 Jie Zhang1†
Jie Zhang1† Weiwen Qiu
Weiwen Qiu Xiong Zhang
Xiong Zhang Renshi Xu
Renshi Xu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 06 August 2021
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.679204
This article is part of the Research Topic New Insights into the Molecular Mechanism of Amyotrophic Lateral Sclerosis Pathogenesis View all 5 articles
The pathogenesis of sporadic amyotrophic lateral sclerosis (sALS) remains unknown; however, recent research suggests that genetic factors may play an important role. This study aimed at investigating possible genetic risk factors for the pathogenesis of sALS. In our previous study, we conducted a genome-wide association study (GWAS) in 250 sALS patients and 250 control participants of Han ancestry from mainland China (HACM) and retrospectively analyzed the previously reported candidate loci related with sALS including our GWAS investigated results. In this study, twenty-seven candidate loci that were most likely associated with sALS were selected for further analysis in an independent case/control population of 239 sALS patients and 261 control subjects of HACM ethnicity using sequenom massARRAY methodology and DNA sequencing. We discovered that the polymorphism rs2619566 located within the contactin-4 (CNTN4) gene, rs10260404 in the dipeptidyl-peptidase 6 (DPP6) gene, and rs79609816 in the inositol polyphosphate-5-phosphatase B (INPP5B) gene were strongly associated with sALS in subjects of HACM ethnicity. Subjects harboring the minor C allele of rs2619566 and the minor T allele of rs79609816 exhibited an increased risk for sALS development, while carriers of the minor C allele of rs10260404 showed a decreased risk of sALS development compared to the subjects of other genotypes. The polymorphisms of rs2619566, rs10260404, and rs79609816 may change or affect the splicing, transcription, and translation of CNTN4, DPP6, and INPP5B genes and may play roles in the pathogenesis of sALS roles in the pathogenesis of sALS.
Amyotrophic lateral sclerosis (ALS) is the most common adult-onset motor neuron disease. It is characterized by the progressive neuronal loss and degeneration of upper motor neurons and lower motor neurons. The death of motor neurons causes a loss of the ability of central nervous system (CNS) to control voluntary muscle movements, contributing to the development of progressive atrophy of voluntary muscles, eventually resulting in death of the patient due to respiratory failure in the later stages of the disease. Most ALS patients succumb to the disease within 3–5 years after disease onset. Although the patient may or may not present with a family history of the disease, ALS is divided into familial ALS (fALS) and sporadic ALS (sALS) (1). Several genes and/or loci associated with risk of fALS development have been identified in the recent years. Although certain genetic loci have been associated with sALS risk, the exact genetic mechanism for sALS has not been elucidated, and sALS is hypothesized to possess a more complex pathogenesis. The reasons for progressive and selective motor neuron death occurring in sALS remain elusive. The pathogenesis of sALS remains an enigma (Ludolph et al., 2012).
The recently acknowledged pathogenesis of sALS is mainly focused on environmental and genetic factors. Potential environmental factors predisposing to sALS development include viral and bacterial infections (Sher, 2017; Yu and Pamphlett, 2017; Xue et al., 2018), organophosphate, organochlorine (Su et al., 2016; Riancho et al., 2018; Lian et al., 2019), and heavy metal exposure (Callaghan et al., 2011; Garzillo et al., 2015; Peters et al., 2017; Riancho et al., 2018), intense physical activity (Tsitkanou et al., 2019), smoking, electromagnetic fields, electric shocks, cyanotoxins, and military service (Vinceti et al., 2017; Swash and Eisen, 2020). However, none of the known environmental risk factors has been conclusively determined, and no firm conclusions have been deduced thus far (Longinetti and Fang, 2019). If environmental factors are truly a causal risk factor in sALS development, the genetic susceptibility would be expected to increase the risk of sALS development due to exposure to environmental agents (Nowicka et al., 2019). Therefore, genetic factors have garnered considerable attention in the study of sALS pathogenesis since the discovery of SOD1 mutations in sALS. During the last decade, the evolution of molecular genetic technologies has rapidly advanced our knowledge concerning the genetic pathogenesis of sALS. The development of fALS has been attributed to mutations in at least 24 different genes. Certain mutations responsible for fALS development have been identified also in patients with sALS (Chia et al., 2018; Mathis et al., 2019). Recent large-scale parallel sequencing technologies have facilitated disease-gene discovery, rare variants in more than 50 genes have now been identified to be associated with sALS. Thus, sALS has been considered a complex gene-related disease (Van Doormaal et al., 2017).
Genetic testing may aid the exploration of mutations in ALS-related genes. However, the loci that are most likely to be affected in individual patients with sALS cannot be easily predicted (Yousefian-Jazi et al., 2020). Although a series of possible ALS-related genes and mutant loci have been successively reported in the literature, currently there is no single gene or mutant locus that can completely explain the pathogenesis of ALS (Morgan and Orrell, 2016; Yousefian-Jazi et al., 2020). Therefore, it has been suggested that the sALS pathogenesis is associated with multiple genes and/or mutant loci. To this end, the investigation of additional ALS-related genes and/or mutant loci is extremely important.
In this study, we selected the 27 loci that were most likely to be associated with sALS development based on the results of the present study including our GWAS study of 250 sALS and 250 controls as well as other related previous studies (Van Es et al., 2007, 2008; Laaksovirta et al., 2010; Fogh et al., 2014; Van Doormaal et al., 2014; Xie et al., 2014), and we further analyzed them in an independent cohort of the 239 individuals with sALS and the 261 controls of Han ancestry from mainland China (HACM). We discovered that the polymorphism of the rs2619566 in contactin 4 (CNTN4), the rs10260404 in dipeptidyl-peptidase 6 (DPP6), and the rs79609816 in inositol polyphosphate-5-phosphatases B (INPP5B) were markedly associated with sALS development in the subjects of HACM ethnicity. These results provide evidence to a certain extent for the further elucidation of the pathogenesis of sALS.
The sALS dataset was developed based on the data obtained by combining the participants from two affiliated university hospitals, namely The First Affiliated Hospital of Nanchang University and the Affiliated Guangdong General Hospital of Nan fang Medical University. All sALS and control subjects were recruited from HACM in the southern regions of China (Jiangxi and Guangdong Province). A signed informed consent was obtained from all participants in the study. sALS diagnosis was performed according to the El Escorial criteria of the World Federation of Neurology (Brooks et al., 2000). All subjects were subjected to the same evaluation, which included a medical history, a Mini Mental State Exam, a review of the family history of ALS, related disorders in first-degree relatives, toxicant exposure associated with sALS development, biochemical tests, and brain and spinal magnetic resonance imaging to exclude the presence of other neurological diseases that might mimic the clinical presentation of ALS (e.g., tumors, demyelination disorders, hydrocephalus, cervical myelopathy, and others).
The studied populations were composed of a total of 489 sALS cases and 511 controls, of which 250 sALS cases and 250 controls were included in the genome-wide association study (GWAS) analysis (Xie et al., 2014), and 239 sALS cases and 261 controls were included in the polymorphism analysis of the 27 most likely candidate loci for sALS development. The male/female ratio was 143/96 for the sALS cases, and was 148/113 for the controls. The mean (range) age was 47 (45–65) years for the sALS cases, and was 65.7 (65–75) years for the controls. Gender was a significant variable because sALS seemed to affect more men than women, and our data exhibited a significant gender disparity. The control subjects were older than the patients, and this aspect was included in the study design to minimize the chance that the control subjects were too young to have developed the disease. Nevertheless, we controlled for age at enrollment to avoid confounding by age-related factors, and the early- and late-onset sALS patients were excluded. The entire clinical disease course spanned across 3–5 years, which precluded the development of rapid and slow progressive sALS, and the cases with atypical sALS clinical manifestations were removed. Thus, sALS patients with typical age, clinical course, and phenotype were enrolled in this study. Based on the current studied information, the genetic pathogenesis about sALS should be involved in some complex and multiple genes and mutations, isn’t sole gene or mutation to contribute to sALS. Therefore, we didn’t perform the genetic testing of the known ALS-related genes to exclude whether or not the presently known genes and mutations existed in our sALS patients and control subjects.
In our previous pooling GWAS performed by inclusion of 250 sALS patients and 250 control subjects from HACM, we revealed that the 7 loci, namely rs79609816 and rs62172104 in INPP5B, rs9825420 in ITGA9, rs2685056 in ALCAM, rs7117082 in OPCML, rs9329300 in PFKP, and rs11061269 in GPR133, were strongly associated with sALS development in subjects of the HACM origin (the significance threshold was p < 5.8 × 10−8), and these loci were not reported previously (Xie et al., 2014). Moreover, we retrospectively analyzed the previously reported candidate loci related with pathogenesis of sALS, and found that 20 SNPs, namely rs62484656 in CSMD1, rs17722673 in HECW1, rs882467 in DPP6, rs3812208 in ATXN1, rs28461450 in LIPC, rs79591932 in RBMS1, rs9907321 in SLC39A11, rs4964009 in ITPR2, rs34517760 in SOD1, rs13065219 in CNTN4, rs10260404 in DPP6, rs697739 in ATXN1, rs3825776 in LIPC, rs10192369 in RBMS1, rs8066857 in SLC39A11, rs2306677 in ITPR2, rs13048019 in SOD1, rs2619566 in CNTN4, rs16856202 in DISC1, and rs34517613 in KRT18P55, were susceptibility loci for sALS, which were strongly associated with sALS development in the previous studies (Van Es et al., 2007, 2008; Laaksovirta et al., 2010; Fogh et al., 2014; Van Doormaal et al., 2014; Xie et al., 2014). Therefore, in this study, 27 SNPs were selected to further ascertain their association with the development of sALS (Table 1).
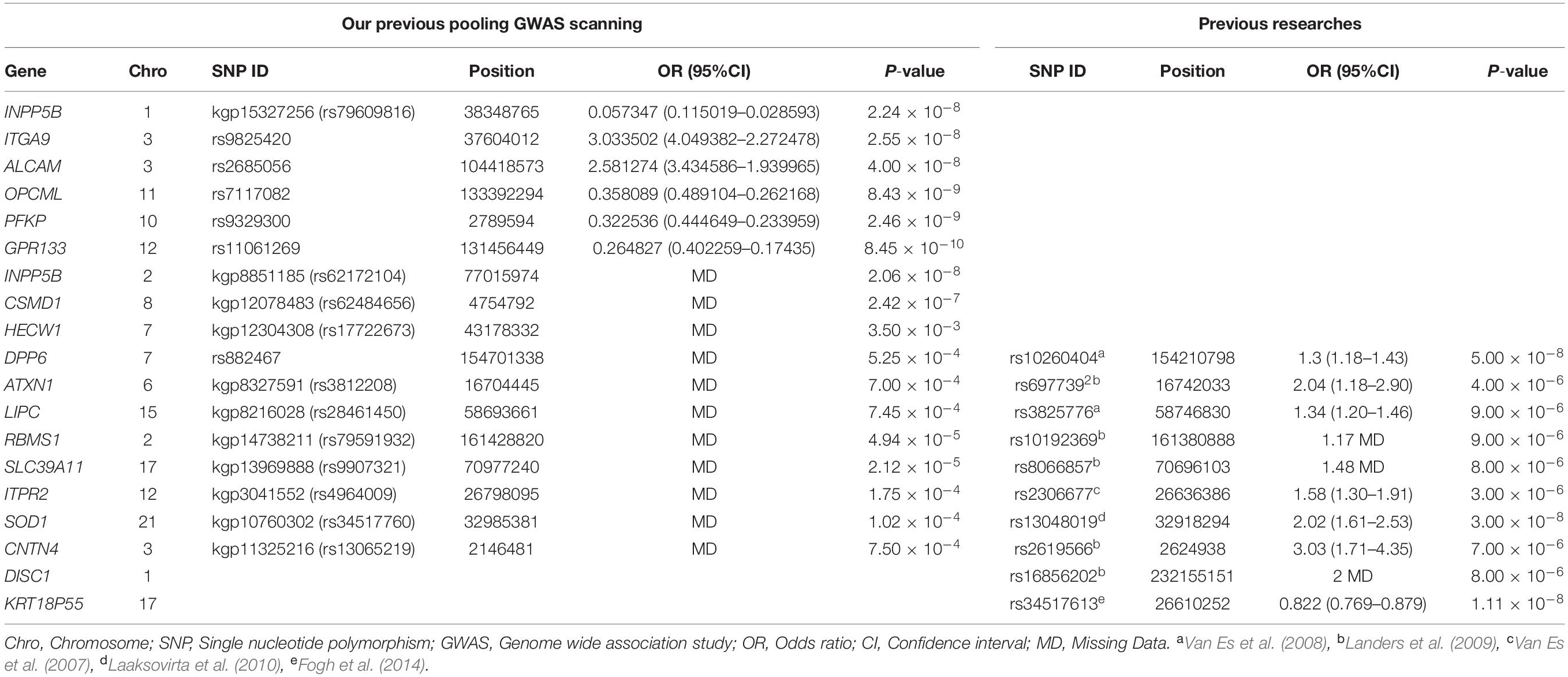
Table 1. All SNPs were chosen in the association study.
Genotyping was performed using the sequenom massARRAY platform (Sequenom, San Diego, California, United States) according to the manufacturer’s instructions. The selected 27 SNPs were genotyped as part of a sequenom plex, which enabled high-throughput multiplexing of the assays into a single well. For quality control, 5% of the samples were subjected to the repeated genotyping, and the results showed 100% consistency.
Amplification instrument: ABI GeneAmp® 9700 384 dual, mechanical arm, massARRAY nanodispenser RS1000. Analyzer: massARRAY compact system. Reagents: The Complete genotyping reagent kit for massARRAY® compact 384.
A PCR cocktail solution was prepared by combining 1.8 μL ddH2O, 0.5 μL 10 × PCR Buffer, 0.1 μL dNTPs, 0.2 μL PCR enzyme (5 U/μL), 1 μL primer mix (0.5 μM), 0.4 μL 25 mM MgCl2, and 1 μL DNA template, to obtain a total volume of 5 μL. One-μL volume of the appropriate genomic DNA (5–10 ng/μL) was added into each well of a 384-well microtiter plate (Marsh Biomedical Products, Inc. #SP 0401 Sequenom). Four-μL volume of the PCR cocktail solution was dispensed into each well of the 384-well microtiter plate, followed by centrifugation of the microtiter plate at 1,000 RPM for 1 min. Subsequently, contents in the microtiter plate were gently mixed and were re-centrifuged before conduction of PCR. PCR was conducted using the 384-well microtiter plate as per the following amplification conditions: 94°C denaturation for 20 s, 56°C annealing for 30 s, 72°C extension for 1 min, for a total of 45 cycles. The primers described in Table 2 were used for conducting the PCR reactions for this study. This general PCR protocol using a 384-well microtiter plate was applied to the different PCR amplifications performed in this study.
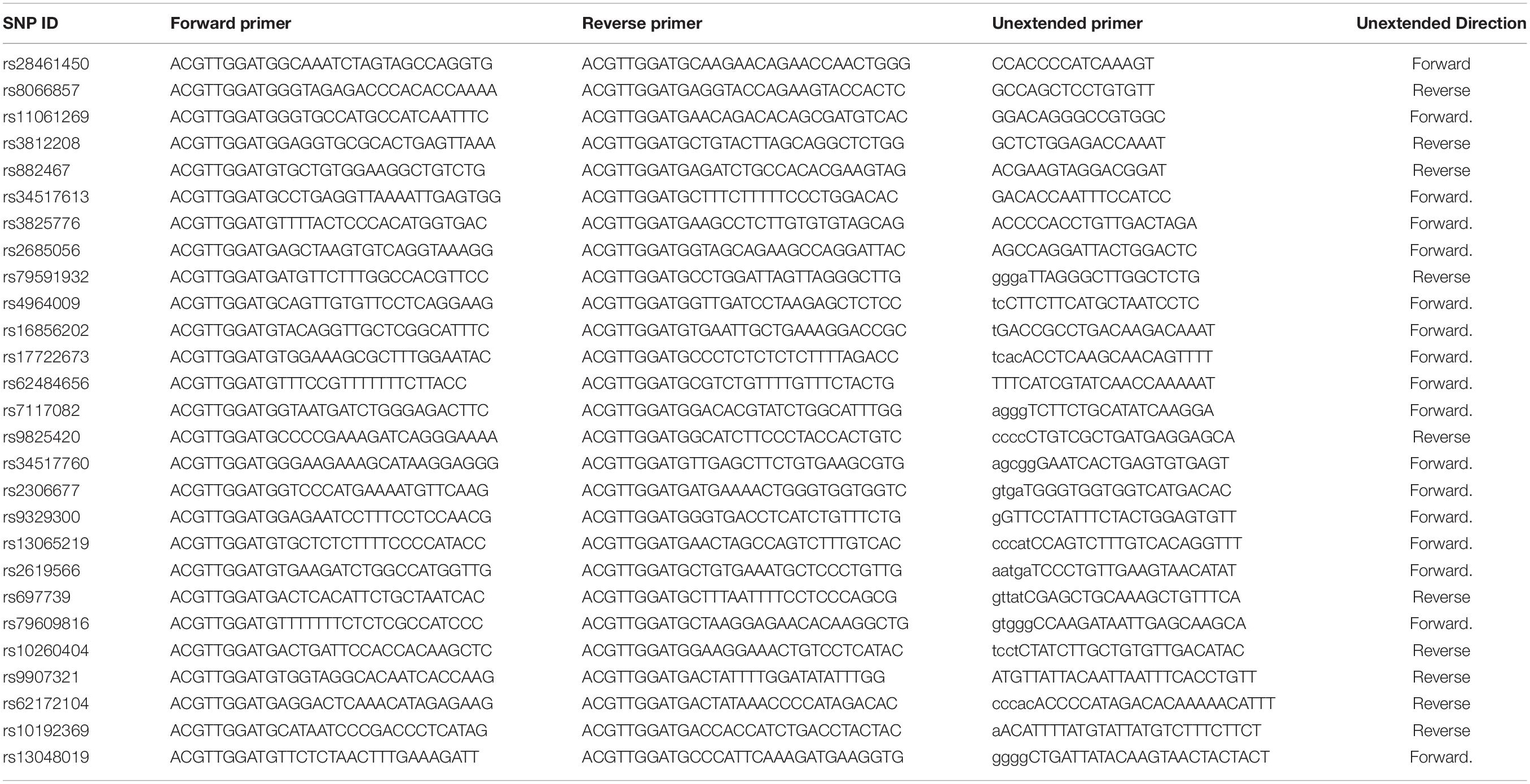
Table 2. Primer sequences genotyped by sequenom.
The SAP enzyme solution was prepared in a 1.5-mL tube as by combining 1.53 μL RNase-free ddH2O, 0.3 μL SAP enzyme, and 0.17 μL SAP buffer, in a total volume of 2 μL. The 1.5-mL tube containing the SAP enzyme solution was subjected to vortexing for 5 s, and was then subjected to centrifugation for 10 s at 5,000 RPM. Subsequently, 2 μL of the SAP enzyme solution was added to each well in the 384-well sample microtiter plate containing the PCR products, and the plate was sealed using a plate-sealing film. The 384-well microtiter plate was then subjected to centrifugation at 1,000 RPM for 1 min and incubated at 37°C for 40 min and at 85°C for 5 min.
The high plex iPLEX gold reaction cocktail solution was prepared in a 1.5-mL tube by combining 0.619 μL RNase-free ddH2O, 0.2 μL iPLEX Buffer Plus, 0.2 μL iPLEX termination mix, 0.94 μL iPLEX extend primer mix, and 0.041 μL iPLEX enzyme. A 384-well sample microtiter plate was centrifuged at 1,000 RPM for 1 min, after which 2 μL of high Plex iLEX gold reaction solution was added to each well, followed by the addition of 7 μL of PCR/SAP reaction solution, for a total of 9 μL volume per well. The 384-well sample microtiter plate with plate was sealed with sealing film, and centrifuged at 1,000 RPM for 1 min. The 384-well microtiter plate containing the samples was then subjected to a thermocycling reaction according to the following conditions: 94°C for 30 s, 94°C for 5 s, 52°C for 5 s, 80°C for 5 s, for 5 cycles, a total of 45 cycles, followed by incubation at 72°C for 3 min.
The cleanup of high plex iPLEX gold reaction products involved the spreading of clean resin onto the 384-well plate containing the iPLEX products, addition of nanopure water to each well, rotation of the plate, and centrifugation of the 384-well plate.
The ACQUIRE module controlled the massARRAY analyzer to acquire spectra from each SpectroCHIP.
For each SNPs, Sanger sequencing of a subset of samples was performed using ABI3500 (ABI3730xl; Applied Biosystems, Inc. CA) to confirm the genotyping results of sequenom massARRAY. Their nucleotide variants were analyzed using the DNASTARLaser gene software (Version v7.1) and compared with the DNA sequence information obtained from NCBI GenBank.
The functional predictions for the intronic C/T polymorphism in the CNTN4 gene (SNP rs2619566), the intronic C/T polymorphism in the DPP6 gene (SNP rs10260404), and the intronic T/A polymorphism in the INPP5B gene (SNP rs79609816) were further analyzed. The prediction of binding sites and transcription factor binding sites was performed using the NHRscan (Sandelin and Wasserman, 2005) and Mscan (Alkema et al., 2004), respectively, the prediction of secondary structures was performed using Mfold (Zuker, 2003), and the prediction of binding sites for miRNA and long non-coding RNA (lncRNA) fragments was performed using Ensemble1 (version GRCh38). The 100-bp sequence both upstream and downstream of each SNP position was analyzed for the presence of the predicted binding sites for transcription factors using NHRscan. The 100-bp sequence both upstream and downstream of each SNP was also analyzed for prediction of the secondary structure. The 50-bp sequence upstream and downstream of each SNP was analyzed by sequence formatting, and was then aligned by using BLAST with the Ensemble non-coding RNA sequence to explore miRNA- and lncRNA-binding sites.
Statistical analysis was performed using the SPSS (Version 19.0) statistical software (SPSS, Chicago, IL, United States). The Hardy-Weinberg equilibrium (HWE) was first evaluated in the healthy controls. Pearson chi-square tests were used to compare the frequencies of alleles and genotypes in cases and controls. Minor allele frequency (MAF) and odds ratios (OR) with 95% confidence intervals (95% CI) were estimated to determine the role of each SNP in sALS risk. Two-tailed p < 0.05 were considered as statistically significant.
In this study, we first performed a pooling GWAS for 250 sALS and 250 controls selected from the HACM population to screen for possible variant loci associated with the pathogenesis of sALS. The results showed that 7 novel SNPs (the rs79609816 and rs62172104 in INPP5B, the rs9825420 in ITGA9, the rs2685056 in ALCAM, the rs7117082 in OPCML, the rs9329300 in PFKP, and the rs11061269 in GPR133) were strongly associated with sALS in the HACM population (Xie et al., 2014). Secondly, we explored previously reported candidate loci associated with the pathogenesis of sALS, and revealed that 20 SNPs (the rs62484656 in CSMD1, the rs17722673 in HECW1, the rs882467 in DPP6, the rs3812208 in ATXN1, the rs28461450 in LIPC, the rs79591932 in RBMS1, the rs9907321 in SLC39A11, the rs4964009 in ITPR2, the rs34517760 in SOD1, the rs13065219 in CNTN4, the rs10260404 in DPP6, the rs697739 in ATXN1, the rs3825776 in LIPC, the rs10192369 in RBMS1, the rs8066857 in SLC39A11, the rs2306677 in ITPR2, the rs13048019 in SOD1, the rs2619566 in CNTN4, the rs16856202 in DISC1, and the rs34517613 in KRT18P55) were potentially associated with sALS (Table 1; Van Es et al., 2007, 2008; Laaksovirta et al., 2010; Fogh et al., 2014; Van Doormaal et al., 2014).
To further assess the association with sALS for the 27 candidate loci identified by GWAS, we performed sequenom massARRAY and DNA sequencing analyses using an independent cohort of 239 sALS cases and the 261 controls of Chinese ethnicity (Table 1). The following three novel SNPs were identified: the rs2619566 in the CNTN4 gene (Chr 3:2583254) (Figure 1A), rs10260404 in the DPP6 gene (Chr 7:154513713) (Figure 1B), and rs79609816 in INPP5B gene (Chr 1:37883093) (Figure 1C). The information of the 3 SNPs has been summarized in Tables 3, 4. All 3 SNPs were intronic polymorphisms. The minor allele frequencies (MAFs) of rs2619566 in the CNTN4 gene and rs79609816 in the INPP5B gene were higher in the sALS patients (44.9 and 8.6%, respectively) than those in the controls (35.6 and 3.1%, respectively). The minor allele of rs2619566 (OR = 1.476, 95% CI = 1.143–1.906, p = 0.003) and rs79609816 (OR = 2.981, 95%CI = 1.649–5.387, p = 0.0003) significantly increased the risk of sALS development in HACM, suggesting these two polymorphisms might represent genetic susceptibility factors. Subjects harboring the minor C allele (CC + CT) of rs2619566 (p = 0.0003) and the minor T allele (TT + TA) of rs79609816 (p = 0.0016) exhibited an increased risk of developing sALS in comparison with the other genotypes (Table 3). The MAFs of rs10260404 in the DPP6 gene were lower in the sALS patients (13.2%) than those in the controls (19.4%). The minor allele of rs10260404 (OR = 0.635, 95% CI = 0.450–0.895, p = 0.009) significantly decreased the risk of developing sALS in HACM, which represented a protective genetic factor. The carriers with the minor C allele (CC + CT) (p = 0.0284) had a significantly decreased risk of developing sALS in the HACM population (Table 3).
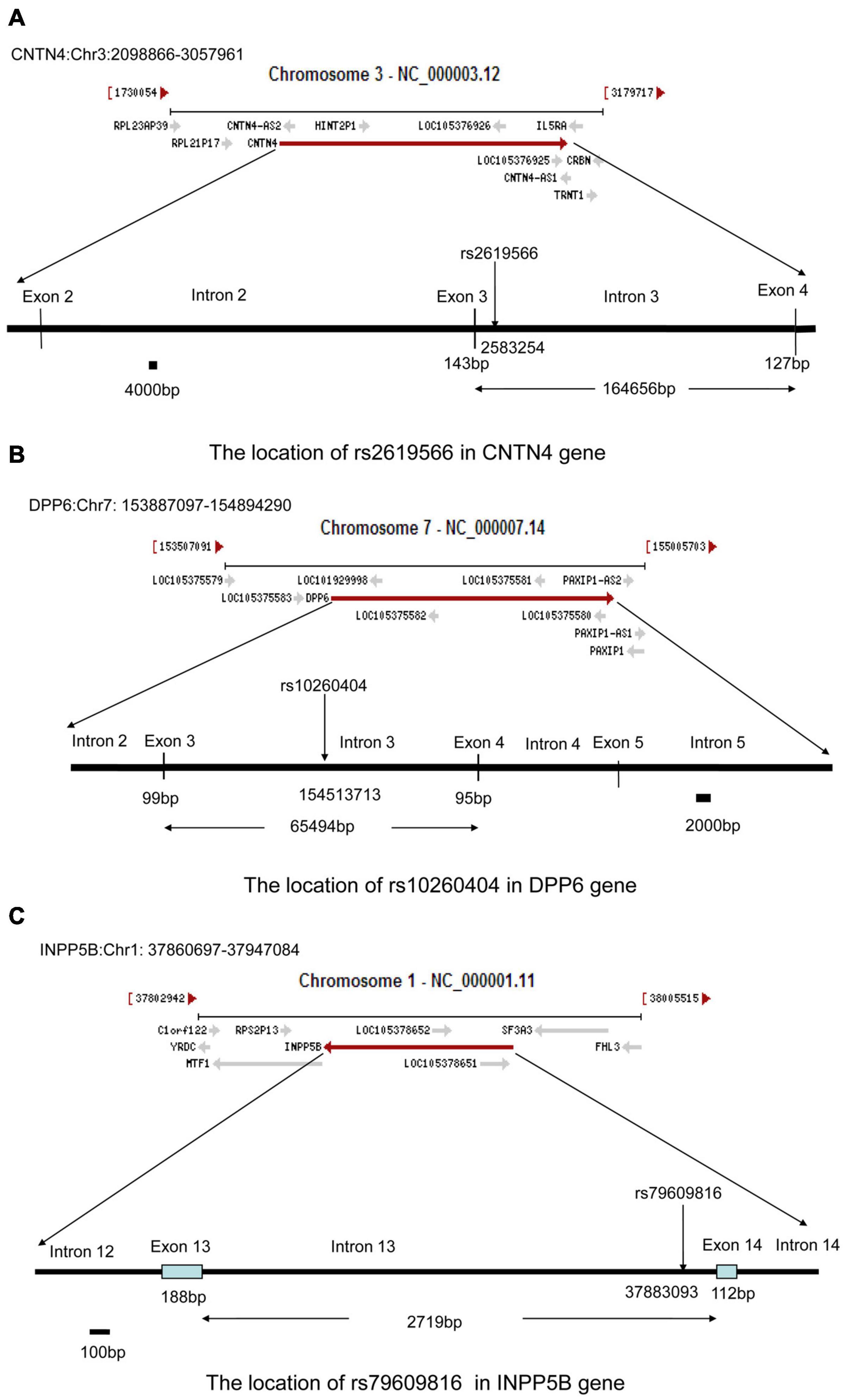
Figure 1. Locations of rs2619566 in the CNTN4 gene, rs10260404 in the DPP6 gene, and rs79609816 in the INPP5B gene. (A) The rs2619566 is located upstream of intron 3, adjoined to exon 3, at position 2583254 of the CNTN4 gene on chromosome 3. (B) The rs10260404 is located in the middle of intron 3, at position 154513713 of the DPP6 gene on chromosome 7. (C) The rs79609816 is located downstream of intron 13, adjoined to exon 14, at position 37883093 of the INPP5B gene on chromosome 1.
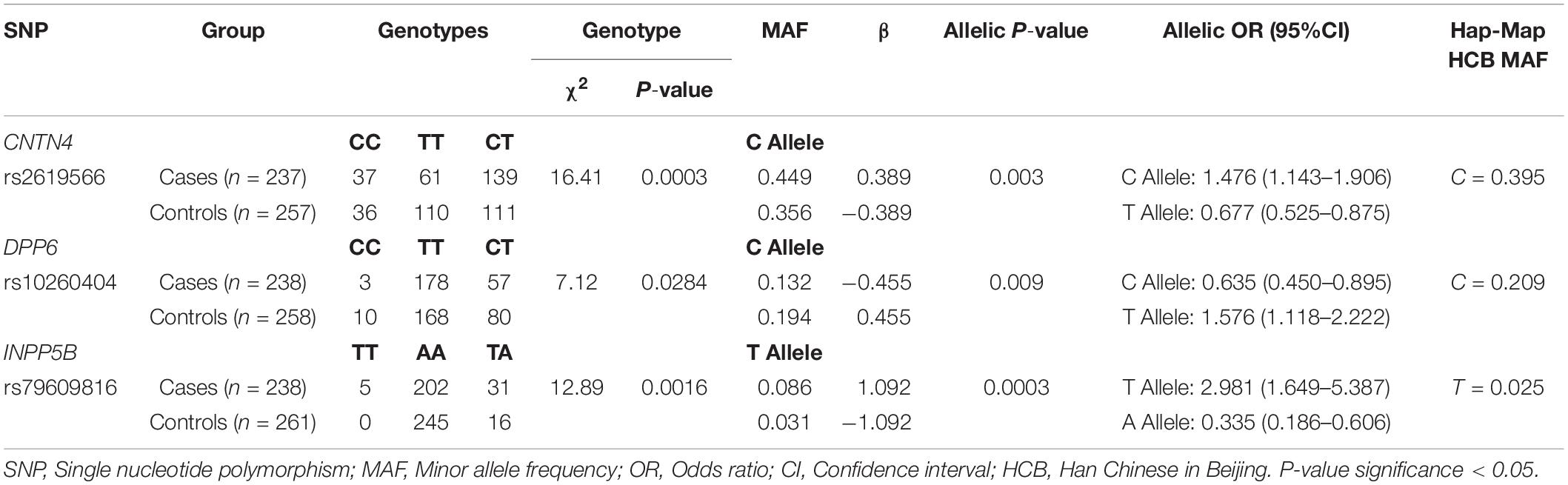
Table 3. Three SNPs shown the nominal significance at P < 0.05 in this study.
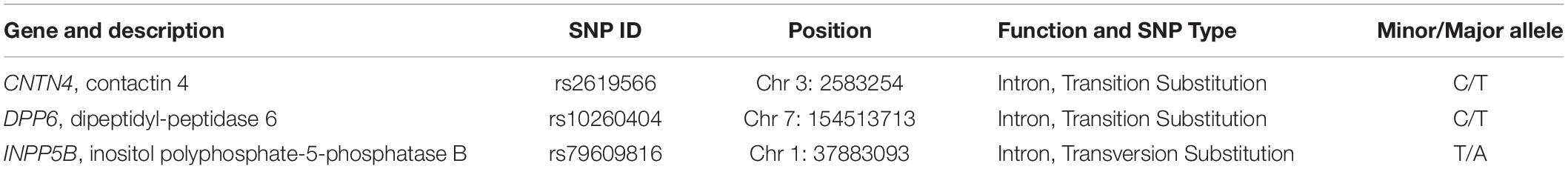
Table 4. Information of three SNPs associated with sALS.
After performing experiments based on the sequenom technology, we randomly selected a few samples from the selected samples to confirm the genotypes for each positive SNP using sanger sequencing performed using ABI3500. The finding exactly coincided with the results obtained from analysis using the sequenom massARRAY technology (Figure 2). The rs2619566 polymorphism in the CNTN4 gene was a T > C variation (Figure 3A). The rs10260404 polymorphism in the DPP6 gene was a T > C variation (Figure 3B). The rs79609816 polymorphism in the INPP5B gene was an A > T variation (Figure 3C).
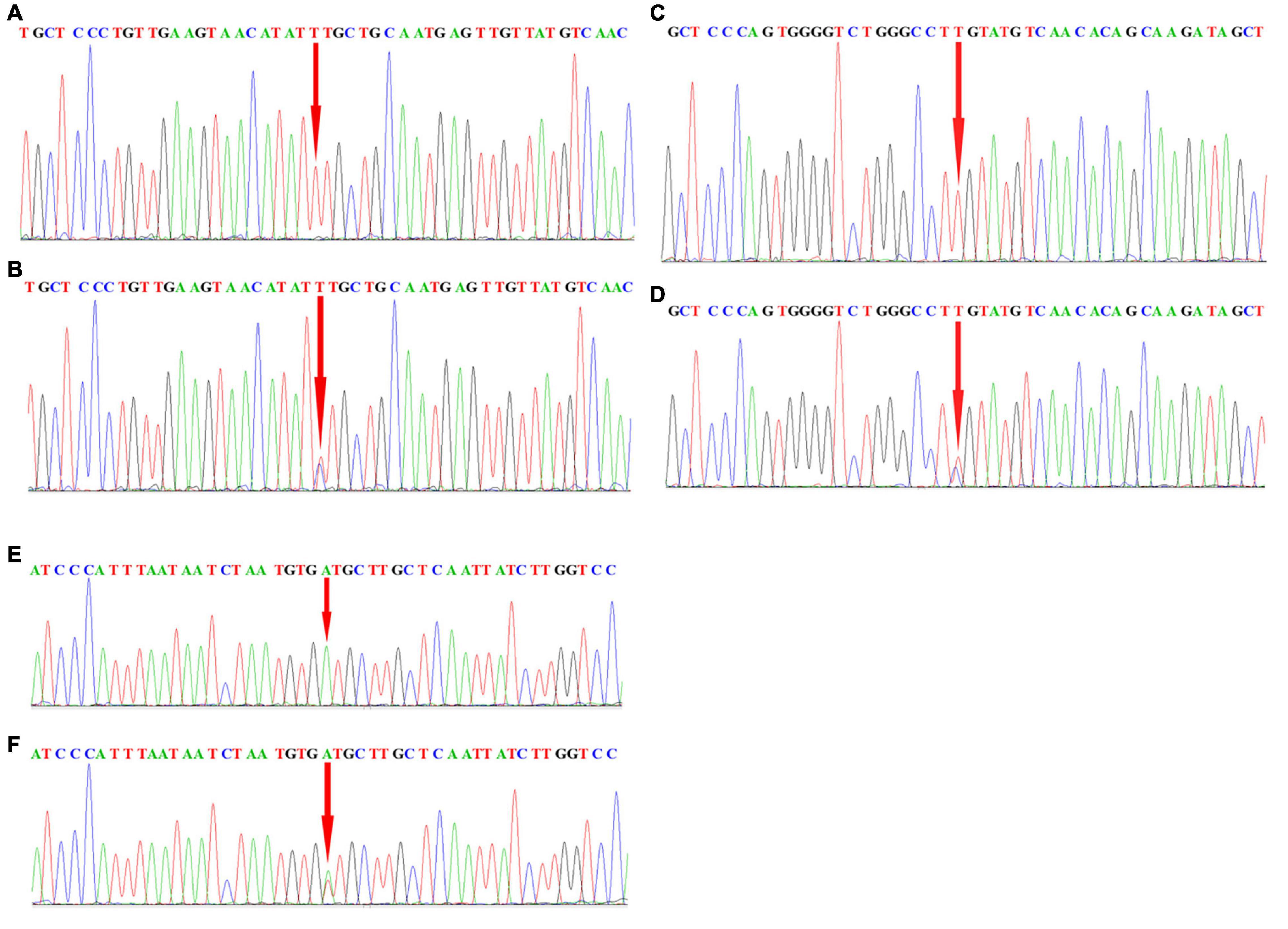
Figure 2. Sequencing data showing rs2619566 in CNTN4, rs10260404 in DPP6, and rs79609816 in INPP5B. (A,B) Peak graph showing the rs2619566 T > C variation in the CNTN4 gene. (C,D) Peak graph showing the rs10260404 T > C variation in the DPP6 gene. (E,F) Peak graph showing the rs79609816 A > T variation in the INPP5B gene. The variant loci are indicated by using a red arrow.
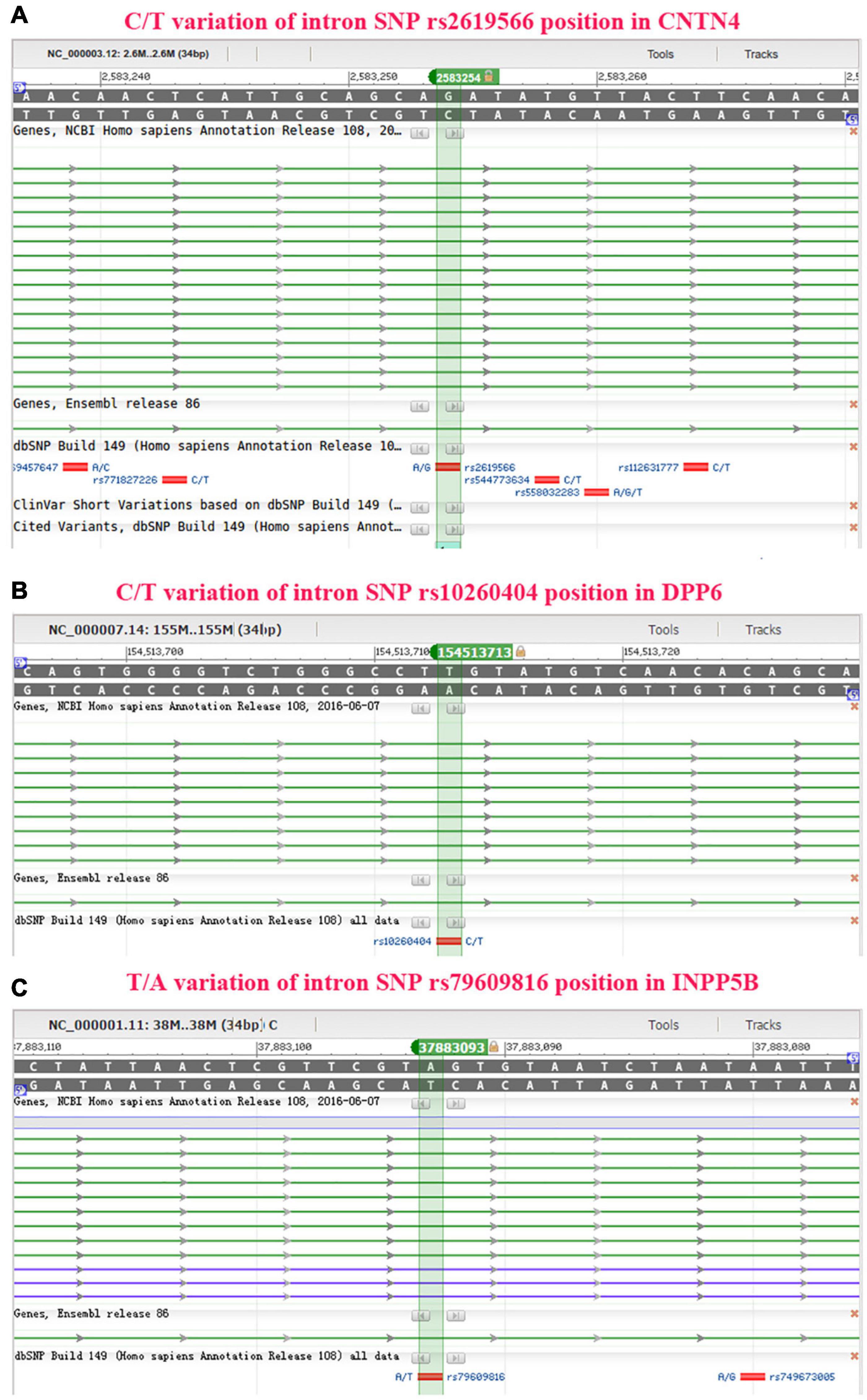
Figure 3. The rs2619566 variants in the CNTN4 gene, rs10260404 variants in the DPP6 gene, and rs79609816 variants in the INPP5B gene. (A) The rs2619566 T > C SNP site is shown in the CNTN4 gene. (B) The rs10260404 T > C SNP site is shown in the DPP6 gene. (C) The rs79609816 A > T SNP site is shown in the INPP5B gene. The variant loci are marked by using a green column.
The IR0 element with the sequence CAGATATGTTAC at positions 99–110 of the CNTN4 gene (Figures 4A,B), the IR0 element with the sequence GGGTCTGGGCCT at positions 89–100, the ER6 element with the sequence GTATGTCAACACAGCAAG at positions 102–119 of the DPP6 gene (Figures 4C,D), and the ER8 element with the sequence GCATCACATTAGATTATTAA at positions 98–117 of the INPP5B gene (Figures 4E,F) were predicted to be binding fragments. The SNP in the CNTN4 gene associated with sALS was located at the third nucleotide position in the binding fragment of the CNTN4 gene (Figures 4A,B). The SNP for the DPP6 gene was located in the middle of the two binding fragments identified in the DPP6 gene (Figures 4C,D), while the SNP for INPP5B was located at the fourth nucleotide position of the binding fragment of the INPP5B gene (Figures 4E,F).

Figure 4. Binding site prediction using NHRscan. (A) The IR0 CAGATATGTTAC sequence at positions 99–110 of the CNTN4 gene is predicted as a binding element, and the SNP is located at the third nucleotide position of the binding fragment. The sites marked using a blue frame indicate the predicted binding fragments. (B) Statistical information about the predicted binding sites of the CNTN4 gene. (C) The IR0 GGGTCTGGGCCT sequence at positions 89–100 and the ER6 GTATGTCAACACAGCAAG sequence at positions 102–119 of the DPP6 gene are predicted as binding fragments, and the SNP is located in the middle of the two binding fragments in the DPP6 gene. The sites marked using a blue frame indicate the predicted binding fragments. (D) Statistical information about the predicted binding sites in the DPP6 gene. (E) The ER8 GCATCACATTAGATTATTAA sequence at positions 98–117 of the INPP5B gene is predicted as a binding fragment, and the SNP is located at the fourth nucleotide position of the binding fragment. The sites marked using a blue frame indicate the predicted binding fragments. (F) Statistical information about the predicted binding sites in the INPP5B gene.
The sites between 793 and 800 bp, and between 751 and 758 bp in the CNTN4 gene (Figure 5A), the sites between 440 and 421 bp, and between 312 and 293 bp in the DPP6 gene (Figure 5B), and the sites between 338 and 323 bp, and between 163 and 156 bp in the INPP5B gene (Figure 5C) away from the upstream of binding site were hypothesized to be binding sites for transcription factors.
Figure 5. Transcription factor prediction using Mscan. The prediction based on NHRscan and Mscan provided consistent results. (A) Transcription factor binding site located between 793 and 800 bp, and between 751 and 758 bp away from the upstream of binding site in CNTN4. (B) Transcription factor binding site located between 440 and 421 bp, and between 312 and 293 bp away from the upstream of binding site in DPP6. (C) Transcription factor binding site located between 338 and 323 bp, and between 163 and 156 bp away from the upstream of the binding site in the INPP5B gene.
As shown in Figures 6A–C, all secondary structures of the binding site regions in the CNTN4, DPP6, and INPP5B genes formed a hairpin-like structure. No binding sites for miRNA were found after conduction of the miRNA analysis. During the analysis for lncRNA-binding site, the following three sites were predicted to be the possible candidates for lncRNA binding: theENST00000562617 site between 25 and 38 bp upstream of the SNP site, the ENST00000432505 site between 16 and 28 bp away from the downstream, and the ENST00000436078 site between 20 and 32 bp from the SNP in the CNTN4 gene (Table 5). The ENST00000450077 element between 9 and 21 bp downstream of the SNP site, ENST00000453348 between 28 and 16 bp away from the upstream, ENST00000452622 between 21 and 10 bp at the upstream, ENST00000577700 between 7 and 5 bp at the upstream, and ENST00000418297 between 5 and 21 bp at the downstream in DPP6 were predicted to be the possible binding candidates of lncRNAs (Table 6). The ENST00000451362 element between 11 and 23 bp downstream of the SNP site, ENST00000591702,ENST00000401018 and ENST00000433505 between 45 and 32 bp away from the upstream of the variation site in INPP5B were predicted to be the possible binding candidates for lncRNAs (Table 7).

Table 5. List of CNTN4 possible binding candidates of lncRNAs.
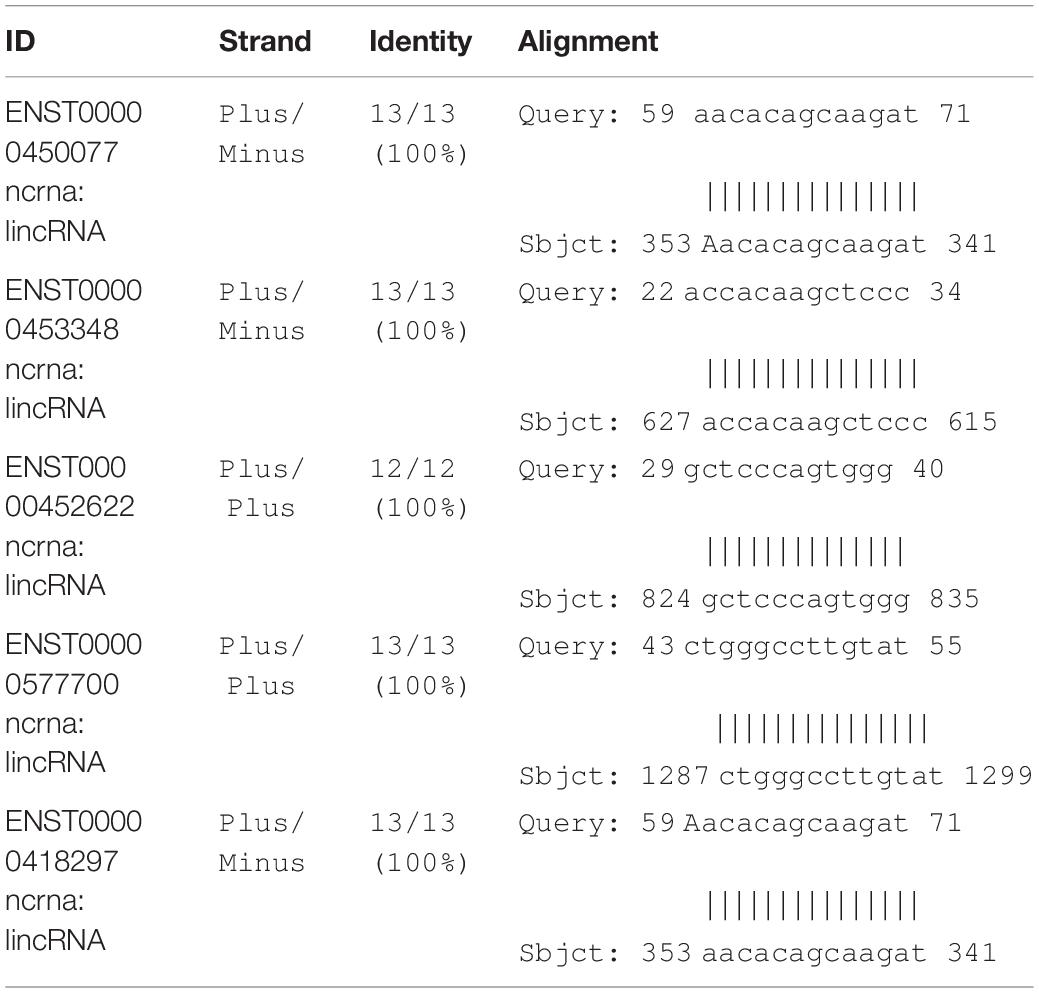
Table 6. List of DDP6 possible binding candidates of lncRNAs.
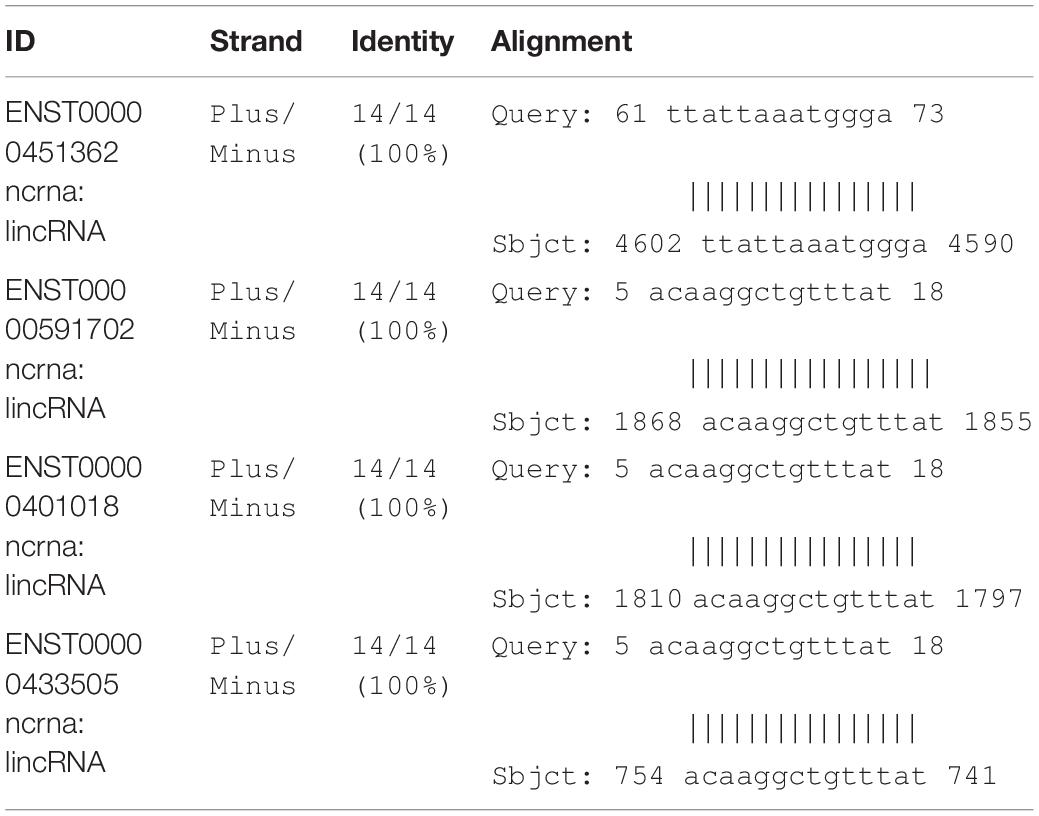
Table 7. List of INPP5B possible binding candidates of lncRNAs.
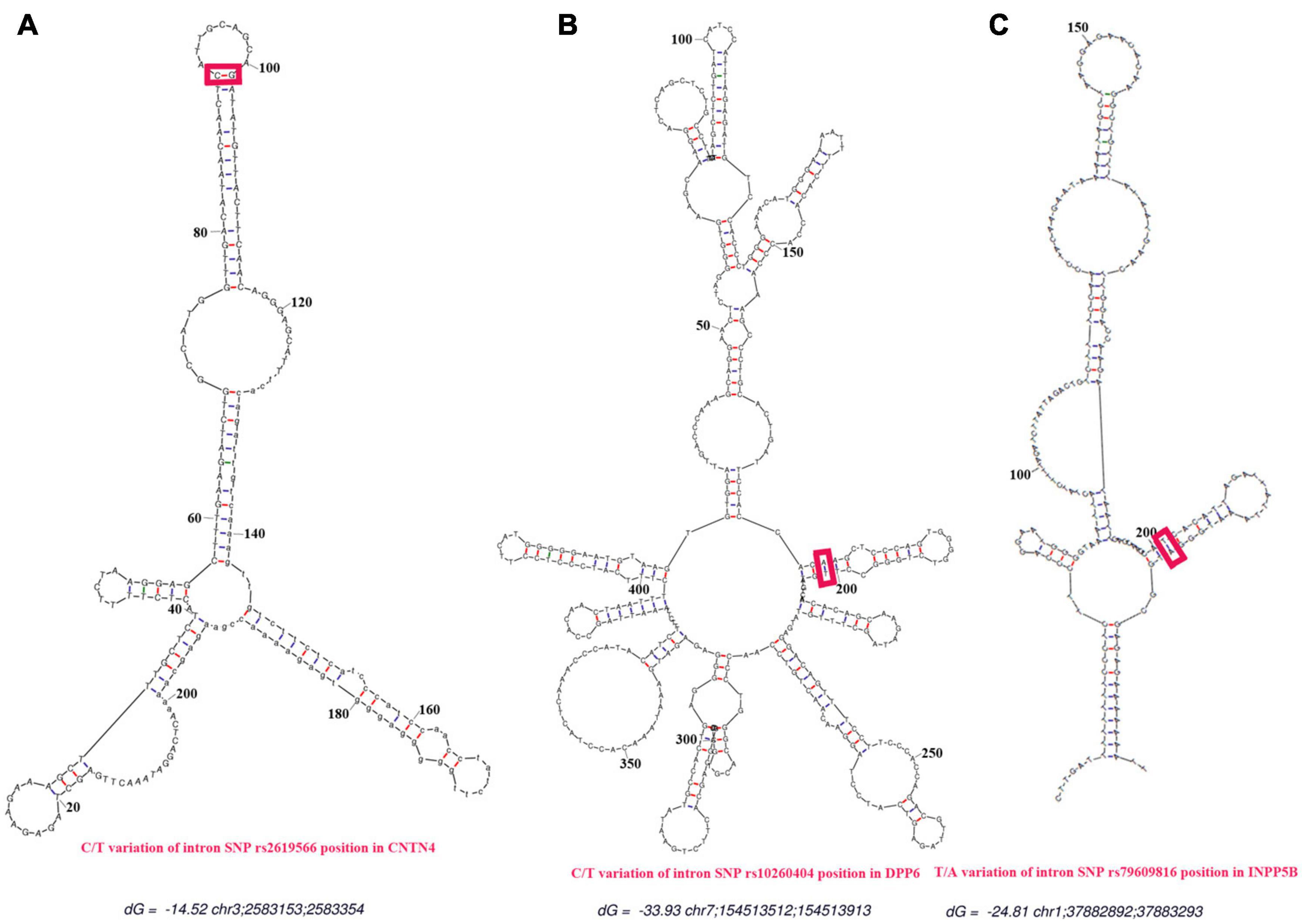
Figure 6. Prediction of secondary structures. The red line represents the predicted binding sites for proteins. The red frames indicate the positions of the SNPs. (A) Secondary structure for the rs2619566 T > C variation in the CNTN4 gene. (B) Secondary structure for the rs10260404 T > C variation in the DPP6 gene. (C) Secondary structure for the rs79609816 A > T variation in the INPP5B gene. All secondary structures of the binding regions in the CNTN4, DPP6, and INPP5B genes were predicted as hairpin-like structures.
Presently, the pathogenesis of sALS is not well understood, while the results of recent investigations suggest that genetic factors may play an important role. To better understand the extent by which genetic factors contribute to the risk of sALS development, it is important to find possible genes or loci that contribute to sALS susceptibility. We conducted a two-stage study in the HACM population consisting of a total of 489 sALS cases and 511 controls, excluding the rapid and slow progressing sALS cases, the early- and late-onset cases, and the sALS cases of atypical clinical manifestations. In the first stage, we performed a pooling GWAS involving 250 sALS cases and 250 controls from the HACM population to screen for possible loci associated with sALS and identified 7 SNPs that showed the most remarkable association with sALS in the HACM population (Xie et al., 2014). Furthermore, we explored previously reported candidate loci associated with the pathogenesis of sALS, and revealed that 20 SNPs were potentially associated with sALS (Table 1; Van Es et al., 2007, 2008; Laaksovirta et al., 2010; Fogh et al., 2014; Van Doormaal et al., 2014). In the second stage, we performed a sequenom massARRAY and DNA sequencing analysis using samples of an independent cohort of 239 sALS cases and 261 controls of HACM ethnicity in order to further identify the relationship with sALS risk for the above-mentioned 27 candidate loci in this study. The results revealed that the rs2619566 in CNTN4, the rs10260404 in DPP6, and the rs79609816 in INPP5B were markedly associated with sALS in the HACM population.
Furthermore, we analyzed the allele and genotype frequencies in sALS cases and controls for the 3 SNPs associated with sALS in the HACM population. All 3 SNPs were intronic polymorphisms. The MAFs of the rs2619566 in the CNTN4 gene and the rs79609816 in the INPP5B gene were significantly higher in the sALS patients than those in the controls. The minor allele frequency of the rs2619566 and the rs79609816 significantly increased the risk of sALS development in the HACM population; the subjects harboring the minor allele C (CC + CT) of rs2619566 and the minor allele T (TT + TA) of rs79609816 exhibited an increased risk of sALS development in comparison with subjects of other genotypes, which indicated that these genotypes were susceptibility factors. The minor allele of rs10260404 in the DPP6 gene significantly decreased the risk of sALS development in HACM, and the carriers with the minor C allele (CC + CT) of rs10260404 showed a decreased risk of sALS development, thus indicating that the minor allele might be a protective factor (Table 3).
Additionally, we conducted functional predictions for the 3 SNPs in the CNTN4, DPP6, and INPP5B genes. The results of this functional prediction analysis showed that the CNTN4, DPP6, and INPP5B polymorphic regions (3 SNPs) might be binding sites for transcription factors. Possible binding sites for transcription factors were identified at regions more than 700 bp upstream of the CNTN4 SNPs, more than 400 bp upstream of the DPP6 SNPs, and more than 300 bp upstream of the INPP5B SNPs. The region in the vicinity of more than 20 bp of the CNTN4 and DPP6 SNPs, and the vicinity of more than 30 bp of the INPP5B SNPs were predicted to be lncRNA non-coding regions, which might play an important role in the regulation of binding proteins (Figures 4–6 and Tables 5–7). These alterations in the CNTN4, DPP6, and INPP5B genes might change or affect their splicing, transcription, and translation, might lead to the generation of abnormal functional and/or structural proteins, and might affect the development of sALS.
CNTN4 is also known as the AXCAM or BIG-2 gene, and the gene encodes a member of the contactin family of immunoglobulins. CNTNs are axon-associated cell adhesion molecules that demonstrate certain important functions in neuronal network formation and plasticity. The encoded protein is a glycosylphosphatidylinositol-anchored neuronal membrane protein that may play a role in the formation of axon connections in the developing nervous system. The alternative splice results in the generation of multiple transcript variants (Cottrell et al., 2011; Cuoco et al., 2011; Mikulska et al., 2011; Guo et al., 2012; Kaurani et al., 2014). Additionally, the encoded protein of the gene also participates in the function of axon guidance, axonal fasciculation, axonogenesis, brain development, negative regulation of neuronal differentiation, nervous system development, neuronal cell-cell adhesion, neuronal projection development, and regulation of synaptic plasticity (Fernandez et al., 2004; Oguro-Ando et al., 2017).
CNTN4 alteration may contribute toward the infliction of damage of the neuronal axons, as well as affect neuron differentiation, adhesion, projections, and synaptic plasticity. In our study, we found that the subjects harboring the minor C allele (CC + CT) of the rs2619566 polymorphism in the CNTN4 gene had a significantly increased risk of developing sALS. The pathogenesis mechanisms related to sALS development may rely on the CNTN4 gene harboring the minor C allele (CC + CT) that results in rs2619566 changes or affects splicing, transcription, or translation of the CNTN4 gene, thereby generating an abnormal CNTN4 protein, which induces abnormalities in differentiation, adhesion, projection, synaptic plasticity, axonal guidance, and fasciculation of motor neurons, contributing to the development of lesions and disturbance in neuronal network formation and plasticity, subsequently resulting in the development of sALS (Figure 7A).
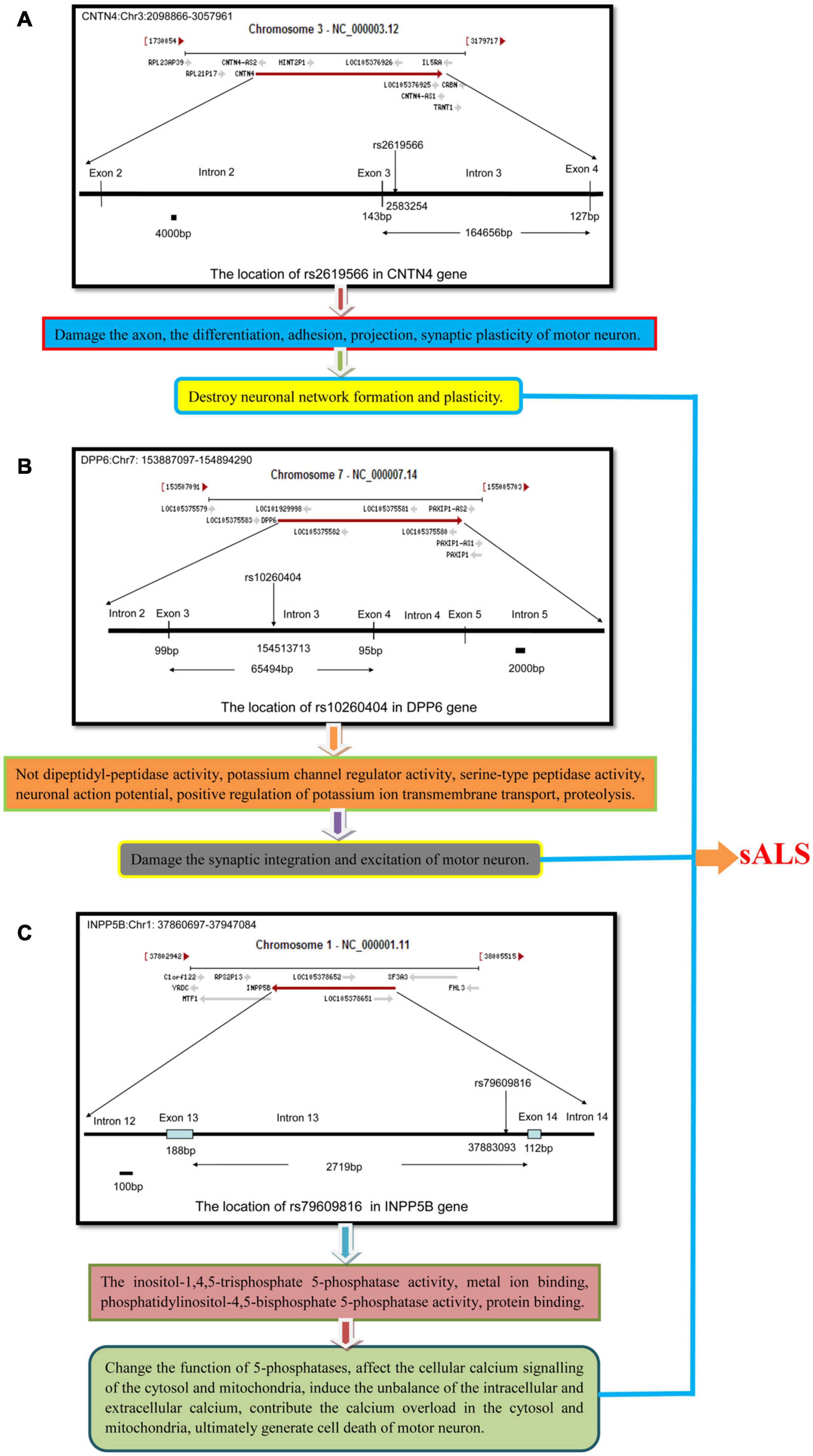
Figure 7. The schematic diagram about the mechanisms that the polymorphisms of rs2619566 in CNTN4, rs10260404 in DPP6 and rs79609816 in INPP5B might result in sALS from HACM. The pathogenesis of sALS might be a mechanism of multiple genes and loci participation through a series of complex pathophysiologic pathways. (A) The polymorphisms of rs2619566 in CNTN4 might be associated with destroy of neuronal network formation and plasticity. (B) The polymorphisms of rs10260404 in DPP6 might be related to damaging synaptic integration and excitation of neuron. (C) The polymorphisms of rs79609816 in INPP5B might induce the calcium disorder in cytosol and mitochondria, contributed to neuron death through a series of pathophysiological processes.
DPP6 also is known as VF2 DPPX and MRD33. This gene encodes a single-pass type II membrane protein that is a member of the peptidase S9B family of serine proteases. This protein has no detectable protease activity, and this is most likely attributed to the absence of the conserved serine residue normally present in the catalytic domain of serine proteases. However, it does bind specific voltage-gated potassium channels and alters their expression and biophysical properties. Variations in this gene may be associated with susceptibility to sALS (Cronin et al., 2008, 2009; Del Bo et al., 2008; Kwee et al., 2012). Only 9 published studies have reported on the association between DPP6 and sALS thus far, and of these 9 studies, only 2 independent studies were conducted in the Chinese population (Li et al., 2009; Chen et al., 2012), and both studies suggested the absence of association of the rs10260404 variant in DPP6 with sALS development in the Chinese population. However, the other 7 studies performed in European populations including Italian, Dutch, Polish, and Irish, and Americans, reported results that were dramatically different; 5 of the 7 studies suggested that DPP6 was a candidate gene for sALS, and reported several candidate loci associated with sALS (Cronin et al., 2008, 2009; Del Bo et al., 2008; Kwee et al., 2012), while 2 of the 7 studies indicated that mutations in these genes were unlikely to be associated with sALS (Chiò et al., 2009; Daoud et al., 2010). Two independent studies suggested the absence of association of the rs10260404 variant in DPP6 with sALS development in the Chinese population (Li et al., 2009; Chen et al., 2012). Actually, in our previous GWAS study, there was no correlation between the rs10260404 variant in DPP6 and sALS yet (Xie et al., 2014). But in this validation study, it showed that the rs10260404 variant in DPP6 was associated with sALS in Chinese Han populations. The contrary conclusions might be due to the differences of their ethnicity or population background or sample size. The further study need conduct in the larger sample size and different ethnicity and population background.
An alternative splicing of DPP6 results in the generation of multiple transcript variants (Lin et al., 2014). DPP6 is critical for the synaptic integration and excitation of neurons (Wolf et al., 2014). The main functions of DPP6 include the not dipeptidyl-peptidase activity, potassium channel regulator activity, serine-type peptidase activity (Yokotani et al., 1993), neuronal action potential regulation, positive regulation of potassium ion transmembrane transport, and proteolysis (Soh and Goldstein, 2008). Our results revealed that carriers of the minor allele C (CC + CT) of the rs10260404 polymorphism in the DPP6 gene had an decreased risk of developing sALS, suggesting that presence of the minor C allele might change or affect the splicing, transcription, or translation of the DPP6 gene, which might be responsible for an decreased risk of sALS development. According to the existing knowledge on the DPP6 function, the protective pathogenesis associated with sALS might be attributable to an abnormal alteration of the protein activity due to the rs10260404 polymorphism-associated changes improving the synaptic integration and excitation of motor neurons. Meanwhile, the known functions of DPP6 might be involved in the process, subsequently contributing to the decreased risk of sALS (Figure 7B).
INPP5B is also known as 5PTase. This gene encodes a member of a family of inositol polyphosphate-5-phosphatases (5-phosphatases) (Ross et al., 1991). The enzyme functions in the regulation of calcium signaling by inactivating inositol phosphates (IPs). The encoded protein is localized in the cytosol and mitochondria, and establishes association with membranes through an isoprenyl modification near the C-terminus. Cellular calcium signaling is controlled by the production of IPs and by phospholipase C in response to extracellular signals. The IPs signaling molecules are inactivated by activities of a family of 5-phosphatases. Several alternatively spliced transcript variants of this gene have been described, but the full-length transcript features of a few of these variants have not been determined. The main functions of INPP5B include inositol-1,4,5-trisphosphate 5-phosphatase activity, metal ion binding, phosphatidylinositol-4,5-bisphosphate 5-phosphatase activity, and protein binding (Jefferson and Majerus, 1995; Noakes et al., 2011). In the results of the present study, the MAFs for the rs79609816 polymorphism in the INPP5B gene were higher in the sALS patients than those in the controls, and subjects harboring the minor allele T (TT + TA) had a significantly higher risk of developing sALS. We hypothesize that the minor allele T of the INPP5B rs79609816 polymorphism may affect the splicing, transcription, or translation of the INPP5B gene, that may further generate an abnormal INPP5B protein, which results in an increased risk for sALS development and represents a susceptibility factor for this disease. The alteration due to the INPP5B rs79609816 variant might change the function of 5-phosphatases, affect cellular calcium signaling in the cytosol and mitochondria, induce an imbalance in the intracellular and extracellular calcium levels, contribute to calcium overload in the cytosol and mitochondria, and ultimately result in the death of motor neurons (Figure 7C). In conclusion, our study found that 3 polymorphisms, namely the CNTN4 rs2619566, the DPP6 rs10260404, and the rs79609816, were significantly associated with sALS development in the HACM population (Figure 7).
Based on the currently known functions of the CNTN4, DPP6, and INPP5B genes, it is possible to postulate that CNTN4 rs2619566 variants may inflict damage on the neuronal network formation and plasticity of motor neurons, that DPP6 rs10260404 variants may improve the synaptic integration and excitation of motor neurons, and that INPP5B rs79609816 variants may destroy cellular calcium signaling of the cytosol and mitochondria in motor neurons, ultimately influence the sALS development. In the process, the function of axon guidance, axonal fasciculation, axonogenesis, negative regulation of neuron differentiation, nervous system development, neuron cell-cell adhesion, neuron projection development, regulation of synaptic plasticity, not dipeptidyl-peptidase activity, potassium channel regulator activity, serine-type peptidase activity, neuronal action potential, positive regulation of potassium ion transmembrane transport, proteolysis, inositol-1,4,5-trisphosphate-5-phosphatase activity, metal ion binding, phosphatidylinositol-4,5-bisphosphate 5-phosphatase activity, and protein binding may all directly or indirectly participate in the development of sALS (Figure 7).
Our data are consistent with those obtained using a model of sALS pathogenesis that involves a series of complex pathophysiologic processes attributed to the presence of multiple genes and loci. These polymorphisms might change or affect splicing, transcription, and translation, resulting in the production of abnormal CNTN4, DPP6, and INPP5B proteins, which might inflict damage or disrupt or improve neuronal network formation and plasticity, the synaptic integration and excitation, and the cellular calcium signaling of the cytosol and mitochondria of motor neurons, ultimately influencing the development of sALS. Our study further suggests that multiple genes and loci participate in the susceptibility to sALS. Our results provide valuable data for conducting further studies on the elucidation of the pathogenesis of sALS.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
The study was approved by the Institutional Review Board of the Hospital Human Ethics Committee of The First Affiliated Hospital of Nanchang University and was conducted in accordance with the approved guidelines and regulations. The patients/participants provided their written informed consent to participate in this study.
JZ and RX conceived, designed, and performed the experiments, analyzed the data, and wrote the manuscript. XZ, YD, and HN conceived and designed the experiments. JZ, WQ, and FH performed the experiments, analyzed the data, and were the jointed first authors. XZ, YD, HN, and RX contributed reagents, materials, tools, and services and were the co-corresponding authors. RX was the corresponding author. All authors were involved in the drafting, critical revision, read, and final approval of the manuscript for publication and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved and contributed significantly to this research and in the preparation of the manuscript.
We thank the National Natural Science Foundation of China (81960244, 81160161, and 81360198), Education Department of Jiangxi Province (GJJ170042, GJJ13198, and GJJ170021), Jiangxi Provincial Department of Science and Technology [20192BAB205044, (2014)-47, 20142BBG70062, and 20171BAB215022], Health and Family Planning Commission of Jiangxi Province (20191018 and 20181019), Jiangxi Provincial Department of Science and Technology Gan Po Elite 555 [Jiangxi Finance Elite Education Refers to (2015) 108], and the Natural Science Foundation of Guangdong Province (2019A1515011341) for extending financial support for this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are grateful to the patients who generously contributed their samples and their time necessary for execution of this study. We are also thankful to Gene for Health Biotech (Shanghai) Co., Ltd. for providing assistance for the bioinformatics analysis. We would like to thank Editage for English language editing.
sALS, Sporadic Amyotrophic Lateral Sclerosis; HACM, Han Ancestry of Chinese Mainland; CNS, Central Nervous System; fALS, Familial ALS; CNTN4, Contactin 4; DPP6, Dipeptidyl-Peptidase 6; INPP5B, Inositol Polyphosphate-5-Phosphatases B/2; SNPs, Single Nucleotide Polymorphisms; GWAS, Genome-Wide Association Study; PCR, Polymerase Chain Reaction; SAP, Shrimp Alkaline Phosphatase; MAF, Minor Allele Frequency; OR, Odds Ratios; CI, Confidence Intervals.
Alkema, W. B., Johansson, O., Lagergren, J., and Wasserman, W. W. (2004). MSCAN: identification of functional clusters of transcription factor binding sites. Nucleic Acids Res. 32, W195–W198.
Brooks, B. R., Miller, R. G., Swash, M., Munsat, T. L., and World Federation of Neurology Research Group on Motor Neuron Diseases. (2000). El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 1, 293–299. doi: 10.1080/146608200300079536
Callaghan, B., Feldman, D., Gruis, K., and Feldman, E. (2011). The association of exposure to lead, mercury, and selenium and the development of amyotrophic lateral sclerosis and the epigenetic implications. Neurodegener. Dis. 8, 1–8. doi: 10.1159/000315405
Chen, Y., Zeng, Y., Huang, R., Yang, Y., Chen, K., Song, W., et al. (2012). No association of five candidate genetic variants with amyotrophic lateral sclerosis in a Chinese population. Neurobiol. Aging 33:2721.e3-5.
Chia, R., Chiò, A., and Traynor, B. J. (2018). Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 17, 94–102.
Chiò, A., Schymick, J. C., Restagno, G., Scholz, S. W., Lombardo, F., Lai, S. L., et al. (2009). A two-stage genome-wide association study of sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 18, 1524–1532.
Cottrell, C. E., Bir, N., Varga, E., Alvarez, C. E., Bouyain, S., and Zernzach, R. (2011). Contactin 4 as an autism susceptibility locus. Autism Res. 4, 189–199. doi: 10.1002/aur.184
Cronin, S., Berger, S., Ding, J., Schymick, J. C., Washecka, N., Hernandez, D. G., et al. (2008). A genome wide association study of sporadic ALS in a homogenous Irish population. Hum. Mol. Genet. 17, 768–774. doi: 10.1093/hmg/ddm361
Cronin, S., Tomik, B., Bradley, D. G., Slowik, A., and Hardiman, O. (2009). Screening for replication of genome-wide SNP associations in sporadic ALS. Eur. J. Hum. Genet. 17, 213–218. doi: 10.1038/ejhg.2008.194
Cuoco, C., Ronchetto, P., Gimelli, S., Béna, F., Divizia, M. T., Lerone, M., et al. (2011). Microarray based analysis of an inherited terminal 3p26.3 deletion, containing only the CHL1 gene, from a normal father to his two affected children. Orphanet. J. Rare Dis. 6:12. doi: 10.1186/1750-1172-6-12
Daoud, H., Valdmanis, P. N., Dion, P. A., and Rouleau, G. A. (2010). Analysis of DPP6 and FGGY as candidate genes for amyotrophic lateral sclerosis. Amyotroph. Lateral. Scler. 11, 389–391. doi: 10.3109/17482960903358857
Del, Bo, R., Ghezzi, S., Corti, S., Santoro, D., Prelle, A., et al. (2008). DPP6 gene variability confers increased risk of developing sporadic amyotrophic lateral sclerosis in Italian patients. J. Neurol. Neurosurg. Psychiatry. 79:1085. doi: 10.1136/jnnp.2008.149146
Fernandez, T., Morgan, T., Davis, N., Klin, A., Morris, A., Farhi, A., et al. (2004). Disruption of contactin 4 (CNTN4) results in developmental delay and other features of 3p deletion syndrome. Am. J. Hum. Genet. 74, 1286–1293. doi: 10.1086/421474
Fogh, I., Ratti, A., Gellera, C., Lin, K., Tiloca, C., Moskvina, V., et al. (2014). A genome-wide association meta-analysis identifies a novel locus at 17q11.2 associated with sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 23, 2220–2231.
Garzillo, E. M., Miraglia, N., and Pedata, P. (2015). Amyotrophic lateral sclerosis and exposure to metals and other occupational/ environmental hazardous materials: state of the art. G. Ital. Med. Lav. Ergon. 37, 8–19.
Guo, H., Xun, G., Peng, Y., Xiang, X., Xiong, Z., Zhang, L., et al. (2012). Disruption of Contactin 4 in two subjects with autism in Chinese population. Gene 505, 201–205. doi: 10.1016/j.gene.2012.06.051
Jefferson, A. B., and Majerus, P. W. (1995). Properties of type II inositol polyphosphate 5 phosphatase. J. Biol. Chem. 270, 9370–9377. doi: 10.1074/jbc.270.16.9370
Kaurani, L., Vishal, M., Kumar, D., Sharma, A., Mehani, B., Sharma, C., et al. (2014). Gene-rich large deletions are overrepresented in POAG patients of Indian and Caucasian origins. Invest. Ophthalmol. Vis. Sci. 55, 3258–3264. doi: 10.1167/iovs.14-14339
Kwee, L. C., Liu, Y., Haynes, C., Gibson, J. R., Stone, A., Schichman, S. A., et al. (2012). A high-density genome-wide association screen of sporadic ALS in US veterans. PLoS One 7:e32768. doi: 10.1371/journal.pone.0032768
Laaksovirta, H., Peuralinna, T., Schymick, J. C., Scholz, S. W., Lai, S. L., Myllykangas, L., et al. (2010). Chromosome 9p21 in amyotrophic lateral sclerosis in finland: a genome-wide association study. Lancet Neurol. 9, 978–985. doi: 10.1016/s1474-4422(10)70184-8
Landers, J. E., Melki, J., Meininger, V., Glass, J. D., van den Berg, L. H., van Es, M. A., et al., et al. (2009). Reduced expression of the Kinesin-Associated Protein 3 (KIFAP3) gene increases survival in sporadic amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U.S.A. 106, 9004–9009. doi: 10.1073/pnas.0812937106
Li, X. G., Zhang, J. H., Xie, M. Q., Liu, M. S., Li, B. H., Zhao, Y. H., et al. (2009). Association between DPP6 polymorphism and the risk of sporadic amyotrophic lateral sclerosis in Chinese patients. Chin. Med. J. 122, 2989–2992.
Lian, L., Liu, M., Cui, L., Guan, Y., Liu, T., Cui, B., et al. (2019). Environmental risk factors and amyotrophic lateral sclerosis (ALS): a case-control study of ALS in China. J. Clin. Neurosci. 66, 12–18. doi: 10.1016/j.jocn.2019.05.036
Lin, L., Long, L. K., Hatch, M. M., and Hoffman, D. A. (2014). DPP6 domains responsible for its localization and function. J. Biol. Chem. 289, 32153–32165. doi: 10.1074/jbc.m114.578070
Longinetti, E., and Fang, F. (2019). Epidemiology of amyotrophic lateral sclerosis: an update of recent literature. Curr. Opin. Neurol. 32, 771–776. doi: 10.1097/wco.0000000000000730
Ludolph, A. C., Brettschneider, J., and Weishaupt, J. H. (2012). Amyotrophic lateral sclerosis. Curr. Opin. Neurol. 25, 530–535.
Mathis, S., Goizet, C., Soulages, A., Vallat, J. M., and Le, and Masson, G. (2019). Genetics of amyotrophic lateral sclerosis: a review. J. Neurol. Sci. 399, 217–226.
Mikulska, K., Pepłowski, L., and Nowak, W. (2011). Nanomechanics of Ig-like domains of human contactin (BIG-2). J. Mol. Model. 17, 2313–2323. doi: 10.1007/s00894-011-1010-y
Morgan, S., and Orrell, R. W. (2016). Pathogenesis of amyotrophic lateral sclerosis. Br. Med. Bull. 119, 87–98.
Noakes, C. J., Lee, G., and Lowe, M. (2011). The PH domain proteins IPIP27A and B link OCRL1 to receptor recycling in the endocytic pathway. Mol. Biol. Cell. 22, 606–623. doi: 10.1091/mbc.e10-08-0730
Nowicka, N., Juranek, J., Juranek, J. K., and Wojtkiewicz, J. (2019). Risk factors and emerging therapies in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 20:2616. doi: 10.3390/ijms20112616
Oguro-Ando, A., Zuko, A., Kleijer, K. T. E., and Burbach, J. P. H. (2017). A current view on contactin-4, -5, and -6: implications in neurodevelopmental disorders. Mol. Cell Neurosci. 81, 72–83. doi: 10.1016/j.mcn.2016.12.004
Peters, T. L., Kamel, F., Lundholm, C., Maria, F., Weibull, C. E., Dale, P. S., et al. (2017). Occupational exposures and the risk of amyotrophic lateral sclerosis. Occup. Environ. Med. 74, 87–92.
Riancho, J., Bosque-Varela, P., Perez-Pereda, S., Povedano, M., de Munaín, A. L., and Santurtun, A. (2018). The increasing importance of environmental conditions in amyotrophic lateral sclerosis. Int. J. Biometeorol. 62, 1361–1374.
Ross, T. S., Jefferson, A. B., Mitchell, C. A., and Majerus, P. W. (1991). Cloning and expression of human 75-kDa inositol polyphosphate-5-phosphatase. J. Biol. Chem. 266, 20283–20289. doi: 10.1016/s0021-9258(18)54920-6
Sandelin, A., and Wasserman, W. W. (2005). Prediction of nuclear hormone receptor response elements. Mol. Endocrinol. 19, 595–606. doi: 10.1210/me.2004-0101
Sher, R. B. (2017). The interaction of genetics and environmental toxicants in amyotrophic lateral sclerosis: results from animal models. Neural. Regen. Res. 12, 902–905. doi: 10.4103/1673-5374.208564
Soh, H., and Goldstein, S. A. (2008). ISA channel complexes include four subunits each of DPP6 and Kv4.2. J. Biol. Chem. 283, 15072–15077. doi: 10.1074/jbc.m706964200
Su, F. C., Goutman, S. A., Chernyak, S., Mukherjee, B., Callaghan, B. C., Batterman, S., et al. (2016). Association of environmental toxins with amyotrophic lateralsclerosis. JAMA Neurol. 73, 803–811. doi: 10.1001/jamaneurol.2016.0594
Swash, M., and Eisen, A. (2020). Hypothesis: amyotrophic lateral sclerosis and environmental pollutants. Muscle Nerve 62, 187–191. doi: 10.1002/mus.26855
Tsitkanou, S., Della Gatta, P., Foletta, V., and Russell, A. (2019). The role of exercise as a non-pharmacological therapeutic approach for amyotrophic lateral sclerosis: beneficial or detrimental? Front. Neurol. 10:783.
Van Doormaal, P. T. C., Ticozzi, N., Weishaupt, J. H., Kenna, K., Diekstra, F. P., Verde, F., et al. (2017). The role of de novo mutations in the development of amyotrophic lateral sclerosis. Hum. Mutat. 38, 1534–1541.
Van Doormaal, P. T., Ticozzi, N., and Gellera, C. (2014). Analysis of the KIFAP3 gene in amyotrophic lateral sclerosis: a multicenter survival study. Neurobiol. Aging 35, 2420.e13–14.
Van Es, M. A., Van Vught, P. W., Blauw, H. M., Franke, L., Saris, C. G., Andersen, P. M., et al. (2007). ITPR2 as a susceptibility gene in sporadic amyotrophic lateral sclerosis: a genome-wide association study. Lancet Neurol. 6, 869–877.
Van Es, M. A., Van Vught, P. W., Blauw, H. M., Franke, L., Saris, C. G., and Van den Bosch, L. (2008). Genetic variation in DPP6 is associated with susceptibility to amyotrophic lateral sclerosis. Nat. Genet. 40, 29–31.
Vinceti, M., Filippini, T., Violi, F., Rothman, K. J., Costanzini, S., Malagoli, C., et al. (2017). Pesticide exposure assessed through agricultural crop proximity and risk of amyotrophic lateral sclerosis. Environ. Health 16:91.
Wolf, E. J., Rasmusson, A. M., Mitchell, K. S., Logue, M. W., Baldwin, C. T., and Miller, M. W. (2014). A genome-wide association study of clinical symptoms of dissociation in a trauma-exposed sample. Depress. Anxiety 31, 352–360. doi: 10.1002/da.22260
Xie, T., Deng, L., Mei, P., Zhou, Y., Wang, B., and Zhang, J. (2014). Genome-wide association study combining pathway analysis for typical sporadic amyotrophic lateral sclerosis in Chinese Han populations. Neurobiol. Aging 35, 1778.e9–1778.e23.
Xue, Y. C., Feuer, R., Cashman, N., and Luo, H. (2018). Enteroviral infection: the forgotten link to amyotrophic lateral sclerosis? Front. Mol. Neurosci. 11:63.
Yokotani, N., Doi, K., Wenthold, R. J., and Wada, K. (1993). Non-conservation of a catalytic residue in a dipeptidyl aminopeptidase IV-related protein encoded by a gene on human chromosome 7. Hum. Mol. Genet. 2, 1037–1039. doi: 10.1093/hmg/2.7.1037
Yousefian-Jazi, A., Seol, Y. H., Kim, J., Ryu, H. L., Lee, J., and Ryu, H. (2020). Pathogenic genome signatures that damage motor neurons in amyotrophic lateral sclerosis. Cells 9:2687. doi: 10.3390/cells9122687
Yu, B., and Pamphlett, R. (2017). Environmental insults: critical triggers for amyotrophic lateral sclerosis. Transl. Neurodegener. 6:15.
Keywords: genetics, single nucleotide polymorphism, pathogenesis, amyotrophic lateral sclerosis, Chinese Han ancestry population
Citation: Zhang J, Qiu W, Hu F, Zhang X, Deng Y, Nie H and Xu R (2021) The rs2619566, rs10260404, and rs79609816 Polymorphisms Are Associated With Sporadic Amyotrophic Lateral Sclerosis in Individuals of Han Ancestry From Mainland China. Front. Genet. 12:679204. doi: 10.3389/fgene.2021.679204
Received: 15 April 2021; Accepted: 25 June 2021;
Published: 06 August 2021.
Edited by:
Sokol V. Todi, Wayne State University, United StatesReviewed by:
Ana Westenberger, University of Lübeck, GermanyCopyright © 2021 Zhang, Qiu, Hu, Zhang, Deng, Nie and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiong Zhang, eGlvbmc3MTVAMTI2LmNvbQ==; Youqing Deng, eW91cWluZzE5NjlAMTI2LmNvbQ==; Hongbing Nie, bmllaG9uZ2JpbmdAaG90bWFpbC5jb20=; Renshi Xu, MTM3NjcwMTU3NzBAMTYzLmNvbQ==; eHVyZW5zaGlAbmN1LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.