 Bo Yang1
Bo Yang1 Hong Liu2Qin-Wen Xu1Yi-Fan Sun1Sui Xu3Hao Zhang1
Hong Liu2Qin-Wen Xu1Yi-Fan Sun1Sui Xu3Hao Zhang1 Jian-Xia Tang2,3
Jian-Xia Tang2,3 Guo-Ding Zhu2,3
Guo-Ding Zhu2,3 Yao-Bao Liu2,3
Yao-Bao Liu2,3 Jun Cao1,2,3*
Jun Cao1,2,3* Yang Cheng1*
Yang Cheng1*- 1Laboratory of Pathogen Infection and Immunity, Department of Public Health and Preventive Medicine, Wuxi School of Medicine, Jiangnan University, Wuxi, China
- 2Center for Global Health, School of Public Health, Nanjing Medical University, Nanjing, China
- 3Key Laboratory of National Health and Family Planning Commission on Parasitic Disease Control and Prevention, Jiangsu Provincial Key Laboratory on Parasite and Vector Control Technology, Jiangsu Institute of Parasite Diseases, Wuxi, China
Plasmodium falciparum surface-related antigen (SRA) is located on the surfaces of gametocyte and merozoite and has the structural and functional characteristics of potential targets for multistage vaccine development. However, little information is available regarding the genetic polymorphism of pfsra. To determine the extent of genetic variation about P. falciparum by characterizing the sra sequence, 74 P. falciparum samples were collected from migrant workers who returned to China from 12 countries of Africa between 2015 and 2019. The full length of the sra gene was amplified and sequenced. The average pairwise nucleotide diversities (π) of P. falciparum sra gene was 0.00132, and the haplotype diversity (Hd) was 0.770. The average number of nucleotide differences (k) for pfsra was 3.049. The ratio of non-synonymous (dN) to synonymous (dS) substitutions across sites (dN/dS) was 1.365. Amino acid substitutions of P. falciparum SRA could be categorized into 35 unique amino acid variants. Neutrality tests showed that the polymorphism of PfSRA was maintained by positive diversifying selection, which indicated its role as a potential target of protective immune responses and a vaccine candidate. Overall, the ability of the N-terminal of PfSRA antibodies to evoke inhibition of merozoite invasion of erythrocytes and conserved amino acid at low genetic diversity suggest that the N-terminal of PfSRA could be evaluated as a vaccine candidate against P. falciparum infection.
Introduction
Malaria has been a major global health concern of humans throughout history and is a leading cause of disease and death across many tropical and subtropical countries. In 2019, an estimated 229 million malaria cases and 409,000 malaria-caused deaths globally were reported (WHO, 2020). Among the five species of Plasmodium that infect humans, P. falciparum infection causes the highest mortality and morbidity and the most serious clinical symptoms (Buffet et al., 2011).
The resurgence and spread of antimalarial drug resistance (Ménard et al., 2015; WWARN, 2015) along with vector resistance to insecticides (Dhiman and Veer, 2014; Strode et al., 2014) have the potential to reduce the impact of existing malaria control strategies and make vaccines a public health priority. Although extensive studies have been conducted on several blood-stage antigens, few have shown the quality required for a candidate vaccine. In a systematic screen of uncharacterized P. falciparum proteins for potential blood-stage vaccine candidates, using data from transcriptome studies of P. falciparum, data-mining analysis of the genes with peak mRNA expression levels in late schizogony was performed (Bozdech et al., 2003; Le Roch et al., 2003) and another study on the prediction of PfSUB-1 protease specificity (Gilson et al., 2006). The results showed that P. falciparum surface-related antigen (PfSRA) emerged as the top hit with both signal peptide and a predicted glycosylphosphatidylinositol (GPI) attachment site. PfSRA is localized on the surfaces of both gametocytes and merozoites. The processed 32-kDa PfSRA protein fragment binds normal human erythrocytes. Immunoepidemiological studies in malaria-infected populations suggest the presence of naturally acquired protective antibodies against PfSRA. Parasite growth inhibition assays indicated that the antibodies against PfSRA could potently inhibit the invasion of merozoite on erythrocytes. Overall, the structural and functional characteristics of PfSRA indicate that it would be a promising vaccine target (Amlabu et al., 2018).
The low protective efficacy of vaccines against clinical malaria has been in part limited by extensive genetic diversity, which enables parasites to evade human immune responses and may lead to vaccine failure (Takala et al., 2002). However, the evidence for pfsra genetic diversity is limited. Therefore, it is necessary to study the characteristics of pfsra toward finding suitable vaccine candidates and understanding its population genetic structure. Accordingly, this study analyzed the full-length sequence of sra from the P. falciparum collected from infected migrant workers returning to the Jiangsu Province from Africa. We determined the nucleotide divergence and polymorphisms level of sra sequences to trace signatures of selection and to determine the extent of genetic variation in P. falciparum by characterizing the sra sequence at the nucleotide and protein levels.
Materials and Methods
Study Areas and Blood Samples Collection
The samples of P. falciparum were obtained from febrile patients in Jiangsu Province, China, from 2015 to 2019, who had returned from working in tropical regions of sub-Saharan Africa endemic for malaria (Chu et al., 2018). A total of 74 P. falciparum-infected blood samples were collected from 12 countries. The subjects were identified for mono-infection of P. falciparum by microscopic examination of blood smears stained with Giemsa. The isolates were identified by specific polymerase chain reaction (PCR).
Amplification and Sequencing Analysis of pfsra
The full-length nucleotide sequences of sra from P. falciparum were divided into four fragments and amplified by PCR with primers designed as pfsra-1-Forward (5′-ATG TTT CTA AGT TCT AAG AAA AGA A-3′) and pfsra-1-Reverse (5′-AAA GGA ATC TGT CTC ATT ATT TGT T-3′), pfsra-2-Forward (5′-GAT AAT GAA GAA ACA GAA GAT ATT G-3′), and pfsra-2-Reverse (5′-ATC TAA TAG TTG TAT ATA AGC ATA TTT ATT AAC-3′), pfsra-3-Forward (5′-AAT AAG AAT TCA AAT CAA TCA TAT AAT T-3′) and pfsra-3-Reverse (5′-ATA ATA TTT CCT CAC AAT TTT TAC ATG-3′), and pfsra-4-Forward (5′-GTA CCT GCC AAA ATT AAA TAT ATA GAA-3′) and pfsra-4-Reverse (5′-TTA ATA TAT CGA AAT AAA TAT CAT AAG-3′), respectively. The pfsra (PlasmoDB, PF3D7_1431400) sequence from the Plasmodium Genomics Resource database was used as the reference gene sequence. The PCR amplification reactions were performed in a volume of 50 μl including 100 ng of genomic DNA, 0.2 μM each of the forward and reverse primers, 0.2 mM deoxynucleoside triphosphate, 2.5 units of DNA polymerase in 1 × FastPfu buffer (TransStart® FastPfu DNA polymerase, Beijing, China), and nuclease-free water up to 50 μl. The PCR amplification of pfsra genes was carried out in Mastercycler (Eppendorf, Hamburg, Germany). Amplification was performed as follows: denaturation at 95°C for 2 min, 35 cycles of 95°C for 20 s, 50°C for 20 s, and 65°C for 1 min, and final extension at 65°C for 5 min. The PCR products were analyzed using 1% agarose gel electrophoresis, stained with SuperStain (CWBIO, Jiangsu, China), and visualized by ultraviolet transilluminator (Bio-Rad ChemiDoc MP, Hercules, United States). The lengths of the PCR products were estimated based on their mobility relative to a standard DNA marker (TransGen Biotech, Beijing, China). Sequencing reactions were performed using GENEWIZ (Suzhou, China) with an ABI 3730xl DNA Analyzer (Thermo Fisher Scientific, Waltham, United States). All 74 samples generated a single amplification fragment of the expected size, and direct Sanger DNA sequencing of the forward and reverse directions was conducted to ensure the accuracy of the obtained sequences.
Sequence Alignment and Genetic Data Analysis
The geographical distribution map of P. falciparum samples was constructed by Arcgis10.2 software (ESRI, 2011). In order to evaluate diversity, pfsra sequence was used as template and aligned using GeneDoc2.7.0.1 The primary structure of the PfSRA protein was demonstrated by UniProt.2 The nucleotide sequences of pfsra were translated into the deduced amino acid (aa) sequences by DNASTAR (Burland, 2000). The predicted amino acid sequences of PfSRA from the PCR sequenced genomic fragments were aligned with the sequence of P. falciparum genome strain 3D7 by the MUSCLE algorithms in the MEGA 7.0 program (Kumar et al., 2016). A logo plot for each pfsra population was constructed to analyze the polymorphic characteristics of PfSRA by the WebLogo program.3 In addition, a codon-based test of purifying selection was analyzed by MEGA 7.0 program (Kumar et al., 2016). The non-synonymous mutations (dN), synonymous mutations (dS), and the dN/dS ratio from MEGA 7.0 were tested and compared by the Z-test (p < 0.05) using Nei and Gojobori’s method, corrected by Jukes and Cantor and 1,000 bootstrap replications (Nei and Gojobori, 1986). Under purifying selection, dN will be less than dS (dN/dS < 1), while when the positive selection is more advantageous, dN will exceed dS (dN/dS > 1).
The average pairwise nucleotide diversity (π), number of haplotypes (H), and haplotype diversity (Hd) were calculated by DnaSP v6 (Rozas et al., 2017). The nucleotide diversity was analyzed by DnaSP v6 with a window length of 100 base pairs (bp) and a step size of 25 bp. In addition, the neutrality tests (Tajima’s D, Fu and Li’s D∗, and Fu and Li’s F∗) implemented in DnaSP v6 software were utilized to measure the departure of the neutral mode prediction of molecular evolution (Fu and Li, 1993; Tajima, 1993). In order to determine the evolutionary relationship of the aligned sequences, based on nucleotide sequences, the phylogenetic tree of sra was constructed with the neighbor-joining method in MEGA 7.0. The sra sequences of diverse malaria parasites species included the SRA haplotypes of plasmodium from humans, non-human primate, avian, and murine malaria, which were obtained from the PlasmoDB and NCBI databases.
Results
Geographical Origin of P. falciparum
A total of 74 clinical isolates of P. falciparum showed the geographical distribution in 12 sub-Saharan Africa countries. These isolates were mainly from the west coast of Africa, including Angola (n = 16, 21.6%), Nigeria and Equatorial Guinea (n = 13, 17.6%), and the Republic of the Congo (n = 9, 12.2%) (Figure 1). Of the 74 sequencing samples, 3 were from Eastern Africa (Uganda), 22 were from Western Africa (Sierra Leone, Côte d’Ivoire, Ghana, and Nigeria), 31 were from Central Africa (Cameroon, Equatorial Guinea, Gabon, Republic of the Congo, and Democratic Republic of the Congo), and 18 were from Southern Africa (Zambia and Angola). Overall, 74 cases of P. falciparum infection were identified in our study (Table 1). Full details for the isolates were provided (Supplementary Table 1).

Figure 1. Origins of malaria cases were imported from 12 countries of sub-Saharan Africa to the Jiangsu Province of China. (A) A map of Africa showing the countries of origin of P. falciparum isolates. (B) The total number of genotyped samples per region and percentage of samples.
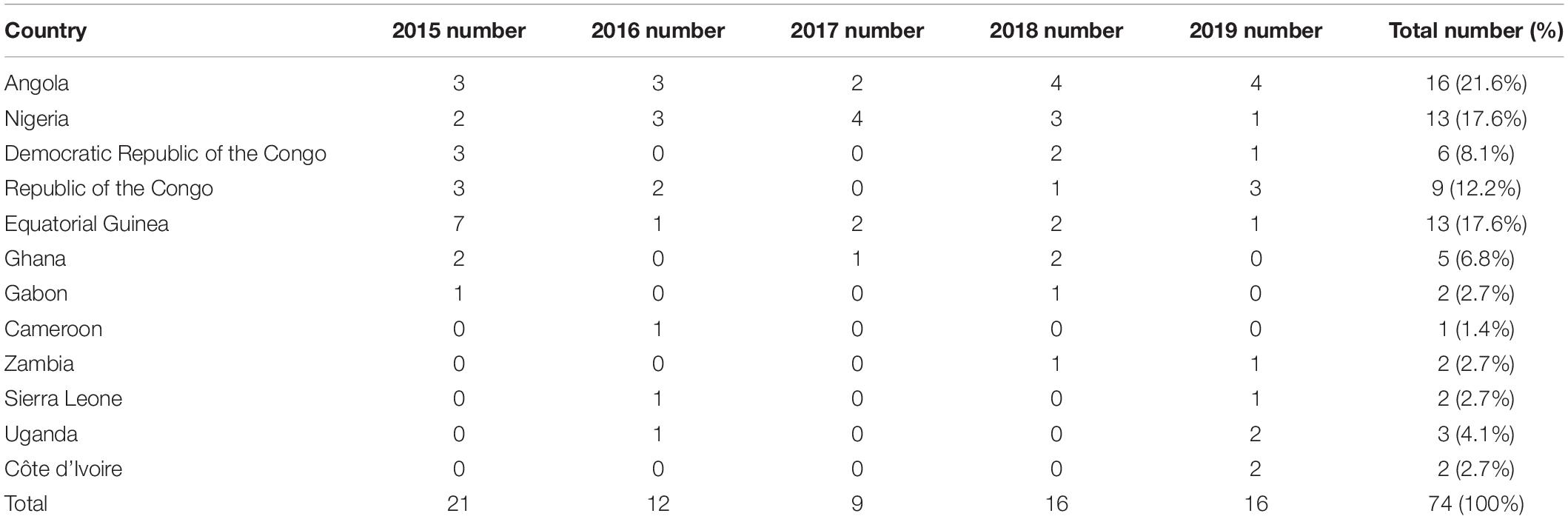
Table 1. Origin of imported Plasmodium falciparum in 2015–2019.
Characterization of PfSRA
The length of SRA encoded by the pfsra full-length was 990 (aa), beginning with the predicted 24-aa signal peptide sequence (aa 1–24) and ending with a GPI-anchor (aa 969–990). Other specific regions, such as two coiled-coil regions, were also identified in the predicted protein primary structure of P. falciparum (aa 413–433 and 437–457) (Figure 2A). The full-length nucleotide sequences of sra from P. falciparum was 3,149 bp and was amplified by PCR using primer 1 (1–25 bp), primer 2 (924–948 bp), primer 3 (801–826 bp), primer 4 (1,768–1,800 bp), primer 5 (1,702–1,729 bp), primer 6 (2,675–2,701 bp), primer 7 (2,602–2,628 bp), and primer 8 (3,123–3,149 bp) (Figure 2B).

Figure 2. Predicted P. falciparum SRA protein primary structure and primer design diagram. (A) Diagram of PfSRA protein primary structure. (B) Diagram of pfsra primers design. (C) Amplification of four fragments of pfsra by PCR. Abbreviations: M, DNA marker; 1, the first fragment of pfsra (948 bp); 2, the second fragment of pfsra (1 kb); 3, the third fragment of pfsra (1 kb); 4, the fourth fragment of pfsra (548 bp).
Nucleotide Polymorphism of pfsra
The sra genes of 74 P. falciparum isolates were successfully amplified by PCR, corresponding to nucleotides 1–948, 801–1,800, 1,702–2,701, and 2,602–3,149, respectively, and a single PCR product with an expected size of 948 bp (pfsra1), 1 kb (pfsra2 and pfsra3), and 548 bp (pfsra4) (Figure 2C). The direct sequencing of the purified PCR fragments indicated that there were no superimposed signals on the electropherograms of pfsra. Compared with the reference 3D7 strain, 74 isolates (100%) showed non-synonymous mutation. Overall, 63 single nucleotide polymorphisms (SNPs) were found in 74 isolates with the average π value of 0.00132 for pfsra. The sliding method plot using DnaSP v6 with a window length of 100 bp and a step size of 25 bp showed that the π value of pfsra is in the range of 0–0.01023. The conservative regions of 0–0.6 and 0.8–1.6 kb were observed in pfsra with π values of 0 approximately (Figure 3). The average number of nucleotide differences (k) of pfsra was 3.049. Nucleotide diversity of pfsra was categorized into 35 distinct haplotypes, and the estimated Hd was 0.770 (Table 2). For amino acid, the frequencies and types of mutation in the full-length of PfSRA (aa 1–990) were briefly presented in Figure 4. The C-terminal fragments of the PfSRA (aa 585–591, aa 879–900) showed relatively high polymorphism. More detailed amino acid comparison results are reported in Supplementary Figure 1.

Figure 3. Sliding window plot analyses show the sequence diversity (π) and Tajima’s D. (A) Sequence diversity of PfSRA. (B) Tajima’s D of PfSRA.
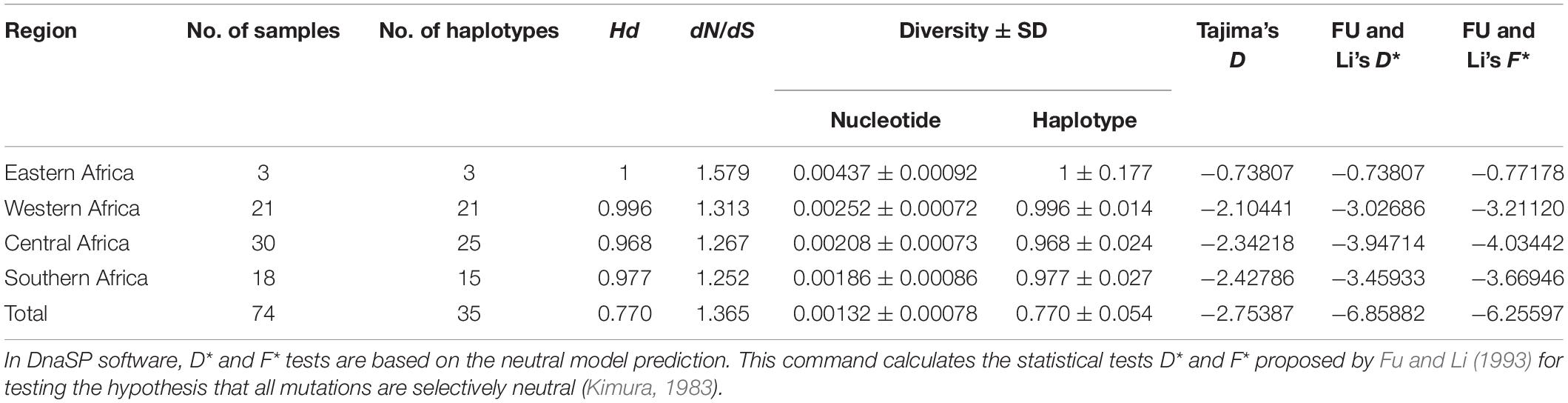
Table 2. Estimates of number of haplotypes, haplotype diversity, nucleotide diversity, and neutrality indices of pfsra.
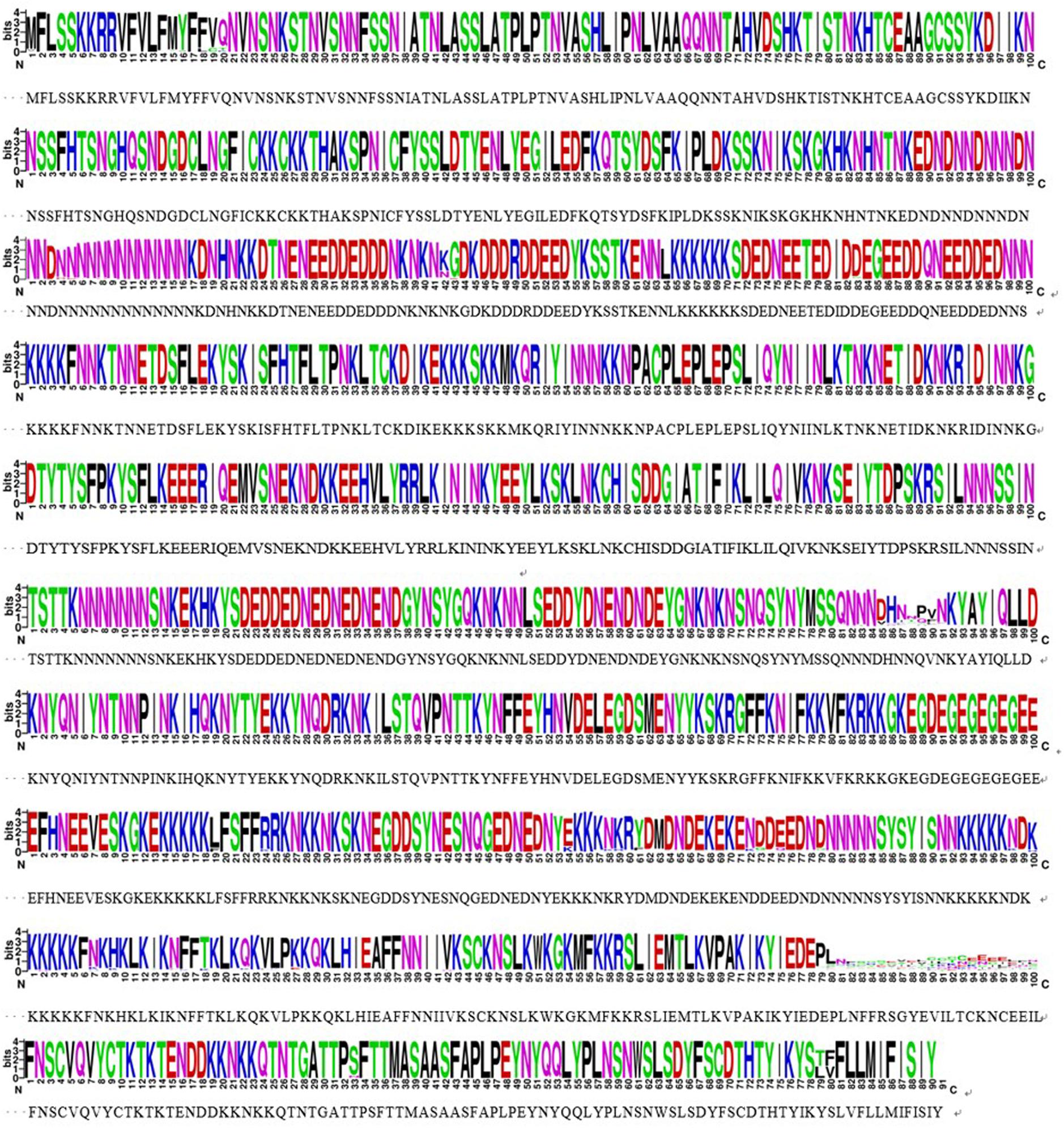
Figure 4. Conservative locus analysis of the PfSRA amino acid sequences defined by WEBLOGO. Each logo consists of stacks of symbols, and each position in the sequence corresponds to stacks of symbols. The height within the stack of each individual amino acid abbreviation indicates its relative frequency at that specific position.
Genetic Population Structure of pfsra
Based on the average values of dS and dN, the population genetic structure of the P. falciparum samples was analyzed using the sra gene polymorphisms in the codon-based purifying selection test. Results showed that there was diversifying selection or positive selection in P. falciparum sra population (dS − dN = −0.00075). In addition, the mean ratio of across sites non-synonymous (dN) to synonymous (dS) substitutions (dN/dS) was 1.365, and most of the nucleotide substitutions detected were non-synonymous, which also showed that the genetic variations of pfsra were maintained by positive selection. Tajima’s D and Fu and Li’s D∗ and F∗ tests rejected a neutral polymorphism occurrence model with values of pfsra (Tajima’s D = −2.75387, p < 0.05, Fu and Li’s D∗ = −6.85882, p < 0.05, and Fu and Li’s F∗ = −6.25597, p < 0.05) (Table 2). Full details for all study countries were provided (Supplementary Table 2).
Phylogenetic Analysis of sra
As predicted based on the signature of positive selection and the level of genetic diversity described above, the phylogenetic relationship among 35 distinct haplotypes was detected in the pfsra sequences (1 was from Eastern Africa; 9 were from Western Africa; 14 were from Central Africa; and 11 were from Southern Africa) (Figure 5). The phylogenetic tree of 11 alleles of sra gene of 11 species of human and non-human Plasmodium primates was constructed by the neighbor-joining method (Supplementary Figure 2). Supplementary Table 3 provides the sra gene ID number and gene length of other malaria parasite species.
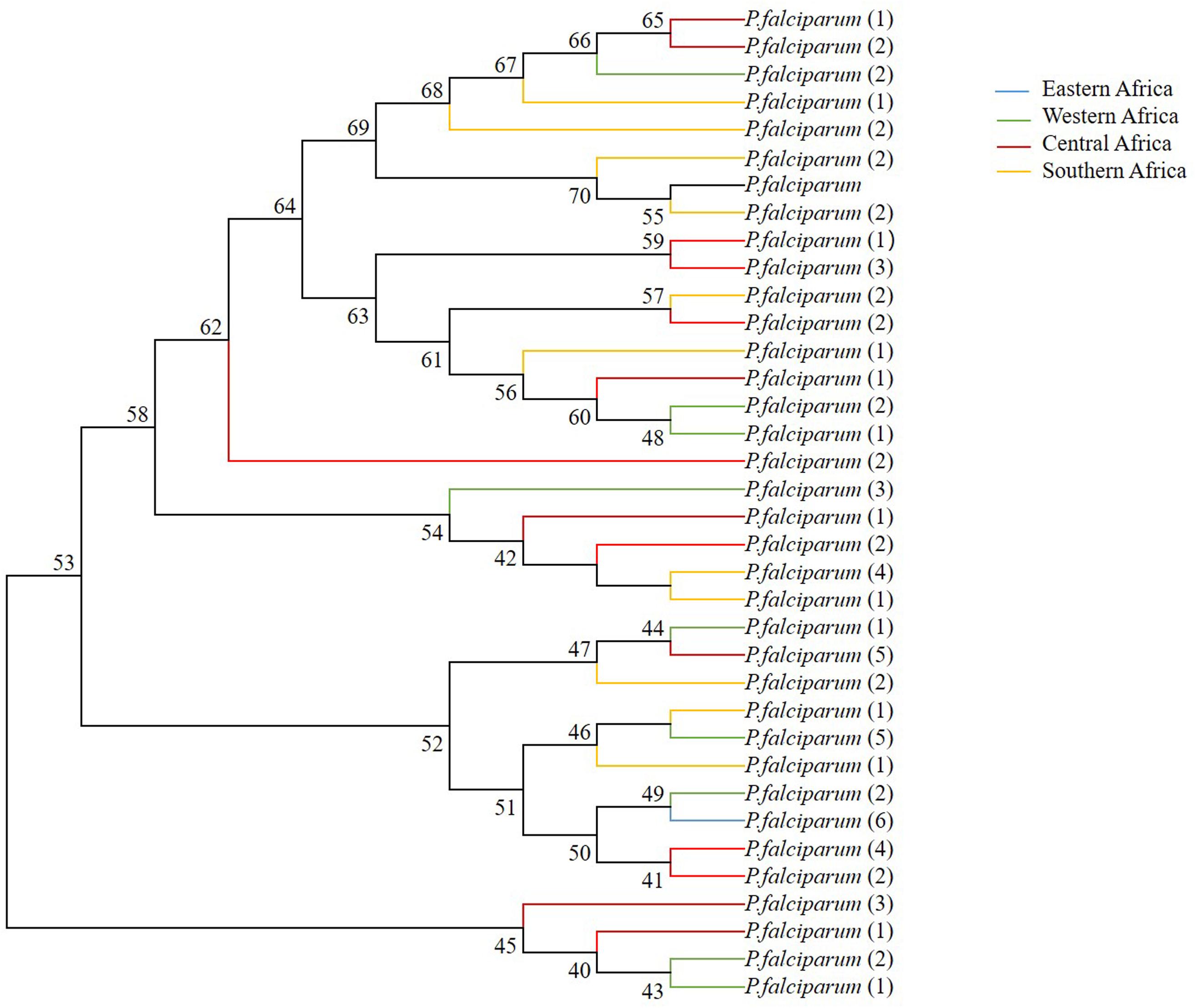
Figure 5. Phylogenetic relationship of sra full-length genes within pfsra sequences based on the neighbor-joining method. Numbers at nodes show bootstrap values.
Discussion
Apart from the complex life cycle of the malaria parasite involving the mosquito vector and human host, the malaria parasite exhibits extensive antigenicity and genetically diverse stages that may pose an adverse obstacle to malarial control strategies. Thus, a deeper understanding of patterns and mechanisms of sequence variation and genetic recombination may contribute to the design of a vaccine that represents the global repertoire of polymorphic malaria surface antigens (Bharti et al., 2012). Systematic screens for uncharacterized P. falciparum invasion-related proteins evaluated PfSRA as one of the top hits that emerged; it contains coiled-coil domains known to be less polymorphic (Villard et al., 2007; Kulangara et al., 2009; Amlabu et al., 2018). Our investigation into the extent of sequence variation is consistent with this. In addition, coiled-coil domains form a stable structure, which elicit functional antibodies, thus blocking the related domains in many organisms and were considered to be the basis for the chemical synthesis of three PfSRA peptides designed to generate antibodies (Tripet et al., 2006; Gustchina et al., 2013; Jiang et al., 2016; Amlabu et al., 2018). Furthermore, these domains have been evaluated as potential targets for immunotherapy such as peptide-based vaccine strategies (Stoute et al., 1995; Demangel et al., 1998; Adda et al., 1999). We analyzed the full-length of pfsra (74 isolates) and found that the C-terminal fragments of the pfsra (π = 0.00198) show polymorphism probably due to selection pressure. Comparatively, the N-terminal of sra is a relatively conserved sequence (π = 0.00083).
A previous study suggested that people infected with malaria have naturally acquired antibodies against PfSRA and PfSRA N-terminal antibodies could partially inhibit merozoite invasion of erythrocytes by parasite growth inhibition assays (Amlabu et al., 2018). Now, the evidence of relatively conservative N-terminus might raise the possibility that it has the potential to be a candidate for anti-malarial vaccine. The ratio of non-synonymous (dN) to synonymous (dS) substitutions across sites was used as an index to evaluate selection pressure; dN/dS > 1 indicates diversifying positive selection. Further neutrality tests were carried out to determine the types and characteristics of natural selection on the pfsra. Statistically significant negative values of neutrality tests suggest an excess of rare polymorphisms in the population and provide evidence of purifying or directional (positive) selection (Fu and Li, 1993; Akey et al., 2004). The phylogenetic tree of 11 alleles of sra gene of 11 species showed that pfsra and other species occupied distinct bifurcating branches, supporting an ancient divergence times of the malarial parasite lineage.
The nucleotide diversity of pfsra in Southern Africa (π = 0.00186 ± SD 0.00086), Central Africa (π = 0.00208 ± SD 0.00073), and Western Africa (π = 0.00252 ± SD 0.00072) was lower than that in Eastern Africa (π = 0.00437 ± SD 0.00092), which may be related to the higher transmission rate of P. falciparum in Eastern Africa. Furthermore, more samples are needed in future research to support our findings and to control the limitations of small sample size (large confidence interval) in a single area. A previous study had also shown that P. falciparum has a spectrum of population structure: linkage “equilibrium,” low levels of differentiation and high diversity in regions with high levels of transmission (Anderson et al., 2000; Huang et al., 2020). Mutation, recombination, gene flow, and natural selection may contribute to the genetic diversity of malaria parasites (Cole-Tobian and King, 2003).
In the analysis of pfsra full-length, there were abundant polymorphisms found. Samples from the four Africa regions showed their own distinct diversity patterns. Interestingly, two larger-size parasite population (Western Africa and Central Africa) showed more polymorphisms compared to those in Eastern Africa and Southern Africa. Some mutations showed the regional differences based on the geographical isolation effect; for example, the 15th amino acid mutant (M15Y) only occurred in Central Africa; K333E was only found in Western Africa. These phenomena indicate that it is necessary to continuously monitor these regional characteristic mutations in order to explore their association with regional malaria epidemics. Overall, apart from the conserved N-terminus, the composition of PfSRA vaccine should consider the high-frequency alleles instead of the C-terminus of wild-type ones (Conway, 2007).
Epidemiological studies have indicated that the level of heterologous mating in malaria populations is positively correlated with the prevalence of mixed allele infections and transmission rates (Chenet et al., 2008). The generation of relevant genetic, immunologic, and epidemiologic data for the sra gene is necessary, especially in areas with low malaria endemicity. Even in geographical areas with low transmission, the development of vaccine strategies should include results of diversity analysis. The uneven geographical distribution of alleles may jeopardize the development and use of vaccines targeting specific variable site, as local variation may not be taken into account in vaccine design (Cole-Tobian and King, 2003). The study of different genes and their alleles is helpful for us to understand the trends of genetic variation and if alleles could render vaccine ineffective. Given the genetic diversity found in the region, an alternative to improve the vaccine effectiveness is to create a construct with the most common region-specific alleles (Cole-Tobian and King, 2003; Chenet et al., 2008).
Conclusion
The C-terminal fragments of the sra gene of P. falciparum showed polymorphism due to positive diversifying selection, which would hinder SRA-based vaccine development. Comparatively, in addition to the coiled-coil domains that have been evaluated as potential targets of peptide-based vaccines previously, the conserved N-terminal of pfsra is also a promising vaccine candidate against P. falciparum infection.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics Statement
This study was approved by the Ethics Committee of Jiangsu Institute of Parasitic Diseases (JIPD) (IRB00004221), Wuxi, China. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
BY and YC conceptualized the study, wrote the manuscript and contributed to the interpretation of the data. BY, HL, Q-WX, SX, HZ, J-XT, and G-DZ collected and analyzed the samples. Y-FS performed statistical and bioinformatics analysis. BY, YC, Y-BL, and JC revised the manuscript critically for important intellectual content. All the authors contributed to this article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (81871681 and 81971967), the Jiangsu Provincial Department of Science and Technology (BM2018020), the Jiangnan University Student Innovation Training Program (1285210232175260), the National First-class Discipline Program of Food Science and Technology (JUFSTR20180101), and the Jiangsu Provincial Project of Invigorating Health Care through Science, Technology and Education.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank all participants in this study, doctors, and local health departments for their support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.688606/full#supplementary-material
Supplementary Figure 1 | Amino acid sequence alignment of P. falciparum SRA. Gray area indicates that amino acid is identical across all aligned sequences.
Supplementary Figure 2 | Neighbor-joining tree of 11 unique alleles encoded by sra from 11 Plasmodium parasite species. Numbers at nodes show bootstrap values.
Supplementary Table 1 | Country of origin and parasitemia of P. falciparum samples.
Supplementary Table 2 | Neutrality tests of pfsra among 74 P. falciparum isolates.
Supplementary Table 3 | The sra Gene ID number and gene length of other Plasmodium species.
Footnotes
- ^ https://github.com/karlnicholas/GeneDoc
- ^ https://www.uniprot.org/uniprot/Q8ILF1
- ^ https://weblogo.berkeley.edu/logo.cgi
References
Adda, C. G., Tilley, L., Anders, R. F., and Foley, M. (1999). Isolation of peptides that mimic epitopes on a malarial antigen from random peptide libraries displayed on phage. Infect. Immun. 67, 4679–4688. doi: 10.1128/Iai.67.9.4679-4688.1999
Akey, J. M., Eberle, M. A., Rieder, M. J., Carlson, C. S., Shriver, M. D., Nickerson, D. A., et al. (2004). Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol. 2:e286. doi: 10.1371/journal.pbio.0020286
Amlabu, E., Mensah-Brown, H., Nyarko, P. B., Akuh, O. A., Opoku, G., Ilani, P., et al. (2018). Functional characterization of Plasmodium falciparum surface-related antigen as a potential blood-stage vaccine target. J. Infect. Dis. 218, 778–790. doi: 10.1093/infdis/jiy222
Anderson, T. J., Haubold, B., Williams, J. T., Estrada-Franco, J. G., Richardson, L., Mollinedo, R., et al. (2000). Microsatellite markers reveal a spectrum of population structures in the malaria parasite Plasmodium falciparum. Mol. Biol. Evol. 17, 1467–1482. doi: 10.1093/oxfordjournals.molbev.a026247
Bharti, P. K., Shukla, M. M., Sharma, Y. D., and Singh, N. (2012). Genetic diversity in the block 2 region of the merozoite surface protein-1 of Plasmodium falciparum in central India. Malar. J. 11:78. doi: 10.1186/1475-2875-11-78
Bozdech, Z., Llinás, M., Pulliam, B. L., Wong, E. D., Zhu, J., and DeRisi, J. L. (2003). The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 1:E5. doi: 10.1371/journal.pbio.0000005
Buffet, P. A., Safeukui, I., Deplaine, G., Brousse, V., Prendki, V., Thellier, M., et al. (2011). The pathogenesis of Plasmodium falciparum malaria in humans: insights from splenic physiology. Blood 117, 381–392. doi: 10.1182/blood-2010-04-202911
Burland, T. G. (2000). DNASTAR’s lasergene sequence analysis software. Methods Mol. Biol. 132, 71–91. doi: 10.1385/1-59259-192-2:71
Chenet, S. M., Branch, O. H., Escalante, A. A., Lucas, C. M., and Bacon, D. J. (2008). Genetic diversity of vaccine candidate antigens in Plasmodium falciparum isolates from the Amazon basin of Peru. Malar. J. 7:93. doi: 10.1186/1475-2875-7-93
Chu, R., Zhang, X., Xu, S., Chen, L., Tang, J., Li, Y., et al. (2018). Limited genetic diversity of N-terminal of merozoite surface protein-1 (MSP-1) in Plasmodium ovale curtisi and P. ovale wallikeri imported from Africa to China. Parasit. Vectors 11:596. doi: 10.1186/s13071-018-3174-0
Cole-Tobian, J., and King, C. L. (2003). Diversity and natural selection in Plasmodium vivax Duffy binding protein gene. Mol. Biochem. Parasitol. 127, 121–132. doi: 10.1016/s0166-6851(02)00327-4
Conway, D. J. (2007). Molecular epidemiology of malaria. Clin. Microbiol. Rev. 20, 188–204. doi: 10.1128/CMR.00021-06
Demangel, C., Rouyre, S., Alzari, P. M., Nato, F., Longacre, S., Lafaye, P., et al. (1998). Phage-displayed mimotopes elicit monoclonal antibodies specific for a malaria vaccine candidate. Biol. Chem. 379, 65–70. doi: 10.1515/bchm.1998.379.1.65
Dhiman, S., and Veer, V. (2014). Culminating anti-malaria efforts at long lasting insecticidal net? J. Infect. Public Health 7, 457–464. doi: 10.1016/j.jiph.2014.06.002
Fu, Y. X., and Li, W. H. (1993). Statistical tests of neutrality of mutations. Genetics 133, 693–709. doi: 10.1093/genetics/133.3.693
Gilson, P. R., Nebl, T., Vukcevic, D., Moritz, R. L., Sargeant, T., Speed, T. P., et al. (2006). Identification and stoichiometry of glycosylphosphatidylinositol-anchored membrane proteins of the human malaria parasite Plasmodium falciparum. Mol. Cell Proteomics 5, 1286–1299. doi: 10.1074/mcp.M600035-MCP200
Gustchina, E., Li, M., Ghirlando, R., Schuck, P., Louis, J. M., Pierson, J., et al. (2013). Complexes of neutralizing and non-neutralizing affinity matured Fabs with a mimetic of the internal trimeric coiled-coil of HIV-1 gp41. PLoS One 8:e78187. doi: 10.1371/journal.pone.0078187
Huang, H. Y., Liang, X. Y., Lin, L. Y., Chen, J. T., Ehapo, C. S., Eyi, U. M., et al. (2020). Genetic polymorphism of Plasmodium falciparum circumsporozoite protein on Bioko Island, equatorial guinea and global comparative analysis. Malar. J. 19:245. doi: 10.1186/s12936-020-03315-4
Jiang, Z., Gera, L., Mant, C. T., Hirsch, B., Yan, Z., Shortt, J. A., et al. (2016). Platform technology to generate broadly cross-reactive antibodies to alpha-helical epitopes in hemagglutinin proteins from influenza A viruses. Biopolymers 106, 144–159. doi: 10.1002/bip.22808
Kimura, M. (1983). The Neutral Theory of Molecular Evolution. Cambridge, MA: Cambridge University Press.
Kulangara, C., Kajava, A. V., Corradin, G., and Felger, I. (2009). Sequence conservation in Plasmodium falciparum alpha-helical coiled coil domains proposed for vaccine development. PLoS One 4:e5419. doi: 10.1371/journal.pone.0005419
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Le Roch, K. G., Zhou, Y., Blair, P. L., Grainger, M., Moch, J. K., Haynes, J. D., et al. (2003). Discovery of gene function by expression profiling of the malaria parasite life cycle. Science 301, 1503–1508. doi: 10.1126/science.1087025
Ménard, S., Ben Haddou, T., Ramadani, A. P., Ariey, F., Iriart, X., Beghain, J., et al. (2015). Induction of multidrug tolerance in Plasmodium falciparum by extended artemisinin pressure. Emerg. Infect. Dis. 21, 1733–1741. doi: 10.3201/eid2110.150682
Nei, M., and Gojobori, T. (1986). Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol. 3, 418–426. doi: 10.1093/oxfordjournals.molbev.a040410
Rozas, J., Ferrer-Mata, A., Sánchez-DelBarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Stoute, J. A., Ballou, W. R., Kolodny, N., Deal, C. D., Wirtz, R. A., and Lindler, L. E. (1995). Induction of humoral immune-response against Plasmodium-Falciparum sporozoites by immunization with a synthetic peptide mimotope whose sequence was derived from screening a filamentous phage epitope library. Infect. Immun. 63, 934–939. doi: 10.1128/Iai.63.3.934-939.1995
Strode, C., Donegan, S., Garner, P., Enayati, A. A., and Hemingway, J. (2014). The impact of pyrethroid resistance on the efficacy of insecticide-treated bed nets against African anopheline mosquitoes: systematic review and meta-analysis. PLoS Med. 11:e1001619. doi: 10.1371/journal.pmed.1001619
Tajima, F. (1993). Simple methods for testing the molecular evolutionary clock hypothesis. Genetics 135, 599–607. doi: 10.1093/genetics/135.2.599
Takala, S., Branch, O., Escalante, A. A., Kariuki, S., Wootton, J., and Lal, A. A. (2002). Evidence for intragenic recombination in Plasmodium falciparum: identification of a novel allele family in block 2 of merozoite surface protein-1: Asembo Bay area cohort project XIV. Mol. Biochem. Parasitol. 125, 163–171. doi: 10.1016/S0166-6851(02)00237-2
Tripet, B., Kao, D. J., Jeffers, S. A., Holmes, K. V., and Hodges, R. S. (2006). Template-based coiled-coil antigens elicit neutralizing antibodies to the SARS-coronavirus. J. Struct. Biol. 155, 176–194. doi: 10.1016/j.jsb.2006.03.019
Villard, V., Agak, G. W., Frank, G., Jafarshad, A., Servis, C., Nébié, I., et al. (2007). Rapid identification of malaria vaccine candidates based on alpha-helical coiled coil protein motif. PLoS One 2:e645. doi: 10.1371/journal.pone.0000645
WHO (2020). World Malaria Report. Available online at: http://www.who.int/publications/i/item/9789240015791 (accessed November 30, 2020).
Keywords: Plasmodium falciparum, PfSRA, genetic diversity, imported malaria cases, DNA sequencing, Jiangsu Province
Citation: Yang B, Liu H, Xu Q-W, Sun Y-F, Xu S, Zhang H, Tang J-X, Zhu G-D, Liu Y-B, Cao J and Cheng Y (2021) Genetic Diversity Analysis of Surface-Related Antigen (SRA) in Plasmodium falciparum Imported From Africa to China. Front. Genet. 12:688606. doi: 10.3389/fgene.2021.688606
Received: 31 March 2021; Accepted: 01 July 2021;
Published: 05 August 2021.
Edited by:
Matthew Adekunle Adeleke, University of KwaZulu-Natal, South AfricaReviewed by:
Francis Ntumngia, University of South Florida, United StatesIkhide G. Imumorin, Georgia Institute of Technology, United States
Copyright © 2021 Yang, Liu, Xu, Sun, Xu, Zhang, Tang, Zhu, Liu, Cao and Cheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Cao, Y2FvanVuY25AaG90bWFpbC5jb20=; Yang Cheng, d29lcnNlbmdAMTI2LmNvbQ==