding MYB61 (RchiOBHm_Chr3g0458721) slightly increased at DT1 and then decreased by 6.10-fold. After rewatering, most gene expression did not change significantly compared with CK, only a small amount of gene expression up-regulated (Supplementary Table 11).
Among the three drought treatments of the bHLH families, the numbers of genes up-regulated in one period were more than the numbers of genes down-regulated. The number of down-regulated genes in two periods was more than that. Most genes in the bHLH family up-regulated during early drought treatment of DT1. While a large number of genes significantly down-regulated during the middle and later drought periods (DT2 and DT3). Among these, there was a slight upward adjustment, up-regulated by 1.02- to 4.66-fold at DT1. Nevertheless, there were 37 genes down-regulated at DT2 or DT3. One gene encoding bHLH72 (RchiOBHm_Chr7g0182341) down-regulated 6.94-fold. A gene bHLH121 (RchiOBHm_Chr6g0245181) that belongs to the bHLH family has no significant change in gene expression during drought treatment but appeared to be up-regulated after rewatering (Supplementary Table 11).
Among AP2/DREB and bZIP transcription factors, more genes exhibited down-regulation under drought treatment. Nineteen members of bZIPs were found to be responsive to drought stress, while 12 down-regulated from 1.24- to 4.80-fold (Supplementary Table 11). Five genes encoding DREBs showed down-regulation that exceeded 6.60-fold at DT1. The most significant down-regulation occurred by DREB1D (RchiOBHm_Chr7g0199351), which down-regulated 9.64-fold (Supplementary Table 11).
Co-expression Network Construction and Identification of WGCNA Modules
Gene co-expression network gene clustering and module cutting combined genes with similar expression patterns on the same branch. Each branch represented a co-expression module with different colors representing different modules. The unexpressed genes in more than half of the samples were filtered, the expression patterns of 30,012 genes obtained from transcriptome sequencing were performed by WGCNA. A total of 18 modules were identified according to the similarity of expression patterns (Figure 5A). Cluster analysis was performed to evaluate genes in the modules and were observed through heat maps (Figure 5B). Furthermore, to identify modules that were significantly associated with different drought stress, the module-trait correlation relationships were constructed (Figure 5C). There were four modules including blue, navajowhite1, salmon4, and coral2 were closely associated, with traits related to four treatments (0.99, 0.99, 0.87, and 0.76, respectively). The expression pattern of the eigengenes represents the gene expression profile of the entire module, and the expression patterns of the four selected modules were analyzed (Figure 6).
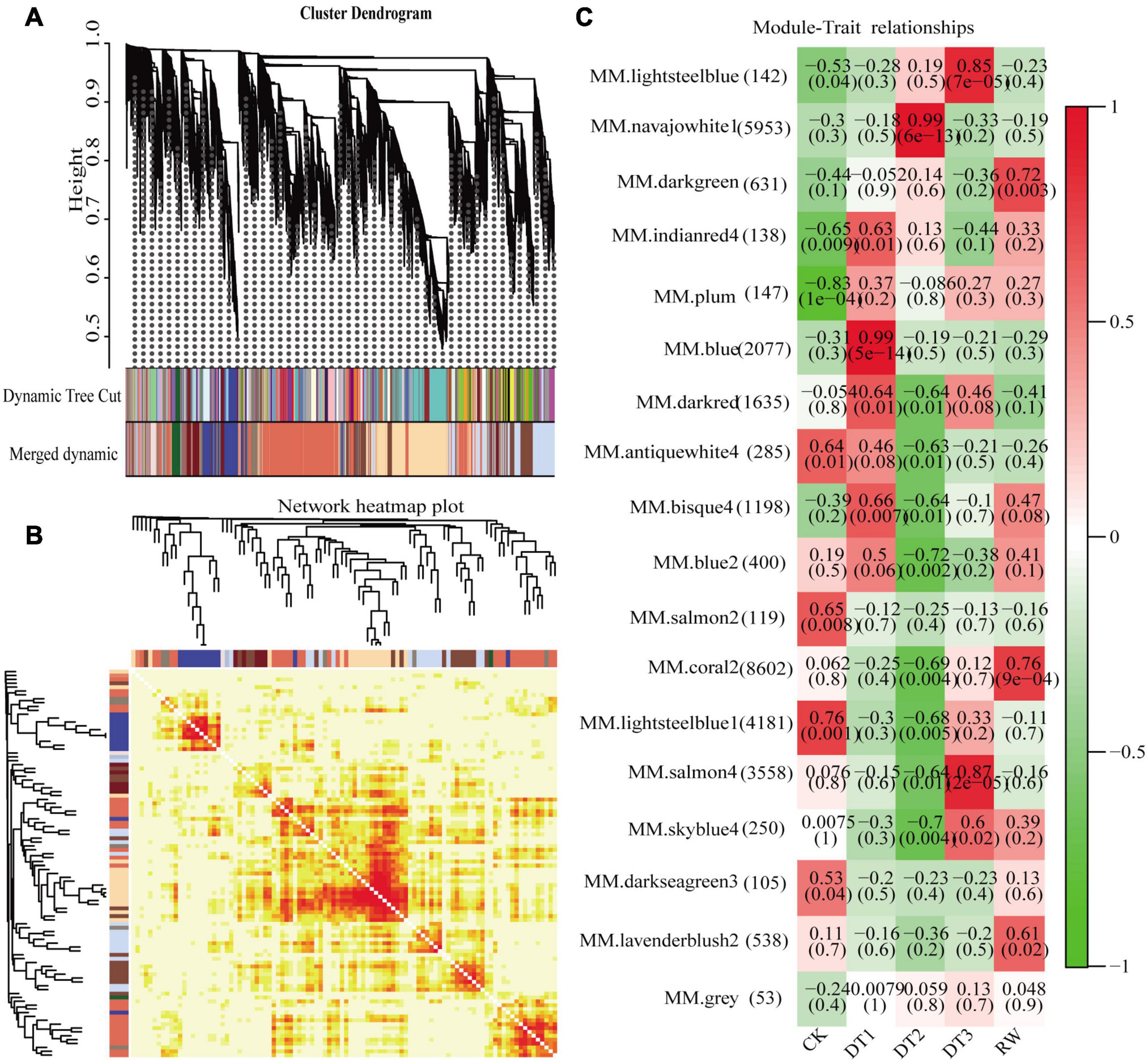
Figure 5. Identification of DEGs by WGCNA. (A) Module level clustering diagram. (B) Network heatmap plot. (C) Module-trait associations. (Each column corresponds to different processing conditions, and each row corresponds to the characteristic gene of the module. The correlation between two is indicated in cell by Pearson correlation coefficient and p value in parentheses. Cell color ranges from red (high positive correlation) to green (high negative correlation), and the number of genes contained in each module is in left parentheses).
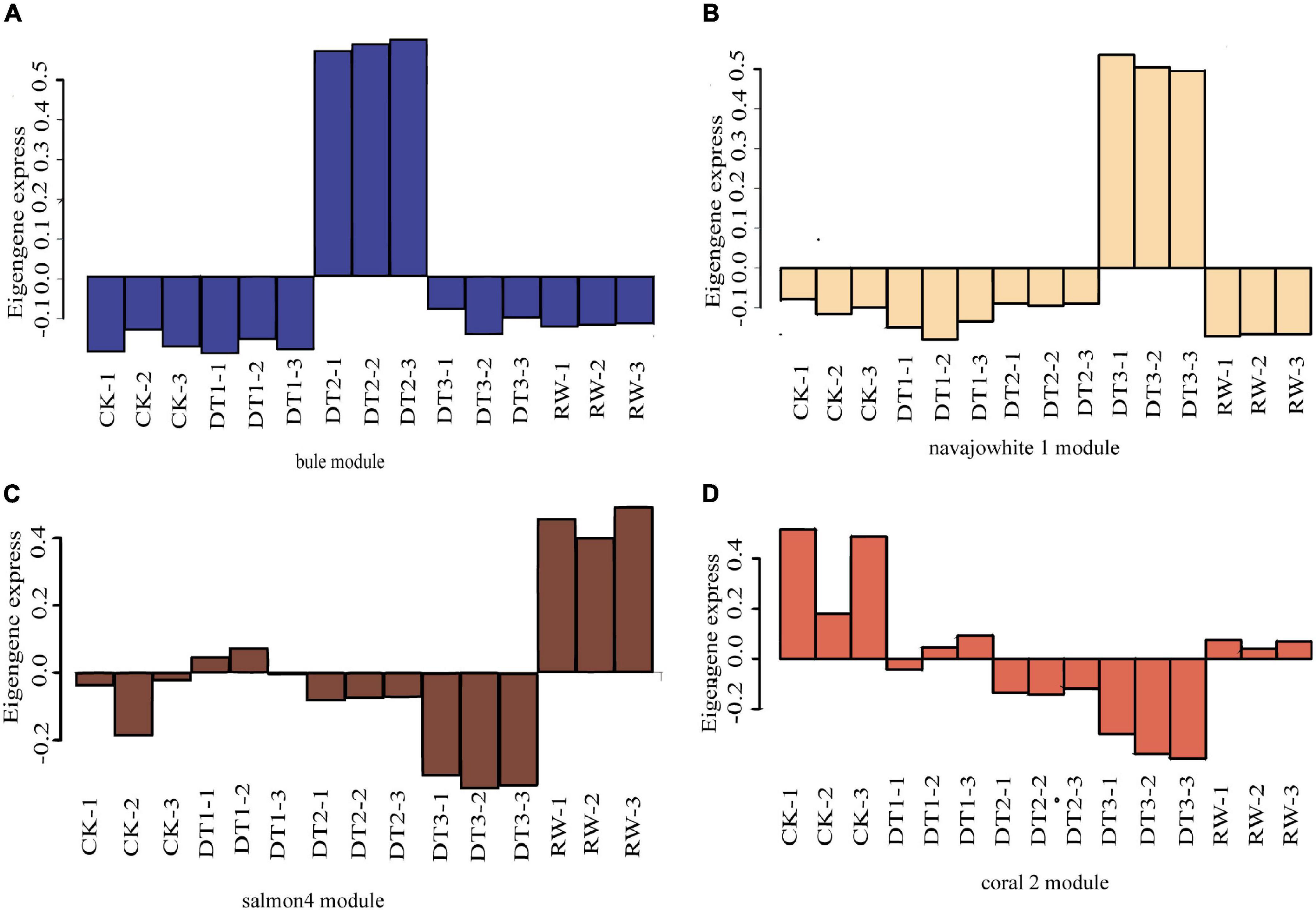
Figure 6. The expression pattern of the co-expressed genes in the representative module. (A) Blue module. (B) Navajowhite1 module. (C) Salmon4 module. (D) Coral2 module.
Four modules of blue, navajowhite1, salmon4, and coral2, were annotated through GO and KEGG analysis. The blue module of DT1 was most significant in GO analysis, which was mainly enriched in signal transduction and stress response (Supplementary Table 13). The results of KEGG analysis were similar, mainly focusing on signal transduction and synthesis of metabolites (Supplementary Table 13). The navajowhite1 module showed significant rich GO terms, indicating many genes were enriched in various stress responses. Those that were mainly enriched in ‘response to stimulus’ (GO:0050896), ‘response to chemical’ (GO:0042221), ‘response to water’ (GO:0009415), and ‘response to water deprivation’ (GO:0009414) were also significantly enriched (Figure 7A). KEGG enrichment analysis displayed the main enrichment in metabolic pathways (Figure 7B). In the salmon4 module, GO terms and KEGG pathways were enriched in metabolic progress, which indicates that the rose produces metabolites in the later drought stage to resist drought stress (Supplementary Tables 13, 14). However, the process of compound metabolisms was focused on the coral2 module (Supplementary Tables 13, 14). The GO terms about stress response are mainly concentrated in navajowhite1 and blue module. As a result, navajowhite1 and blue module genes may play a critical role in the process of rose drought stress and rewatering.
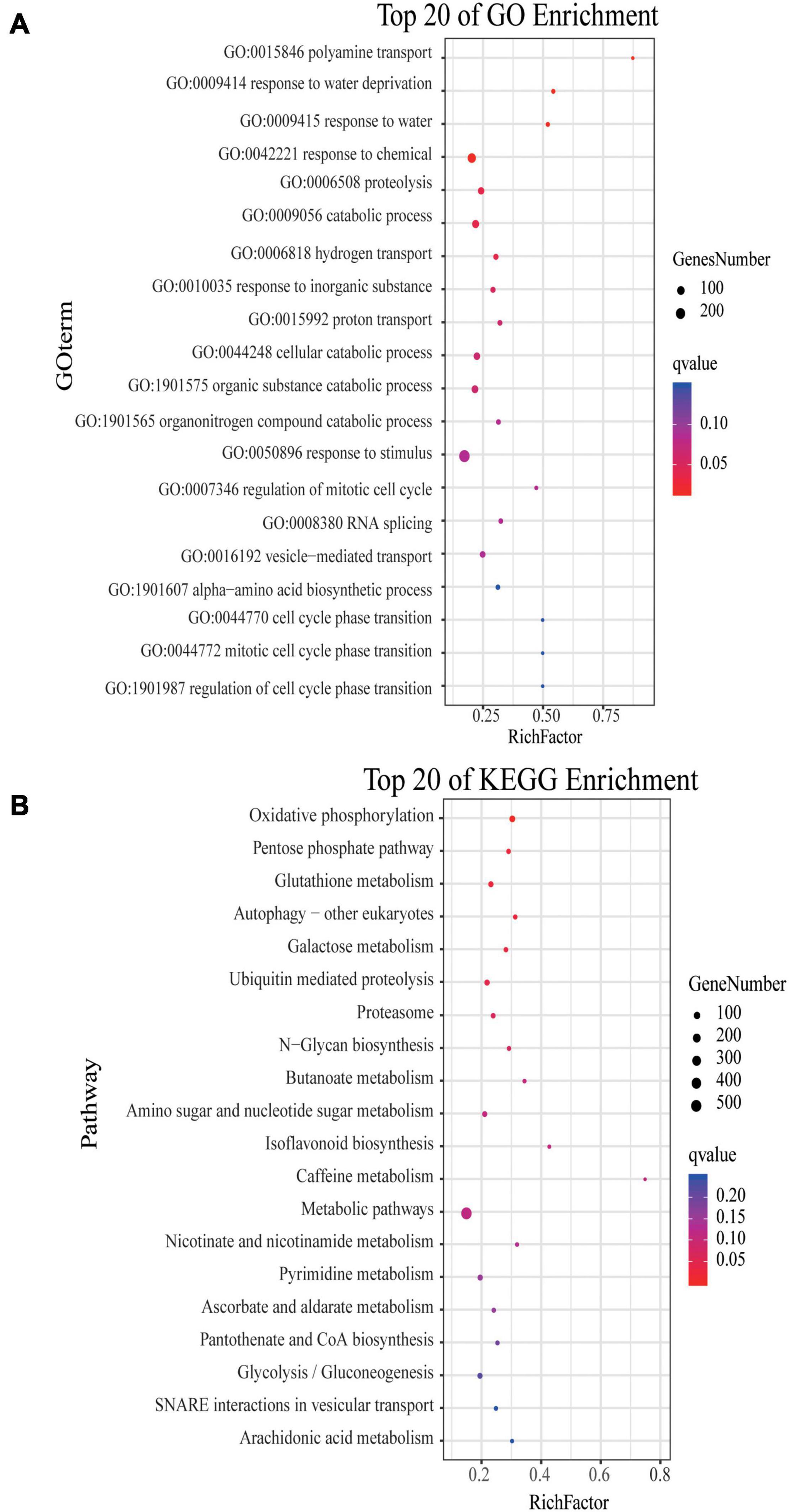
Figure 7. GO and KEGG analysis of DEGs in the navajowhite1 module. (A) The top 20 of GO enrichment. (B) The top 20 of KEGG enrichment. (The q-value ranges from 0 to 0.05. The closer the q-value is to 0, the more significant the enrichment is. Genes number is the number of genes enriched in pathways).
Identification of Hub TFs and Network Construction
Transcription factors are an essential class of regulatory proteins in biological processes. This study analyzed the TFs in each module. Although there was a difference in the distribution of TFs in different modules, it is mainly concentrated in WRKY, NAC, MYB, ERF, bHLH, bZIP, C2H2, and other transcription factor families. According to previous reports, these TFs were involved in the process of plant stress regulation (Erpen et al., 2018). TFs of navajowhite1 and the blue module were analyzed, the highly connected examples that were used as core genes were screened out. Many DEGs Identified in the navajowhite1 module were annotated as TFs, including several WRKY, MYB, NAC, ERF, ARF, and bHLH TFs (Figure 8A). Based on the hub gene correlation network and their high connectivity, 22 of these TFs were selected: two genes (RchiOBHm_Chr5g0034761 and RchiOBHm_Chr6g0308271) encoding NAC072, two genes (RchiOBHm_Chr2g0167461 and RchiOBHm_Chr7g0181811) encoding NAC100, NAC087 (RchiOBHm_Chr3g0497471), MYB33 RchiOBHm_Chr7g0228621), MYB75 (RchiOBHm_Chr2g0116041), MYB102 (RchiOBHm_Chr5 g0006031), MYB78 (RchiOBHm_Chr4g0426181), WRKY3 (RchiOBHm_Chr4g0439041), WRKY4 (RchiOBHm_Chr2 g0166991), WRKY6 (RchiOBHm_Chr5g0040801), WRKY27 (RchiOBHm_Chr1g0378621), WRKY28 (RchiOBHm_ Chr2g0151681), WRKY71 (RchiOBHm_Chr5g0013131), two genes (RchiOBHm_Chr4g0429851 and RchiOBHm_ Chr2g0175911) encoding WRKY75, ERF113 (RchiOBHm_Chr4g0428891), ARF1 (RchiOBHm_Chr7g021 9771), ARF5 (RchiOBHm_Chr6g0302551), ARF8 (RchiOBHm_Chr3g0487771), and the bHLH3 (RchiOBHm_Chr6g0288981) (Figure 8B). The experiment showed the expression of these genes was significantly upregulated at DT3 except for WRKY6, bHLH3, ARF1, and ARF8 (Figure 8B). In the blue module, 20 TFs were identified as hub genes including ERFs, bHLHs, WRKYs, MYBs, and NACs (Figure 8C). ERF109 and bHLH162 have higher connectivity among them (Figure 8C). The expression of all 20 TFs increased in DT2 (Figure 8C).
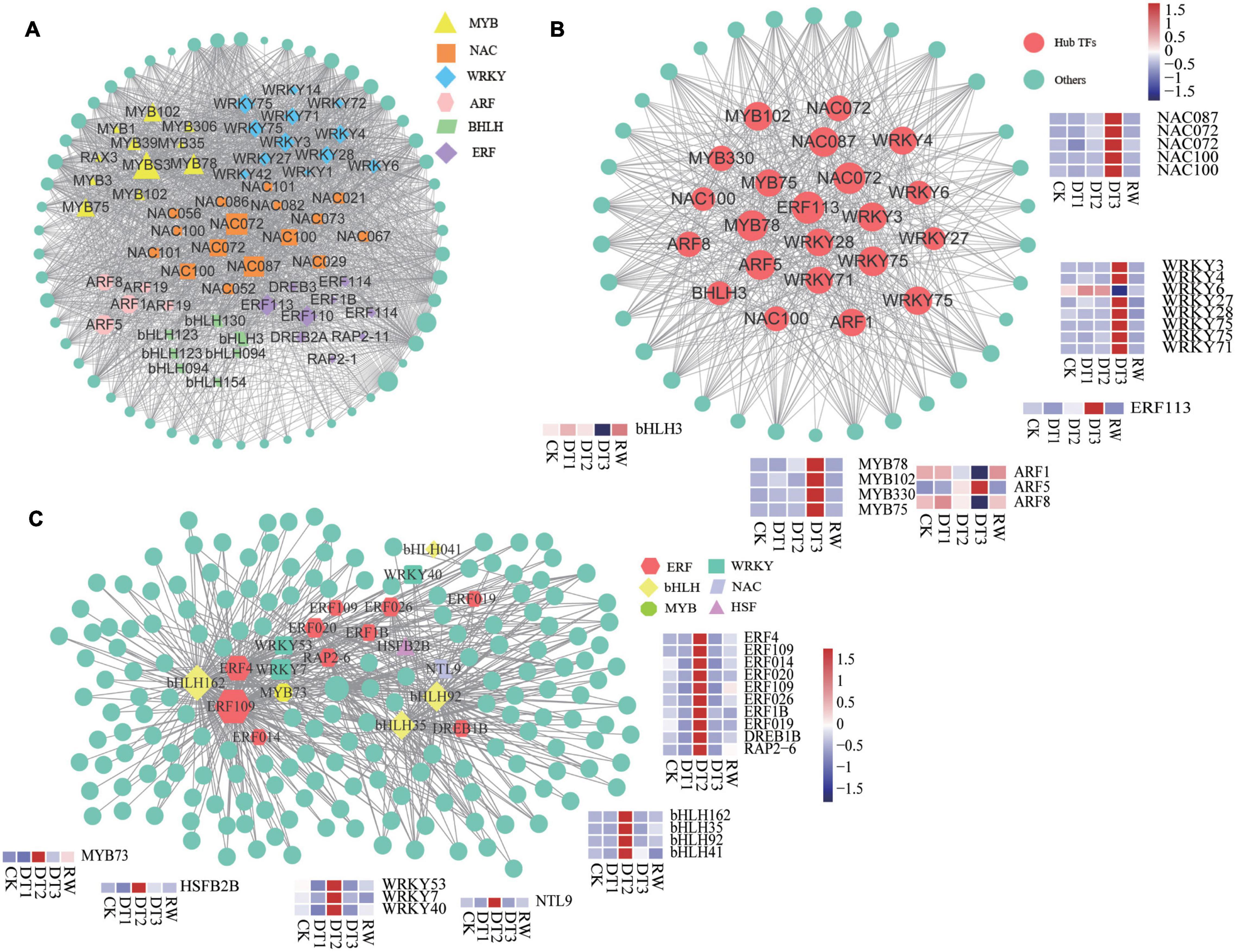
Figure 8. Identification and selection of vital transcription factors in navajowhite1 and blue module. (A) Network analysis of TFs in navajowhite1 module. (B) Network of the top 22 hub TFs and related genes in the navajowhite1 module. (C) Network analysis of hub TFs in the blue module.
Validation of the DEGs by qRT-PCR
To verify the accuracy and reproducibility of the transcriptome analysis, 13 DEGs were selected to analyze the transcript abundance using qRT-PCR, including seven randomly selected transcription factors from the navajowhite1 module, four TFs, and two ABA synthesis-related DEGs. The results showed that the expression profiles detected by qRT-PCR were positively correlated with the RNA-Seq results (Figure 9). Therefore, the reliability of the RNA-seq data was confirmed by the consistency between the qRT-PCR results and RNA-seq analyses.
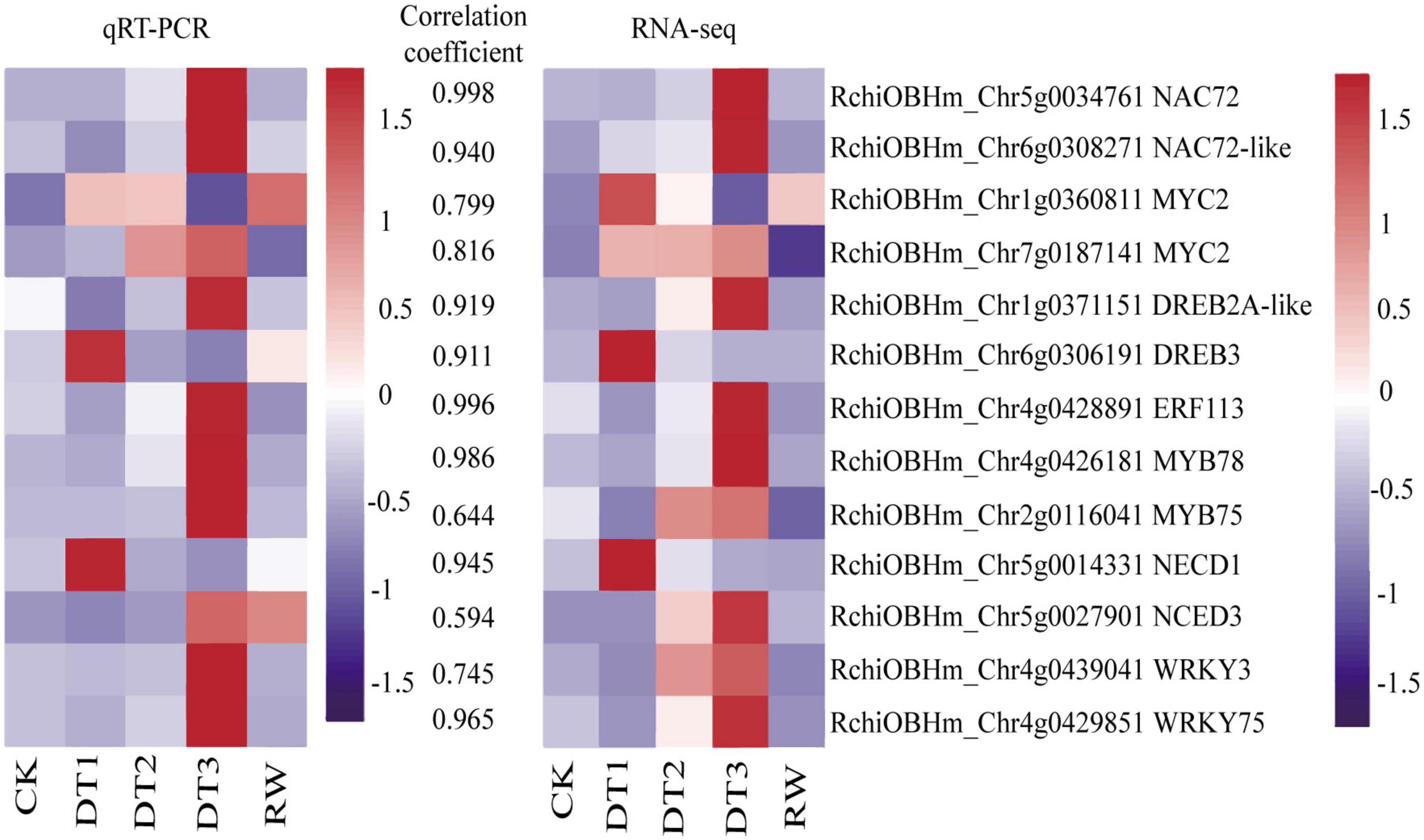
Figure 9. Heatmaps of the validation and correlation analysis of 13 selected DEGs. The values between the two heatmaps represent the correlation coefficients of qRT-PCR and RNA-seq values from each gene.
Discussion
TFs Involved in the Drought Stress Response
Weighted gene co-expression network analysis is an analysis method for analyzing the gene expression patterns of multiple samples. Genes with similar expression patterns can be clustered, moreover, the relationship between modules and specific traits can be analyzed (Zhang and Horvath, 2005). WGCNA analysis can present the interaction relationship between genes, and identify the hub genes at the center of the regulatory network. It also can use the module eigengenes values to perform correlation analysis with specific traits, more accurately (Silva et al., 2019). More and more studies use WGCNA to analyze key genes in plants under abiotic stress (Shao et al., 2021). In this study, through the analysis of WGCNA and transcription regulation modules, the highly connected TFs in modules were selected. Numerous TFs were obtained in the transcriptome sequencing in this study including the NACs, WRKYs, MYBs, AP2/ERFs, bHLHs, and so on. Many studies have shown that NACs play positive roles in the regulation of plant stress resistance. In particular, Arabidopsis (Tran et al., 2004), wheat (Mao et al., 2014; Zhang et al., 2015), and rice (Hong et al., 2016; Sung et al., 2018) have received the most research attention on NACs, which found that overexpression of NACs transcription factor family genes can significantly enhance the tolerance of transgenic plants to multiple stresses. In addition, rose (Rosa hybrida) RhNAC3 can improve rose petals dehydration tolerance, and can also improve the drought tolerance of transgenic Arabidopsis (Jiang et al., 2014). In the study of rose (Rosa chinensis ‘Slater’s crimson China’) heat resistance, it was found that both NAC27 and NAC72 up-regulated (Li et al., 2019b). NAC27 and NAC72 were also involved in ABA signal transduction under heat stress of Rhododendron hainanensis (Zhao et al., 2018). Overexpression of NAC72 in Arabidopsis thaliana can increase the drought resistance of plants, and also proved that it mediates the regulation of ABA-responsive genes (Li et al., 2016). Gene encoding NAC072 with high connectivity in the navajowhite1 module was selected, indicating that it plays an important role in the regulation of rose drought stress. In agreement with previous studies, it can be concluded that NAC072 may be involved in ABA signal transduction under drought stress in R. chinensis. Furthermore, the role of the core gene in the navajowhite1 module NAC087 and NAC100 in drought also requires further consideration.
The WRKY genes play pivotal roles in stress responses. Many researchers have demonstrated that WRKYs have crucial biological functions in the response of plants to different kinds of biotic and abiotic stresses (Jiang et al., 2017). Studies on Arabidopsis, rice, and soybean have shown the importance of the WRKYs in response to drought stress. For example, AtWRKY57 (Jiang et al., 2012), AtABO3 (Ren et al., 2010), OsWRKY47 (Raineri et al., 2015), and GmWRKY54 (Wei et al., 2019) can improve the drought tolerance of plants. Likewise, VaWRKY14 responds to drought and cold stress, and hence the overexpression of VaWRKY14 can enhance the drought tolerance of transgenic Arabidopsis (Zhang et al., 2018). However, some WRKYs can act as a negative regulator of abiotic stress in plants. For instance, GhWRKY33 (Wang et al., 2019b) and SlWRKY81 (Ahammed et al., 2020; Li et al., 2020) reduce the drought tolerance of transgenic plants. In the analysis, eight WRKY genes including WRKY75, WRKY28, and WRKY27 were found to have high connectivity in the navajowhite1 module, indicating that they may be involved in drought response. Other research has found that PagWRKY75 was down-regulated in the early stages of salt and osmotic stress, transgenic poplar lines overexpressing PagWRKY75 were more sensitive to salt and osmotic (Zhao et al., 2019b). It can be assumed that WRKY75 plays an essential regulatory role in abiotic stress, but the function of WRKY75 in drought stress screened in this study requires further verification.
APETALA2/ethylene-responsive factor (AP2/ERF) TFs have been found to regulate plants and to enable them to be resistant to abiotic stress (Li et al., 2021b). In this study, AP2/ERF TFs were observed that may be involved in the regulation of rose drought stress. One gene encoding ERF109 identified in a blue module appeared to play an important role in roses in resisting drought stress, the function of which requires further study. The study showed that PtrERF109 is positively regulated with Poncirus trifoliata to resist the cold (Wang et al., 2019a). Moreover, DREB2A, DREB3, and DREB1B were found in this study. More and more studies have shown the function of DREBs in resisting abiotic stress in plants. Overexpression of StDREB2 improved the drought resistance of cotton (El-Esawi and Alayafi, 2019). However, RhDREB2B is a negative regulator to resist abiotic stress (Li et al., 2021b).
The function of bHLHs in resisting abiotic stress in Arabidopsis thaliana has also been studied (Liu et al., 2014; Qiu et al., 2020). bHLH162 and bHLH35 have been identified in this study, providing new candidate genes for roses to resist drought stress. Meanwhile, MYBs including MYBS3, MYB75, and MYB78 require further investigation. Particularly for rice, OcMYBS3 is essential for resisting cold stress (Su et al., 2010).
Phytohormone Signals Under Drought Stress
Phytohormone plays a key role in abiotic stress responses and coordinates various signal transduction pathways. In this study, the gene expression involved in several plant hormone-related signal transduction pathways changed significantly after drought treatment, indicating that plant hormones may play a crucial role in the response of the rose to drought stress. ABA acts as an endogenous messenger in a plant’s abiotic stress responses (Teng et al., 2014). Drought results in a substantial increase in plant ABA levels, accompanied by a major changes in gene expression and adaptive physiological responses. NCED is a key rate-limiting enzyme in the ABA synthesis pathway (Schwartz et al., 1997; Iuchi, 2002). Currently, some NCED3 genes have been isolated and identified, corresponding gene function studies have also shown that NCED3 genes play an important role in enhancing plant drought resistance (Li et al., 2019a). The study found that an NCED3 gene was up-regulated by 2.70-fold at DT1. Interestingly, NCED6 down-regulated by 9.40-fold in DT3. Lefebvre et al. (2006) state that AtNCED6 is involved in ABA biosynthesis during seed development. The role of NCED6 in drought stress still requires further study. Another ABA synthesis related to gene aldehyde oxidase (AAO3) and zeaxanthin cyclooxygenase (ZEP) were identified, both of which were significantly down-regulated in DT3. These results suggested that the synthesis of ABA may increase during the early drought stage and decrease during the later drought stage. Therefore, ABA-mediated signal transduction may be involved in the drought response of R. chinensis.
There is more and more evidence suggesting that the phytohormone ethylene is essential for regulating various developmental processes and stress responses of plants (Pei et al., 2017). Mcmichael et al. (1972) reported that the release of ethylene may increase under drought conditions. Studies on pineapples have shown that drought-stressed plants produce significantly less ethylene in leaf and stem tissues compared to control plants (Min and Bartholomew, 2005). These conflicting results may be due to the differences in distinct species. In general, the production of ethylene can be affected by changing ACC biosynthesis under drought stress (Larrainzar et al., 2014). In plants, ACC synthase (ACS) and ACC oxidase (ACO) are the two most essential enzymes in the ethylene biosynthetic pathway (Kende, 1989). In the study, the expression of three ACS genes was suppressed under drought stress, the expression of one ACS gene up-regulated, while the expression of two ACO genes was also up-regulated. It is not yet possible to speculate whether the ethylene content has increased or decreased under drought conditions. Ethylene response factors (ERFs) are downstream components of the ethylene signaling pathway (Guo and Ecker, 2004). Experiments showed that ERFs play important roles in plant abiotic stress response (Pan et al., 2012; Yu et al., 2017). A large number of ERFs exhibit different expressions under different drought levels. Their role under drought stress needs further study.
Jasmonates (JAs), a class of oxygenated lipid derivatives, are phytohormones necessary for plant growth and environmental adaptation (Wasternack and Hause, 2013). MYCs proteins are one of the JASMONATE-ZIM (JAZ) target proteins and a key transcription factor regulating the corresponding genes downstream of JA. According to the reports, MYC2, MYC3, and MYC4 are the central nodes of the JA signal regulation (Chico et al., 2020). Lipoxygenase (LOX), allene oxide synthase (AOS), allene oxide cyclase (AOC), and oxophytodienoate reductase (OPR) are important enzyme genes in the JA synthesis pathway (Devoto and Turner, 2005). The study has observed that RhHB1 binds to the RhLOX4 promoter to suppress its expression in the cut rose (Rosa hybrida). As a result, dehydration tolerance decreases (Fan et al., 2020). These genes were identified, while most genes were up-regulated under drought stress. Other studies have shown that JA was observed to increase in Pinus pinaster (Pedranzani et al., 2007) plants and Oryza sativa (rice) (Du et al., 2013) leaves exposed to drought conditions. Based on these results, it can be conjectured that the JA pathway plays a role in the response to drought stress in R. chinensis.
Moreover, recent studies have indicated that the role of brassinosteroid (BR) in plant response to drought stress (Nolan et al., 2017a, b, 2020). In Fabregas et al.’s (2018) study, the overexpression of BRL3, a vascular-rich member of the BR family, can confer Arabidopsis drought resistance without compromising normal plant growth. In this study, the expression of two BRL genes increased under drought stress, and hence BRLs may play positive parts in the response to drought. In addition, BR and ABA regulated the drought resistance of plants. Indeed, the molecular basis of the antagonistic effect of the BR-ABA pathway has been defined (Wang et al., 2018; Nolan et al., 2020). The role of ABA in promoting the response to drought stress suggests that BRs will inhibit drought stress responses. Yet, only the role of hormone series resistance has been explored in model plants. This study also laid the foundation for the interaction of hormones in the drought resistance of rose.
Signaling Pathway Mediates Drought-Stress Responses
When plants are subjected to adversity stress due to changes in the external environment, they sense adversity stimuli from outside, recognize and transduce between cells and cells, and then transmit them to downstream genes, thereby causing downstream affect gene expression to resist adversity. Calcium ions are the second messengers of the cell, which mediates the calcium signal plays an essential role in the plant response to external stimuli (Kudla et al., 2010; Reddy et al., 2011). As a second messenger, calcium ion can not only maintain the stability of cell structure, but also play a vital role in signal transmission and participate in plant stress response (Xiong et al., 2002; Shinozaki et al., 2003). The three main families of calcium sensors in Ca2+ signaling in plants are calmodulin (CaM), calcineurin B-like (CBL), and calcium-dependent protein kinases (CDPKs) (Ranty et al., 2006; Boudsocq and Sheen, 2013). Analysis of the CaM/CMLs identified in this study revealed that five genes up-regulated, two up-regulated and then down-regulated, and fifteen down-regulated under drought stress. AtCML8, AtCML13, AtCML18 and AtCML25 down-regulated by salt damage and drought (McCormack et al., 2005). ShCML44 up-regulated under cold, drought, and salt damage (Munir et al., 2016). In contrast, the gene encoding CML44 down-regulated and the gene encoding CML8 up-regulated under drought induction in this study. CDPKs/CPKs are involved in multiple stress signaling pathways. In Arabidopsis, Overexpressed AtCPK6 confers drought tolerance (Xu et al., 2010), while AtCPK21 is a negative regulator of osmotic response (Franz et al., 2011). In this study, six genes encoding putative Ca2+-dependent protein kinases up-regulated during drought stress, and four of them showed high expression levels during the DT2 period. These results indicate that the CDPK pathway was significantly activated in DT2. CBLs communicate with downstream CIPKs for signal transmission. The CBL-CIPK pathways also participate in plant abiotic stress response (Xiang et al., 2007). In the current research, three genes encoding CBLs-CIPKs significantly down-regulated in DT3, which indicates that the CBL-CIPK pathway may contribute to regulating a later drought response in R. chinensis. In summary, the above results suggest that Ca2+ mediated signaling pathway has key functions in the response of R. chinensis to drought stress.
Plant’s sense and mediate various intracellular and extracellular signals through cell surface receptor kinases after stimulating by the external environment. Studies have shown that receptor-like protein kinases (RLKs) play a vital role in plant growth and stress adaptation (Wang et al., 2007). Leucine-rich repeat receptor protein kinases (LRR-RLKs), a class of single transmembrane proteins, are the largest family of plant receptor protein kinases (Gou et al., 2010). The LRR-RLKs genes identified by the study were 1 up-regulated and 10 down-regulated under drought stress, which was similar to the expression of LRR-RLKs genes (3 up-regulated, 38 down-regulated) in chrysanthemum under dehydration stress (Xu et al., 2013). Additionally, multiple MAPKs were observed during the plant response to abiotic stresses such as salt, drought, cold, and heat, mainly rapid activation of MPK3, MPK4, and MPK6 (Zhu, 2016). The results in this study are similar, as gene encoding MPK3 significantly down-regulated on DT1, indicating that MPK3 responded quickly when drought stress occurred.
MAPK cascade signals can also transmit reactive oxygen species (ROS) signals to downstream targets. When plants were subjected to environmental stress, ROS generated signals in the plant, triggering a series of changes. Plants also have an antioxidant system protecting against poisonous oxygen (Jaspers and KangasjcTrvi, 2010). Abiotic stress can cause plants to produce excessive ROS which can cause damage to plants. Plants have efficient enzymes including SOD, POD, APX, GPX, and so on. The antioxidant defense mechanisms can protect plants from oxidative stress (Gill and Tuteja, 2010). In this study, two genes encoding APX were up-regulated and four were down-regulated. Similarly, in the study of Citrullus lanatus, the changes of APX genes are different under drought stress (Goitseone et al., 2018). The post-harvest petals of cut rose (Rosa hybrida cv. Samantha) under water deficit stress reported that the Transcription level of RhAPX may be involved in the response of water deficit stress (Jin et al., 2006). Interestingly, three genes encoding SOD were identified, all of which were reduced under drought stress. When SOD activity was measured, the level of SOD activity increased as the degree of drought stress increased. The expression pattern of SOD genes in this study is similar to the expression pattern of the SmFSD2 in drought. The SmFSD2 did not change significantly over 3 days, but decreased to the lowest value at 9 days (Shafi et al., 2019). These results indicate that the ROS signaling pathway may be involved in the regulation of R. chinensis response to drought stress.
Metabolism in Response to Drought Stress in R. chinensis
Primary and secondary metabolites act as signaling molecules or protective agents when plants respond to adversity stress. The primary metabolic processes were involved in the response to the drought stress of R. chinensis, producing a series of osmotic protective agents. Such as sugars, starches, amino acids, and lipids. Accumulation of sugars in various plants is related to high tolerance to drought stress (Ali et al., 2019), which is consistent with the observation of the experiment. Most genes related to carbohydrate synthesis including fructose, glucose, mannose, sucrose, and trehalose were induced and significantly up-regulated under drought stress. The gene encoding trehalose phosphate synthase (TPP1) down-regulated under drought stress. Among the 11 OsTPS genes in the rice (Oryza sativa) genome, only OsTPS1 has TPS activity (Zang et al., 2011). Overexpression of OsTPS1 can improve the tolerance of rice seedlings to drought stress. At the same time, it also causes the expression of some stress-related genes to be up-regulated and the phenotype has no obvious change (Li et al., 2011). Trehalose may involve in the process of rose’s resistance to drought. The expression levels of the four genes encoding starch synthase significantly reduced at DT3, with no significant change in expression levels in the remaining periods compared with the control. It is speculated that the starch content may decrease in a later drought.
Lipids are essential components of cells and organelles. A series of genes that synthesize lipid biosynthesis have been identified in drought treated R. chinensis plants. Long-chain acyl-CoA synthetases (LACS) are indispensable in the pathway of lipids synthesis and degradation of higher plants. There are nine genes in the LACSs family in Arabidopsis, among which LACS1 and LACS2 are involved in the synthesis of wax and cutin together, with an overlap in function (Lü et al., 2009; Weng et al., 2010). Drought stress can cause rapid accumulation of wax on plant surface (Kosma et al., 2009). In this study, six genes encoding LACS2 were induced under drought, while five of them significantly down-regulated at the DT3. Studies on bananas, Agave sisalana (Sarwar et al., 2019), and Poa pratensis (Ni et al., 2016) have reported that waxy synthetic transcripts significantly down-regulated under drought stress. These results confirm that wax may play key roles in regulating the drought resistance of R. chinensis. In brief, it can be speculated that the wax content may be reduced in the later period of drought stress. In addition, the gene encoding GDSL esterase/lipase was significantly induced at the beginning of drought stress. Studies on the GDSL esterase/lipase gene of Arabidopsis thaliana revealed that this gene helps plants resist abiotic stress (Lai et al., 2017), and is induced in M. wufengensis by cold stress (Deng et al., 2019). In summary, lipid metabolism is crucial for R. chinensis adaptation to drought stress.
Furthermore, as part of the adaptation mechanism to the environment of plants, secondary metabolism is sensitive to both biological and abiotic stresses (Hartmann, 2007). It is known that the phenylpropane pathway has high responsiveness to different abiotic stresses such as injury, drought, and low or high temperature. White grapes (Vitis vinifera L.) cope with drought by stimulating the phenylpropane pathway, which reflects the result of this study. The experiment showed that most of the secondary metabolites are derived from phenylpropane biosynthesis and flavonoid biosynthesis pathways. The genes that encode phenylalanine ammonia lyase (PAL) and coumaric acid-CoA ligase (4CL) were identified. They are all related to enzyme genes in the phenylpropane metabolism pathway. They significantly up-regulated at DT1, indicating that the phenylpropane metabolism pathway was induced under drought stress. The transcription of PAL of Caragana korshinskii increased under field and laboratory drought conditions (Liu et al., 2019). These results also confirm the results of the present report. Another major derivative of the phenylpropane pathway is flavonoids, which play a key role in adversity stress. Flavonoids with free radical scavenging activity alleviate oxidative and drought stress in Arabidopsis thaliana (Nakabayashi et al., 2014). This study reported that genes encoding flavonoid synthase (FLS), chalcone isomerase (CHS), and anthocyanin synthase (ANS) were induced under drought. Flavonoid products may affect the regulation of R. chinensis by drought stress.
Model Construction of Rose Drought Response
This study aimed to enhance understanding of the mechanisms through which R. chinensis responds to drought and identify the TFs related to drought tolerance. Figure 10 summarizes the model of R. chinensis drought response. According to the transcriptome data, signal receptors, such as ion channel proteins sense external drought stimuli and transmit through Ca2+, ROS, and phytohormone signaling transduction. The signal transmits protein kinases and protein phosphorylases, which can activate the transcription factors. The activation of TFs triggers downstream drought-responsive gene transcription such as lipid metabolism, carbohydrate metabolism, and secondary metabolism to regulate cell homeostasis. The hub TFs involved in roses’ drought response and rewatering were identified through WGCNA, providing resources for further research on the drought control network of roses. The new findings of this study, on the relationship between unreported TFs and drought resistance regulation, still require further experimental explore.
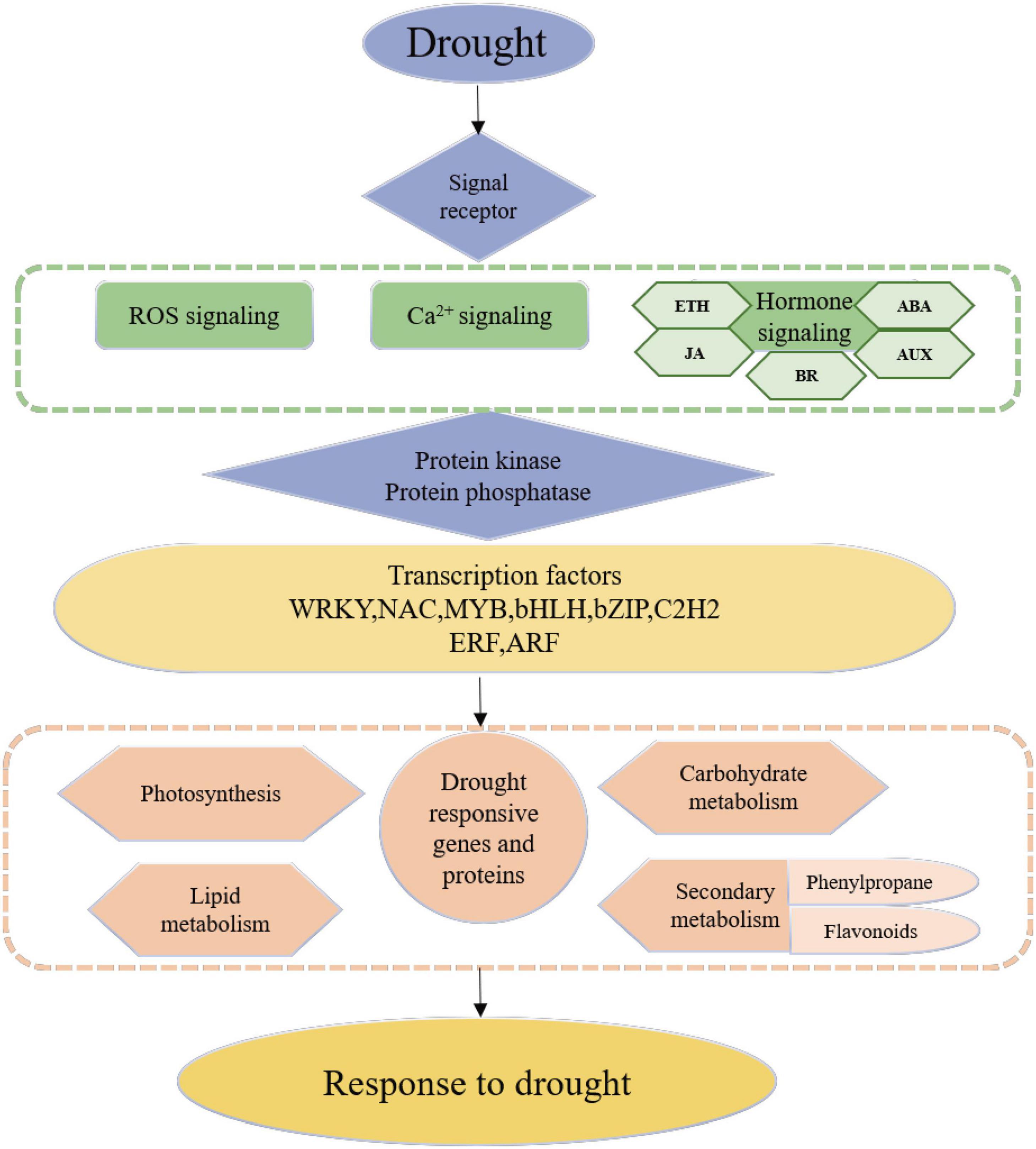
Figure 10. The hypothetical model of Rosa chinensis ‘Old Blush’ in response to drought stress.
Data Availability Statement
The datasets presented in this study can be found in the NCBI repositories (BioProject ID: PRJNA722055).
Author Contributions
YL, SZ, XJ, and HF conceived and designed the research. XJ performed the experiments, analyzed the data, and wrote the manuscript. YB and NJ cultivated and provided the rose plants. All authors have read and agreed to the published version of the manuscript.
Funding
Research was funded by Beijing Municipal Science and Technology Project (z191100008519005) and Chongming District, Shanghai Agricultural Science and Technology Innovation Project (2019CNKC-03).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.690264/full#supplementary-material
Supplementary Figure 1 | The distribution of cleaned RNA-Seq reads mapped to the rose reference genome. (A) Percent of mapped to genome regions of CK groups. (B) Percent of mapped to genome regions of DT1 groups. (C) Percent of mapped to genome regions of DT2 groups. (D) Percent of mapped to genome regions of DT3 groups. (E) Percent of mapped to genome regions of RW groups.
Supplementary Table 1 | Top 20 enriched GO terms of up-regulated DEGs.
Supplementary Table 2 | Top 20 enriched GO terms of down-regulated DEGs.
Supplementary Table 3 | Significantly enriched gene pathways involving DEGs under the drought stress treatment.
Supplementary Table 4 | Protein kinases-related genes that were differentially expressed in response to drought treatment.
Supplementary Table 5 | Calcium-related genes that were differentially expressed in response to drought treatment.
Supplementary Table 6 | ROS producing and scavenging system-related genes that were differentially expressed in response to drought treatment.
Supplementary Table 7 | Differently expressed related to hormone metabolism and signaling in response to drought treatment.
Supplementary Table 8 | Starch and sucrose metabolism-related genes that were differentially expressed in response to drought treatment.
Supplementary Table 9 | Lipid metabolism process-related genes that were differentially expressed in response to drought treatment.
Supplementary Table 10 | Photosynthesis-related genes that were differentially expressed in response to drought treatment.
Supplementary Table 11 | Transcription factor families.
Supplementary Table 12 | The primers sequences of selected genes used in qRT-PCR.
Supplementary Table 13 | Top 20 enriched GO terms of modules.
Supplementary Table 14 | Top 20 enriched KEGG pathway of modules.
References
Adamipour, N., Khosh-Khui, M., Salehi, H., Razi, H., Karami, A., and Moghadam, A. (2020). Metabolic and genes expression analyses involved in proline metabolism of two rose species under drought stress. Plant Physiol. Biochem. 155, 105–113. doi: 10.1016/j.plaphy.2020.07.028
Ahammed, G. J., Li, X., Yang, Y., Liu, C., Zhou, G., Wan, H., et al. (2020). Tomato WRKY81 acts as a negative regulator for drought tolerance by modulating guard cell H2O2-mediated stomatal closure. Environ. Exp. Bot. 171:103960. doi: 10.1016/j.envexpbot.2019.103960
Ali, Q., Shahid, S., Ali, S., Javed, M. T., and Haider, M. Z. (2019). Trehalose Metabolism in Plants under Abiotic Stresses: Profiling and CounterAction. Boca Raton, FL: CRC Press.
Al-Yasi, H., Attia, H., Alamer, K., Hassan, F., Ali, E., Elshazly, S., et al. (2020). Impact of drought on growth, photosynthesis, osmotic adjustment, and cell wall elasticity in Damask rose. Plant Physiol. Biochem. 150, 133–139. doi: 10.1016/j.plaphy.2020.02.038
Ashburner, A., Ball, C. A., Blake, J. A., and Botstein, D. (2000). Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29.
Boudsocq, M., and Sheen, J. (2013). CDPKs in immune and stress signaling. Trends Plant Sci. 18, 30–40. doi: 10.1016/j.tplants.2012.08.008
Chaves, M. M., Maroco, J. P., and Pereira, J. S. (2003). Understanding plant responses to drought - From genes to the whole plant. Funct. Plant Biol. 30, 239–264. doi: 10.1071/fp02076
Chico, J. M., Lechner, E., Fernandez-Barbero, G., Canibano, E., Garcia-Casado, G., Franco-Zorrilla, J. M., et al. (2020). CUL3(BPM) E3 ubiquitin ligases regulate MYC2, MYC3, and MYC4 stability and JA responses. Proc. Natl. Acad. Sci. U S A. 117, 6205–6215. doi: 10.1073/pnas.1912199117
Dai, F., Zhang, C., Jiang, X., Kang, M., Yin, X., Lu, P., et al. (2012). RhNAC2 and RhEXPA4 are involved in the regulation of dehydration tolerance during the expansion of rose petals. Plant Physiol. 160, 2064–2082. doi: 10.1104/pp.112.207720
David Gamboa-Tuz, S., Pereira-Santana, A., Alejandro Zamora-Briseno, J., Castano, E., Espadas-Gil, F., Tonatiuh Ayala-Sumuano, J., et al. (2018). Transcriptomics and co-expression networks reveal tissue-specific responses and regulatory hubs under mild and severe drought in papaya (Carica papaya L.). Sci. Rep. 8:14539. doi: 10.1038/s41598-018-32904-2
Deng, S., Ma, J., Zhang, L., Chen, F., Sang, Z., Jia, Z., et al. (2019). De novo transcriptome sequencing and gene expression profiling of Magnolia wufengensis in response to cold stress. BMC Plant Biol. 19:321. doi: 10.1186/s12870-019-1933-5
Devoto, A., and Turner, J. G. (2005). Jasmonate-regulated Arabidopsis stress signalling network. Physiol. Plant. 123, 161–172. doi: 10.1111/j.1399-3054.2004.00418.x
Dhriti, S., and Ashverya, L. (2015). Transcriptional regulation of drought response: a tortuous network of transcriptional factors. Front. Plant Sci. 6:895. doi: 10.3389/fpls.2015.00895
Ding, A., Li, S., Li, W., Hao, Q., Wan, X., Wang, K., et al. (2019). RhNAC31, a novel rose NAC transcription factor, enhances tolerance to multiple abiotic stresses in Arabidopsis. Acta Physiol. Plantarum 41:75. doi: 10.1007/s11738-019-2866-1
Du, H., Liu, H., and Xiong, L. (2013). Endogenous auxin and jasmonic acid levels are differentially modulated by abiotic stresses in rice. Front. Plant Sci. 4:397. doi: 10.3389/fpls.2013.00397
El-Esawi, M. A., and Alayafi, A. A. (2019). Overexpression of StDREB2 transcription factor enhances drought stress tolerance in cotton (Gossypium barbadense L.). Genes 10:142. doi: 10.3390/genes10020142
Erpen, L., Devi, H. S., Grosser, J. W., and Dutt, D. (2018). Potential use of the DREB/ERF, MYB, NAC and WRKY transcription factors to improve abiotic and biotic stress in transgenic plants. Plant Cell Tiss Org. Cult. 132, 1–25. doi: 10.1007/s11240-017-1320-6
Fabregas, N., Lozano-Elena, F., Blasco-Escamez, D., Tohge, T., Martinez-Andujar, C., Albacete, A., et al. (2018). Overexpression of the vascular brassinosteroid receptor BRL3 confers drought resistance without penalizing plant growth. Nat. Commun. 9:4680. doi: 10.1038/s41467-018-06861-3
Fan, Y., Liu, J., Zou, J., Zhang, X., Jiang, L., Liu, K., et al. (2020). The RhHB1/RhLOX4 module affects the dehydration tolerance of rose flowers (Rosa hybrida) by fine-tuning jasmonic acid levels. Hortic. Res. 7:74. doi: 10.1038/s41438-020-0299-z
Fang, H., Wang, H. L., Li, H. G., Su, Y., and Shuang, L. (2018). PeCHYR1, a ubiquitin E3 ligase from Populus euphratica, enhances drought tolerance via ABA-induced stomatal closure by ROS production in Populus. Plant Biotechnol. J. 16, 1514–1528. doi: 10.1111/pbi.12893
Franco-Zorrilla, J. M., Lopez-Vidriero, I., Carrasco, J. L., Godoy, M., Vera, P., and Solano, R. (2014). DNA-binding specificities of plant transcription factors and their potential to define target genes. Proc. Natl. Acad. Sci. U S A. 111, 2367–2372. doi: 10.1073/pnas.1316278111
Franz, S., Ehlert, B., Liese, A., Kurth, J., Cazale, A. C., and Romeis, T. (2011). Calcium-dependent protein kinase CPK21 functions in abiotic stress response in Arabidopsis thaliana. Mol. Plant 4, 83–96. doi: 10.1093/mp/ssq064
Gadzinowska, J., Ostrowska, A., Hura, K., Dziurka, M., Pawlowska, B., and Hura, T. (2019). Physiological traits determining high adaptation potential of sweet briar (Rosa rubiginosa L.) at early stage of growth to dry lands. Sci. Rep. 9:19390. doi: 10.1038/s41598-019-56060-3
Gill, S. S., and Tuteja, N. (2010). Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 48, 909–930. doi: 10.1016/j.plaphy.2010.08.016
Goitseone, M., Hisashi, T., and Kinya, A. (2018). The cDNA structures and expression profile of the ascorbate peroxidase gene family during drought stress in wild watermelon. J. Agricult. Sci. 10:56. doi: 10.5539/jas.v10n8p56
Gou, X., He, K., Yang, H., Yuan, T., Lin, H., Clouse, S. D., et al. (2010). Genome-wide cloning and sequence analysis of leucine-rich repeat receptor-like protein kinase genes in Arabidopsis thaliana. BMC Genomics 11:19. doi: 10.1186/1471-2164-11-19
Guo, H., and Ecker, J. (2004). The ethylene signaling pathway: new insights. Curr. Opin. Plant Biol. 7, 40–49. doi: 10.1016/j.pbi.2003.11.011
Hartmann, T. (2007). From waste products to ecochemicals: fifty years research of plant secondary metabolism. Phytochemistry 68, 2831–2846. doi: 10.1016/j.phytochem.2007.09.017
Heath, R. L., and Packer, K. (1968). Leaf senescense: correlated with increased levels of membrane permeability and lipid peroxidation, and decreased levels of superoxide dismutase and catalase. J. Exp. Bot. 32, 93–101. doi: 10.1093/jxb/32.1.93
Hong, Y., Zhang, H., Lei, H., Li, D., and Song, F. (2016). Overexpression of a stress-responsive NAC transcription factor gene ONAC022 improves drought and salt tolerance in rice. Front. Plant Sci. 7:4. doi: 10.3389/fpls.2016.00004
Huang, X., Hou, L., Meng, J., You, H., Li, Z., Gong, Z., et al. (2018). The antagonistic action of abscisic acid and cytokinin signaling mediates drought stress response in Arabidopsis. Mol. Plant 11, 970–982. doi: 10.1016/j.molp.2018.05.001
Huang, Z. H., Ma, A. J., Yang, S. S., Liu, X. F., Zhao, T. T., Zhang, J. S., et al. (2020). Transcriptome analysis and weighted gene co-expression network reveals potential genes responses to heat stress in turbot Scophthalmus maximus. Comp. Biochem. Physiol. D-Genom. Proteom. 33:100632. doi: 10.1016/j.cbd.2019.100632
Iuchi, S. (2002). Regulation of drought tolerance by gene manipulation of 9-cis-epoxycarotenoid dioxygenase, a key enzyme in abscisic acid biosynthesis in Arabidopsis (vol 27, pg 325, 2001). Plant J. 30, 611–611.
Jaspers, P., and KangasjcTrvi, J. (2010). Reactive oxygen species in abiotic stress signaling. Physiol. Plant 138, 405–413. doi: 10.1111/j.1399-3054.2009.01321.x
Jiang, J. J., Ma, S. H., Ye, N. H., Cao, J. S., and Zhang, J. (2017). WRKY transcription factors in plant responses to stresses. J. Integr. Plant Biol. 59, 86–101. doi: 10.1111/jipb.12513
Jiang, X., Li, S., Ding, A., Zhang, Z., Hao, Q., Wang, K., et al. (2018). The novel rose MYB transcription factor RhMYB96 enhances salt tolerance in transgenic Arabidopsis. Plant Mol. Biol. Rep. 36, 406–417. doi: 10.1007/s11105-018-1094-y
Jiang, X. Q., Zhang, C. Q., Lu, P. T., Jiang, G. M., Liu, X. W., Dai, F. W., et al. (2014). RhNAC3, a stress- associated NAC transcription factor, has a role in dehydration tolerance through regulating osmotic stress- related genes in rose petals. Plant Biotechnol. J. 12, 38–48. doi: 10.1111/pbi.12114
Jiang, Y., Liang, G., and Yu, D. (2012). Activated expression of WRKY57 confers drought tolerance in Arabidopsis. Mol. Plant 5, 1375–1388. doi: 10.1093/mp/sss080
Jin, J., Shan, N., Ma, N., Bai, J., and Gao, J. (2006). Regulation of ascorbate peroxidase at the transcript level is involved in tolerance to postharvest water deficit stress in the cut rose (Rosa hybrida L.) cv. Samantha. Postharvest Biol. Technol. 40, 236–243. doi: 10.1016/j.postharvbio.2006.01.014
Joshi, R., Wani, S. H., Singh, B., Bohra, A., Dar, Z. A., Lone, A. A., et al. (2016). Transcription factors and plants response to drought stress: current understanding and future directions. Front. Plant Sci. 7:1029. doi: 10.3389/fpls.2016.01029
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Klie, M., and Debener, T. (2011). Identification of superior reference genes for data normalisation of expression studies via quantitative PCR in hybrid roses (Rosa hybrida). BMC Res. Notes 4:518. doi: 10.1186/1756-0500-4-518
Kosma, D. K., Bourdenx, B., Bernard, A., Parsons, E. P., Lu, S., Joubes, J., et al. (2009). The impact of water deficiency on leaf cuticle lipids of Arabidopsis. Plant Physiol. 151, 1918–1929. doi: 10.1104/pp.109.141911
Kudla, J., Batistic, O., and Hashimoto, K. (2010). Calcium signals: the lead currency of plant information processing. Plant Cell 22, 541–563. doi: 10.1105/tpc.109.072686
Lai, C. P., Huang, L. M., Chen, L. F. O., Chan, M. T., and Shaw, J. F. (2017). Genome-wide analysis of GDSL-type esterases/lipases in Arabidopsis. Plant Mol. Biol. 95, 181–197. doi: 10.1007/s11103-017-0648-y
Langfelder, P., and Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559. doi: 10.1186/1471-2105-9-559
Larrainzar, E., Molenaar, J. A., Wienkoop, S., Gil-Quintana, E., Alibert, B., Limami, A. M., et al. (2014). Drought stress provokes the down-regulation of methionine and ethylene biosynthesis pathways in Medicago truncatula roots and nodules. Plant Cell Environ. 37, 2051–2063. doi: 10.1111/pce.12285
Lefebvre, V., North, H., Frey, A., Sotta, B., Seo, M., Okamoto, M., et al. (2006). Functional analysis of Arabidopsis NCED6 and NCED9 genes indicates that ABA synthesized in the endosperm is involved in the induction of seed dormancy. Plant J. 45, 309–319. doi: 10.1111/j.1365-313X.2005.02622.x
Li, H. W., Zang, B. S., Deng, X. W., and Wang, X. P. (2011). Overexpression of the trehalose-6-phosphate synthase gene OsTPS1 enhances abiotic stress tolerance in rice. Planta 234, 1007–1018. doi: 10.1007/s00425-011-1458-0
Li, T., Sun, J., Li, C., Lu, Z., and Xia, J. (2019a). Cloning and expression analysis of the FvNCED3 gene and its promoter from ash (Fraxinus velutina). J. Forestry Res. 30, 471–482. doi: 10.1007/s11676-018-0632-7
Li, Z. Q., Xing, W., Luo, P., Zhang, F. J., Jin, X. L., and Zhang, M. H. (2019b). Comparative transcriptome analysis of Rosa chinensis ‘Slaters’ crimson China’ provides insights into the crucial factors and signaling pathways in heat stress response. Plant Physiol. Biochem. 142, 312–331. doi: 10.1016/j.plaphy.2019.07.002
Li, Z. Y., Chen, W., Zhang, C., Du, C. X., Shao, G. Y., Cui, Y. Y., et al. (2019c). RcMYBPA2 of Rosa chinensis functions in proanthocyanidin biosynthesis and enhances abiotic stress tolerance in transgenic tobacco. Plant Cell Tissue Organ. Culture 137, 441–454. doi: 10.1007/s11240-019-01580-z
Li, W., Fu, L., Geng, Z., Zhao, X., Liu, Q., and Jiang, X. (2021a). Physiological characteristic changes and full-length transcriptome of rose (Rosa chinensis) roots and leaves in response to drought stress. Plant Cell Physiol. 61, 2153–2166. doi: 10.1093/pcp/pcaa137
Li, W., Geng, Z., Zhang, C., Wang, K., and Jiang, X. (2021b). Whole-genome characterization of Rosa chinensis AP2/ERF transcription factors and analysis of negative regulator RcDREB2B in Arabidopsis. BMC Genomics 22:90. doi: 10.1186/s12864-021-07396-6
Li, X., Li, X., Li, M., Yan, Y., Xu, L., and Ling, L. (2016). Dual function of NAC072 in ABF3-mediated ABA-responsive gene regulation in Arabidopsis. Front. Plant Sci. 7:1075. doi: 10.3389/fpls.2016.01075
Li, X., Wan, H., Zhou, G., Cheng, Y., and Ahammed, G. J. (2020). SlWRKY81 reduces drought tolerance by attenuating proline biosynthesis in tomato. Sci. Horticult. 270:109444. doi: 10.1016/j.scienta.2020.109444
Liu, C., Mao, B., Ou, S., Wang, W., Liu, L., Wu, Y., et al. (2018). Correction to: OsbZIP71, a bZIP transcription factor, confers salinity and drought tolerance in rice. Plant Mol. Biol. 97, 467–468. doi: 10.1007/s11103-018-0745-6
Liu, F., Xie, L., Yao, Z., Zhou, Y., and Gong, C. (2019). Caragana korshinskii phenylalanine ammonialyase is up-regulated in the phenylpropanoid biosynthesis pathway in response to drought stress. Biotechnol. Biotechnol. Equipment 33, 842–854. doi: 10.1080/13102818.2019.1623718
Liu, S., Zenda, T., Dong, A., Yang, Y., Wang, N., and Duan, H. (2021). Global transcriptome and weighted gene co-expression network analyses of growth-stage-specific drought stress responses in maize. Front. Genet. 12:645443. doi: 10.3389/fgene.2021.645443
Liu, W., Tai, H., Li, S., Gao, W., Zhao, M., Xie, C., et al. (2014). bHLH122 is important for drought and osmotic stress resistance in Arabidopsis and in the repression of ABA catabolism. New Phytol. 201, 1192–1204. doi: 10.1111/nph.12607
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lu, C. F., Pu, Y., Liu, Y. T., Li, Y. J., Qu, J. P., Huang, H., et al. (2019). Comparative transcriptomics and weighted gene co-expression correlation network analysis (WGCNA) reveal potential regulation mechanism of carotenoid accumulation in Chrysanthemum x morifolium. Plant Physiol. Biochem. 142, 415–428. doi: 10.1016/j.plaphy.2019.07.023
Lü, S., Song, T., Kosma, D. K., Parsons, E. P., Rowland, O., and Jenks, M. A. (2009). Arabidopsis CER8 encodes LONG-CHAIN ACYL-COA SYNTHETASE 1 (LACS1) that has overlapping functions with LACS2 in plant wax and cutin synthesis. Plant J. 59, 553–564. doi: 10.1111/j.1365-313x.2009.03892.x
Mao, X., Chen, S., Li, A., Zhai, C., Jing, R., and Turgay, U. (2014). Novel NAC transcription factor TaNAC67 confers enhanced multi-abiotic stress tolerances in Arabidopsis. PLoS One 9:e84359. doi: 10.1371/journal.pone.0084359
McCormack, E., Tsai, Y. C., and Braam, J. (2005). Handling calcium signaling: Arabidopsis CaMs and CMLs. Trends Plant Sci. 10, 383–389. doi: 10.1016/j.tplants.2005.07.001
Mcmichael, B. L., Jordan, W. R., and Powell, R. D. (1972). An effect of water stress on ethylene production by intact cotton petioles. Plant Physiol. 49, 658–660. doi: 10.1104/pp.49.4.658
Min, X. J., and Bartholomew, D. P. (2005). Effects of flooding and drought on ethylene metabolism, titratable acidity and fruiting of pineapple. Acta Horticulturae 666, 135–148. doi: 10.17660/actahortic.2005.666.13
Mordor Intelligence (2020). Netherlands Floriculture Market-Growth, Trends, COVID-19 Impact, and Forecasts (2021-2026). Hyderabad: Mordor Intelligence.
Munir, S., Liu, H., Xing, Y., Hussain, S., Ouyang, B., Zhang, Y., et al. (2016). Overexpression of calmodulin-like (ShCML44) stress-responsive gene from Solanum habrochaites enhances tolerance to multiple abiotic stresses. Sci. Rep. 6:31772.
Nakabayashi, R., Yonekura-Sakakibara, K., Urano, K., Suzuki, M., Yamada, Y., Nishizawa, T., et al. (2014). Enhancement of oxidative and drought tolerance in Arabidopsis by overaccumulation of antioxidant flavonoids. Plant J. 77, 367–379. doi: 10.1111/tpj.12388
Ni, Y., Guo, N., Zhao, Q., and Guo, Y. (2016). Identification of candidate genes involved in wax deposition in Poa pratensis by RNA-seq. BMC Genom. 17:314. doi: 10.1186/s12864-016-2641-2
Niu, J., Zhang, S., Liu, S., Ma, H., Chen, J., Shen, Q., et al. (2018). The compensation effects of physiology and yield in cotton after drought stress. J. Plant Physiol. 22, 30–48. doi: 10.1016/j.jplph.2018.03.001
Niu, Y., Wang, Y., Li, P., Zhang, F., Liu, H., and Zheng, G. (2013). Drought stress induces oxidative stress and the antioxidant defense system in ascorbate-deficient vtc1 mutants of Arabidopsis thaliana. Acta Physiol. Plantarum 35, 1189–1200. doi: 10.1007/s11738-012-1158-9
Nolan, T., Chen, J., and Yin, Y. (2017a). Cross-talk of brassinosteroid signaling in controlling growth and stress responses. Biochem. J. 474, 2641–2661. doi: 10.1042/BCJ20160633
Nolan, T. M., Brennan, Yang, M. R., Chen, J. N., and Zhang, M. C. (2017b). Selective autophagy of BES1 Mediated by DSK2 balances plant growth and survival. Dev. Cell 41, 33–46.e7.
Nolan, T. M., Vukasinovic, N., Liu, D., Russinova, E., and Yin, Y. (2020). Brassinosteroids: multidimensional regulators of plant growth, development, and stress responses. Plant Cell 32, 295–318. doi: 10.1105/tpc.19.00335
Ogata, H., Goto, S., Sato, K., Fujibuchi, W., Bono, H., and Kanehisa, M. (1999). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27, 29–34.
Pan, Y., Seymour, G. B., Lu, C., Hu, Z., Chen, X., and Chen, G. (2012). An ethylene response factor (ERF5) promoting adaptation to drought and salt tolerance in tomato. Plant Cell Rep. 31, 349–360. doi: 10.1007/s00299-011-1170-3
Pedranzani, H., Sierra-De-Grado, R., Vigliocco, A., Miersch, O., and Abdala, G. (2007). Cold and water stresses produce changes in endogenous jasmonates in two populations of Pinus pinaster Ait. Plant Growth Regulation 52, 111–116. doi: 10.1007/s10725-007-9166-2
Pei, H., Wang, H., Wang, L., Zheng, F., and Dong, C. H. (2017). Regulatory Function of Ethylene in Plant Responses to Drought, Cold, and Salt Stresses. Hoboken, NJ: John Wiley & Sons Inc.
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., and Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protocols 11, 1650–1667. doi: 10.1038/nprot.2016.095
Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T., and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. doi: 10.1038/nbt.3122
Qiu, J. R., Huang, Z., Xiang, X. Y., Xu, W. X., Wang, J. T., Chen, J., et al. (2020). MfbHLH38, a Myrothamnus flabellifolia bHLH transcription factor, confers tolerance to drought and salinity stresses in Arabidopsis. BMC Plant Biol. 20:542. doi: 10.1186/s12870-020-02732-6
Raineri, J., Wang, S., Peleg, Z., Blumwald, E., and Chan, R. L. (2015). The rice transcription factor OsWRKY47 is a positive regulator of the response to water deficit stress. Plant Mol. Biol. 88, 401–413. doi: 10.1007/s11103-015-0329-7
Ranty, B., Aldon, D., and Galaud, J. P. (2006). Plant calmodulins and calmodulin-related proteins: multifaceted relays to decode calcium signals. Plant Signal. Behav. 1, 96–104. doi: 10.4161/psb.1.3.2998
Raymond, O., Gouzy, J., Just, J., Badouin, H., Verdenaud, M., Lemainque, A., et al. (2018). The Rosa genome provides new insights into the domestication of modern roses. Nat. Genet. 50, 772–777. doi: 10.1038/s41588-018-0110-3
Reddy, A. S. N., Ali, G. S., Celesnik, H., and Day, I. S. (2011). Coping with stresses: roles of calcium- and calcium/calmodulin-regulated gene expression. Plant Cell 23, 2010–2032. doi: 10.1105/tpc.111.084988
Ren, X., Chen, Z., Liu, Y., Zhang, H., Zhang, M., Liu, Q., et al. (2010). ABO3, a WRKY transcription factor, mediates plant responses to abscisic acid and drought tolerance in Arabidopsis. Plant J. Cell Mol. Biol. 63, 417–429. doi: 10.1111/j.1365-313x.2010.04248.x
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Sarwar, M. B., Ahmad, Z., Rashid, B., Hassan, S., Gregersen, P. L., and Leyva, et al. (2019). De novo assembly of Agave sisalana transcriptome in response to drought stress provides insight into the tolerance mechanisms. Sci. Rep. 9:396.
Schwartz, S. H., Tan, B. C., Gage, D. A., Zeevaart, J. A., and McCarty, D. R. (1997). Specific oxidative cleavage of carotenoids by VP14 of maize. Science 276, 1872–1874. doi: 10.1126/science.276.5320.1872
Shafi, A., Gill, T., Zahoor, I., Ahuja, P. S., Sreenivasulu, Y., Kumar, S., et al. (2019). Ectopic expression of SOD and APX genes in Arabidopsis alters metabolic pools and genes related to secondary cell wall cellulose biosynthesis and improve salt tolerance. Mol. Biol. Rep. 46, 1985–2002. doi: 10.1007/s11033-019-04648-3
Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Shao, A., Wang, W., Fan, S., Xu, X., Yin, Y., Erick, A., et al. (2021). Comprehensive transcriptional analysis reveals salt stress-regulated key pathways, hub genes and time-specific responsive gene categories in common bermudagrass (Cynodon dactylon (L.) Pers.) roots. BMC Plant Biol. 21:175. doi: 10.1186/s12870-021-02939-1
Shinozaki, K., Yamaguchi-Shinozaki, K., and Seki, M. (2003). Regulatory network of gene expression in the drought and cold stress responses. Curr. Opin. Plant Biol. 6:410. doi: 10.1016/s1369-5266(03)00092-x
Silva, K., Singh, J., Bednarek, R., Fei, Z., and Khan, A. (2019). Differential gene regulatory pathways and co-expression networks associated with fire blight infection in apple (Malus × domestica). Horticulture Res. 6:35.
Su, C.-F., Wang, Y.-C., Hsieh, T.-H., Lu, C.-A., Tseng, T.-H., and Yu, S.-M. (2010). A Novel MYBS3-Dependent Pathway Confers Cold Tolerance in Rice. Plant Physiol. 153, 145–158. doi: 10.1104/pp.110.153015
Sung, S. J., Nuri, O., Joong, C. P., Shic, K. Y., Do, C. Y., and Ju-Kon, K. (2018). Overexpression of OsNAC14 improves drought tolerance in rice. Front. Plant Sci. 9:310. doi: 10.3389/fpls.2018.00310
Teng, K., Li, J., Liu, L., Han, Y., Du, Y., Zhang, J., et al. (2014). Exogenous ABA induces drought tolerance in upland rice: the role of chloroplast and ABA biosynthesis-related gene expression on photosystem II during PEG stress. Acta Physiologiae Plantarum 36, 2219–2227. doi: 10.1007/s11738-014-1599-4
Tran, L. S., Nakashima, K., Sakuma, Y., Simpson, S. D., Fujita, Y., Maruyama, K., et al. (2004). Isolation and functional analysis of Arabidopsis stress-inducible NAC transcription factors that bind to a drought-responsive cis-element in the early responsive to dehydration stress 1 promoter. Plant Cell 16, 2481–2498. doi: 10.1105/tpc.104.022699
Valliyodan, B., and Nguyen, H. T. (2006). Understanding regulatory networks and engineering for enhanced drought tolerance in plants. Curr. Opin. Plant Biol. 9, 189–195. doi: 10.1016/j.pbi.2006.01.019
Wang, H., Chevalier, D., Larue, C., Ki, S., and John, C. (2007). The protein phosphatases and protein kinases of Arabidopsis thaliana. Arabidopsis Book 5:e0106.
Wang, H., Tang, J., Liu, J., Hu, J., Liu, J., Chen, Y., et al. (2018). Abscisic acid signaling inhibits brassinosteroid signaling through dampening the dephosphorylation of BIN2 by ABI1 and ABI2. Mol. Plant 11, 315–325. doi: 10.1016/j.molp.2017.12.013
Wang, M., Dai, W. S., Du, J., Ming, R. H., Dahro, B., and Liu, J. H. (2019a). ERF109 of trifoliate orange (Poncirus trifoliata (L.) Raf.) contributes to cold tolerance by directly regulating expression of Prx1 involved in antioxidative process. Plant Biotechnol. J. 17, 1316–1332. doi: 10.1111/pbi.13056
Wang, N.-N., Xu, S.-W., Sun, Y.-L., Liu, D., Zhou, L., Li, Y., et al. (2019b). The cotton WRKY transcription factor (GhWRKY33) reduces transgenic Arabidopsis resistance to drought stress. Sci. Rep. 9:724.
Wasternack, C., and Hause, B. (2013). Jasmonates: biosynthesis, perception, signal transduction and action in plant stress response, growth and development. an update to the 2007 review in annals of botany. Ann. Bot. 11, 1021–1058. doi: 10.1093/aob/mct067
Wei, W., Liang, D. W., Bian, X. H., Shen, M., Xiao, J. H., and Zhang, W. K. (2019). Gm WRKY 54 improves drought tolerance through activating genes in ABA and Ca 2+ signaling pathways in transgenic soybean. Plant J. 100, 384–398. doi: 10.1111/tpj.14449
Weng, H., Molina, I., Shockey, J., and Browse, J. (2010). Organ fusion and defective cuticle function in a lacs1lacs2 double mutant of Arabidopsis. Planta 231, 1089–1100. doi: 10.1007/s00425-010-1110-4
Xiang, Y., Huang, Y., and Xiong, L. (2007). Characterization of stress-responsive CIPK genes in rice for stress tolerance improvement. Plant Physiol. 144, 1416–1428. doi: 10.1104/pp.107.101295
Xiong, L., Schumaker, K. S., and Zhu, J. K. (2002). Cell signaling during cold. Drought, and Salt Stress. Plant Cell 14(Suppl.), S165–S183.
Xu, J., Tian, Y. S., Peng, R. H., Xiong, A. S., Zhu, B., Jin, X. F., et al. (2010). AtCPK6, a functionally redundant and positive regulator involved in salt/drought stress tolerance in Arabidopsis. Planta 231, 1251–1260. doi: 10.1007/s00425-010-1122-0
Xu, Y. J., Gao, S., Yang, Y. J., Huang, M. Y., Cheng, L. N., Wei, Q., et al. (2013). Transcriptome sequencing and whole genome expression profiling of chrysanthemum under dehydration stress. BMC Genom. 14:662. doi: 10.1186/1471-2164-14-662
Yu, Y., Yang, D., Zhou, S., Gu, J., Wang, F., Dong, J., et al. (2017). The ethylene response factor OsERF109 negatively affects ethylene biosynthesis and drought tolerance in rice. Protoplasma 254, 401–408. doi: 10.1007/s00709-016-0960-4
Zandalinas, S. I., Damián, B., Vicent, A., and Aurelio, G. C. (2017). Modulation of antioxidant defense system is associated with combined drought and heat stress tolerance in citrus. Front. Plant Sci. 8:953. doi: 10.3389/fpls.2017.00953
Zang, B., Li, H., Li, W., Deng, X. W., and Wang, X. (2011). Analysis of trehalose-6-phosphate synthase (TPS) gene family suggests the formation of TPS complexes in rice. Plant Mol. Biol. 76, 507–522. doi: 10.1007/s11103-011-9781-1
Zhang, B., and Horvath, S. (2005). A general framework for weighted gene co-expression network analysis. Statist. Appl. Genet. Mol. Biol. 4:Article17.
Zhang, L., Cheng, J., Sun, X., Zhao, T., Li, M., Wang, Q., et al. (2018). Overexpression of VaWRKY14 increases drought tolerance in Arabidopsis by modulating the expression of stress-related genes. Plant Cell Rep. 37, 1159–1172. doi: 10.1007/s00299-018-2302-9
Zhang, L., Zhang, L., Xia, C., Zhao, G., Jia, J., and Kong, X. (2015). The novel wheat transcription factor TaNAC47 enhances multiple abiotic stress tolerances in transgenic plants. Front. Plant Sci. 6:1174. doi: 10.3389/fpls.2015.01174
Zhang, X., Lu, S. C., Jiang, C. H., Wang, Y. F., Lv, B., Shen, J. B., et al. (2014). RcLEA, a late embryogenesis abundant protein gene isolated from Rosa chinensis, confers tolerance to Escherichia coli and Arabidopsis thaliana and stabilizes enzyme activity under diverse stresses. Plant Mol. Biol. 85, 333–347. doi: 10.1007/s11103-014-0192-y
Zhao, D. Q., Zhang, X. Y., Fang, Z. W., Wu, Y. Q., and Tao, J. (2019a). Physiological and transcriptomic analysis of tree peony (Paeonia section Moutan DC.) in response to drought stress. Forests 10:135. doi: 10.3390/f10020135
Zhao, K., Zhang, D., Lv, K., Zhang, X., and Jiang, T. (2019b). Functional characterization of poplar WRKY75 in salt and osmotic tolerance. Plant Sci. 289:110259. doi: 10.1016/j.plantsci.2019.110259
Zhao, Y., Yu, W., Hu, X., Shi, Y., Liu, Y., Zhong, Y., et al. (2018). Physiological and transcriptomic analysis revealed the involvement of crucial factors in heat stress response of Rhododendron hainanense. Gene 660, 109–119. doi: 10.1016/j.gene.2018.03.082
Zhou, A., Ma, H., Liu, E., Jiang, T., Shuang, F., Gong, S., et al. (2017). Transcriptome sequencing of dianthus spiculifolius and analysis of the genes involved in responses to combined cold and drought stress. Int. J. Mol. Sci. 18:849. doi: 10.3390/ijms18040849
Zhou, R., Yu, X., Zhao, T., Ottosen, C. O., Rosenqvist, E., and Wu, Z. (2019). Physiological analysis and transcriptome sequencing reveal the effects of combined cold and drought on tomato leaf. BMC Plant Biol. 19:377. doi: 10.1186/s12870-019-1982-9
Zhu, J. K. (2002). Salt and drought stress signal transduction in plants. Annu. Rev. Plant Biol. 53, 247–273. doi: 10.1146/annurev.arplant.53.091401.143329
Keywords: drought stress, comparative transcriptome analysis, weighted gene co-expression network analysis, rose, transcription factors
Citation: Jia X, Feng H, Bu Y, Ji N, Lyu Y and Zhao S (2021) Comparative Transcriptome and Weighted Gene Co-expression Network Analysis Identify Key Transcription Factors of Rosa chinensis ‘Old Blush’ After Exposure to a Gradual Drought Stress Followed by Recovery. Front. Genet. 12:690264. doi: 10.3389/fgene.2021.690264
Received: 02 April 2021; Accepted: 24 May 2021;
Published: 15 July 2021.
Edited by:
Shang-Qian Xie, Hainan University, ChinaReviewed by:
Jingang Wang, Northeast Agricultural University, ChinaWaltram Ravelombola, Texas A&M University, United States
Copyright © 2021 Jia, Feng, Bu, Ji, Lyu and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yingmin Lyu, bHV5aW5nbWluQGJqZnUuZWR1LmNu; Shiwei Zhao, emhhb3NoaXdlaUBiZWlqaW5nYmcuY29t