 Elizabeth G. Woo
Elizabeth G. Woo Nahid Tayebi
Nahid Tayebi Ellen Sidransky
Ellen Sidransky- Medical Genetics Branch, National Human Genome Research Institute, National Institutes of Health, Bethesda, MD, United States
Introduction
Gaucher disease (GD) is a rare autosomal recessive lysosomal storage disorder caused by pathologic variants in GBA1, the gene encoding the enzyme glucocerebrosidase. Deficiency of glucocerebrosidase leads to the accumulation of the substrates glucocerebroside and glucosylsphingosine in macrophages and neuronal cells. Variants in GBA1 are a significant genetic risk factor for Define- Parkinson disease (PD) (Sidransky et al., 2009). Both patients with GD and heterozygous carriers are at an increased risk of developing PD, although the exact mechanism of this association is not fully understood (Avenali et al., 2020). An estimated 5–20% of patients with PD carry a GBA1 mutation, although the frequency varies between populations (Gan-Or et al., 2015). The association of GBA1 variants with this common complex disorder has led to an increasing interest in analyzing GBA1 in larger cohorts using next-generation sequencing (NGS) methods (Nalls et al., 2014; Gorostidi et al., 2016; Blauwendraat et al., 2018; Stoker et al., 2020). Challenges arise in using NGS for GBA1 analysis, however, due to the downstream, highly homologous pseudogene.
GBA1 is located in a gene-rich region on chromosome 1q21 that encompasses seven genes and two pseudogenes within an 85-kb region (Winfield et al., 1997). GBA1 contains 11 exons and 10 introns over a length of 7.6 kb. Located 16 kb downstream is the shorter (5.7 kb), non-processed pseudogene, GBAP1 (Horowitz et al., 1989). In the coding regions, GBAP1 is 96% homologous to the functional gene, increasing to around 98% in the region between intron 8 and the 3′ UTR. However, GBAP1 lacks a 55-bp segment in exon 9, which is the major exonic difference between the two sequences (Walley and Harris, 1993; Beutler et al., 1995; Tayebi et al., 1996b). GBA1 has several Alu intronic insertions that are not present in GBAP1, indicating that evolutionarily, the gene duplication occurred prior to the integration of the Alu sequences (Horowitz et al., 1989). The closest downstream gene, located just beyond GBAP1 is metaxin (MTX1), encoding for part of the mitochondrial outer membrane import complex protein 1, which is transcribed convergently to GBA1 (Long et al., 1996). MTX1 also has a pseudogene located in the 16-kb region between GBA1 and GBAP1 (Figure 1A). Several hundred mutations have been identified in GBA1 including missense mutations, deletions, insertions, splice site mutations, and complex recombinant alleles (Hruska et al., 2008).
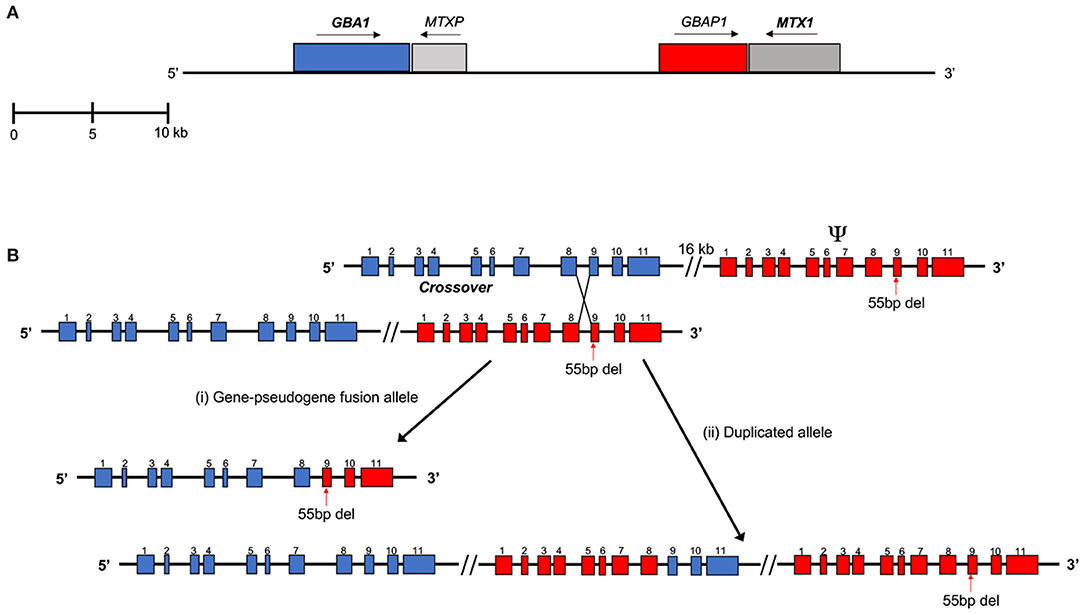
Figure 1. (A) Scaled representation of GBA1, MTX1, and their highly homologous pseudogenes. MTXP lies in the 16 kb region between GBA1 and GBAP1. MTX1 and MTXP are transcribed in the direction opposite to that of GBA1 and GBAP1. (B) Example of a reciprocal crossover event, resulting in a (i) gene-pseudogene fusion allele and a (ii) duplicated allele. These recombinant alleles are difficult to detect using next-generation sequencing methods. Sequence reads containing the pseudogene-derived segment (red) of the recombinant alleles may align to the pseudogene instead. This can lead to false negatives and reduce the read depth of GBA1. In this example, reads containing the 55 bp deletion from the recombinant alleles may align preferentially to the pseudogene (Adapted in part from Tayebi et al., 2003).
The presence of these pseudogenes in this region increases the risk for recombination and the generation of complex alleles, particularly due to the close physical proximity and high homology between GBA1 and GBAP1 (Chen et al., 1997). Recombinant alleles can arise from both reciprocal and non-reciprocal recombination, resulting in gene fusions, duplications, and gene conversion alleles (Cormand et al., 2000; Tayebi et al., 2003). These genes can serve as a model to demonstrate the effect of unequal pairing and crossover resulting in recombinant alleles in the genome. Identifying complex alleles poses a significant challenge when relying on NGS analysis.
GBA1 Genotyping by Conventional Means
Previously, PCR-based screening techniques were often used targeting specific panels of 7–10 common GBA1 pathologic variants (Park et al., 2002). These variants were predicted to account for over 90% of mutant alleles among Ashkenazi Jews, a population with a carrier rate of ~1 in 14 (Do et al., 2019). However, relying on such panels can miss other mutant alleles, as demonstrated by a 2019 study that fully sequenced GBA1 in an Ashkenazi Jewish Parkinson disease patient cohort (Ruskey et al., 2019). Sanger sequencing substantially increased variant detection, even in this group, particularly for the common E326K (p.E365K) variant, known to be present in ~1% of the general population (Lau et al., 1999; Park et al., 2002). This highlights the benefit of sequencing the full GBA1 gene to detect variants that could be missed using targeted genotyping of selected pathogenic variants. When sequencing GBA1, specific primers are required to ensure that functional GBA1 is being amplified and sequenced rather than the pseudogene (Dandana et al., 2016). For example, primers targeted to the 55-bp pseudogene gap in exon 9 can erroneously misreport patients heteroallelic for a recombination in that region as homozygosity for the second allele (Tayebi et al., 1996b).
Next-Generation Sequencing of GBA1
Next-generation sequencing is able to generate vast amounts of sequence data and has been applied to identify variants in GBA1. The use of whole-exome and whole-genome sequencing has increased the discovery rate of these variants. For example, a recent large-scale NGS strategy identified 18 novel GBA1 variants, as well as a rarely reported complex allele likely to be a Dutch founder allele (Zampieri et al., 2017; den Heijer et al., 2020). One Gaucher diagnostics laboratory relying on NGS run by Centogene (Rostock, Germany) provides an on-line list of their identified GBA1 variants (CentoLSD™), which now includes over 600 different GBA1 variants with differing predicted clinical significance (https://www.centogene.com/centolsd.html). Many NGS protocols have utilized short-read sequencing, which can provide data at a relatively low cost and high accuracy but may be less reliable in cases where there are structural variants and repetitive or highly homologous regions (Mandelker et al., 2016; Mantere et al., 2019). If the functional gene is not specifically amplified during library preparation, reads generated by the pseudogene could align with the functional gene, leading to false-positive results, while reads belonging to the functional gene might align to the pseudogene, reducing read depth and mapping quality (Claes and De Leeneer, 2014). Long range-PCR (LR-PCR) can help to selectively amplify the functional gene (Tayebi et al., 1996a; Jeong et al., 2011). Even after selectively amplifying the functional gene, challenges still remain in the alignment phase, particularly when recombinant alleles contain regions identical to the pseudogene. In an Illumina short-read NGS method for sequencing GBA1, Zampieri et al. initially aligned reads to the whole genome, but missed almost all recombinant alleles, which had aligned preferentially to GBAP1 (Zampieri et al., 2017). After aligning to GBA1 rather than the whole genome, they were able to identify recombinant variants that had initially been false negatives, except for the 55-bp deletion mutation. Alignment against the whole genome did not significantly affect the mapping quality and coverage of missense mutations unrelated to recombination. Care should therefore be taken not only in the library preparation, but also in data analysis steps of NGS pipelines to sequence GBA1.
The introduction of long-read sequencing technology in recent years addressed some limitations of short-read sequencing (Goodwin et al., 2016). The ability to read longer sequences can help to differentiate highly homologous regions. One such technology is the Oxford Nanopore system, which analyzes a single DNA molecule as it passes through a pore (Logsdon et al., 2020). Resulting disruptions to the current are analyzed in real-time to determine the sequence. This strategy has been applied for the detection of GBA1 variants using the Oxford Nanopore MinION (Leija-Salazar et al., 2019). The authors were able to detect the 55-bp exonic deletion, but not the more common recombinant allele RecNciI. Another study aimed to expand upon this protocol by refining and applying it to the discovery of GBA1 variants in a PD longitudinal cohort from New Zealand (Graham et al., 2020). They validated the protocol of Leija-Salazar et al. for genotyping GBA1 and updated the software pipeline, improving accuracy and reducing the computational workload, but were unable to detect any recombinant gene conversion alleles or deletions of >50 bp.
Challenges in Detecting Recombinant Alleles
Accurate and comprehensive NGS analysis of GBA1 is complicated by complex gene-pseudogene rearrangements and recombination. Recombinant alleles make up a significant proportion of GBA1 mutations and sites of recombination events have been identified between intron 2 to exon 11 (Hruska et al., 2008). With the high exonic homology between GBA1 and its pseudogene, there is an increased likelihood of both reciprocal and non-reciprocal recombination (Tayebi et al., 2003). These can result in alleles with multiple exonic point mismatches, such as in RecNciI. In this allele, the site of crossover of chromosomal mispairing between gene and pseudogene occurs within intron 9 and continues to the 3'-UTR of the gene, introducing three exon 10 nucleotide mismatches (Latham et al., 1990). Another frequently described reciprocal recombinant allele, RecTL, covers the pseudogene sequence from intron 8 or the beginning of exon 9 [Figure 1B(i)] (Zimran and Horowitz, 1994). Reciprocal crossover can also result in a partial duplication of the gene and pseudogene sequence [Figure 1B(ii)]. The non-reciprocal exchange of homologous sequence, also known as gene conversion, may also occur. One example is an allele that includes only the 55-bp deletion in exon 9. This is a small, converted gene sequence that resembles the pseudogene. Some polymorphic sequences in GBAP1 (Martinez-Arias et al., 2001) also been observed. Mostly, homologous recombination events occur in regions of high sequence homology between the gene and pseudogene, such as in introns 8 and 9. Several instances of recombination have occurred as a result of crossover between MTX and its pseudogene as well (Tayebi et al., 2003). Quantitative real-time PCR and Southern blot analysis have been used to identify fusions and duplications in patients with recombinant GBA1 alleles (Velayati et al., 2011).
The complexity of GBA1 gene rearrangements cannot be sufficiently captured using most current NGS methods. Several recent studies using NGS technology without Sanger sequencing validation have not reported the presence of recombinant alleles, including a recent study performed on more than 3,000 PD cases (den Heijer et al., 2020). Thus, it is likely that the results presented may be an underestimation (Zampieri et al., 2017).
Non-Conventional Inheritance
Gaucher disease is an autosomal recessive disorder that usually results from the inheritance of a mutant allele from each parent. Increasing evidence suggests this may not always be the case, and that there are important exceptions to the traditional Mendelian pattern of inheritance, such as new mutations or uniparental disomy (Nakka et al., 2019). There have been several reported cases of unrelated patients with type 2 GD with de novo or germline mutations on the maternal allele (Saranjam et al., 2013; Hagege et al., 2017). Uniparental disomy of chromosome 1 was reported in a proband with concurrent type 3 GD and Charcot-Marie-Tooth disease, who was homozygous for both L444P (p.L483P) in GBA1 and S78L in MPZ (Benko et al., 2008). There have also been cases of patients with all types of GD carrying more than one GBA1 mutation on the same allele (Hassan et al., 2018). For example, a common mutant allele in Greece and the Balkans includes both H255Q (p.H255Q) and D409H (p.D448H) together in cis (Vithayathil et al., 2009). These examples of non-conventional inheritance underscore the importance of careful and comprehensive examination of entire coding regions for accurate genotyping. This has important implications for diagnostics and establishing genotype-phenotype correlations. Current NGS technology, however, does not always accurately identify all nucleotide changes and recombinant alleles. As a result, NGS is currently limited in its clinical diagnostic capacity for comprehensive GBA1 screening, and for now, Sanger sequencing should be used for the most accurate genotyping.
Discussion
Next-generation sequencing allows for unbiased simultaneous analysis of many genes. In addition, it makes it more feasible to analyze specific genes in larger cohorts for the study of common diseases like Parkinson disease. With the inclusion of GBA1 in many Parkinson disease NGS analyses, it is important to consider the effects of the nearby homologous pseudogene. Recombinant alleles in GBA1 have been identified in patients with GD and with PD that might be missed when relying on NGS analysis alone without Sanger sequencing validation.
Challenges exist in short-read NGS methods for sequencing highly homologous regions. The shorter reads cannot be mapped uniquely to the reference genome, especially in cases where there are recombinant alleles aligning to the homologous region. A computational custom scaffold-based approach was recently introduced to improve the detection and phasing of targeted complex variants using short reads (Zeng et al., 2020). This method was able to detect a 55-bp deletion in GBA1 confirmed by Sanger sequencing. In addition, there is evidence to suggest that the choice of polymerase used could be a factor in the accuracy of NGS variant identification. A recent study performed a large-scale screening of GBA1 based on an NGS protocol and found a significant number of false negatives due to a polymerase-dependent allelic imbalance (den Heijer et al., 2021). After performing a structured assessment of varied PCR conditions, they found that changing the polymerase used led to the resolution of these false negatives. Allele frequency was unaffected by a change in the other conditions. This raises the possibility that current estimates of variant frequency in populations could be underestimates, due to polymerase-dependent false negatives as well.
Long-read NGS has many advantages, one of which is an improved ability to discriminate functional genes such as GBA1 from their pseudogenes. An additional advantage is the ability to phase mutations and assign haplotypes. It can also detect intronic SNPs. There are still some limitations, including high costs, low throughput, and high per-base error rates. Long-read NGS also has a limited ability to accurately resolve homopolymers and to detect small insertions and deletions. Importantly, it is still unable to consistently detect recombinant alleles.
The most accurate method for detecting all GBA1 variants and recombination remains Sanger sequencing. Without validation by Sanger sequencing, the frequencies of GBA1 variants based on NGS analysis may be underestimated, particularly for complex recombinant alleles. Real-time PCR can also be used to identify recombinant alleles (Velayati et al., 2011). NGS pipelines should be carefully designed in order to capture variants should from the functional gene rather than the pseudogene and attempt to include complex alleles. While long-read sequencing shows promise for increased accuracy of GBA1 NGS analysis, it is still currently limited in its capacity to recognize recombinant alleles. The shortcomings identified will likely also be pertinent for the analysis of other genes with highly homologous pseudogenes. As sequencing technology continues to rapidly progress, we will likely continue to see improved detection of GBA1 variants. This has exciting potential for clinical diagnostics and studies of large patient cohorts.
Author Contributions
EW: drafted the manuscript and figure. NT: organized the topic, reviewed the literature, and edited the manuscript. ES: conceived of the topic and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Intramural Research Programs of the National Human Genome Research Institute and the National Institutes of Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Avenali, M., Blandini, F., and Cerri, S. (2020). Glucocerebrosidase defects as a major risk factor for Parkinson's Disease. Front. Aging Neurosci. 12:97. doi: 10.3389/fnagi.2020.00097
Benko, W. S., Hruska, K. S., Nagan, N., Goker-Alpan, O., Hart, P. S., Schiffmann, R., et al. (2008). Uniparental disomy of chromosome 1 causing concurrent Charcot-Marie-Tooth and Gaucher disease Type 3. Neurology 70, 976–978. doi: 10.1212/01.wnl.0000305963.37449.32
Beutler, E., Gelbart, T., Demina, A., Zimran, A., and LeCoutre, P. (1995). Five new Gaucher disease mutations. Blood Cells Mol. Dis. 21, 20–24. doi: 10.1006/bcmd.1995.0004
Blauwendraat, C., Bras, J. M., Nalls, M. A., Lewis, P. A., Hernandez, D. G., Singleton, A. B., et al. (2018). Coding variation in GBA explains the majority of the SYT11-GBA Parkinson's disease GWAS locus. Mov. Disord. 33, 1821–1823. doi: 10.1002/mds.103
Chen, K. S., Manian, P., Koeuth, T., Potocki, L., Zhao, Q., Chinault, A. C., et al. (1997). Homologous recombination of a flanking repeat gene cluster is a mechanism for a common contiguous gene deletion syndrome. Nat. Genet. 17, 154–163. doi: 10.1038/ng1097-154
Claes, K. B., and De Leeneer, K. (2014). Dealing with pseudogenes in molecular diagnostics in the next-generation sequencing era. Methods Mol. Biol. 1167, 303–315. doi: 10.1007/978-1-4939-0835-6_21
Cormand, B., Diaz, A., Grinberg, D., Chabas, A., and Vilageliu, L. (2000). A new gene-pseudogene fusion allele due to a recombination in intron 2 of the glucocerebrosidase gene causes Gaucher disease. Blood Cells Mol. Dis. 26, 409–416. doi: 10.1006/bcmd.2000.0317
Dandana, A., Ben Khelifa, S., Chahed, H., Miled, A., and Ferchichi, S. (2016). Gaucher disease: clinical. biological and therapeutic aspects. Pathobiology 83, 13–23. doi: 10.1159/000440865
den Heijer, J. M., Cullen, V. C., Quadri, M., Schmitz, A., Hilt, D. C., Lansbury, P., et al. (2020). A large-scale full GBA1 gene screening in Parkinson's disease in the Netherlands. Mov. Disord. 35, 1667–1674. doi: 10.1002/mds.28112
den Heijer, J. M., Schmitz, A., Lansbury, P., Cullen, V. C., Hilt, D. C., Bonifati, V., et al. (2021). False negatives in GBA1 sequencing due to polymerase dependent allelic imbalance. Sci. Rep. 11:161. doi: 10.1038/s41598-020-80564-y
Do, J., McKinney, C., Sharma, P., and Sidransky, E. (2019). Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 14:36. doi: 10.1186/s13024-019-0336-2
Gan-Or, Z., Amshalom, I., Kilarski, L. L., Bar-Shira, A., Gana-Weisz, M., and Mirelman, A.. (2015). Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 84, 880–887. doi: 10.1212/WNL.0000000000001315
Goodwin, S., McPherson, J. D., and McCombie, W. R. (2016). Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 17, 333–351. doi: 10.1038/nrg.2016.49
Gorostidi, A., Marti-Masso, J. F., Bergareche, A., Rodriguez-Oroz, M. C., Lopez de Munain, A., and Ruiz-Martinez, J. (2016). Genetic mutation analysis of Parkinson's disease patients using multigene next-generation sequencing panels. Mol. Diagn. Ther. 20, 481–491. doi: 10.1007/s40291-016-0216-1
Graham, O. E. E., Pitcher, T. L., Liau, Y., Miller, A. L., Dalrymple-Alford, J. C., Anderson, T. J., et al. (2020). Nanopore sequencing of the glucocerebrosidase (GBA) gene in a New Zealand Parkinson's disease cohort. Parkinsonism Relat. Disord. 70, 36–41. doi: 10.1016/j.parkreldis.2019.11.022
Hagege, E., Grey, R. J., Lopez, G., Roshan Lal, T., Sidransky, E., and Tayebi, N. (2017). Type 2 Gaucher disease in an infant despite a normal maternal glucocerebrosidase gene. Am. J. Med. Genet. A. 173, 3211–3215. doi: 10.1002/ajmg.a.38487
Hassan, S., Lopez, G., Stubblefield, B. K., Tayebi, N., and Sidransky, E. (2018). Alleles with more than one mutation can complicate genotype/phenotype studies in Mendelian disorders: lessons from Gaucher disease. Mol. Genet. Metab. 125, 1–3. doi: 10.1016/j.ymgme.2018.06.013
Horowitz, M., Wilder, S., Horowitz, Z., Reiner, O., Gelbart, T., and Beutler, E. (1989). The human glucocerebrosidase gene and pseudogene: structure and evolution. Genomics 4, 87–96. doi: 10.1016/0888-7543(89)90319-4
Hruska, K. S., LaMarca, M. E., Scott, C. R., and Sidransky, E. (2008). Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 29, 567–583. doi: 10.1002/humu.20676
Jeong, S. Y., Kim, S. J., Yang, J. A., Hong, J. H., Lee, S. J., and Kim, H. J. (2011). Identification of a novel recombinant mutation in Korean patients with Gaucher disease using a long-range PCR approach. J. Hum. Genet. 56, 469–471. doi: 10.1038/jhg.2011.37
Latham, T., Grabowski, G. A., Theophilus, B. D., and Smith, F. I. (1990). Complex alleles of the acid beta-glucosidase gene in Gaucher disease. Am. J. Hum. Genet. 47, 79–86.
Lau, E. K., Tayebi, N., Ingraham, L. J., Winfield, S. L., Koprivica, V., Stone, D. L., et al. (1999). Two novel polymorphic sequences in the glucocerebrosidase gene region enhance mutational screening and founder effect studies of patients with Gaucher disease. Hum. Genet. 104, 293–300. doi: 10.1007/s004390050957
Leija-Salazar, M., Sedlazeck, F. J., Toffoli, M., Mullin, S., Mokretar, K., Athanasopoulou, M., et al. (2019). Evaluation of the detection of GBA missense mutations and other variants using the Oxford Nanopore MinION. Mol. Genet. Genomic Med. 7:e564. doi: 10.1002/mgg3.564
Logsdon, G. A., Vollger, M. R., and Eichler, E. E. (2020). Long-read human genome sequencing and its applications. Nat. Rev. Genet. 21, 597–614. doi: 10.1038/s41576-020-0236-x
Long, G. L., Winfield, S., Adolph, K. W., Ginns, E. I., and Bornstein, P. (1996). Structure and organization of the human metaxin gene (MTX) and pseudogene. Genomics 33, 177–184. doi: 10.1006/geno.1996.0181
Mandelker, D., Schmidt, R. J., Ankala, A., McDonald Gibson, K., Bowser, M., Sharma, H., et al. (2016). Navigating highly homologous genes in a molecular diagnostic setting: a resource for clinical next-generation sequencing. Genet. Med. 18, 1282–1289. doi: 10.1038/gim.2016.58
Mantere, T., Kersten, S., and Hoischen, A. (2019). Long-read sequencing emerging in medical genetics. Front. Genet. 10:426. doi: 10.3389/fgene.2019.00426
Martinez-Arias, R., Comas, D., Mateu, E., and Bertranpetit, J. (2001). Glucocerebrosidase pseudogene variation and Gaucher disease: recognizing pseudogene tracts in GBA alleles. Hum. Mutat. 17, 191–198. doi: 10.1002/humu.4
Nakka, P., Pattillo Smith, S., O'Donnell-Luria, A. H., McManus, K. F., Team andMe Research, Mountain, J. L., et al. (2019). Characterization of prevalence and health consequences of uniparental disomy in four million individuals from the general population. Am. J. Hum. Genet. 105, 921–932. doi: 10.1101/540955
Nalls, M. A., Pankratz, N., Lill, C. M., Do, C. B., Hernandez, D. G., Saad, M., et al. (2014). Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson's disease. Nat. Genet. 46, 989–993. doi: 10.1038/ng.3043
Park, J. K., Tayebi, N., Stubblefield, B. K., LaMarca, M. E., MacKenzie, J. J., Stone, D. L., et al. (2002). The E326K mutation and Gaucher disease: mutation or polymorphism? Clin. Genet. 61, 32–34. doi: 10.1034/j.1399-0004.2002.610106.x
Ruskey, J. A., Greenbaum, L., Ronciere, L., Alam, A., Spiegelman, D., Liong, C., et al. (2019). Increased yield of full GBA sequencing in Ashkenazi Jews with Parkinson's disease. Eur. J. Med. Genet. 62, 65–69. doi: 10.1016/j.ejmg.2018.05.005
Saranjam, H., Chopra, S. S., Levy, H., Stubblefield, B. K., Maniwang, E., Cohen, I. J., et al. (2013). A germline or de novo mutation in two families with Gaucher disease: implications for recessive disorders. Eur. J. Hum. Genet. 21, 115–117. doi: 10.1038/ejhg.2012.105
Sidransky, E., Nalls, M. A., Aasly, J. O., Aharon-Peretz, G., Annesi, E. R., Barbosa, E. R., et al. (2009). Multi-center analysis of glucocerebrosidase mutations in Parkinson disease. N. Engl. J. Med. 361, 1651–1661. doi: 10.1056/NEJMoa0901281
Stoker, T. B., Camacho, M., Winder-Rhodes, S., Liu, G., Scherzer, C. R., Foltynie, T., et al. (2020). Impact of GBA1 variants on long-term clinical progression and mortality in incident Parkinson's disease. J. Neurol. Neurosurg. Psychiatry 91, 695–702. doi: 10.1136/jnnp-2020-322857
Tayebi, N., Cushner, S., and Sidransky, E. (1996a). Differentiation of the glucocerebrosidase gene from pseudogene by long-template PCR: implications for Gaucher disease. Am. J. Hum. Genet. 59, 740–741.
Tayebi, N., Stern, H., Dymarskaia, I., Herman, J., and Sidransky, E. (1996b). 55-base pair deletion in certain patients with Gaucher disease complicates screening for common Gaucher alleles. Am. J. Med. Genet. 66, 316–319.
Tayebi, N., Stubblefield, B. K., Park, J. K., Orvisky, E., Walker, J. M., LaMarca, M. E., et al. (2003). Reciprocal and nonreciprocal recombination at the glucocerebrosidase gene region: implications for complexity in Gaucher disease. Am. J. Hum. Genet. 72, 519–534. doi: 10.1086/367850
Velayati, A., Knight, M. A., Stubblefield, B. K., Sidransky, E., and Tayebi, N. (2011). Identification of recombinant alleles using quantitative real-time PCR implications for Gaucher disease. J. Mol. Diagn. 13, 401–405. doi: 10.1016/j.jmoldx.2011.02.005
Vithayathil, J., Gibney, G., Baxevanis, A. D., Stubblefield, B. K., Sidransky, E., and Tayebi, N. (2009). Glucocerebrosidase mutation H255Q appears to be exclusively in cis with D409H: structural implications. Clin. Genet. 75, 503–504. doi: 10.1111/j.1399-0004.2009.01163.x
Walley, A. J., and Harris, A. (1993). A novel point mutation (D380A) and a rare deletion (1255del55) in the glucocerebrosidase gene causing Gaucher's disease. Hum. Mol. Genet. 2, 1737–1738. doi: 10.1093/hmg/2.10.1737
Winfield, S. L., Tayebi, N., Martin, B. M., Ginns, E. I., and Sidransky, E. (1997). Identification of three additional genes contiguous to the glucocerebrosidase locus on chromosome 1q21: implications for Gaucher disease. Genome Res. 7, 1020–1026. doi: 10.1101/gr.7.10.1020
Zampieri, S., Cattarossi, S., Bembi, B., and Dardis, A. (2017). GBA analysis in next-generation era: pitfalls, challenges, possible solutions. J. Mol. Diagn. 19, 733–741. doi: 10.1016/j.jmoldx.2017.05.005
Zeng, Q., Leach, N. T., Zhou, Z., Zhu, H., Smith, J. A., Rosenblum, L. S., et al. (2020). A customized scaffolds approach for the detection and phasing of complex variants by next-generation sequencing. Sci. Rep. 10:15060. doi: 10.1038/s41598-020-71471-3
Keywords: next-generation sequencing, glucocerebrosidase, Gaucher disease, GBA1, pseudogene, genotyping
Citation: Woo EG, Tayebi N and Sidransky E (2021) Next-Generation Sequencing Analysis of GBA1: The Challenge of Detecting Complex Recombinant Alleles. Front. Genet. 12:684067. doi: 10.3389/fgene.2021.684067
Received: 22 March 2021; Accepted: 27 May 2021;
Published: 21 June 2021.
Edited by:
María L. Couce, Complejo Hospitalario Universitario de Santiago, SpainReviewed by:
Said El Shamieh, Beirut Arab University, LebanonCopyright © 2021 Woo, Tayebi and Sidransky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ellen Sidransky, c2lkcmFuc2VAbWFpbC5uaWguZ292