 Sandra Hoffmann1,2
Sandra Hoffmann1,2 Ralph Roeth1,3Sabrina Diebold4Jasmin Gogel1David Hassel5
Ralph Roeth1,3Sabrina Diebold4Jasmin Gogel1David Hassel5 Steffen Just4
Steffen Just4 Gudrun A. Rappold1,2*
Gudrun A. Rappold1,2*- 1Department of Human Molecular Genetics, Institute of Human Genetics, University Hospital Heidelberg, Heidelberg, Germany
- 2DZHK (German Centre for Cardiovascular Research), Partner Site Heidelberg/Mannheim, Heidelberg, Germany
- 3nCounter Core Facility, Institute of Human Genetics, University of Heidelberg, Heidelberg, Germany
- 4Clinic for Internal Medicine II - Molecular Cardiology, University Hospital Ulm, Ulm, Germany
- 5Department of Internal Medicine III ‐ Cardiology, University Hospital Heidelberg, Heidelberg, Germany
SHOX deficiency causes a spectrum of clinical phenotypes related to skeletal dysplasia and short stature, including Léri-Weill dyschondrosteosis, Langer mesomelic dysplasia, Turner syndrome, and idiopathic short stature. SHOX controls chondrocyte proliferation and differentiation, bone maturation, and cellular growth arrest and apoptosis via transcriptional regulation of its direct target genes NPPB, FGFR3, and CTGF. However, our understanding of SHOX-related pathways is still incomplete. To elucidate the underlying molecular mechanisms and to better understand the broad phenotypic spectrum of SHOX deficiency, we aimed to identify novel SHOX targets. We analyzed differentially expressed genes in SHOX-overexpressing human fibroblasts (NHDF), and confirmed the known SHOX target genes NPPB and FGFR among the most strongly regulated genes, together with 143 novel candidates. Altogether, 23 genes were selected for further validation, first by whole-body characterization in developing shox-deficient zebrafish embryos, followed by tissue-specific expression analysis in three shox-expressing zebrafish tissues: head (including brain, pharyngeal arches, eye, and olfactory epithelium), heart, and pectoral fins. Most genes were physiologically relevant in the pectoral fins, while only few genes were also significantly regulated in head and heart tissue. Interestingly, multiple sox family members (sox5, sox6, sox8, and sox18) were significantly dysregulated in shox-deficient pectoral fins together with other genes (nppa, nppc, cdkn1a, cdkn1ca, cyp26b1, and cy26c1), highlighting an important role for these genes in shox-related growth disorders. Network-based analysis integrating data from the Ingenuity pathways revealed that most of these genes act in a common network. Our results provide novel insights into the genetic pathways and molecular events leading to the clinical manifestation of SHOX deficiency.
Introduction
Linear growth and skeletal development are highly dynamic processes that depend on transcription factors, signaling molecules, and extracellular matrix proteins. Long bones grow by endochondral ossification, a sequential replacement of cartilaginous tissue by bone. The growth plate, located near the ends of the long bones, provides a continuous supply of chondrocytes for endochondral ossification, which is initiated during early fetal development and continues until adolescence (Kronenberg, 2003; Lui et al., 2018). Any failure in this process causes a wide range of skeletal disorders. SHOX is one of several critical factors regulating chondrogenesis in the growth plate (Marchini et al., 2016).
Alterations in SHOX expression impairs human growth and causes a spectrum of clinical phenotypes related to skeletal dysplasia and short stature including Léri-Weill dyschondrosteosis, Langer mesomelic dysplasia, Turner syndrome, and idiopathic short and tall stature (Rao et al., 1997; Belin et al., 1998; Shears et al., 1998; Oliveira and Alves, 2011; Fukami et al., 2016; Marchini et al., 2016; Monzani et al., 2019). However, no genotype-phenotype correlation exists in individuals with SHOX mutation/deletion within the coding region (Binder et al., 2004), and also downstream enhancer deletions do not seem to greatly differ from those with coding mutations/deletions with respect to phenotypic characteristics and severity (Benito-Sanz et al., 2005; Rosilio et al., 2012). Although the penetrance of SHOX deficiency is high, its clinical expression is variable, even among family members (Schiller et al., 2000; Binder, 2011). CYP26C1 has been identified as a modifier of severity in patients with SHOX deficiency, but certainly others will follow that contribute to the variable expressivity (Montalbano et al., 2016).
SHOX is a transcriptional regulator in chondrocyte proliferation and differentiation, bone maturation, cartilage synthesis, and cellular growth arrest and apoptosis via its direct target genes NPPB, FGFR3, and CTGF (Marchini et al., 2004, 2007; Decker et al., 2011; Beiser et al., 2014; Hristov et al., 2014). It also promotes linear growth at the growth plate via interaction with the SOX trio (SOX5, SOX6, and SOX9; Aza-Carmona et al., 2011). Further characterization of SHOX-dependent networks is critical, not only for elucidating SHOX functions and their link to disease, but also for discovering novel therapeutic targets in SHOX-related disorders.
To better understand the disease mechanisms underlying SHOX deficiency, we first performed genome-wide expression profiling in SHOX-overexpressing human fibroblasts to uncover novel SHOX target genes. Considering the complexity of the growth plate and the spatiotemporal expression of SHOX during bone development, we aimed to validate the physiological relevance of the identified genes in an animal model.
A SHOX ortholog is not present in rodents, so zebrafish has been a valuable model for studying the role of SHOX during development. Zebrafish embryos are excellent models for studying bone physiology and pathology (Carnovali et al., 2019) because they are small, have rapid external development, have a transparent larval body, and can easily be manipulated genetically. Another important consideration is that a shox ortholog and conserved non-coding elements (CNEs) are present in the zebrafish genome (Kenyon et al., 2011; Verdin et al., 2015). Several studies have already confirmed that shox deficiency impairs early embryonic growth and bone formation in zebrafish and that shox morphants have a shorter body length and reduced pectoral fin size (Sawada et al., 2015; Montalbano et al., 2016; Yokokura et al., 2017). Therefore, we used zebrafish as a read-out animal model to validate our putative target genes and confirm their physiological relevance. Our study highlights the importance of various gene families in shox-related growth disorders including the cyclin-dependent kinase inhibitors (CKI), cytochrome P450 26 subfamily (CYP26), natriuretic peptides (NP), and SRY-related HMG-box (SOX) genes.
Materials and Methods
Cell Culture
Normal human dermal fibroblasts (NHDF cells; PromoCell: C12300) were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Gibco, Thermo Fisher Scientific), supplemented with 10% fetal bovine serum, penicillin (100 U/ml), and streptomycin (100 μg/ml) in a 5% CO2-humidified incubator at 37°C. 1 × 106 NHDF cells were transfected with 3.5 μg pcDNA4/TO-SHOX-WT using the Amaxa® Human Dermal Fibroblasts Nucleofector® Kit (Lonza) according to the manufacturer’s instructions. In control experiments, NHDF were transfected with 3.5 μg pcDNA4/TO-SHOX-HM, which encodes a homeodomain-mutant of SHOX leading to the amino acid exchange Y141D.
Microarray Analysis
Gene expression profiling was performed using the Sentrix® HumanRef-8 Expression BeadChips from Illumina according to the manufacturer’s protocol. Total RNA from NHDF cells was isolated using Illustra RNAspin Isolation Kit (Cytiva) according to the manufacturer’s instructions. For array hybridization, we prepared biotin-labeled cRNA from 250 μg total RNA according to Illumina’s recommended sample labeling procedure using the MessageAmp™ II aRNA Amplification Kit (Thermo Fisher Scientific). Hybridization was performed at 58°C in GEX-HCB buffer (Illumina) at a concentration of 100 ng cRNA/μl for 20 h. Microarrays were washed twice in E1BC buffer (Illumina) at room temperature for 5 min. After blocking for 5 min in 1% Blocker Casein in PBS, array signals were developed by a 10 min incubation in 1 μg/ml Cy3-streptavidin (Cytiva) solution and 1% blocking solution. After final wash in E1BC, the arrays were dried and scanned using a Beadstation array scanner. Data extraction was done for all beads individually, and outliers were removed if >2.5 median absolute deviation (MAD). All remaining data points were used for the calculation of the mean average signal and SD for each probe. For each analyzed time point, we compared signals of normalized gene expression of SHOX-WT transfected samples to SHOX-HM transfected controls. Differentially expressed genes were defined by a change in expression of more than 3-fold (SHOX-WT/SHOX-HM).
Zebrafish Embryos and Microinjections
Zebrafish (Danio rerio) were maintained as previously described (Westerfield, 2000). For all morpholino injection procedures, the Tg (myl7:GFP) strain was used (Rottbauer et al., 2006). Morpholino-modified antisense oligonucleotides (MO; Gene Tools) were directed against the exon 2 intron 2 junction in shox, causing reduction of pectoral fins as described previously (Montalbano et al., 2016). Shox antisense oligonucleotides or a standard control oligonucleotide (MO control), diluted in 0.2 M KCl, were microinjected into one-cell-stage zebrafish embryos (Just et al., 2011).
nCounter Expression Analysis
Zebrafish tissues were isolated at 55 hours post-fertilization (hpf) and total RNA was extracted with the Direct-zol RNA Microprep Kit (Zymo Research) according to the manufacturer’s instructions. For each experiment, whole zebrafish embryos (10–15), hearts (30–70), heads (~20), or pectoral fins (30–40) were pooled per condition to obtain 50 ng of input material. Three to five independent experiments (n) were performed. mRNA expression levels were measured at the nCounter Core Facility Heidelberg using the nCounter SPRINT Profiler. This RNA quantification technology utilizes a direct digital detection of mRNA molecules and allows multiplexed target measurement in a single reaction with high sensitivity and specificity even with low amounts of input material (50 ng). A detailed probe design is given in Supplementary Table 1. The workflow is described at http://www.nanostring.com/elements/workflow. Background correction and data normalization were performed using the nSolver Analysis Software 4.0 (NanoString Technologies). The most stably expressed genes were chosen for normalization based on the geNorm method.
Statistical Analyses
Statistical analysis was carried out using GraphPad Prism 9 (GraphPad Software, La Jolla, CA, United States). For multiple t-tests, the two-stage step-up method of Benjamini, Krieger, and Yekutieli with a desired false discovery rate (FDR) of 5% was used to correct the values of p. All conditions statistically different from the control were indicated by *p < 0.05, **p < 0.01, and ***p < 0.001.
Ingenuity Pathway Analysis
Ingenuity pathway analysis (IPA) is a web-based software application for the evaluation, integration, and interpretation of omics data (Kramer et al., 2014; QIAGEN Inc.).1 IPA analysis indicates relationships and interactions between genes based on published data. A list of 24 selected genes (including SHOX) was uploaded to perform a pathway and core analysis. The core analysis comprises enrichment pathway analysis to identify significant biological processes and molecular functions in which these genes are involved. These include, for example, differentiation and development of chondrocytes, cartilage and connective tissue, as well as growth failure, short stature, and limb defects.
Databases
DisGeNET database (Pinero et al., 2015).2 This database enables browsing, searching, and analyzing of genetic information linked to human diseases based on expert curated repositories, GWAS catalogues, animal models, and the scientific literature. DisGeNET is a discovery platform containing one of the largest publicly available collections of genes and variants associated to human diseases. The current version of DisGeNET (v7.0) contains 1,134,942 gene-disease associations, between 21,671 genes and 30,170 diseases, disorders, traits, and clinical or abnormal human phenotypes, and 369,554 variant-disease associations, between 194,515 variants and 14,155 diseases, traits, and phenotypes.
Results
SHOX expression is low in tissues and cell lines (Durand et al., 2011) and only detectable at a very low level in mammalian organs during development (Supplementary Figure 1). The highest endogenous expression of SHOX so far was found in primary human fibroblasts (NHDF; Durand et al., 2011) and zebrafish embryos (Sawada et al., 2015; Montalbano et al., 2016). But even in fibroblasts, expression levels are too low to perform reliable knockdown experiments.
To investigate SHOX regulatory pathways and identify novel SHOX targets, we analyzed transcriptional changes following overexpression of SHOX wild type (WT) compared with a functional homeodomain mutant (HM) in NHDF fibroblast cells. SHOX-HM was previously detected in a patient with short stature carrying a c.421T>G missense mutation that led to an amino acid exchange (p.Y141D) in the homeodomain, which impairs SHOX function by reducing DNA-binding and transactivation capacity (Schneider et al., 2005). Therefore, this variant is an ideal negative control for differential expression analysis.
Gene expression profiling was carried out in NHDF cells at three different stages: 6, 12, and 24 h after transfection (Figure 1A). Whole-genome Illumina HumanRef8 expression beadchips (Human Sentrix-8 V2) were used to measure gene expression at each time point. Normalized expression of SHOX-WT was compared with normalized expression of SHOX-HM in the cells and displayed as ratios (Supplementary Tables 2–4). Microarray data of all three time points were then merged to analyze all differentially expressed genes (Supplementary Table 5). Out of 24,500 annotated transcripts targeted by the Illumina expression beadchips, 145 genes were differentially expressed with a more than 3-fold change. Forty-eight genes were downregulated and 97 genes were upregulated upon SHOX overexpression. The top 10 upregulated genes at all three time points are shown in Figure 1B. At the earliest time point (6 h), only one gene (SOX8) was significantly upregulated after SHOX overexpression based on a threshold 3-fold change in expression. At 12 and 24 h after transfection, the top 10 differentially expressed genes were all upregulated more than 3-fold (Figure 1B).
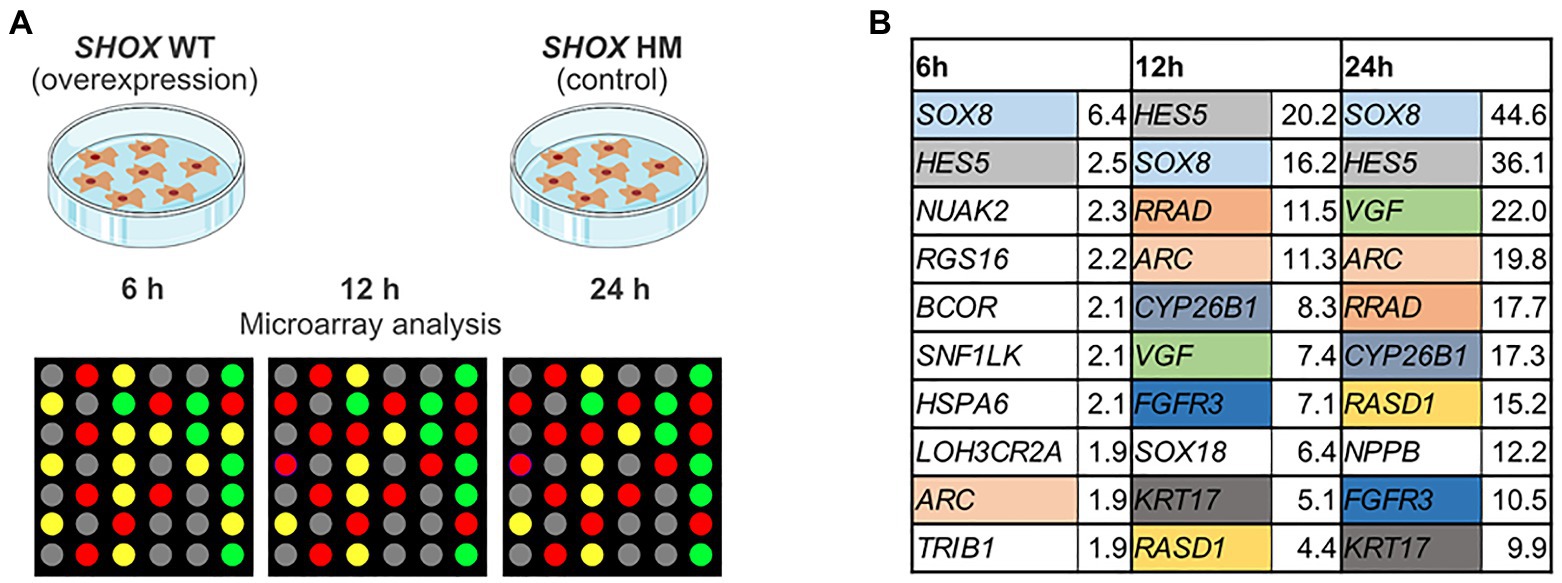
Figure 1. Microarray analysis of SHOX overexpressing wild type (WT) and mutant (HM) normal human dermal fibroblast (NHDF) cells. (A) Overview of experimental design. (B) Top 10 regulated genes in SHOX overexpressing NHDF cells after 6, 12, and 24 h. Identical colors indicate the same genes at different time points. Relative expression ratios are indicated next to the gene’s name.
We identified two known SHOX target genes in the top 10 list – NPPB (Marchini et al., 2007) and FGFR3 (Decker et al., 2011). In addition, we found novel target genes from families already associated with SHOX (Supplementary Table 6). These included SOX8 and SOX18 of the SOX gene family and CYP26B1 of the CYP26 gene family. SOX18, which was in the top 10 list upon 12 h of SHOX expression, fell to position 11 after 24 h despite a 9-fold upregulation. NPPB only appeared in the top 10 list after 24 h of SHOX expression. To further investigate family-based regulations, we selected further gene members (n = 4) of these families for validation, as well as eight genes that were previously associated with SHOX in the literature. The 11 genes from our list of top-regulated genes (Figure 1B) and the additional 12 genes from the literature are summarized in Table 1.
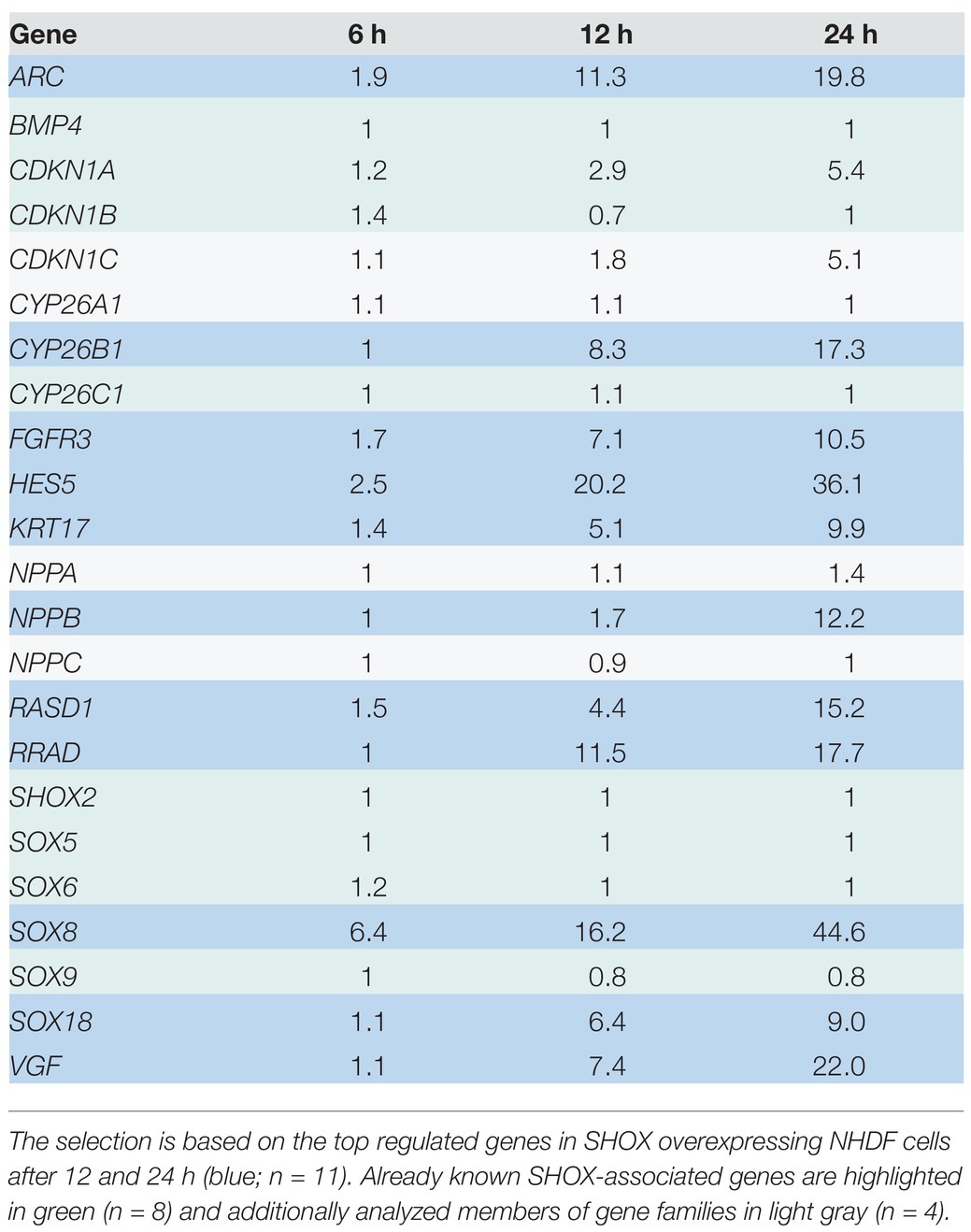
Table 1. Genes selected for validation.
We used zebrafish embryos as an in vivo model to verify our selected putative SHOX-regulated genes. The expression of the zebrafish orthologs (n = 22; ARC does not have an ortholog) was analyzed at 55 hpf after morpholino-mediated knockdown of shox. Differential expression was found for 5/22 analyzed genes (shox2, her15.1, cdkn1a, rasd1, and cyp26c1) after shox knockdown using RNA isolated from whole zebrafish embryos (Figure 2A). Knockdown efficiency was only nominally significant for shox (p = 0.0284; q = 0.0635). To provide a possible explanation for this, we addressed its tissue-specific regulations. Thus, we performed analyses in shox-expressing zebrafish tissues from three different regions: pectoral fins, head (brain, pharyngeal arches, eye, and olfactory epithelium), and heart of shox-deficient and control embryos at 55 hpf (Figures 2B–D).
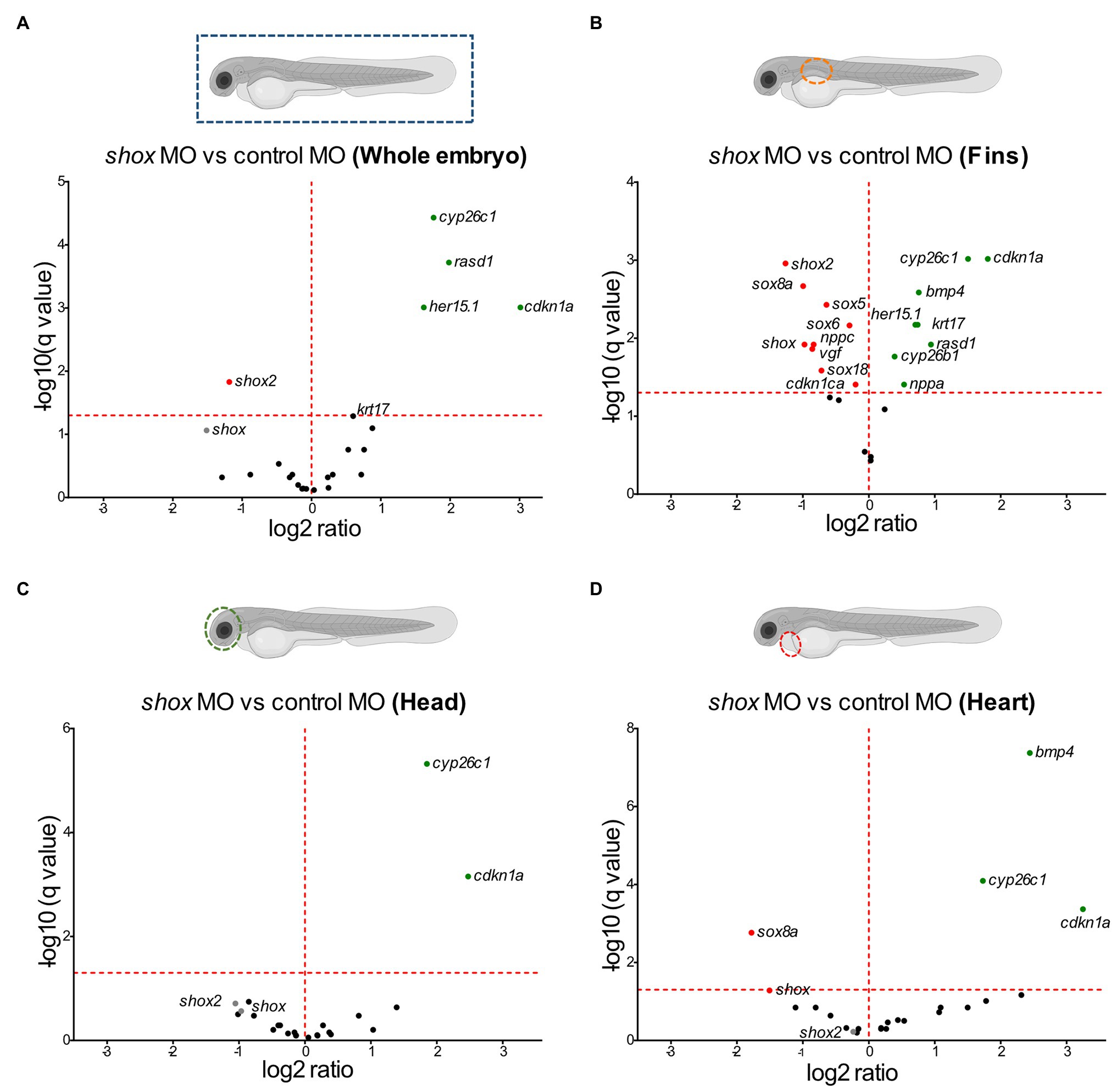
Figure 2. Tissue-specific validation of putative Shox target genes in zebrafish embryos after shox knockdown. Volcano plot presentations of expression ratio of selected shox target genes in (A) whole zebrafish tissue (n = 6 shox MO, n = 6 control MO), (B) fins (n = 3 shox MO, n = 3 control MO), (C) head (n = 4 shox MO, n = 4 control MO), and (D) heart (n = 4 shox MO, n = 5 control MO) at 55 hours post-fertilization (hpf) after morpholino-mediated knockdown of shox compared to controls. Genes above the red horizontal dashed line are significantly regulated. Genes shown to the left of the dashed vertical line are downregulated, those on the right are upregulated. Statistical significance was determined by multiple unpaired t-tests adjusted by the two-stage step-up method of Benjamini, Krieger, and Yekutieli (adjusted values of p = q-values). MO, morpholino.
After shox knockdown, shox was significantly downregulated in the pectoral fin (p = 0.0149; q = 0.0118) but not in the head (p = 0.0648; q = 0.2381), and nominally significant downregulation was detected in the heart (p = 0.0142; q = 0.0566) as measured by nCounter analysis (Figures 2B–D). Almost all analyzed genes (16/22) were significantly dysregulated in the pectoral fins at the developmental time point 55 hpf (Figure 3). Of the top 10 regulated genes in NHDF cells, seven were dysregulated in zebrafish fins (cyp26b1, her15.1, krt17, rasd1, sox8a, sox18, and vgf), although the regulatory direction was not always the same as in NHDF cells. Interestingly, members of the sox and cki family were significantly dysregulated in the pectoral fin (Figure 4), highlighting the important role of these gene families in Shox-related growth defects.
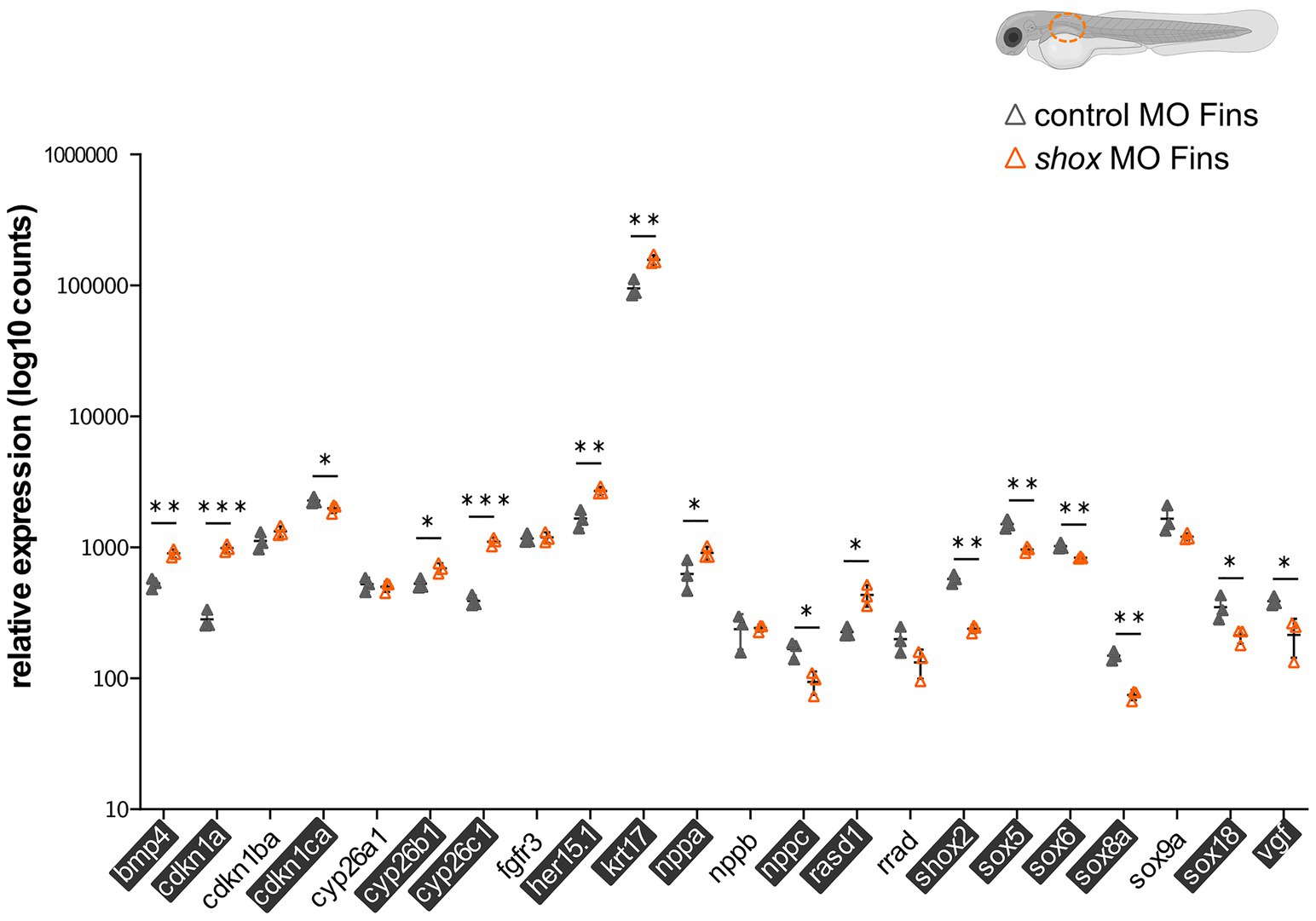
Figure 3. Validation of putative Shox target genes in zebrafish fins. Tissue-specific analysis of all selected shox targets upon morpholino-mediated shox knockdown in zebrafish fins 55 hpf (orange) compared to controls (gray); n = 3 experiments per condition. Significantly regulated genes are highlighted. For ARC, no Zebrafish otholog exists. HES5 corresponds to her15.1 in zebrafish. Statistical significance was determined by multiple unpaired t-tests adjusted by the two-stage step-up method of Benjamini, Krieger, and Yekutieli (adjusted values of p are indicated by *p < 0.05; **p < 0.01; and ***p < 0.001). MO, morpholino; hpf, hours post-fertilization.
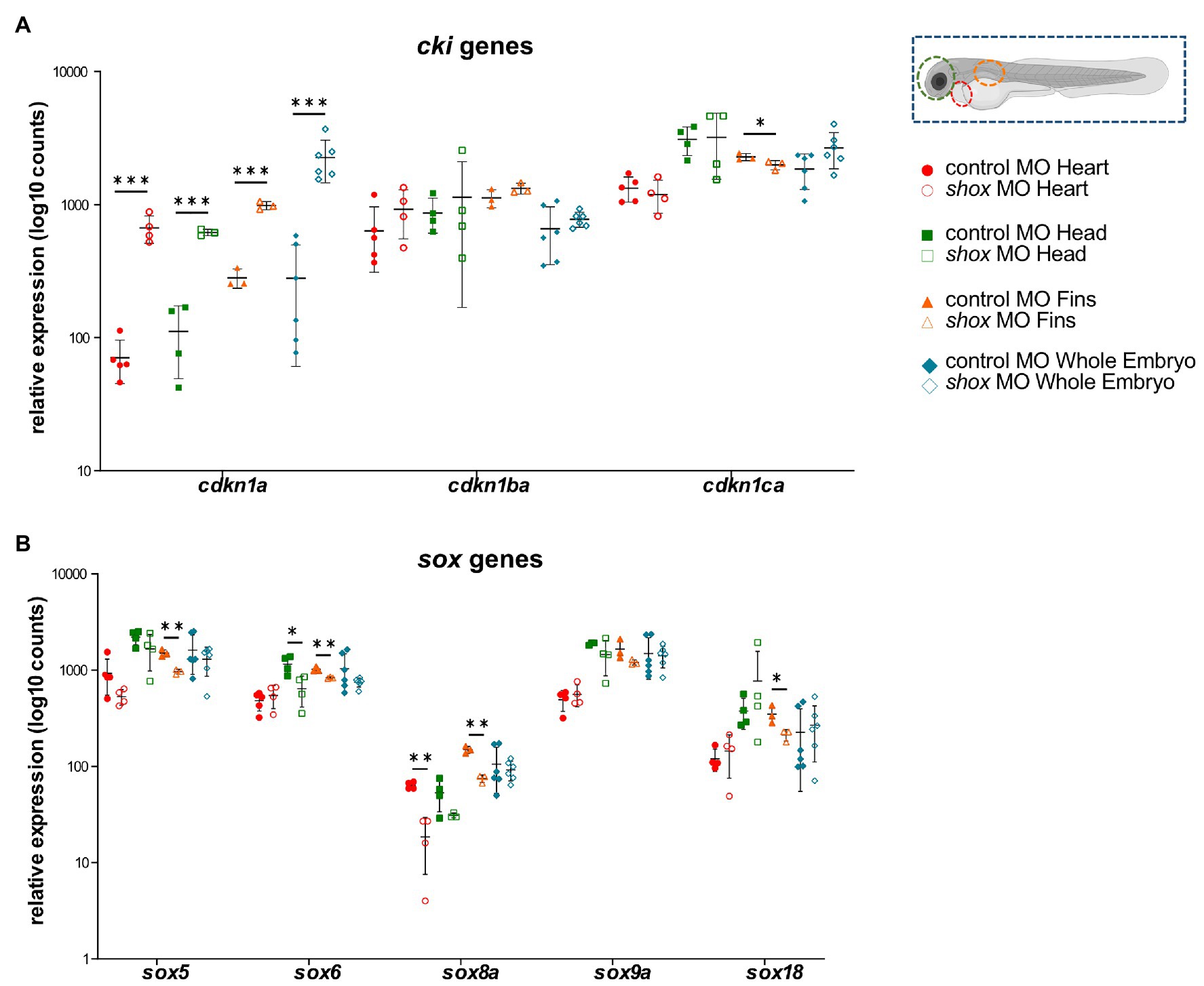
Figure 4. Tissue-specific analysis of gene families upon morpholino-mediated shox knockdown in zebrafish embryos 55 hpf compared to controls. Relative expression levels for (A) the cyclin-dependent kinase inhibitor (cki) family members cdkn1a, cdkn1ba, and cdkn1ca; (B) members of the SRY-related HMG-box (sox) genes sox5, sox6, sox8a, sox9a, and sox18 in heart (red; n = 4–5), head (green; n = 4), fin (orange; n = 3) or whole embryo (blue; n = 6). Statistical significance was determined by multiple unpaired t-tests adjusted by the two-stage step-up method of Benjamini, Krieger, and Yekutieli (adjusted values of p are indicated as *p < 0.05, **p < 0.01, and ***p < 0.001). MO, morpholino.
To identify transcriptional pathways in which the novel SHOX-dependent genes play a role, we performed a functional network analysis, integrating the 23 selected genes with SHOX using the IPA software tool. Our analysis demonstrates that the majority of the newly identified/selected genes are connected within the same network of the known SHOX-regulated genes (except CYP26A1, RASD1, and SOX18). We also identified significant biological processes and molecular functions that involve these genes (Supplementary Table 7). These included differentiation and development of chondrocytes, cartilage, and connective tissue. The most important annotated pathways relevant to SHOX-associated diseases included genes involved in growth failure, short stature, and limb defects (Figure 5A).
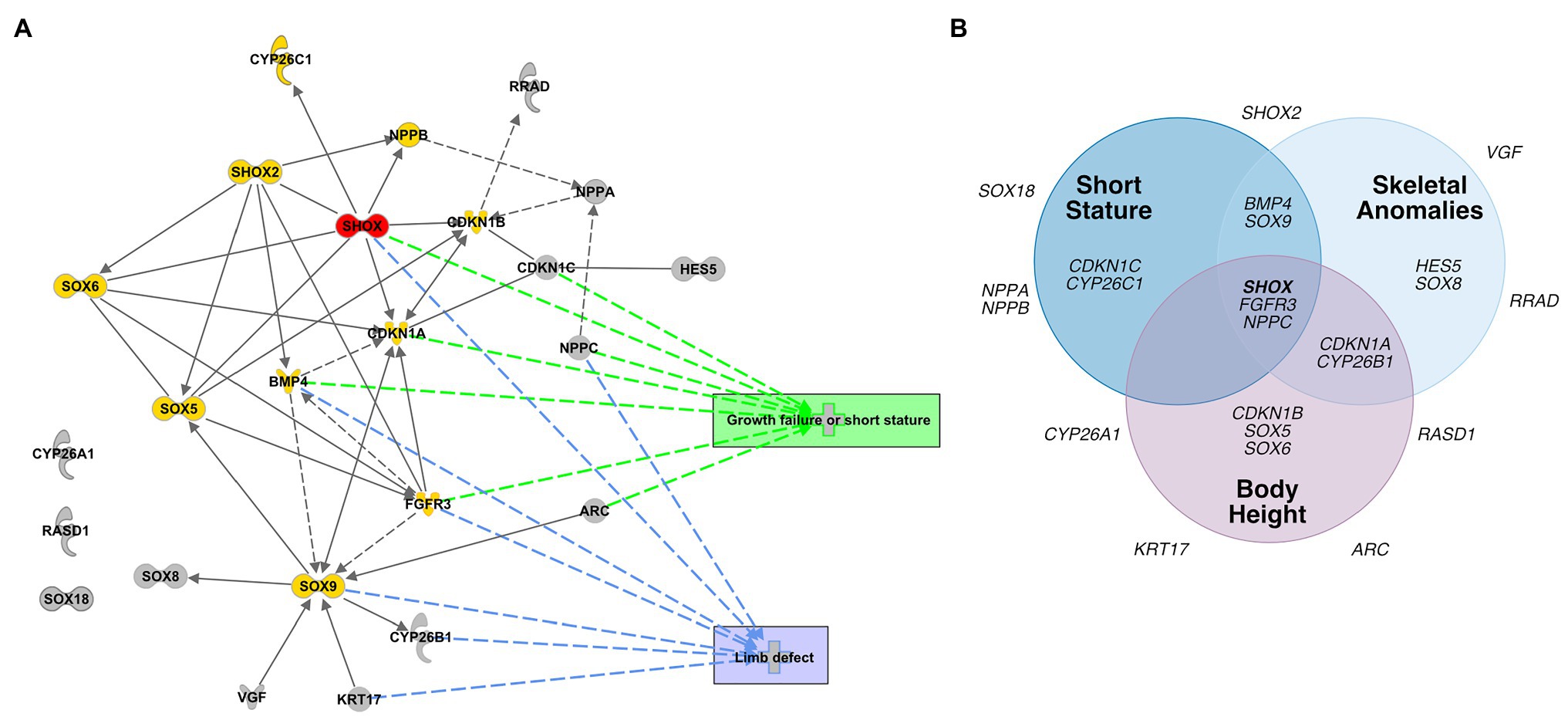
Figure 5. Database analyses of SHOX-regulated genes. (A) Network-based pathway analysis of SHOX-regulated genes using the Ingenuity pathway analysis (IPA) software. SHOX is highlighted in red. Known SHOX-associated genes are shown in yellow. Novel SHOX-regulated genes are shown in gray. Direct regulations are indicated by arrows and interactions as lines; indirect regulations or interactions are presented as dotted arrows/lines, respectively. The most important annotated pathways relevant to SHOX-associated diseases or function are highlighted in green (genes involved in growth failure and short stature) and blue (genes involved in limb defects). (B) Gene-disease association according to DisGeNET. Venn diagram showing associations between SHOX and 23 SHOX-regulated genes with respect to skeletal phenotypes based on the DisGeNET database (https://www.disgenet.org/home/; Pinero et al., 2015).
To predict the pathological relevance of the SHOX-regulated genes, we used the DisGeNET database (see footnote 2; Pinero et al., 2015) to summarize the known disease associations for each of the selected 23 genes (Figure 5B). Thirteen genes were associated with skeletal phenotypes such as short stature, skeletal abnormalities, and height; 10 genes had no such associations to date and, therefore, might represent novel candidates for skeletal disease.
Discussion
Identifying SHOX-regulated genes is essential to understanding how SHOX signaling exerts its diverse effects in individuals with SHOX deficiency. To identify novel target genes, we compared the transcriptional activity of wild type SHOX with a non-functional mutant form of SHOX in NHDF cells by microarray analysis. Although derived from human fibroblasts, NHDF cells do not fully reflect the situation in the growth plate. Despite this limitation, all identified 145 differentially up‐ and downregulated target genes downstream of SHOX can now be further investigated as candidates for various disorders. The proteins encoded by this list of SHOX-regulated genes are implicated in a wide variety of functions, consistent with the diverse effects of SHOX on development (Marchini et al., 2016). These functions include chondrocyte proliferation and differentiation, bone maturation, cartilage synthesis, cellular growth arrest, and apoptosis. Some genes represent known direct targets or have been previously associated with SHOX, but most are unknown SHOX-regulated genes. The known SHOX target genes NPPB and FGFR3 (Marchini et al., 2007; Decker et al., 2011) were among the top regulated genes after 12 and 24 h of overexpression. These findings show that NHDF cells are a valid and valuable model for analyzing the gene networks regulated by SHOX in vitro.
Considering the complexity of bone development and the spatiotemporal expression of SHOX, 11 of the upregulated and 12 additional genes were chosen for in vivo validation in zebrafish. Previous evidence has highlighted the role of SHOX as a transcriptional activator (Rao et al., 2001); therefore, we focused in this study only on those transcripts that were upregulated in NHDF cells and selected these for validation. Shox knockdown in whole zebrafish embryos altered embryonic growth and bone formation as previously described (Sawada et al., 2015; Montalbano et al., 2016; Yokokura et al., 2017). Gene expression analysis in shox-deficient whole zebrafish embryos also demonstrated a significant downregulation of shox2, the only shox paralog, and upregulation of four selected target candidates (cyp26c1, rasd1, her15.1, and cdkn1a). A shox2-associated cardiac phenotype is not present in whole embryos (Blaschke et al., 2007; Hoffmann et al., 2013), despite its strong downregulation (p = 0.0029; q = 0.0010) and known role in the orchestration of heart function.
To uncover any tissue-specific differences in SHOX-mediated gene expression, we performed separate analyses in fin, heart, and head tissue. Some genes (e.g., shox2) were downregulated in fin and head tissue after shox knockdown but not in heart tissue. This explains why we did not observe a cardiac phenotype in these embryos (Figure 2). Bmp4 and sox8a were significantly dysregulated in fin and heart but not in whole zebrafish embryos, indicating the importance of tissue-specific analyses to identify cell-specific effects that may not be detected in whole embryos.
A more specific investigation, particularly in the fins, provided further insight: indeed, 16 of the 22 analyzed target candidates revealed significantly differential expression in zebrafish fins. The most downregulated gene was shox2, while cdkn1a was the most upregulated one. SHOX and SHOX2 proteins are known to interact (Aza-Carmona et al., 2014), but this is the first published evidence that shox2 is a downstream target of Shox in the zebrafish fin. Cdkn1a, on the other hand, is a known cellular mediator of SHOX and is upregulated in SHOX-expressing cells (Marchini et al., 2004); however, we observed an upregulation of cdkn1a after shox knockdown in zebrafish fins. The known SHOX targets NPPB and FGFR3 were dysregulated in NHDF cells but not in zebrafish. Different directions of regulation in different model systems have been observed for SHOX targets. SHOX was shown to positively regulate FGFR3 expression in NHDF cells, confirming our results, but Shox overexpression in limb bud-derived chicken micromass cultures resulted in downregulation of Fgfr3 and may explain the almost mutually exclusive expression patterns of Fgfr3 and Shox in embryonic chicken limbs (Decker et al., 2011). Tissue-specific and cofactor-dependent opposing regulation of genes by specific transcription factors has been reported before (Lambert et al., 2018). Cofactors (coactivator or corepressor) vary in their availability and interaction with transcription factors in different tissues and this variation mediates distinct cell-specific effects. For example, Bmp4 is a direct transcriptional target of Shox2 and is positively regulated during cardiogenesis, whereas an opposite regulation has been described in limb development (Yu et al., 2007; Puskaric et al., 2010; Bobick and Cobb, 2012). This may explain why some genes that are upregulated after SHOX overexpression in NHDF cells are not downregulated after shox knockdown in zebrafish fins. Yet, differential expression was identified for many genes in both zebrafish and human cells, indicating conserved regulatory mechanisms between these two species.
Multiple members of the sex determining region Y-box (sox) family (sox5, sox6, sox8a, and sox18) were shown to be significantly dysregulated in shox-deficient pectoral fins among other genes (nppa, nppc, cdkn1a, cdkn1ca, cyp26b1, and cy26c1), highlighting the important role of various gene family members in Shox-related growth disorders. SHOX is known to interact with the SOX trio (SOX5/SOX6 and SOX9; Aza-Carmona et al., 2011), but we show for the first time that Shox transcriptionally regulates sox5 and sox6 but not sox9. Knockdown of Sox6 downregulates Shox expression in chicken (Lin et al., 2018), suggesting a reciprocal regulation between shox and sox6. Two other SOX family members, SOX8 and SOX18, were among the most upregulated genes in NHDF cells and confirmed to be significantly downregulated in shox-deficient pectoral fins. Among the SOX proteins, SOX9 exhibits the closest similarity to SOX8 (Schepers et al., 2000), but the role of SOX8 during chondrogenesis is not completely understood. SOX8 is highly upregulated during chondrogenesis in vitro, most likely under the control of SOX9 and both together with SOX5 and SOX6 are required for transcription of COL2A1 and many other chondrogenic molecules (Herlofsen et al., 2014). Thus, SOX8/sox8a might promote chondrogenesis via a SHOX/shox network. SOX18 silencing significantly induces cell cycle arrest and apoptosis, and inhibits cell migration, invasion, and cell growth in vitro and in vivo. In contrast, SOX18 is overexpressed in osteosarcoma, and promotes cell proliferation, migration, and invasion, inhibiting cell cycle arrest and apoptosis (Zhu et al., 2018). Given the role of SHOX as a modulator of cell proliferation and apoptosis (Marchini et al., 2004; Hristov et al., 2014), shox-dependent regulation of sox18 might play a role in these processes.
Other interesting gene family members regulated by Shox are the cyclin-dependent kinase inhibitor 1 (cki) genes. Two members, CDKN1A and CDKN1B, have previously been reported as cellular mediators of SHOX. In human osteosarcoma cells, the induction of SHOX expression elevates CDKN1A and CDKN1B levels (Marchini et al., 2004). Together with CDKN1C, this gene family contributes to the regulation of cell cycle progression by regulating the activity of cell cycle kinases during chondrogenic differentiation (Yan et al., 1997; Negishi et al., 2001; Bellosta et al., 2003; Rentsendorj et al., 2010; Cardelli et al., 2013). In shox-deficient zebrafish fins, cdkn1a was strongly upregulated, cdkn1ba was not affected, and the third family member, cdkn1ca, was downregulated. This gene family provides another example of differential regulation depending on the cell and tissue type. CKI family members are well-connected in the SHOX-dependent gene regulatory network (Figure 5A), suggesting a role in chondrocyte proliferation, controlled by SHOX-dependent regulation. FGFR3 signaling induces CDKN1A expression (Su et al., 1997; Nakajima et al., 2003), so may directly link SHOX and CDKN1A.
The three members of the cytochrome P450 family 26, CYP26A1, CYP26B1, and CYP26C1, encode retinoic acid metabolizing enzymes involved in chondrogenesis (Pennimpede et al., 2010; Cunningham and Duester, 2015). CYP26C1 is a genetic modifier of SHOX deficiency and downregulates shox expression in zebrafish (Montalbano et al., 2016). Moreover, CYP26C1 variants cause isolated short stature in the absence of SHOX deficiency (Montalbano et al., 2018). Here, we show that shox knockdown significantly upregulates cyp26c1 in zebrafish fins. This reciprocal regulation might maintain the delicate balance of retinoic acid levels during fin development. In addition, we observed dysregulation of CYP26B1/cyp26b1 in NHDF cells and zebrafish fins, while cyp26a1 was not affected by Shox. Cyp26b1 regulates osteogenesis in the axial skeleton as indicated by the zebrafish stocksteif mutant (Spoorendonk et al., 2008). The detailed relationship between CYP26B1 and SHOX deficiency so far remains unclear.
Our tissue-specific validation approach also indicates new roles for Shox during different embryonic developmental processes, including neuronal development and cardiogenesis, similar to/reminiscent of its paralog Shox2 (Yu et al., 2007; Vickerman et al., 2011; Liu et al., 2012; Ha and Dougherty, 2018; Hu et al., 2018). Shox2-regulated targets, such as Bmp4 and Nppb, are involved in various biological processes such as heart and limb development (Yu et al., 2007; Puskaric et al., 2010; Aza-Carmona et al., 2014; Hoffmann et al., 2017).
The differential expression of sox8a, bmp4, cyp26c1, and cdkn1a in shox-deficient zebrafish hearts was clearly a Shox-specific effect since shox2 expression was unaffected in this tissue, so did not have any indirect regulatory effects. While shox expression has previously been described in the zebrafish heart (Kenyon et al., 2011; Sawada et al., 2015), the role of shox during cardiogenesis remains unclear and requires further analysis in this model.
Shox-regulated gene expression in head tissue of zebrafish embryos also indicated a possible role for SHOX in brain development or neurodevelopmental disease. Microduplications at the pseudoautosomal SHOX locus have already been identified in patients with autism spectrum disorders (ASD) and related neurodevelopmental conditions (Tropeano et al., 2016). It is interesting to note that 17 of the 23 investigated genes are also associated with neuronal phenotypes including neurodegenerative diseases, intellectual disability/ASD, and psychiatric disorders (Supplementary Figure 2).
Evidence from both human and animal models has supported the notion that the genetic network involving SHOX, FGFR3, NPPC, among others, is an important regulator of growth. Mutations in these genes have not only been shown to cause rare skeletal malformations but also to contribute to the common forms of height (Rao et al., 1997; Ogata et al., 2000; Toydemir et al., 2006; Bocciardi et al., 2007; Tassano et al., 2013; Estrada et al., 2021). The target genes identified in this study add new members to this network which may contribute to rare skeletal forms or common forms of height and thus represent novel candidates for tall or short stature or other skeletal anomalies. Damaging mutations in these target genes may explain clinical phenotypes, e.g., short stature. As shown in Figure 5, some of the novel identified SHOX target genes have already been associated with a skeletal phenotype.
In conclusion, we have identified novel SHOX targets, many of which are consistent with known SHOX functions in limb development, and thereby provide novel insights into the genetic pathways leading to the clinical manifestation of SHOX deficiency. In addition, our tissue-specific approach suggests that some target genes may also play a role in other organs, such as heart and brain, indicating a pleiotropic role for SHOX in different phenotypic traits.
Data Availability Statement
The datasets generated and analyzed for this study can be found in the public NCBI GEO database (GSE169732) and can be accessed via https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE169732.
Ethics Statement
Ethical review and approval was not required for the animal study because only zebrafish embryos at early life stages (55 hpf) were used in this study.
Author Contributions
SH and GR designed the study and experimental design and wrote the manuscript. Material preparation, acquisition, and data analysis were performed by SH, RR, SD, JG, DH, and SJ. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the Deutsche Forschungsgemeinschaft (DFG; RA 380/12-1) and University of the Medical Faculty, Heidelberg (9422170). We acknowledge financial support within the funding program Open Access Publishing, by the Baden-Wuerttemberg Ministry of Science, Research and the Arts and by the Ruprecht-Karls-Universität Heidelberg.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are indebted to Sebastian Bender and Stephan Wolf, who started this project using NHDF cells. We thank the nCounter Core Facility Heidelberg (head: Beate Niesler) for technical support and Claire Bacon for reading the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.688808/full#supplementary-material
Footnotes
References
Aza-Carmona, M., Barca-Tierno, V., Hisado-Oliva, A., Belinchon, A., Gorbenko-Del Blanco, D., Rodriguez, J. I., et al. (2014). NPPB and ACAN, two novel SHOX2 transcription targets implicated in skeletal development. PLoS One 9:e83104. doi: 10.1371/journal.pone.0083104
Aza-Carmona, M., Shears, D. J., Yuste-Checa, P., Barca-Tierno, V., Hisado-Oliva, A., Belinchon, A., et al. (2011). SHOX interacts with the chondrogenic transcription factors SOX5 and SOX6 to activate the aggrecan enhancer. Hum. Mol. Genet. 20, 1547–1559. doi: 10.1093/hmg/ddr032
Beiser, K. U., Glaser, A., Kleinschmidt, K., Scholl, I., Roth, R., Li, L., et al. (2014). Identification of novel SHOX target genes in the developing limb using a transgenic mouse model. PLoS One 9:e98543. doi: 10.1371/journal.pone.0098543
Belin, V., Cusin, V., Viot, G., Girlich, D., Toutain, A., Moncla, A., et al. (1998). SHOX mutations in dyschondrosteosis (Leri-Weill syndrome). Nat. Genet. 19, 67–69. doi: 10.1038/ng0198-67
Bellosta, P., Masramon, L., Mansukhani, A., and Basilico, C. (2003). p21(WAF1/CIP1) acts as a brake in osteoblast differentiation. J. Bone Miner. Res. 18, 818–826. doi: 10.1359/jbmr.2003.18.5.818
Benito-Sanz, S., Thomas, N. S., Huber, C., Gorbenko Del Blanco, D., Aza-Carmona, M., Crolla, J. A., et al. (2005). A novel class of Pseudoautosomal region 1 deletions downstream of SHOX is associated with Leri-Weill dyschondrosteosis. Am. J. Hum. Genet. 77, 533–544. doi: 10.1086/449313
Binder, G. (2011). Short stature due to SHOX deficiency: genotype, phenotype, and therapy. Horm. Res. Paediatr. 75, 81–89. doi: 10.1159/000324105
Binder, G., Renz, A., Martinez, A., Keselman, A., Hesse, V., Riedl, S. W., et al. (2004). SHOX haploinsufficiency and Leri-Weill dyschondrosteosis: prevalence and growth failure in relation to mutation, sex, and degree of wrist deformity. J. Clin. Endocrinol. Metab. 89, 4403–4408. doi: 10.1210/jc.2004-0591
Blaschke, R. J., Hahurij, N. D., Kuijper, S., Just, S., Wisse, L. J., Deissler, K., et al. (2007). Targeted mutation reveals essential functions of the homeodomain transcription factor Shox2 in sinoatrial and pacemaking development. Circulation 115, 1830–1838. doi: 10.1161/CIRCULATIONAHA.106.637819
Bobick, B. E., and Cobb, J. (2012). Shox2 regulates progression through chondrogenesis in the mouse proximal limb. J. Cell Sci. 125, 6071–6083. doi: 10.1242/jcs.111997
Bocciardi, R., Giorda, R., Buttgereit, J., Gimelli, S., Divizia, M. T., Beri, S., et al. (2007). Overexpression of the C-type natriuretic peptide (CNP) is associated with overgrowth and bone anomalies in an individual with balanced t(2,7) translocation. Hum. Mutat. 28, 724–731. doi: 10.1002/humu.20511
Cardelli, M., Zirngibl, R. A., Boetto, J. F., Mckenzie, K. P., Troy, T. C., Turksen, K., et al. (2013). Cartilage-specific overexpression of ERRgamma results in Chondrodysplasia and reduced chondrocyte proliferation. PLoS One 8:e81511. doi: 10.1371/journal.pone.0081511
Carnovali, M., Banfi, G., and Mariotti, M. (2019). Zebrafish models of human skeletal disorders: embryo and adult swimming together. Biomed. Res. Int. 2019:1253710. doi: 10.1155/2019/1253710
Cunningham, T. J., and Duester, G. (2015). Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell Biol. 16, 110–123. doi: 10.1038/nrm3932
Decker, E., Durand, C., Bender, S., Rodelsperger, C., Glaser, A., Hecht, J., et al. (2011). FGFR3 is a target of the homeobox transcription factor SHOX in limb development. Hum. Mol. Genet. 20, 1524–1535. doi: 10.1093/hmg/ddr030
Durand, C., Roeth, R., Dweep, H., Vlatkovic, I., Decker, E., Schneider, K. U., et al. (2011). Alternative splicing and nonsense-mediated RNA decay contribute to the regulation of SHOX expression. PLoS One 6:e18115. doi: 10.1371/journal.pone.0018115
Estrada, K., Froelich, S., Wuster, A., Bauer, C. R., Sterling, T., Clark, W. T., et al. (2021). Identifying therapeutic drug targets using bidirectional effect genes. Nat. Commun. 12:2224. doi: 10.1038/s41467-021-21843-8
Fukami, M., Seki, A., and Ogata, T. (2016). SHOX haploinsufficiency as a cause of syndromic and nonsyndromic short stature. Mol. Syndromol. 7, 3–11. doi: 10.1159/000444596
Ha, N. T., and Dougherty, K. J. (2018). Spinal Shox2 interneuron interconnectivity related to function and development. eLife 7:e42519. doi: 10.7554/eLife.42519
Herlofsen, S. R., Hoiby, T., Cacchiarelli, D., Zhang, X., Mikkelsen, T. S., and Brinchmann, J. E. (2014). Brief report: importance of SOX8 for in vitro chondrogenic differentiation of human mesenchymal stromal cells. Stem Cells 32, 1629–1635. doi: 10.1002/stem.1642
Hoffmann, S., Berger, I. M., Glaser, A., Bacon, C., Li, L., Gretz, N., et al. (2013). Islet1 is a direct transcriptional target of the homeodomain transcription factor Shox2 and rescues the Shox2-mediated bradycardia. Basic Res. Cardiol. 108:339. doi: 10.1007/s00395-013-0339-z
Hoffmann, S., Schmitteckert, S., Griesbeck, A., Preiss, H., Sumer, S., Rolletschek, A., et al. (2017). Comparative expression analysis of Shox2-deficient embryonic stem cell-derived sinoatrial node-like cells. Stem Cell Res. 21, 51–57. doi: 10.1016/j.scr.2017.03.018
Hristov, G., Marttila, T., Durand, C., Niesler, B., Rappold, G. A., and Marchini, A. (2014). SHOX triggers the lysosomal pathway of apoptosis via oxidative stress. Hum. Mol. Genet. 23, 1619–1630. doi: 10.1093/hmg/ddt552
Hu, W., Xin, Y., Zhao, Y., and Hu, J. (2018). Shox2: the role in differentiation and development of cardiac conduction system. Tohoku J. Exp. Med. 244, 177–186. doi: 10.1620/tjem.244.177
Just, S., Berger, I. M., Meder, B., Backs, J., Keller, A., Marquart, S., et al. (2011). Protein kinase D2 controls cardiac valve formation in zebrafish by regulating histone deacetylase 5 activity. Circulation 124, 324–334. doi: 10.1161/CIRCULATIONAHA.110.003301
Kenyon, E. J., Mcewen, G. K., Callaway, H., and Elgar, G. (2011). Functional analysis of conserved non-coding regions around the short stature hox gene (shox) in whole zebrafish embryos. PLoS One 6:e21498. doi: 10.1371/journal.pone.0021498
Kramer, A., Green, J., Pollard, J. Jr., and Tugendreich, S. (2014). Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30, 523–530. doi: 10.1093/bioinformatics/btt703
Kronenberg, H. M. (2003). Developmental regulation of the growth plate. Nature 423, 332–336. doi: 10.1038/nature01657
Lambert, S. A., Jolma, A., Campitelli, L. F., Das, P. K., Yin, Y., Albu, M., et al. (2018). The human transcription factors. Cell 175, 598–599. doi: 10.1016/j.cell.2018.01.029
Lin, S., Lin, X., Zhang, Z., Jiang, M., Rao, Y., Nie, Q., et al. (2018). Copy number variation in SOX6 contributes to chicken muscle development. Gene 9:42. doi: 10.3390/genes9010042
Liu, H., Espinoza-Lewis, R. A., Chen, C., Hu, X., Zhang, Y., and Chen, Y. (2012). The role of Shox2 in SAN development and function. Pediatr. Cardiol. 33, 882–889. doi: 10.1007/s00246-012-0179-x
Lui, J. C., Jee, Y. H., Garrison, P., Iben, J. R., Yue, S., Ad, M., et al. (2018). Differential aging of growth plate cartilage underlies differences in bone length and thus helps determine skeletal proportions. PLoS Biol. 16:e2005263. doi: 10.1371/journal.pbio.2005263
Marchini, A., Hacker, B., Marttila, T., Hesse, V., Emons, J., Weiss, B., et al. (2007). BNP is a transcriptional target of the short stature homeobox gene SHOX. Hum. Mol. Genet. 16, 3081–3087. doi: 10.1093/hmg/ddm266
Marchini, A., Marttila, T., Winter, A., Caldeira, S., Malanchi, I., Blaschke, R. J., et al. (2004). The short stature homeodomain protein SHOX induces cellular growth arrest and apoptosis and is expressed in human growth plate chondrocytes. J. Biol. Chem. 279, 37103–37114. doi: 10.1074/jbc.M307006200
Marchini, A., Ogata, T., and Rappold, G. A. (2016). A track record on SHOX: from basic research to complex models and therapy. Endocr. Rev. 37, 417–448. doi: 10.1210/er.2016-1036
Montalbano, A., Juergensen, L., Fukami, M., Thiel, C. T., Hauer, N. H., Roeth, R., et al. (2018). Functional missense and splicing variants in the retinoic acid catabolizing enzyme CYP26C1 in idiopathic short stature. Eur. J. Hum. Genet. 26, 1113–1120. doi: 10.1038/s41431-018-0148-9
Montalbano, A., Juergensen, L., Roeth, R., Weiss, B., Fukami, M., Fricke-Otto, S., et al. (2016). Retinoic acid catabolizing enzyme CYP26C1 is a genetic modifier in SHOX deficiency. EMBO Mol. Med. 8, 1455–1469. doi: 10.15252/emmm.201606623
Monzani, A., Babu, D., Mellone, S., Genoni, G., Fanelli, A., Prodam, F., et al. (2019). Co-occurrence of genomic imbalances on Xp22.1 in the SHOX region and 15q25.2 in a girl with short stature, precocious puberty, urogenital malformations and bone anomalies. BMC Med. Genet. 12:5. doi: 10.1186/s12920-018-0445-8
Nakajima, A., Shimizu, S., Moriya, H., and Yamazaki, M. (2003). Expression of fibroblast growth factor receptor-3 (FGFR3), signal transducer and activator of transcription-1, and cyclin-dependent kinase inhibitor p21 during endochondral ossification: differential role of FGFR3 in skeletal development and fracture repair. Endocrinology 144, 4659–4668. doi: 10.1210/en.2003-0158
Negishi, Y., Ui, N., Nakajima, M., Kawashima, K., Maruyama, K., Takizawa, T., et al. (2001). p21Cip-1/SDI-1/WAF-1 gene is involved in chondrogenic differentiation of ATDC5 cells in vitro. J. Biol. Chem. 276, 33249–33256. doi: 10.1074/jbc.M010127200
Ogata, T., Kosho, T., Wakui, K., Fukushima, Y., Yoshimoto, M., and Miharu, N. (2000). Short stature homeobox-containing gene duplication on the der(X) chromosome in a female with 45,X/46,X, der(X), gonadal dysgenesis, and tall stature. J. Clin. Endocrinol. Metab. 85, 2927–2930. doi: 10.1210/jcem.85.8.6745
Oliveira, C. S., and Alves, C. (2011). The role of the SHOX gene in the pathophysiology of Turner syndrome. Endocrinol. Nutr. 58, 433–442. doi: 10.1016/j.endonu.2011.06.005
Pennimpede, T., Cameron, D. A., Maclean, G. A., Li, H., Abu-Abed, S., and Petkovich, M. (2010). The role of CYP26 enzymes in defining appropriate retinoic acid exposure during embryogenesis. Birth Defects Res. A Clin. Mol. Teratol. 88, 883–894. doi: 10.1002/bdra.20709
Pinero, J., Queralt-Rosinach, N., Bravo, A., Deu-Pons, J., Bauer-Mehren, A., Baron, M., et al. (2015). DisGeNET: a discovery platform for the dynamical exploration of human diseases and their genes. Database 2015:bav028. doi: 10.1093/database/bav028
Puskaric, S., Schmitteckert, S., Mori, A. D., Glaser, A., Schneider, K. U., Bruneau, B. G., et al. (2010). Shox2 mediates Tbx5 activity by regulating Bmp4 in the pacemaker region of the developing heart. Hum. Mol. Genet. 19, 4625–4633. doi: 10.1093/hmg/ddq393
Rao, E., Blaschke, R. J., Marchini, A., Niesler, B., Burnett, M., and Rappold, G. A. (2001). The Leri-Weill and Turner syndrome homeobox gene SHOX encodes a cell-type specific transcriptional activator. Hum. Mol. Genet. 10, 3083–3091. doi: 10.1093/hmg/10.26.3083
Rao, E., Weiss, B., Fukami, M., Rump, A., Niesler, B., Mertz, A., et al. (1997). Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat. Genet. 16, 54–63. doi: 10.1038/ng0597-54
Rentsendorj, A., Mohan, S., Szabo, P., and Mann, J. R. (2010). A genomic imprinting defect in mice traced to a single gene. Genetics 186, 917–927. doi: 10.1534/genetics.110.118802
Rosilio, M., Huber-Lequesne, C., Sapin, H., Carel, J. C., Blum, W. F., and Cormier-Daire, V. (2012). Genotypes and phenotypes of children with SHOX deficiency in France. J. Clin. Endocrinol. Metab. 97, E1257–E1265. doi: 10.1210/jc.2011-3460
Rottbauer, W., Wessels, G., Dahme, T., Just, S., Trano, N., Hassel, D., et al. (2006). Cardiac myosin light chain-2: a novel essential component of thick-myofilament assembly and contractility of the heart. Circ. Res. 99, 323–331. doi: 10.1161/01.RES.0000234807.16034.fe
Sawada, R., Kamei, H., Hakuno, F., Takahashi, S., and Shimizu, T. (2015). In vivo loss of function study reveals the short stature homeobox-containing (shox) gene plays indispensable roles in early embryonic growth and bone formation in zebrafish. Dev. Dyn. 244, 146–156. doi: 10.1002/dvdy.24239
Schepers, G. E., Bullejos, M., Hosking, B. M., and Koopman, P. (2000). Cloning and characterisation of the Sry-related transcription factor gene Sox8. Nucleic Acids Res. 28, 1473–1480. doi: 10.1093/nar/28.6.1473
Schiller, S., Spranger, S., Schechinger, B., Fukami, M., Merker, S., Drop, S. L., et al. (2000). Phenotypic variation and genetic heterogeneity in Leri-Weill syndrome. Eur. J. Hum. Genet. 8, 54–62. doi: 10.1038/sj.ejhg.5200402
Schneider, K. U., Marchini, A., Sabherwal, N., Roth, R., Niesler, B., Marttila, T., et al. (2005). Alteration of DNA binding, dimerization, and nuclear translocation of SHOX homeodomain mutations identified in idiopathic short stature and Leri-Weill dyschondrosteosis. Hum. Mutat. 26, 44–52. doi: 10.1002/humu.20187
Shears, D. J., Vassal, H. J., Goodman, F. R., Palmer, R. W., Reardon, W., Superti-Furga, A., et al. (1998). Mutation and deletion of the pseudoautosomal gene SHOX cause Leri-Weill dyschondrosteosis. Nat. Genet. 19, 70–73. doi: 10.1038/ng0198-70
Spoorendonk, K. M., Peterson-Maduro, J., Renn, J., Trowe, T., Kranenbarg, S., Winkler, C., et al. (2008). Retinoic acid and Cyp26b1 are critical regulators of osteogenesis in the axial skeleton. Development 135, 3765–3774. doi: 10.1242/dev.024034
Su, W. C., Kitagawa, M., Xue, N., Xie, B., Garofalo, S., Cho, J., et al. (1997). Activation of Stat1 by mutant fibroblast growth-factor receptor in thanatophoric dysplasia type II dwarfism. Nature 386, 288–292. doi: 10.1038/386288a0
Tassano, E., Buttgereit, J., Bader, M., Lerone, M., Divizia, M. T., Bocciardi, R., et al. (2013). Genotype-phenotype correlation of 2q37 deletions including NPPC gene associated with skeletal malformations. PLoS One 8:e66048. doi: 10.1371/journal.pone.0066048
Toydemir, R. M., Brassington, A. E., Bayrak-Toydemir, P., Krakowiak, P. A., Jorde, L. B., Whitby, F. G., et al. (2006). A novel mutation in FGFR3 causes camptodactyly, tall stature, and hearing loss (CATSHL) syndrome. Am. J. Hum. Genet. 79, 935–941. doi: 10.1086/508433
Tropeano, M., Howley, D., Gazzellone, M. J., Wilson, C. E., Ahn, J. W., Stavropoulos, D. J., et al. (2016). Microduplications at the pseudoautosomal SHOX locus in autism spectrum disorders and related neurodevelopmental conditions. J. Med. Genet. 53, 536–547. doi: 10.1136/jmedgenet-2015-103621
Verdin, H., Fernandez-Minan, A., Benito-Sanz, S., Janssens, S., Callewaert, B., De Waele, K., et al. (2015). Profiling of conserved non-coding elements upstream of SHOX and functional characterisation of the SHOX cis-regulatory landscape. Sci. Rep. 5:17667. doi: 10.1038/srep17667
Vickerman, L., Neufeld, S., and Cobb, J. (2011). Shox2 function couples neural, muscular and skeletal development in the proximal forelimb. Dev. Biol. 350, 323–336. doi: 10.1016/j.ydbio.2010.11.031
Westerfield, M. (2000). The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio rerio). Eugene: University of Oregon Press.
Yan, Y., Frisen, J., Lee, M. H., Massague, J., and Barbacid, M. (1997). Ablation of the CDK inhibitor p57Kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev. 11, 973–983. doi: 10.1101/gad.11.8.973
Yokokura, T., Kamei, H., Shibano, T., Yamanaka, D., Sawada-Yamaguchi, R., Hakuno, F., et al. (2017). The short-stature homeobox-containing gene (shox/SHOX) is required for the regulation of cell proliferation and bone differentiation in zebrafish embryo and human mesenchymal stem cells. Front. Endocrinol. 8:125. doi: 10.3389/fendo.2017.00125
Yu, L., Liu, H., Yan, M., Yang, J., Long, F., Muneoka, K., et al. (2007). Shox2 is required for chondrocyte proliferation and maturation in proximal limb skeleton. Dev. Biol. 306, 549–559. doi: 10.1016/j.ydbio.2007.03.518
Keywords: short stature, skeletal dysplasia, pectoral fins, zebrafish, SHOX deficiency, skeletal disease associations
Citation: Hoffmann S, Roeth R, Diebold S, Gogel J, Hassel D, Just S and Rappold GA (2021) Identification and Tissue-Specific Characterization of Novel SHOX-Regulated Genes in Zebrafish Highlights SOX Family Members Among Other Genes. Front. Genet. 12:688808. doi: 10.3389/fgene.2021.688808
Edited by:
Liborio Stuppia, University of Studies G. d’Annunzio Chieti and Pescara, ItalyReviewed by:
Tsutomu Ogata, Hamamatsu University School of Medicine, JapanEmanuele Micaglio, IRCCS Policlinico San Donato, Italy
Copyright © 2021 Hoffmann, Roeth, Diebold, Gogel, Hassel, Just and Rappold. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gudrun A. Rappold, Z3VkcnVuLnJhcHBvbGRAbWVkLnVuaS1oZWlkZWxiZXJnLmRl