 Jin Xu
Jin Xu Chen Liu
Chen Liu Yun Song
Yun Song Mingfu Li
Mingfu Li- Institute of Plant Inspection and Quarantine, Chinese Academy of Inspection and Quarantine, Beijing, China
The genus Pennisetum (Poaceae) is both a forage crop and staple food crop in the tropics. In this study, we obtained chloroplast genome sequences of four species of Pennisetum (P. alopecuroides, P. clandestinum, P. glaucum, and P. polystachion) using Illumina sequencing. These chloroplast genomes have circular structures of 136,346–138,119 bp, including a large single-copy region (LSC, 79,380–81,186 bp), a small single-copy region (SSC, 12,212–12,409 bp), and a pair of inverted repeat regions (IRs, 22,284–22,372 bp). The overall GC content of these chloroplast genomes was 38.6–38.7%. The complete chloroplast genomes contained 110 different genes, including 76 protein-coding genes, 30 transfer RNA (tRNA) genes, and four ribosomal RNA (rRNA) genes. Comparative analysis of nucleotide variability identified nine intergenic spacer regions (psbA-matK, matK-rps16, trnN-trnT, trnY-trnD-psbM, petN-trnC, rbcL-psaI, petA-psbJ, psbE-petL, and rpl32-trnL), which may be used as potential DNA barcodes in future species identification and evolutionary analysis of Pennisetum. The phylogenetic analysis revealed a close relationship between P. polystachion and P. glaucum, followed by P. clandestinum and P. alopecuroides. The completed genomes of this study will help facilitate future research on the phylogenetic relationships and evolution of Pennisetum species.
Introduction
The genus Pennisetum L. Rich. belongs to tribe Paniceae of the family Poaceae; it consists of approximately 140 species that are widely distributed in the tropical and subtropical zone regions all over the world. Pennisetum which is divided into three sections Sect. Gymnothrix, Sect. Penicillaria and Sect. Pennisetum is one of the largest genera in the tribe Paniceae (Pilger, 1940; DeLisle, 1963; Brunken, 1977). Pennisetum is an economically important grain crop that is widely cultivated in Africa and Asia (P. glaucum), as well as a pasture crop (P. purpureum, P. ramosum, P. orientale, and P. clandestine) and ornamental plant (P. villosum and P. setaceum) (Martel et al., 1997; Abdi et al., 2019). Many of these species are climate resilient (Kumar et al., 2019), adapted to low-fertility soils, exhibit strong stress resistance, possess high ornamental value, and are perennial in habit. Consequently, Pennisetum (Poaceae) species have become increasingly visible in many countries. This genus is a heterogeneous assemblage of species with different reproductive behaviors (sexual or apomictic), chromosome numbers (x = 5, 7, 8, and 9), ploidy levels (diploid to octoploid), and life cycles (annual, biennial, or perennial) (Martel et al., 1997). The classification and identification of the genus Pennisetum remains a challenge. Pennisetum and Cenchrus are closely related genera of Paniceae, both distributed in tropical and sub-tropical regions. The distinction between Pennisetum and Cenchrus is not clearly defined, and several species that are now included in Cenchrus have previously been assigned to Pennisetum (Donaldio et al., 2009; Chemisquy et al., 2010). Previous studies of Pennisetum have focused on isozymic classification, genetic diversity (Tostain, 1994; Zhang et al., 2016b), and chromosomes (Martel et al., 2004; Techio et al., 2010; Zhang et al., 2015). However, these markers have low variation. Therefore, there is a need to develop effective genetic markers to facilitate the identification, conservation, utilization, and breeding of Pennisetum species.
The chloroplast is a unique organelle that is also the main site of photosynthesis, effectively turning the light of the sun into chemical energy in higher plants and some algae. It has its own independent genome that ranges in size from 120 to 180 kb in higher plants (Li C. et al., 2020; Li D. et al., 2020). It has a simple structure with a low molecular weight and multiple copies (Huang et al., 2020). The structure of the chloroplast genome is circular with quadripartite organization, including a small single-copy region (SSC), a large single-copy region (LSC), and a pair of inverted regions (IRs) (Kim and Lee, 2004; Jansen et al., 2005; Yang et al., 2010; Schwarz et al., 2015; Wang et al., 2016; Mader et al., 2018; Meng et al., 2018). In 1986, the first chloroplast genomes were reported from liverwort (Ohyama et al., 1986) and tobacco (Shinozaki et al., 1986). With the rapid development of next-generation sequencing technologies, such as Roche/454 GS FLX and Illumina GenomeAnalyzer, chloroplast genome sequences can be more efficiently and economically obtained. Approximately 4,100 plant chloroplast genomes have been published and are available in the NCBI database1. The chloroplast genome contains a large number of functional genes, and their application value in the study of species identification and phyletic evolution has gradually been widely accepted by researchers (Kim and Lee, 2004; Raubeson et al., 2007; Wang et al., 2008, 2016; Ma et al., 2017; Chen et al., 2018; Fan et al., 2018).
In this study, we completely sequenced and compared the chloroplast genome of four species of Pennisetum (P. alopecuroides, P. clandestinum, P. glaucum, and P. polystachion) based on next-generation sequencing methods. Our goal was to expand our understanding of the genetic divergence of Pennisetum and identify potential DNA barcodes for identifying Pennisetum species. This study also presents the first sequenced member of the genus Pennisetum. The results will provide basic data for molecular phylogenetic and evolutionary research at the species level.
Materials and Methods
Plant Materials and DNA Extraction
Seeds of P.alopecuroides (Sect. Gymnothrix), P. glaucum (Sect. Penicillaria),P.polystachion (Sect. Pennisetum), and P.clandestinum (Sect. Pennisetum) were acquired from the national medium term genebank of perennial herbage germplasm resources hosted by the Institute of Grassland Research of Chinese Academy of Agricultural Sciences and Professor Li from the National Animal Husbandry Station, under accession number of 01068, 08, HN495, and HN294, respectively. The samples were planted in the laboratory under suitable conditions at the Chinese Academy of Inspection and Quarantine until leaves appeared on the plants. Voucher specimens of each collected species were deposited at the Institute of Plant Quarantine, Chinese Academy of Inspection and Quarantine (Voucher No.2019099PA01, 2019099PG01, 2019099PP01, and 2019099PC01). Total genomic DNA was extracted from the fresh leaf of a single plant using the method of Li et al. (2013). The total DNA quantity was evaluated on 1.0% (w/v) agarose gel electrophoresis SYBR Green I, which can assess the concentration and purity of the DNA samples.
Illumina Sequencing and Assembly
Paired-end (PE) libraries of 350-bp insert sizes were constructed using the Nextera XT DNA library Prep Kit (Illumina Inc., San Diego, CA, United States) according to the manufacturer’s instructions. The Illumina HiSeq X-ten platform was used to perform sequencing, with PE 150-bp reads for each sample.
Raw reads were trimmed using Trimmomatic v0.32, and the resulting clean data were used for assembly and subsequent analysis (Bolger et al., 2014). The clean reads were then assembled into contigs using SPAdes 3.6.1 (Bankevich et al., 2012) with different K-mer parameters. The resulting contigs were selected for chloroplast genome-encoding contigs on BLAST searches. The selected contigs were secondarily assembled using Sequencher 5.4.5 (Gene Codes, Ann Arbor, MI).
Annotation and Comparative Analysis
The assembled chloroplast genome annotation was performed with Plann (Daisie and Quentin, 2015), and the P. glaucum chloroplast genome was used as the reference. The circular chloroplast genome map with structural features was developed using the OrganellarGenomeDRAW (OGDRAW) software (Stephan et al., 2019). CodonW software (Sharp and Li, 1987) was adopted to analyze the relative synonymous codon usage (RSCU). The assembled chloroplast genome sequences of four Pennisetum species were deposited in NCBI under the Genbank accession number MN180104, MW816925-MW816927. Comparative analysis of the complete chloroplast genomes of four Pennisetum species was performed using the mVISTA program (Kelly et al., 2004).
Analysis of Tandem and Simple Sequence Repeats (SSRs)
GMATA (Wang and Wang, 2016) software was used to detect the chloroplast SSRs in the four Pennisetum chloroplast genome sequences, in which the minimum numbers of repeats for mononucleotide, dinucleotides, trinucleotides, tetranucleotides, pentanucleotide, and hexanucleotides were 10, five, four, three, three, and three, respectively. Five types of repeat sequences, namely forward, reverse, complementary, palindromic, and tandem repeats, were identified in the Pennisetum chloroplast genome. Forward, reverse, palindrome, and complementary sequences were identified as described by running the REPuter program (Kurtz et al., 2001) with a minimum repeat size of 30 bp and similarities of 90% or greater sequence identity. Tandem repeats were identified using Tandem Repeats Finder2, with alignment parameters being set to two, seven, and seven for matches, mismatches, and indels, respectively. At the same time, the SSRs of the LSC, SSC, IR, and coding regions, introns, and intergenic regions that correspond to different regions were analyzed.
Sequence Divergence Analysis
The sequences of the Pennisetum chloroplast genomes were aligned using MAFFT v7 (Katoh and Standley, 2013), following which the alignments were inspected and manually adjusted using Se-Al 2.0 (Rambaut, 1996). MEGA 7.0 software was used to calculate the variable and parsimony-informative base sites and the k2p-distances among the chloroplast genomes (Sudhir et al., 2016).
To identify rapidly evolving molecular markers that can be used in further Pennisetum phylogenetic studies, four Pennisetum whole genomes were included in the comparisons to perform sliding window analysis and evaluate the nucleotide variability (Pi) among chloroplast genomes using DnaSP v5.10 software (Librado and Rozas, 2009). The step size was set to 200 bp, and the window length was set to 800 bp.
Phylogenetic Analysis
In this study, a total of 32 species, including 31 from Panicoideae and one outgroup species (Limnopoa meeboldii), were used for assessing phylogenetic relationships by constructing a maximum likelihood tree based on the sequences of 76 protein-coding genes. Of these, 28 chloroplast genomes were downloaded from the NCBI database. The sequences of 76 shared protein-coding genes were extracted from the GenBank formatted file containing all chloroplast genomes using Geneious v11, and alignments of genes were performed using MAFFT v7 (Kearse et al., 2012; Katoh and Standley, 2013), following which they were manually adjusted using Se-Al 2.0 (Rambaut, 1996) as needed. Finally, the data for the chloroplast genome sequences were used to perform phylogenetic analysis among the Pennisetum species.
The concatenated data were analyzed using the maximum likelihood (ML) and the Bayesian inference (BI) methodologies. Prior to ML and BI analyses, the best-fit model of sequence evolution, namely a general time reversible and gamma distribution (GTR + G) model, was selected using ModelFinder v.1.6 (Kalyaanamoorthy et al., 2017) under the Akaike Information Criterion. The ML analyses were performed using RAxML v.8.1 (Stamatakis, 2014) with 1,000 bootstrap replicates. The BI analysis was conducted using MrBayes v.3.2 (Fredrik et al., 2012) using the CIPRES Science Gateway. The Markov chain Monte Carlo (MCMC) algorithm was run for 2 × 5,000,000 generations, with trees sampled every 1,000 generations and the first 25% of generations discarded as burn-in. The remaining trees were used to construct a 50% majority rule consensus tree and estimate posterior probabilities. Posterior probabilities (PP) > 0.95 were considered significant support for a clade.
Results
Chloroplast Genome Organization of Pennisetum
In total, we obtained the chloroplast genomes of four Pennisetum species using the Illumina HiSeq X-ten platform. The coverage of four species were 697 (P. polystachion), 472 (P. glaucum), 230 bp (P. alopecuroides), and 1635 (P. clandestinum), respectively (Supplementary Table 1). All four species of Pennisetum that were sequenced had a typical double-stranded circular DNA molecule with a quadripartite structure. The assembled chloroplast genomes were 136,346 bp (P. polystachion), 138,119 bp (P. glaucum), 138,060 bp (P. alopecuroides), and 138,003 bp (P. clandestinum) in length. They consisted of a pair of IR regions (22,284–22,372 bp) separated by LSC (79,380–81,186 bp) and SSC (12,212–12,409 bp) regions (Figure 1 and Table 1). The size of the P. glaucum chloroplast genome (138,119 bp) was the largest among the four sequenced Pennisetum species, being 1,773 bp longer than that of P. polystachion, 59 bp longer than that of P. alopecuroides, and 116 bp longer than that of P. clandestinum. The total Guanine-Cytosine (GC) content was nearly identical in the chloroplast genomes of the four Pennisetum species. The GC content of both P. alopecuroides and P. glaucum was 38.6%, and that of both P. clandestinum and P. polystachion was 38.7%. Furthermore, the GC content in the IR regions (44.0–44.1%) was noticeably higher than that in the SSC (32.9–33.2%) and LSC (36.5–36.6%) regions in each chloroplast genome.
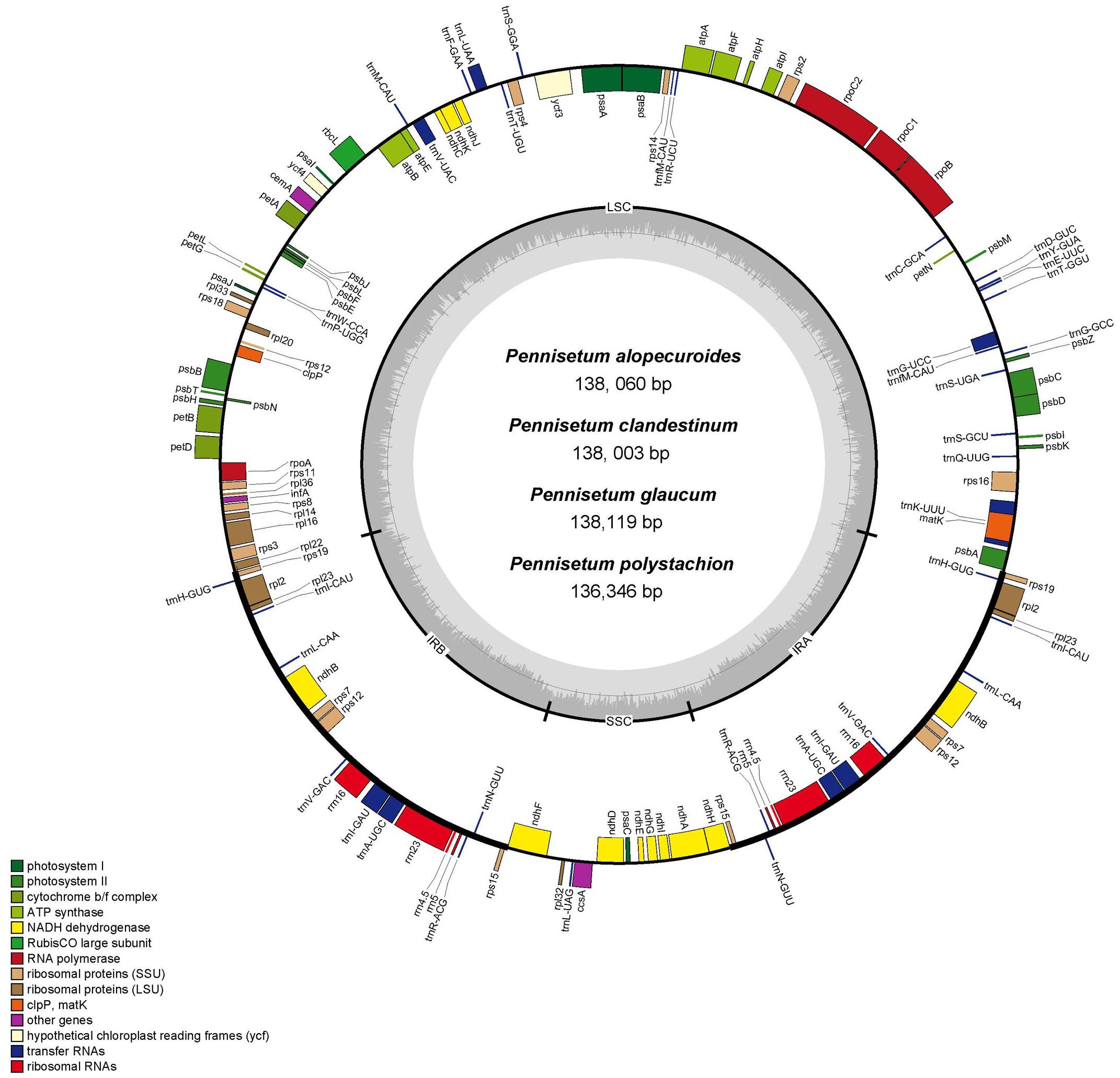
Figure 1. Gene map of the complete chloroplast genome of four Pennisetum species. Genes inside the circle are transcribed clockwise, and those outside are transcribed counterclockwise. The different colors of the blocks represent different functional groups. The darker gray color of the inner circle corresponds to the GC content, and the lighter gray color corresponds to the AT content.
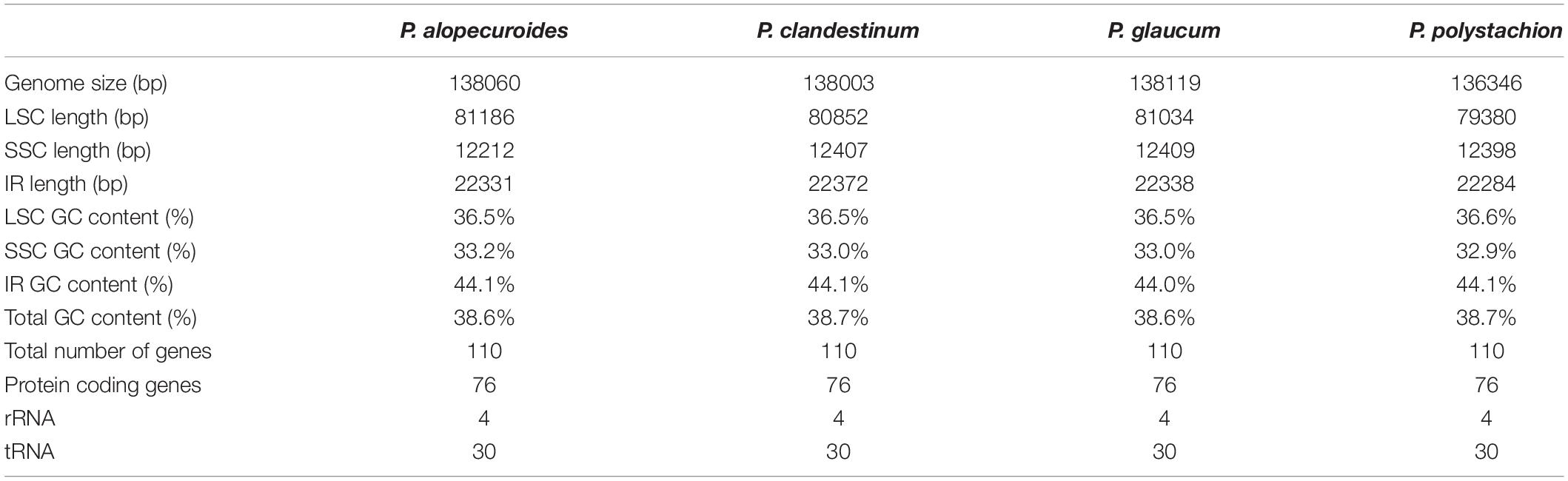
Table 1. Summary statistics for the assembly of the chloroplast genomes of four Pennisetum species.
Each of the analyzed chloroplast genomes contained a total of 110 unique genes, including 76 protein-coding genes, four ribosomal RNA (rRNA) genes, and 30 transfer RNA (tRNA) genes (Table 1). Among them, 19 of these genes were duplicated in the IR regions and contained seven protein-coding genes (rps15,rps12, rps7, ndhB, rpl23, rpl2, and rps19), eight tRNA genes (trnN-GUU, trnA-UGC, trnI-GAU, trnV-GAC, trnL-CAA, trnI-CAU, and trnH-GUG), and four rRNA genes (rrn23, rrn5, rrn4.5, rrn16). In total, 16 genes (ycf3, atpF, petB, petD, ndhA, ndhB, rps12, rpl2, rpl16, trnA-UGC, trnG-GCC, trnI-GAU, trnK-UUU, trnL-UAA, trnV-UAC, and clpP) with one intron were found (Table 2). Among the 16 intron containing genes, four genes (trnA-UGC, trnI-GAU, ndhB, and rpl2) occurred in both IR regions, one gene (ndhA) was in the SSC region, 10 genes (ycf3, atpF, petB, petD, rpl16, trnG-GCC, trnK-UUU, trnL-UAA, trnV-UAC, and clpP) were located in the LSC region, and one gene (rps12) is a trans-spliced gene with a first exon that was located in the LSC region and the other two exons located in both IR regions. The matK was located within the intron of trnK-UUU.
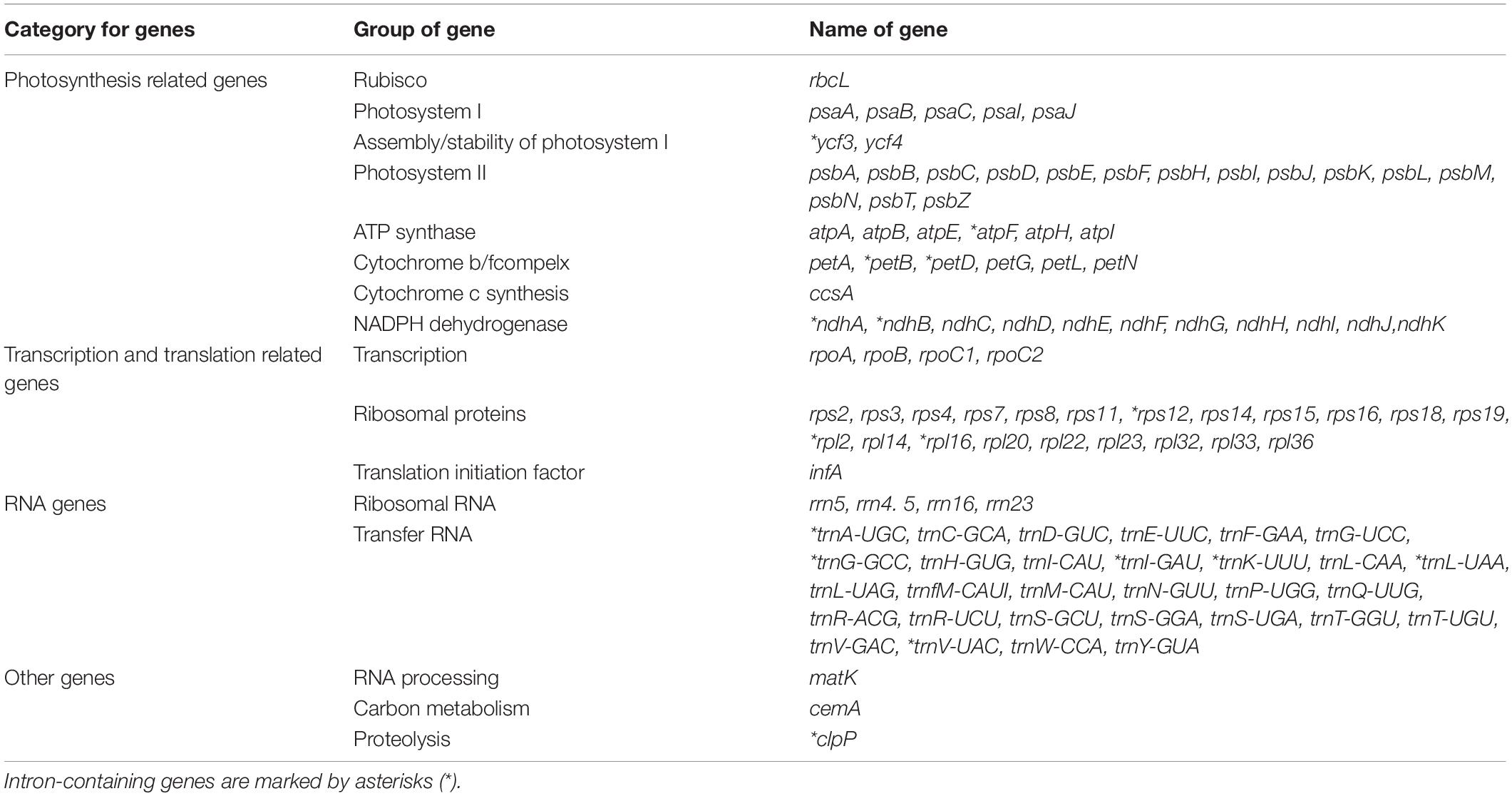
Table 2. List of annotated genes in the chloroplast genome of P. glaucum.
Comparative Analysis of the Chloroplast Genome
In this study, the annotated P. glaucum chloroplast genome was used as a reference in mVISTA for the alignment of the chloroplast genome among the four Pennisetum species and three Pancoideae species (Figure 2). The mVISTA-based identity plot showed conservation in DNA sequence and gene synteny with the whole chloroplast genome and revealed the regions with increased genetic variation. The comparison evidently showed considerable similarities in genome composition and size among the seven species. These species shows a closer relationship because they belongs to the same subfamily Panicoideae. Furthermore, the Pi value was calculated for the chloroplast genome sequences. A total of 139,958 sites, including 1,887 variable sites (1.35%) and 224 parsimony-informative sites (0.16%), were detected across the complete chloroplast genomes (Table 3). We found that the LSC and SSC regions were more variable than the two IR regions, with average Pi values of 0.00161 in the IR regions, 0.00914 in the LSC regions, and 0.01111 in SSC regions. The non-coding region was more variable than the coding region, and the intergenic space was the most variable in the chloroplast genomes.
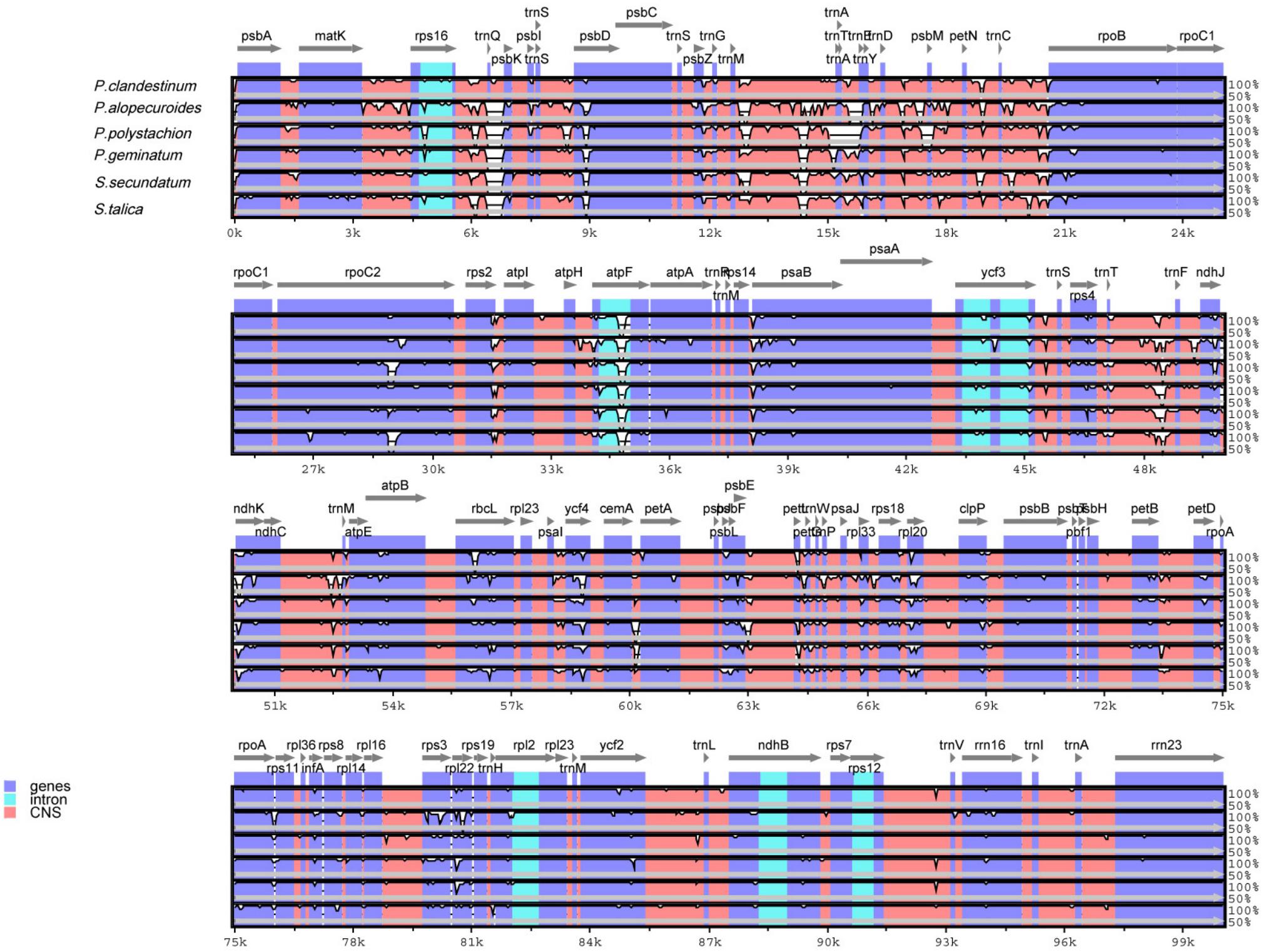
Figure 2. Comparison of chloroplast genomes of four Pennisetum species and other three Paniceae species (P. geminatum, S. secundatum, and S. talica) with P. glaucum as the reference using mVISTA program. The top line shows the genes in order. A cut-off of 70% identity was used for the plots and the Y-scale represents the percent identity between 50 and 100%.
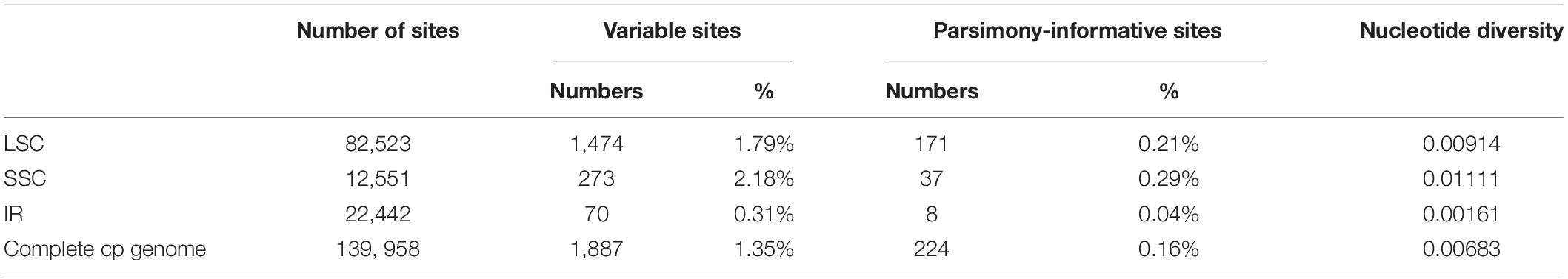
Table 3. Variable sites analyses in Pennisetum chloroplast gemomes.
The bounder positions of chloroplast genomes of the four Pennisetum species were comprehensively compared. The four chloroplast genomes sequenced in this study have identical gene positions in the boundary regions (Figure 3).
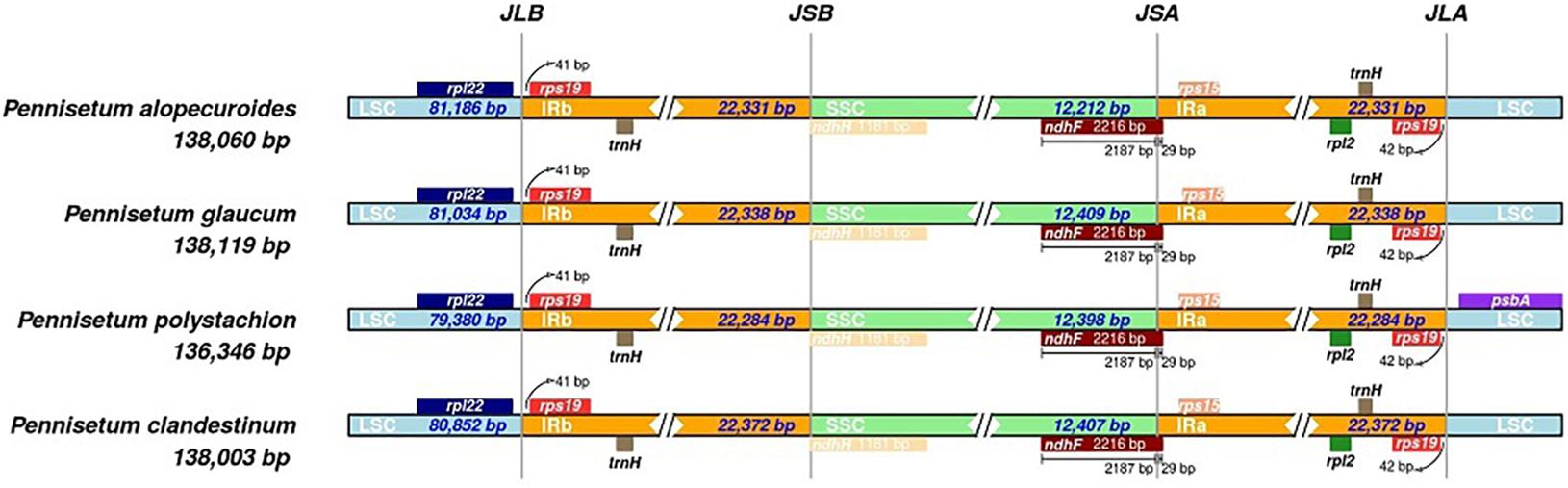
Figure 3. Comparison for border positions of LSC, SSC, and IR regions among four Pennisetum. Genes are denoted by boxes, and the gap between the genes and the boundaries is indicated by the number of bases unless the gene coincides with the boundary. Extensions of genes are also indicated above the boxes.
Sequence Divergence
To detect highly variable regions, we analyzed variable sites in the Pennisetum chloroplast genome by sliding window analysis using the software DnaSP (Figure 4). Nine divergent loci (psbA-matK, matK-rps16, trnN-trnT, trnY-trnD-psbM, petN-trnC, rbcL-psaI, psbE-petL, petA-psbJ, and rpl32-trnL) had a Pi value greater than or equal to 0.02. All of these nine divergent loci were intergenic regions and were present in the LSC region, except for rpl32-trnL, which occurred in the SSC region, with none detected in the IR region. These results also confirmed that the LSC and SSC regions were less conserved than the IR regions.
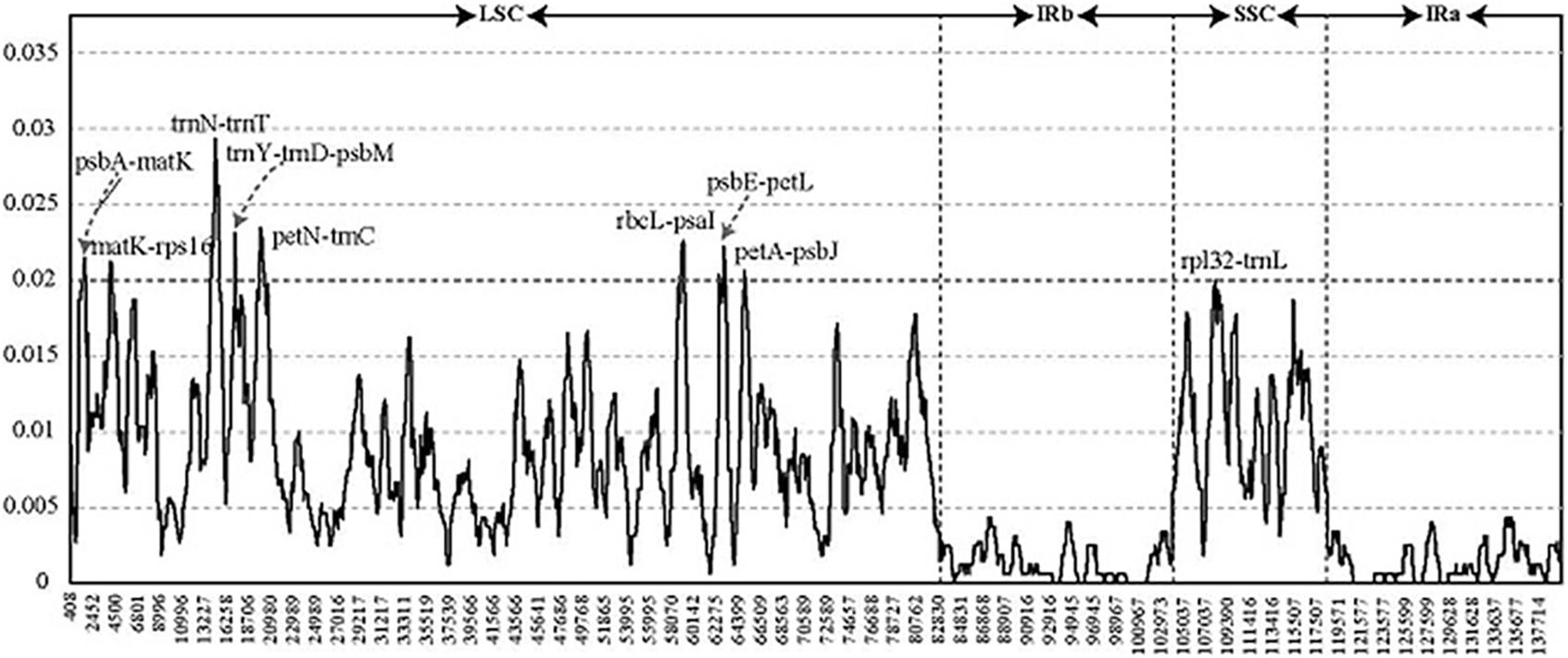
Figure 4. Comparative analysis of the nucleotide diversity values among four Pennisetum chloroplast genomes. X-axis: the position of the midpoint of a window (kb). Y-axis: nucleotide diversity of each window.
We evaluated the marker divergence in this study by comparison with three accepted candidate DNA barcodes (matK, rbcL, and psbA-trnH). We found that the newly identified markers had higher variability than these DNA barcodes (Table 4). The highest variability was detected in the rbcL-psaI region (4.68%), followed by that in the petN-trnC (4.51%), trnY-trnD-psbM (3.87%), trnN-trnT (3.74%), and psbE-petL (3.72%) regions.
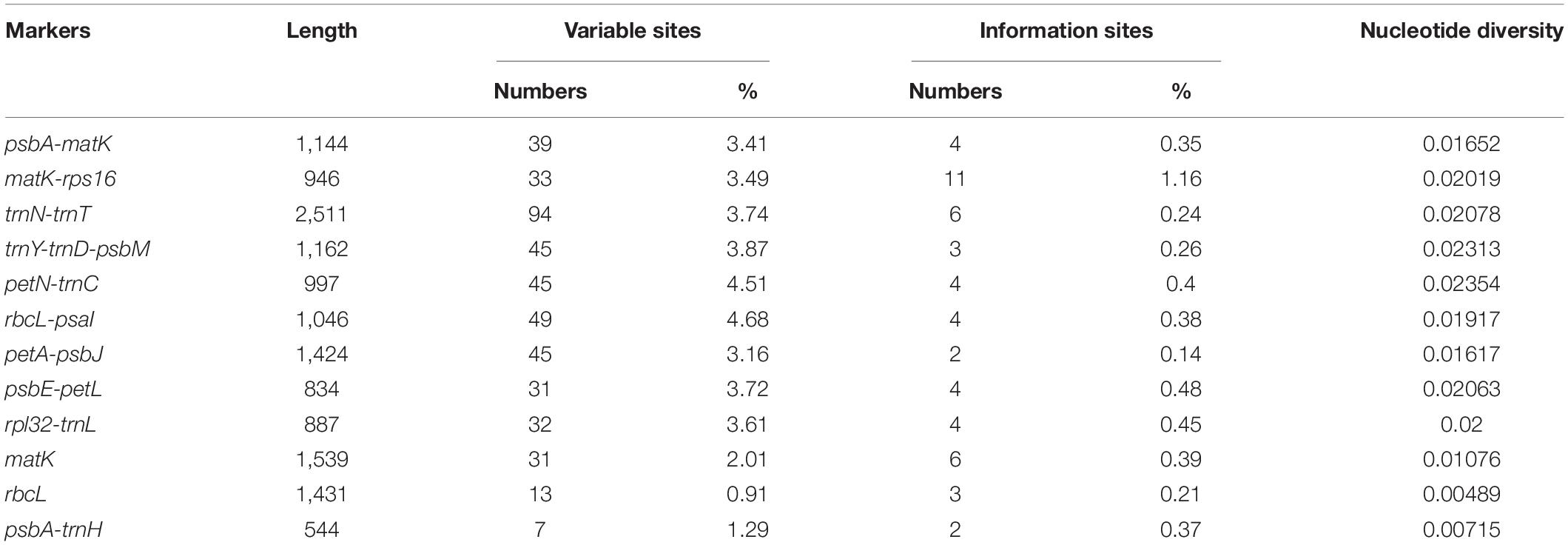
Table 4. Variability of nine novel markers and the three universal chloroplast DNA barcodes in Pennisetum.
SSR Analysis
A total of 226 SSRs were discovered in this study (Figure 5A). The chloroplast genomes of the four species harbored a similar number of SSRs (54, 62, 57, and 53). The number of SSRs was highest in P. clandestinum (62) and lowest in P. polystachion (53). Most of these SSRs were located in the LSC region (Figure 5B). The number of SSRs was similar between the IR and SSC regions. The numbers of mono-, di-, tri-, tetra-, penta-, and hexanucleotides were 148, 21, 4, 45, six, and two, respectively (Figure 5C). Additionally, pentanucleotide SSRs were found in three Pennisetum species, except for P. polystachion. Hexanucleotide SSRs were found in P. glaucum and P. polystachion. Mononucleotide repeats were the most abundant repeats, accounting for 65.5% of the total repeats, while tetranucleotide repeats accounted for 19.9%, and other SSRs accounted for 14.6%. Additionally, the majority of mononucleotide repeats belonged to A/T type (91.2%), and dinucleotide, trinucleotide, tetranucleotide, pentanucleotide, and hexanucleotide SSRs were especially rich in A or T (Figure 5D).
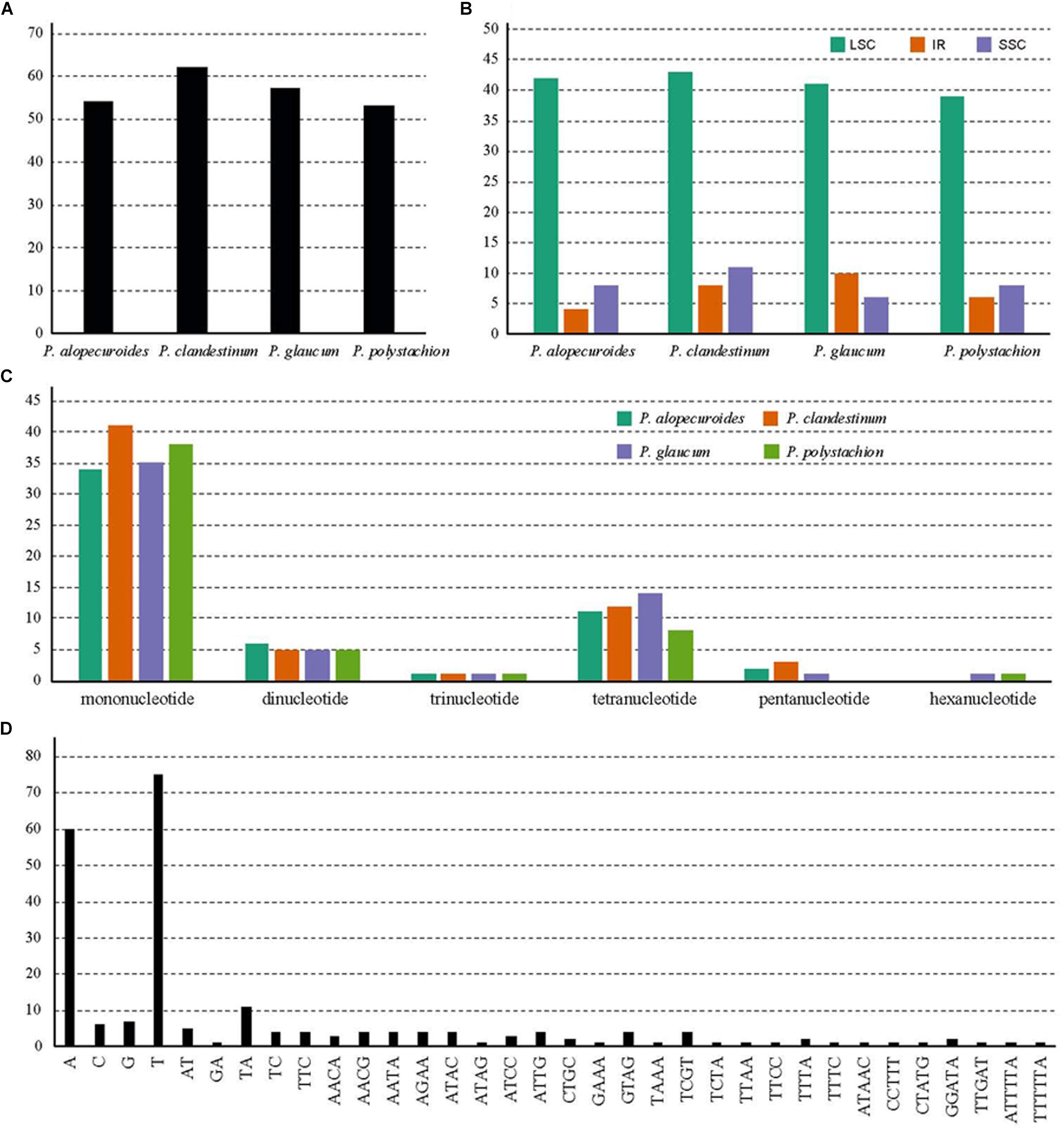
Figure 5. Analysis of simple sequence repeats (SSRs) in four Pennisetum chloroplast genomes. (A) The number of SSRs detected in the four chloroplast genomes; (B) The number of SSRs in large single-copy (LSC), inverted repeat (IR), and small single-copy (SSC) regions in the four chloroplast genomes; (C) The number of SSR types detected in the four sequenced chloroplast genomes; (D) The frequency of identified SSR motifs in different repeat class types.
Codon Usage
The RSCU of the chloroplast genomes of four Pennisetumspecies was calculated using all protein-coding genes. These genes are encoded in 45,448–46,020 codons. Of the 45,448–46,020 codons, leucine (Leu) which encoded by UUA, UUG, CUU, CUC, CUA, and CUG was the most plentiful amino acid, with a frequency of 9.96–10.22% encoded by these codons, comprising 4,557–4,704 of the total number of codons, then serine (Ser) which encoded by UCU, UCC, UCA, UCG, AGU, and AGC was the second abundant amino acid, with a frequency of 9.04–9.36% (4,158–4,253 codons), while tryptophan (Trp) which encoded by UGG was the least frequently encoded amino acid, with a proportion of 1.45–1.5% (665–688 codons) (Figure 6 and Supplementary Table 2). The RSCU can eliminate the influence of amino acid composition on codon usage. Among the codons which the RSCU values are greater than one, the majority of the codons ended with A or U, On the contrary, the codons ended with C or G had RSCU values less than one. This result is consistent with previous studies (Lu et al., 2018; Huang et al., 2020).
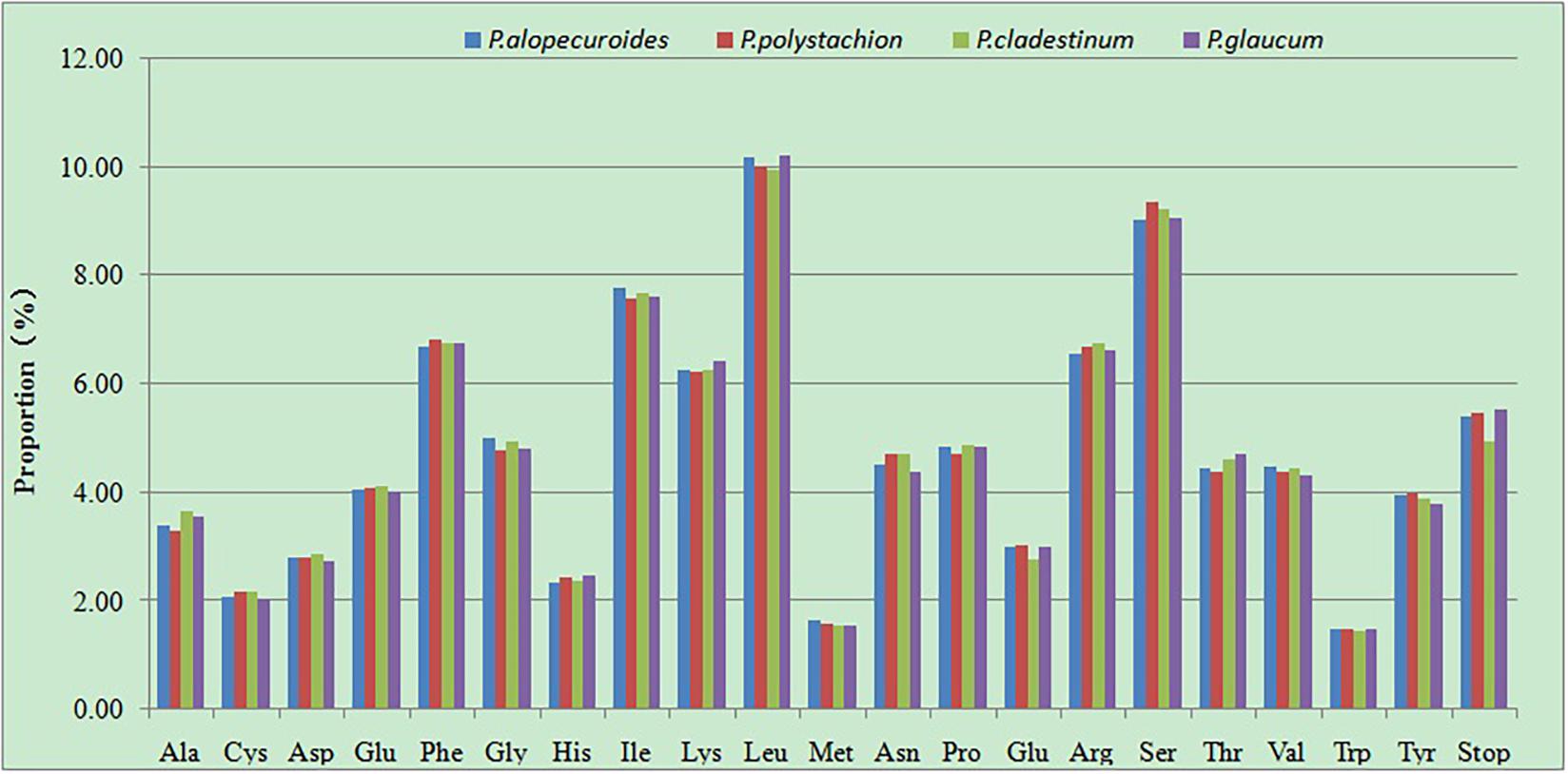
Figure 6. Amino acid proportion in four Pennisetum species protein-coding sequences.
Phylogenetic Analysis
In order to resolve the phylogenetic relationships of Pennisetum, the complete chloroplast genomes of 31 species in the Poaceae subfamily Panicoideae and one outgroup in the Poaceae subfamily Micrairoideae were used to construct phylogenetic trees (Figure 7). The ML tree and BI trees contained 19 of 29 nodes, with ML bootstrap values of 100%, and BI posterior probabilities of 1.0. Both the ML and BI phylogenetic analyses strongly demonstrated Pennisetum as a monophyletic group with 100% bootstrap support and 1.0 posterior probability. Pennisetum polystachion and P. glaucum clustered together with 95% bootstrap support and 1.0 posterior probability, and P. clandestinum and P. alopecuroides clustered together with 100% bootstrap support and 1.0 posterior probability. The clade formed by all four Pennisetum species was most closely related to the species Setaria italica.
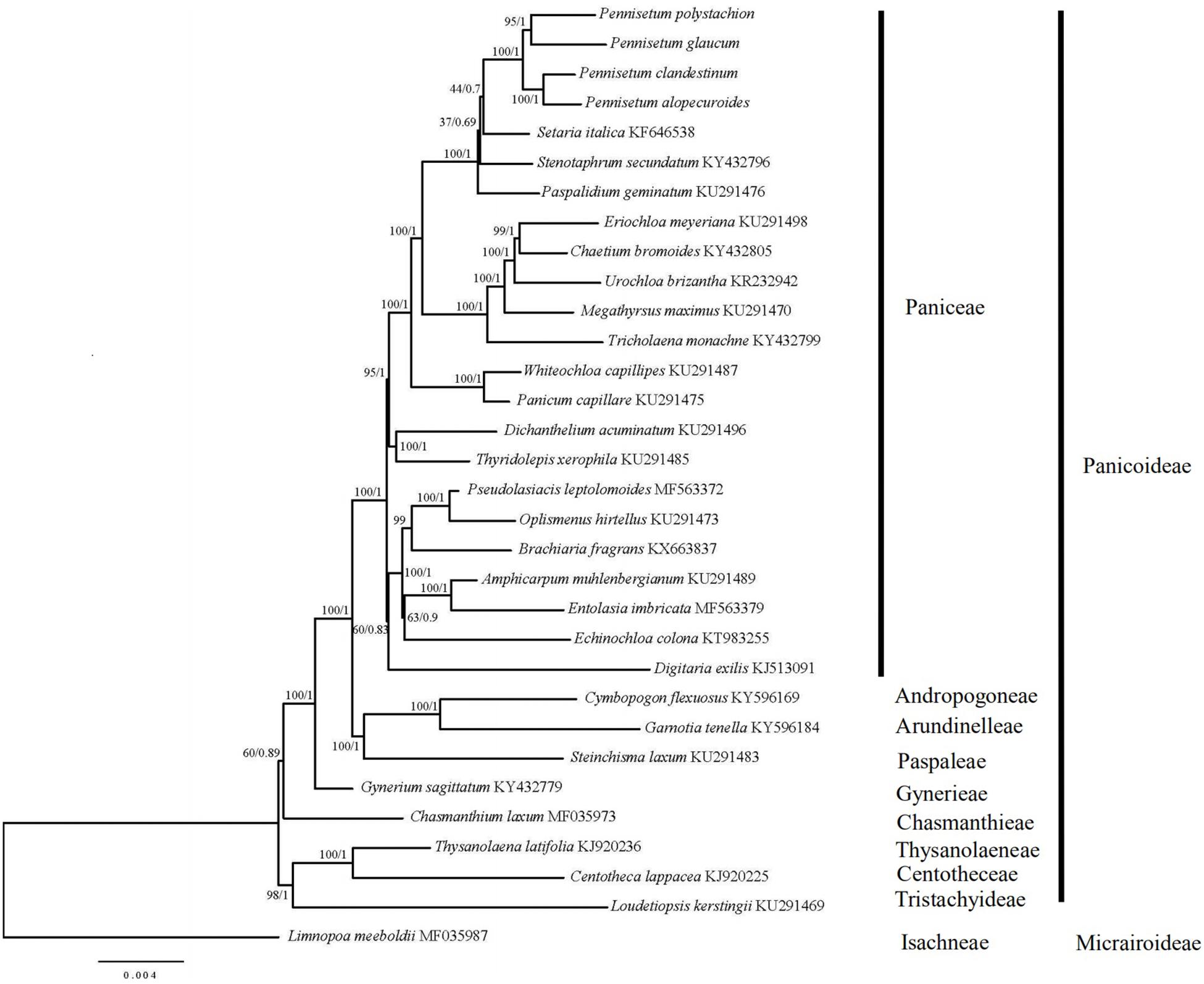
Figure 7. Phylogenetic tree constructed from 32 chloroplast genomes using ML and BI method. The bootstrap values were based on 1000 replicates and were indicated next to the branches.
Discussion
Chloroplast Sequence Variation
In the current study, the chloroplast genome sequences of four species of Pennisetum (P. alopecuroides, P. clandestinum, P. glaucum, and P. polystachion) were sequenced using Illumina sequencing and compared. The results showed that the size of the genome ranged from 136,346 (P. polystachion) to 138,119 bp (P. glaucum), which was longer than the genome of S. italica (Wang and Gao, 2016) and shorter than that of Stenotaphrum secundatum and Paspalidium geminatum (Burke et al., 2016). This length was within the size range of the chloroplast genomes of most angiosperms (Dong et al., 2013). Angiosperm chloroplast genomes usually contain 110–130 genes, with up to 80 protein-coding genes, four rRNA genes, and approximately 30 tRNA genes (Jansen and Ruhlman, 2012). In the current study, all four Pennisetum chloroplast genomes had 110 unique genes consisting of 76 protein-coding genes, four rRNA genes, and 30 tRNA genes. Comparative analysis of the Pennisetum chloroplast genomes using mVISTA revealed the DNA sequence similarities among related species. They were similar in structure, content, and order (Figure 1 and Table 1). The genome size and organization of the intergenic spacers were consistent with the observed variations in the size of the Pennisetum chloroplast genomes. Although the IR regions are more conserved than the SSC and LSC regions in the chloroplast genome, the evolutionary events and variation in the size of the chloroplast genomes in different plants could also be attributed to the expansion and contraction of the border between the SC and IR regions (Raubeson et al., 2007; Wang et al., 2008; Wang and Messing, 2011; Meng et al., 2018; Zhao et al., 2018; Androsiuk et al., 2020). In this study, the genes rps19, ndhF, ndhH, and psbA were located at the juncture of the LSC/IRB, IRB/SSC, SSC/IRA, and IRA/LSC borders. Similar to most angiosperms, IR boundaries were found within ndhF and rps19 genes in the presented chloroplast genomes of Pennisetum.
Molecular Markers
SSRs, known as microsatellites, are significant repetitive elements of the entire genome that consist of only one or a few tandemly repeated nucleotides. They are the same units with different unit numbers located in the homologous regions and play important roles in many applications, including species identification, population genetics, and phylogenetic studies (Yang et al., 2011; Jiao et al., 2012; Zhang et al., 2016a,b; Androsiuk et al., 2020). The SSRs in chloroplast genomes are usually distributed in intergenic regions (Zhao et al., 2015). In the SSR analysis, 226 SSR loci were found in the four Pennisetum chloroplast genomes (Figure 4). Most of these SSR loci were located in the LSC region, followed by the SSC and IR regions. The most abundant were mononucleotide repeats, which contributed to A/T richness. These results are consistent with most reported angiosperms (Cui et al., 2019; Gao et al., 2019; Li W. et al., 2019; Li Y. et al., 2019). Analysis of the SSRs identified in the chloroplast genome of Pennisetum indicated that the chloroplast genome is highly conserved among the four species. The results showed that the genes of the chloroplast genome were largely identical, whereas the intergenic regions were more variable. In general, the chloroplast genome SSRs of the four Pennisetum exhibited abundant variation and are thus likely useful for detecting polymorphisms at the intraspecific level as well as developing markers for Pennisetum species for future evolutionary and genetic diversity studies.
DNA barcoding is a molecular technique for characterizing species of organisms using a short, standardized DNA region. Since it was first proposed in 2003 (Hebert et al., 2003), it has been used for a wide range of purposes, including to identify species (Badotti et al., 2017), to discover new species (Zhao et al., 2011), to support food safety and authenticity of labeling by confirming identity or purity (Galimberti et al., 2012; Huxley-Jones et al., 2012), and to help identify candidate exemplar taxa for comprehensive phylogenetic studies (Hajibabaei et al., 2007). The cytochrome coxidase subunit I (COI) gene has been widely used in previous studies and exhibits a high ability to distinguish between animal species requirements (Hebert et al., 2003). In plants, many loci have been proposed as plant barcodes. The CBOL (Consortium for the Barcode of Life) Plant Working Group as well as previous studies have evaluated and recommended the use of the chloroplast-derived DNA barcode fragments rbcL and matK (Kress and Erickson, 2007; CBOL Plant Working Group, 2009; Kool et al., 2012). In addition, trnH-psbA, the internal spacer region of the chloroplast gene, and the ITS region of the nuclear genome were also investigated (Kress and Erickson, 2007; Chen et al., 2010; Bellanger et al., 2015). Despite the above, the molecular data for Pennisetum are highly limited. In the four Pennisetum species with completely sequenced chloroplast genomes, the genetic variation in the chloroplast regions of matK, rbcL, and psbA-trnH, which are widely used as DNA barcodes in plants, was lower than expected. However, genome-wide comparative analyses based on Pi value supported the identification of nine highly variable regions that could be utilized as a source of potential DNA barcodes for species identification and the reconstruction of phylogenetic relationships within this plant group: psbA-matK, matK-rps16, trnN-trnT, trnY-trnD-psbM, petN-trnC, rbcL-psaI, psbE-petL, petA-psbJ, and rpl32-trnL. Highly Pi values in the psbE-petL, rpl32-trnL, and petA-psbJ regions were also reported in other plants (Dong et al., 2012). The petN-trnC and rbcL-psaI regions have been used as molecular markers for phylogenetic analyses (Morris and Duvall, 2010; Iu Barkalov and Kozyrenko, 2014). Overall, these highly divergent regions presented abundant information for molecular marker development in plant identification and investigating the phylogenetic relationships of Pennisetum.
Phylogenetic Analysis
Chloroplast genome sequences have been successfully used for phylogenetic studies of angiosperms (Jansen et al., 2007; Huang et al., 2014; Kim et al., 2015). The phylogenetic position of Pennisetum in Poaceae was inferred by analyzing the complete chloroplast genome and 76 genes shared among 32 species in Poaceae, for which the full chloroplast genome sequence had been generated and officially published in the database of the NCBI. The ML and BI trees exhibited similar phylogenetic topologies. The phylogenetic trees (ML and BI) demonstrated a significant relationship among Poaceae with high bootstrap values and posterior probabilities (Figure 6). The results showed that the main relationship was the same as other studies among the Family Poaceae (Robert et al., 2015; Soreng et al., 2015). Paniceae, Andropogoneae, Arundinelleae, Paspaleae, Gynerieae, Chasmanthieae, Centotheceae, and Tristachyideae that should placed in Panicoideae lineage clustered together [BP(ML) = 100%, PP = 1.00]. The genus Limnopoa was found to be the earliest diverging lineage. Our research object genus Pennisetum form a high support clade as a paraphyletic group [BP(ML) = 100%, PP = 1.00]. The clade formed by all four Pennisetum species is a sister to the species Setaria italica. And the genus Pennisetum and other 19 genus should be placed in the Paniceae lineage. In the genus Pennisetum, P. polystachion, and P. glaucum could be confidently assigned to one clade [BP(ML) = 95%, PP = 1.00], while P. clandestinum and P. alopecuroides clustered together with high support rate [BP(ML) = 100%, PP = 1.00]. The results are consistent with previous studies (Martel et al., 1997). Our study on the Pennisetum chloroplast genome provides valuable information on these four Pennisetum species and will facilitate future phylogenetic studies as well as other studies of Pennisetum species.
Conclusion
In this study, we sequenced and compared the chloroplast genomes of four Pennisetum species. The chloroplast genomes of the four species were highly conserved with respect to gene orientation, gene content, and GC content. The goal of the present study was to expand our understanding of the diversity of the genus Pennisetum and identify reliable and effective DNA barcodes for Pennisetum species. Several non-coding regions or several intergenic spacer regions (psbA-matK, matK-rps16, trnN-trnT, trnY-trnD-psbM, petN-trnC, rbcL-psaI, petA-psbJ, psbE-petL, and rpl32-trnL) were found to be the most appropriate DNA barcodes for evolutionary and genetic diversity studies of Pennisetum. The molecular data in this study enhance the chloroplast genome resources for the family Poaceae and genus Pennisetum and could be useful for species identification, phylogenic analysis, and evolutionary history analysis.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, MN180104; https://www.ncbi.nlm.nih.gov/genbank/, MW816925; https://www.ncbi.nlm.nih.gov/genbank/, MW816926; and https://www.ncbi.nlm.nih.gov/genbank/, MW816927.
Author Contributions
JX designed the experiment and drafted the manuscript. CL collected the samples and performed the experiment. JX and YS analyzed the data. ML contributed reagents and analysis tools. All of the authors have read and approved the final manuscript.
Funding
The study was financially supported by the National Key Research and Development Program of China (2017YFF0210300 and 2017YFF0210301) and the Basic Scientific Research Foundation of Chinese Academy of Inspection and Quarantine (2018JK010).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank National Medium Forage Bank and Yurong Li from National Animal Husbandry Station for help in specimen collection. We also thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.687844/full#supplementary-material
Footnotes
References
Abdi, S., Dwivedi, A., Kumar, S., and Bhat, V. (2019). Development of EST-SSR markers in Cenchrus ciliaris and their applicability in studying the genetic diversity and cross-species transferability. J. Genet. 98:101. doi: 10.1007/s12041-019-1142-x
Androsiuk, P., Jastrzȩbski, J. P., Paukszto, Ł, Makowczenko, K., Okorski, A., Pszczółkowska, A., et al. (2020). Evolutionary dynamics of the chloroplast genome sequences of six Colobanthus species. Sci. Rep. 10:11522. doi: 10.1038/s41598-020-68563-5
Badotti, F., Oliveira, F. D., Garcia, C. F., Vaz, A., Fonseca, P., Nahum, L. A., et al. (2017). Effectiveness of its and sub-regions as dna barcode markers for the identification of basidiomycota (fungi). BMC Microbiol. 17:42. doi: 10.1186/s12866-017-0958-x
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bellanger, J.-M., Moreau, P.-A., Corriol, G., Bidaud, A., Chalange, R., Dudova, Z., et al. (2015). Plunging hands into the mushroom jar: a phylogenetic framework for Lyophyllaceae (Agaricales. Basidiomycota). Genetica 143, 169–194. doi: 10.1007/s10709-015-9823-8
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brunken, J. N. (1977). A Systematic Study of Pennisetum Sect. Pennisetum (Gramineae). Amer. J. Bot. 64, 161–176. doi: 10.2307/2442104
Burke, S. V., Wysocki, W. P., Zuloaga, F. O., Craine, J. M., and Duvall, M. R. (2016). Evolutionary relationships in panicoid grasses based on plastome phylogenomics (Panicoideae. Poaceae). BMC Plant Biol. 16:140. doi: 10.1186/s12870-016-0823-3
CBOL Plant Working Group (2009). A DNA barcode for land plants. Proc. Natl. Acad. Sci. U. S. A. 106, 12794–12797.
Chemisquy, M. A., Giussani, L. M., Scataglini, M. A., Kellogg, A. E., and Morrone, O. (2010). Phylogenetic studies favour the unification of Pennisetum, Cenchrus and Odontelytrum (Poaceae): a combined nuclear, plastid and morphological analysis, and nomenclatural combinations in Cenchrus. Ann. Bot. 106, 107–130. doi: 10.1093/aob/mcq090
Chen, H., Shao, J., Zhang, H., Jiang, M., Huang, L., Zhang, Z., et al. (2018). Sequencing and analysis of Strobilanthes cusia (ees) Kuntze chloroplast genome revealed the rare simultaneous contraction and expansion of the inverted repeat region in angiosperm. Front. Plant Sci. 9:324. doi: 10.3389/fpls.2018.00324
Chen, S., Yao, H., Han, J., Liu, C., Song, J., Shi, L., et al. (2010). Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PLoS One 5:e8613. doi: 10.1371/journal.pone.0008613
Cui, Y., Nie, L., Sun, W., Xu, Z., Wang, Y., Yu, J., et al. (2019). Comparative and phylogenetic analyses of ginger (Zingiber officinale) in the family Zingiberaceae based on the complete chloroplast genome. Plants 8:283. doi: 10.3390/plants8080283
Daisie, I. H., and Quentin, C. B. C. (2015). Plann: a command-line application for annotating plastome sequences. Appl. Plant Sci. 3:1500026. doi: 10.3732/apps.1500026
DeLisle, D. G. (1963). Taxonomy and distribution of the genus Cenchrus. Iowa State Coll. J. Sci. 37, 259–351.
Donaldio, S., Giussani, L. M., Kellogg, E. A., Zuolaga, F. O., and Morrone, O. (2009). A preliminary molecular phylogeny of Pennisetum and Cenchrus (Poaceae-Paniceae) based on the trnL-F, rpl16 chloroplast markers. Taxon 58, 392–404. doi: 10.1002/tax.582007
Dong, W., Liu, J., Yu, J., Wang, L., and Zhou, S. (2012). Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS One 7:e35071. doi: 10.1371/journal.pone.0035071
Dong, W., Xu, C., Cheng, T., Lin, K., and Zhou, S. (2013). Sequencing angiosperm plastid genomes made easy: a complete set of universal primers and a case study on the phylogeny of Saxifragales. Genome Biol. Evol. 5, 989–997. doi: 10.1093/gbe/evt063
Fan, W., Wu, Y., Yang, J., Shahzad, K., and Li, Z. (2018). Comparative chloroplast genomics of Dipsacales species: insights into sequence variation, adaptive evolution, and phylogenetic relationships. Front. Plant Sci. 9:689. doi: 10.3389/fpls.2018.00689
Fredrik, R., Maxim, T., Paul, V. D. M., Ayres, D. L., Aaron, D., Sebastian, H., et al. (2012). MrBayes 3.2: efcient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Galimberti, A., De Mattia, F., Losa, A., Bruni, I., Federici, S., Casiraghi, M., et al. (2012). DNA barcoding as a new tool for food traceability. Food Res. Int. 50, 55–63. doi: 10.1016/j.foodres.2012.09.036
Gao, C., Deng, Y., and Wang, J. (2019). The complete chloroplast genomes of Echinacanthus species (Acanthaceae): phylogenetic relationships, adaptive evolution, and screening of molecular markers. Front. Plant Sci. 9:1989. doi: 10.3389/fpls.2018.01989
Hajibabaei, M., Singer, G. A. C., Hebert, P. D. N., and Hickey, D. A. (2007). DNA barcoding: how it complements taxonomy, molecular phylogenetics and population genetics. Trends Genet. 23, 167–172. doi: 10.1016/j.tig.2007.02.001
Hebert, P. D. N., Cywinska, A., Ball, S. L., and DeWaard, J. R. (2003). Biological identifcations through DNA barcodes. Proc. R. Soc. Lond. B. 270, 313–321. doi: 10.1098/rspb.2002.2218
Huang, H., Shi, C., Liu, Y., Mao, S., and Gao, L. (2014). Thirteen Camellia chloroplast genome sequences determined by high-throughput sequencing: genome structure and phylogenetic relationships. BMC Evol. Biol. 14:151. doi: 10.1186/1471-2148-14-151
Huang, S., Ge, X., Cano, A., Salazar, B. G. M., and Deng, Y. (2020). Comparative analysis of chloroplast genomes for five Dicliptera species (Acanthaceae): molecular structure, phylogenetic relationships, and adaptive evolution. PeerJ. 8:e8450. doi: 10.7717/peerj.8450
Huxley-Jones, E., Shaw, J. L., Fletcher, C., Parnell, J., and Watts, P. C. (2012). Use of DNA barcoding to reveal species composition of convenience seafood. Conserv. Biol. 26, 367–371. doi: 10.1111/j.1523-1739.2011.01813.x
Iu Barkalov, V., and Kozyrenko, M. M. (2014). Phylogenetic relationships of Salix L. subg. Salix species (Salicaceae) according to sequencing data of intergenic spacers of the chloroplast genome and its rDNA. Russ. J. Genet. 50, 828–837. doi: 10.1134/s1022795414070035
Jansen, R. K., and Ruhlman, T. A. (2012). Plastid Genomes of Seed Plants. Netherlands: Springer, 103–126.
Jansen, R. K., Cai, Z., Raubeson, L. A., Daniell, H., Leebens-Mack, J., Müller, K. F., et al. (2007). Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. U. S. A. 104, 19369–19374. doi: 10.1073/pnas.0709121104
Jansen, R. K., Raubeson, L. A., Boore, J. L., de Pamphilis, C. W., Chumley, T. W., Haberle, R. C., et al. (2005). Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. 395, 348–384. doi: 10.1016/s0076-6879(05)95020-9
Jiao, Y., Jia, H., Li, X., Chai, M., Jia, H., Chen, Z., et al. (2012). Development of simple sequence repeat (SSR) markers from a genome survey of chinese bayberry (Myrica rubra). BMC Genomics 13:201. doi: 10.1186/1471-2164-13-201
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment sofware version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kelly, A. F., Lior, P., Alexander, P., Edward, M. R., and Inna, D. (2004). VISTA:Computational tools for comparative genomics. Nucleic Acid Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Kim, K. J., and Lee, H. L. (2004). Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 11, 247–261. doi: 10.1093/dnares/11.4.247
Kim, K., Lee, S.-C., Lee, J., Yu, Y., Yang, K., Choi, B.-S., et al. (2015). Complete chloroplast and ribosomal sequences for 30 accessions elucidate evolution of Oryza AA genome species. Sci. Rep. 5:15655. doi: 10.1038/srep15655
Kool, A., De Boer, H. J., Krüger, A., Rydberg, A., Abbad, A., Bjorkörk, L., et al. (2012). Molecular identication of commercialized medicinal plants in southern Morocco. PLoS One 7:e39459. doi: 10.1371/journal.pone.0039459
Kress, W. J., and Erickson, D. L. (2007). A two-locus global DNA barcode for land plants: the coding rbcL gene complements the non-coding trnH-psbA spacer region. PLoS One 2:e508. doi: 10.1371/journal.pone.0000508
Kumar, S., Saxena, S., Rai, A., Radhakrishna, A., and Kaushal, P. (2019). Ecological, genetic, and reproductive features of Cenchrus species indicate evolutionary superiority of apomixis under environmental stresses. Ecol. Indic. 105, 126–136. doi: 10.1016/j.ecolind.2019.05.036
Kurtz, S., Choudhuri, J. V., Enno, O., Chris, S., Jens, S., and Robert, G. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Li, C., Zhao, Y., Xu, Z., Yang, G., Peng, J., and Peng, X. (2020). Initial characterization of the chloroplast genome of Vicia sepium, an important wild resource plant, and related inferences about its evolution. Front. Genet. 11:73. doi: 10.3389/fgene.2020.00073
Li, D., Zhu, G., Xu, Y., Ye, Y., and Liu, J. (2020). Complete chloroplast genomes of three medicinal Alpinia species: genome organization, comparative analyses and phylogenetic relationships in family Zingiberaceae. Plants 9:286. doi: 10.3390/plants9020286
Li, J., Wang, S., Yu, J., Wang, L., and Zhou, S. (2013). A modified method of plant DNA extract. Chin. Bull. Bot. 48, 72–78.
Li, W., Zhang, C., Guo, X., Liu, Q., and Wang, K. (2019). Complete chloroplast genome of Camellia japonica genome structures, comparative and phylogenetic analysis. PLoS One 14:e0216645. doi: 10.1371/journal.pone.0216645
Li, Y., Sylvester, S. P., Li, M., Zhang, C., Li, X., Duan, Y., et al. (2019). The complete plastid genome of Magnolia zenii and genetic comparison to Magnoliaceae species. Molecules 24:261. doi: 10.3390/molecules24020261
Librado, P., and Rozas, J. (2009). DnaSP v5: a sofware for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lu, Q. X., Ye, W. Q., Lu, R. S., Xu, W. Q., and Qiu, Y. X. (2018). Phylogenomic and comparative analyses of complete plastomes of Croomia and Stemona (Stemonaceae). Int. J. Mol. Sci. 19:2383. doi: 10.3390/ijms19082383
Ma, Q., Li, S., Bi, C., Hao, Z., Sun, C., and Ye, N. (2017). Complete chloroplast genome sequence of a major economic species. Ziziphus jujuba (Rhamnaceae). Curr. Genet. 63, 117–129. doi: 10.1007/s00294-016-0612-4
Mader, M., Pakull, B., Blanc-Jolivet, C., Paulini-Drewes, M., Bouda, Z., Degen, B., et al. (2018). Complete chloroplast genome sequences of four Meliaceae species and comparative analyses. Int. J. Mol. Sci. 19:701. doi: 10.3390/ijms19030701
Martel, E., Nay, D., Siljak-Yakoviev, S., Brown, S., and Sarr, A. (1997). Genome size variation and basic chromosome number in pearl millet and fourteen related Pennisetum species. J. Hered. 88, 139–143. doi: 10.1093/oxfordjournals.jhered.a023072
Martel, E., Poncet, V., Lamy, F., Siljak-Yakovlev, S., Lejeune, B., and Sarr, A. (2004). Chromosome evolution of Pennisetum species (Poaceae): implications of its phylogeny. Plant Sys. Evol. 249, 139–149. doi: 10.1007/s00606-004-0191-6
Meng, X., Xian, Y., Xiang, L., Zhang, D., Shi, Y., Wu, M., et al. (2018). Complete chloroplast genomes from Sanguisorba: identity and variation among four species. Molecules 23:2137. doi: 10.3390/molecules23092137
Morris, L. M., and Duvall, M. R. (2010). The chloroplast genome of Anomochloa marantoidea (Anomochlooideae. Poaceae) comprises a mixture of grass-like and unique features. Am. J. Bot. 97, 620–627. doi: 10.3732/ajb.0900226
Ohyama, K., Fukuzawa, H., Kohchi, T., Shirai, H., Sano, T., Sano, S., et al. (1986). Chloroplast gene organization deduced from complete sequence of liverwort Marchantia polymorpha chloroplast DNA. Nature 322, 572–574. doi: 10.1038/322572a0
Pilger, R. (1940). “Gramineae,” in Die naturlichen Pflanzenfamilien, eds A. Engler and K. PrantI (Leipzig: Wilhelm Engelman).
Raubeson, L. A., Peery, R., Chumley, T. W., Dziubek, C., Fourcade, H. M., Boore, J. L., et al. (2007). Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics 8:174. doi: 10.1186/1471-2164-8-174
Schwarz, E. N., Ruhlman, T. A., Sabir, J. S. M., Hajrah, N. H., Alharbi, N. S., Almalki, A. L., et al. (2015). Plastid genome sequences of legumes reveal parallel inversions and multiple losses of rps16 in papilionoids. J. Sys. Evol. 53, 458–468. doi: 10.1111/jse.12179
Sharp, P. M., and Li, W. H. (1987). The codon Adaptation Index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15, 1281–1295. doi: 10.1002/ajpa.21279
Shinozaki, K., Ohme, M., Tanaka, M., Wakasugi, T., Hayashida, N., Matsubayashi, T., et al. (1986). The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. Plant Mol. Biol. Report. 5, 2043–2049. doi: 10.1002/j.1460-2075.1986.tb04464.x
Soreng, R. J., Peterson, P. M., Romaschenko, K., Davidse, G., Zuloaga, F. O., Judziewicz, E. J., et al. (2015). A worldwide phylogenetic classification of the Poaceae (Gramineae). J. Sys. Evol. 53, 117–137. doi: 10.1111/jse.12150
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stephan, G., Pascal, L., and Ralph, B. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1093/nar/gkz238
Sudhir, K., Glen, S., and Koichiro, T. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Techio, V. H., Davide, L. C., Cagliari, A., Barbosa, S., and Pereira, A. V. (2010). Karyotipic asymmetry of both wild and cultivated species of Pennisetum. Bragantia 69, 273–279. doi: 10.1590/S0006-87052010000200003
Tostain, S. (1994). Isozymic classification of pearl millet (Pennisetum glaucum. Poaceae) landraces from Niger (West Africa). Plant Sys. Evol. 193, 81–93. doi: 10.1007/BF00983542
Wang, L., Wuyun, T., Du, H., Wang, D., and Cao, D. (2016). Complete chloroplast genome sequences of Eucommia ulmoides: genome structure and evolution. Tree Genet. Genomes 12:12. doi: 10.1007/s11295-016-0970-6
Wang, R., Cheng, C., Chang, C., Wu, C., Su, T., and Chaw, S. (2008). Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol. Biol. 8:36. doi: 10.1186/1471-2148-8-36
Wang, S., and Gao, L. (2016). The complete chloroplast genome of an irreplaceable dietary and model crop, foxtail millet (Setaria italica). Mitochondrial DNA 27, 1–2. doi: 10.3109/19401736.2015.1089562
Wang, W., and Messing, J. (2011). High-throughput sequencing of three Lemnoideae (duckweeds) chloroplast genomes from total DNA. PLoS One 6:e24670. doi: 10.1371/journal.pone.0024670
Wang, X., and Wang, L. (2016). GMATA: an integrated sofware package for genome-scale SSR mining, marker development and viewing. Front. Plant Sci. 7:1350. doi: 10.3389/fpls.2016.01350
Yang, A., Zhang, J., Yao, X., and Huang, H. (2011). Chloroplast microsatellite markers in Liriodendron tulipifera (Magnoliaceae) and cross-species amplifcation in L. chinense. Am. J. Bot. 98, e123–e126. doi: 10.3732/ajb.1000532
Yang, M., Zhang, X., Liu, G., Yin, Y., Chen, K., Yun, Q., et al. (2010). The complete chloroplast genome sequence of date palm (Phoenix dactylifera L.). PLoS One 5:e12762. doi: 10.1371/journal.pone.0012762
Zhang, Y., Du, L., Liu, A., Chen, J., Wu, L., Hu, W., et al. (2016a). The complete chloroplast genome sequences of five Epimedium species: lights into phylogenetic and taxonomic analyses. Front. Plant Sci. 7:306. doi: 10.3389/fpls.2016.00306
Zhang, Y., Yuan, X., Teng, W., Chen, C., and Wu, J. (2016b). Identification and phylogenetic classification of Pennisetum (Poaceae) ornamental grasses based on ssr locus polymorphisms. Plant Mol. Biol. Report. 34, 1–12. doi: 10.1007/s11105-016-0990-2
Zhang, Y., Yuan, X., Teng, W., Chen, C., Liu, H., and Wu, J. (2015). Karyotype diversity analysis and nuclear genome size estimation for Pennisetum Rich. (Poaceae) ornamental grasses reveal genetic relationship and chromosomal evolution. Sci. Hortic. 193, 22–31. doi: 10.1016/j.scienta.2015.06.018
Zhao, J. T., Xu, Y., Xi, L. J., Yang, J. W., Chen, H. W., and Zhang, J. (2018). Characterization of the chloroplast genome sequence of Acer miaotaiense: comparative and phylogenetic analyses. Molecules 23:1740. doi: 10.3390/molecules23071740
Zhao, P., Luo, J., Zhuang, W., Liu, X., and Wu, B. (2011). DNA barcoding of the fungal genus Neonectria and the discovery of two new species. Sci. China Life Sci. 54, 664–674. doi: 10.1007/s11427-011-4184-8
Keywords: chloroplast genome, phylogeny, comparative analysis, Pennisetum species, important pasture
Citation: Xu J, Liu C, Song Y and Li M (2021) Comparative Analysis of the Chloroplast Genome for Four Pennisetum Species: Molecular Structure and Phylogenetic Relationships. Front. Genet. 12:687844. doi: 10.3389/fgene.2021.687844
Received: 30 March 2021; Accepted: 21 June 2021;
Published: 27 July 2021.
Edited by:
Zefeng Yang, Yangzhou University, ChinaReviewed by:
Xingchun Wang, Shanxi Agricultural University, ChinaSeung-Chul Kim, Sungkyunkwan University, South Korea
Copyright © 2021 Xu, Liu, Song and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mingfu Li, bGltZjlAc2luYS5jb20=