
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 08 July 2021
Sec. Computational Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.674783
This article is part of the Research TopicBioinformatics of Genome Regulation, Volume IIView all 18 articles
 Victoria A. Scobeyeva1,2*
Victoria A. Scobeyeva1,2* Ilya V. Artyushin3
Ilya V. Artyushin3 Anastasiya A. Krinitsina4
Anastasiya A. Krinitsina4 Pavel A. Nikitin5Maxim I. Antipin6Sergei V. Kuptsov6
Pavel A. Nikitin5Maxim I. Antipin6Sergei V. Kuptsov6 Maxim S. Belenikin2
Maxim S. Belenikin2 Denis O. Omelchenko7
Denis O. Omelchenko7 Maria D. Logacheva8Evgenii A. Konorov9
Maria D. Logacheva8Evgenii A. Konorov9 Andrey E. Samoilov10Anna S. Speranskaya4,10
Andrey E. Samoilov10Anna S. Speranskaya4,10Amaryllidaceae is a large family with more than 1,600 species, belonging to 75 genera. The largest genus—Allium—is vast, comprising about a thousand species. Allium species (as well as other members of the Amaryllidaceae) are widespread and diversified, they are adapted to a wide range of habitats from shady forests to open habitats like meadows, steppes, and deserts. The genes present in chloroplast genomes (plastomes) play fundamental roles for the photosynthetic plants. Plastome traits could thus be associated with geophysical abiotic characteristics of habitats. Most chloroplast genes are highly conserved and are used as phylogenetic markers for many families of vascular plants. Nevertheless, some studies revealed signatures of positive selection in chloroplast genes of many plant families including Amaryllidaceae. We have sequenced plastomes of the following nine Allium (tribe Allieae of Allioideae) species: A. zebdanense, A. moly, A. victorialis, A. macleanii, A. nutans, A. obliquum, A. schoenoprasum, A. pskemense, A. platyspathum, A. fistulosum, A. semenovii, and Nothoscordum bivalve (tribe Leucocoryneae of Allioideae). We compared our data with previously published plastomes and provided our interpretation of Allium plastome genes’ annotations because we found some noteworthy inconsistencies with annotations previously reported. For Allium species we estimated the integral evolutionary rate, counted SNPs and indels per nucleotide position as well as compared pseudogenization events in species of three main phylogenetic lines of genus Allium to estimate whether they are potentially important for plant physiology or just follow the phylogenetic pattern. During examination of the 38 species of Allium and the 11 of other Amaryllidaceae species we found that rps16, rps2, infA, ccsA genes have lost their functionality multiple times in different species (regularly evolutionary events), while the pseudogenization of other genes was stochastic events. We found that the “normal” or “pseudo” state of rps16, rps2, infA, ccsA genes correlates well with the evolutionary line of genus the species belongs to. The positive selection in various NADH dehydrogenase (ndh) genes as well as in matK, accD, and some others were found. Taking into account known mechanisms of coping with excessive light by cyclic electron transport, we can hypothesize that adaptive evolution in genes, coding subunits of NADH-plastoquinone oxidoreductase could be driven by abiotic factors of alpine habitats, especially by intensive light and UV radiation.
Vascular plants inhabit various ecology niches which may be distinguished by sets of environmental factors (e.g., irradiation level, atmospheric and soil humidity, temperature), all of them may affect photosynthesis. Mountain altitudinal gradient as well as redundantly lit or (as opp.) constantly shadowed habitats seem to be the most powerful “natural geophysical” pressure for evolutionary modification of genes of photosynthetic apparatus. The genes (approximately 120–130 genes) present in chloroplast genomes (plastomes) encode the core proteins of photosynthetic complexes: Photosystem I (psaA, B, C, I, and J) and Photosystem II (psbA-F, H-N, T, and Z), Cytochrome b6f (petA, B, D, G, L, and N)[Hu et al., 2015), NADH dehydrogenase (ndhA-K), ATP synthase (atpA, B, E, F, H, and I), the large RUBISCO subunit (rbcL), chloroplast ribosomal proteins of large and small subunits (rpl and rps), polymerase subunits (rpoA, B, C1, and C2), ATP-dependent protease (clpP), cytochrome c biogenesis (ccsA), membrane protein (cemA), translation initiation factor I (infA), maturase (matK) and some proteins of known or unknown functions of (ycf1_short, ycf1_long, ycf2, ycf3, ycf4), as well as four ribosomal RNAs and various tRNAs (Wicke et al., 2011; Daniell et al., 2016). The core set of plastom’s genes is retained from cyanobacteria ancestors; most of them are required for the light reactions of photosynthesis or functions connected with transcription and translation (Sanchez-Puerta et al., 2005; Green, 2011). The evolution of plastome genes could be under pressure of geophysical abiotic factors of plant habitats (Hanaoka et al., 2012; Hu et al., 2015; Zhang et al., 2016; Macadlo et al., 2020). Among geophysical abiotic factors species vertical range limits seem to be more niche-dependent than horizontal (Hargreaves et al., 2014) and vertical limits are often species niche limits (Lee-Yaw et al., 2016).
Species adaptations to diverse environments are accompanied by mutations under positive selection which may be confirmed using analysis of Kn/Ks ratio. Numerous genes were proved to be under positive selection in taxons which are myco-heterotrophic plants, and also in plants that are partly or entirely non-photosynthetic (Zeng et al., 2017; Dong et al., 2018). The sets of genes which are under positive selection in plants with adaptation to various ecological niches seem to be incoherent. Positive selection in some genes, e.g., rbcL, is widespread in most lineages of land plants (Yao et al., 2019). Some authors propose they are associated with adaptation to various ecological niches, including dry or wet habitats (Kapralov and Filatov, 2006), high altitudes in mountain regions (Hu et al., 2015). matK is reported to be a highly variable sequence which was often used for phylogeny analysis of many plant taxons (Hilu et al., 2003; Tamura et al., 2004). Different sets of genes are found under positive selection in species of various taxonomic groups. For example, in Oryza species (Poaceae, Monocotyledones) which were adapted to either shady or sunny environments, a set of positively selected genes (besides rbcL and matK) was found: accD, ndhD, ndhF, ndhH, psaA, psbB, psbD, psbH, rpl16, rpoA, rpoC2, and ycf68 (Gao et al., 2019). A completely different set of genes (ycf1, ycf2, rps14, rps15, and rps16) was found to be under selection in Cardamine and Nasturtium species (Brassicaceae) (Yan et al., 2019). Another analysis of adaptive evolution in Brassicaceae plastomes in general and in the Cardamine genus in particular resulted in detection of signatures of positive selection in the following genes (besides rbcL and matK): ycf1, rpoC2, rpl14, petD, ndhF, ccsA, accD, and rpl20 at a significant level (Hu et al., 2015). Presumably, detected signals of positive selection in genes may reflect specific adaptations of species to a particular habitat. For example, in Brassicaceae it could possibly be a consequence of adaptation to high altitude environments (Hu et al., 2015).
Furthermore, extensive structural changes, such as large inversions, deletions, loss of functionality (pseudogenization or complete loss) of genes are regularly observed in plastomes of certain plant species or groups of species (various taxa of higher rank) (Haberle et al., 2008; Guisinger et al., 2011; Schwarz et al., 2015; Hsu et al., 2016; Fonseca and Lohmann, 2017). For example, the loss of all ndh (NADH dehydrogenase subunit complex) genes is apparently a result of convergent evolution during the land adaptation of photosynthesis in neighboring clades of different taxa. The ndh genes loss in plastomes is usually linked to heterotrophic type of nutrition, like in Orchidaceae (Lin et al., 2015; Lin et al., 2017) or Lentibulariaceae (Silva et al., 2016). However, the NDH complex may also be lost in photoautotrophic plant species, as it was discovered for a wide spectrum of angiosperms and gymnosperms (de Pamphilis and Palmer, 1990; Haberhausen and Zetsche, 1994; Wakasugi et al., 1994; Martín and Sabater, 2010; Blazier et al., 2011; Omelchenko et al., 2020). Presumably, the loss of ndh genes is linked with inability to tolerate light intensity stress (Omelchenko et al., 2020). But for most plastome genes implications of their loss on the physiological and ecological functionality of plants remain inexplicable and are interpreted as random evolution events or just stated as fact. For example, rpl23 has been found to be a pseudogene in monocots (Ogihara et al., 1991; Morton and Clegg, 1993; Omelchenko et al., 2020) and in all species of Caryophyllales (eudicots) (Raman and Park, 2015), while a wide range of species belonging to other clades of eudicots (e.g., rosids, asterids) have no traces of pseudogenization of rpl23 (Raman and Park, 2015).
Amaryllidaceae is a large family with more than 1,600 species, divided into three subfamilies: Agapanthoideae, Amaryllidoideae, and Allioideae. The genus Allium (Allioideae) is vast, being one of the largest monocotyledonous genera and comprising more than 800 species (Fritsch et al., 2010; Fritsch, 2012; Shah, 2014; Maragheh et al., 2019) or about 973 species according to WCSP database (Govaerts and Fritsch, 2021). Allium species form 15 subgenera that are grouped into three intrageneric evolutionary lineages according to molecular data analysis of plastid and nuclear DNA barcode sequences (Friesen et al., 2006; Li et al., 2010; Wheeler et al., 2013). Besides the Allium genus (comprising monogeneric tribe Allieae), Allioideae has 17 more genera belonging to another three tribes (Gilliesieae, Leucocoryneae, and Tulbaghieae) (Chase et al., 2009; Sassone et al., 2014). Allium species (as well as other members of the Amaryllidaceae) are widespread and diversified, they are adapted to a wide range of habitats from shady forests to open habitats like meadows, steppes and deserts and highlands and are therefore well suited as study objects in studies of plants’ adaptations to various sun irradiation levels and other geophysical factors (Vvedensky, 1935; Wendelbo, 1971). Complete plastome sequences were first sequenced and arranged for the most economically important species of Allium: onion, A. cepa (von Kohn et al., 2013), garlic, A. sativum (Filyushin et al., 2016), edible species A. ursinum and A. paradoxum (Omelchenko et al., 2020). Another wide spectrum of wild and cultivated Allium species plastomes were sequenced and partially analyzed (Filyushin et al., 2018, 2019; Xie et al., 2019, 2020; Liu et al., 2020; Namgung et al., 2021).
According to data available, plastomes of most plant species contain the same number of tRNA genes (30 in total, of them 9 are represented by two copies in IR) and rRNA genes (eight genes, all four are represented by two copies in IR), while the rest 79 genes encode proteins (Wicke et al., 2011; Wambugu et al., 2015). Investigated Allium species had similar number, arrangement and orientation of genes (Filyushin et al., 2016, 2018; Huo et al., 2019; Yusupov et al., 2020). Only A. paradoxum demonstrated notable alterations, such as large 4,825 bp long local inversion in the SSC region and elimination or pseudogenization of the whole ndh gene family, as well as large number of other genes: rps16, rps2, rpl22, petD, infA, rpl23, rps3 (Omelchenko et al., 2020). Recently positive selection pressure was calculated in subgenus Anguinum of Allium and three genes were found with Kn/Ks >1 (accD, rps14, rpl33) (Jin et al., 2019). Soon after this an analysis of average Kn/Ks in a total of 39 complete chloroplast genomes of Allium was performed and quite unexpectedly it revealed an absolutely different set of positively selected genes (with Kn/Ks > 1): psbC, rps11, and psaI (Xie et al., 2020). These inconsistent data prompted us to choose a simple and easy to analyze ecological trait—the highest tolerable altitude of the species. High altitude habitats have high levels of solar radiation, often low humidity and other special meteorological conditions, influencing the whole plant physiology (Gale, 2004). We took the highest tolerable altitude of the Allium species as a selective factor and performed an evolutionary analysis of plastome genes in species with contrasting altitude limits (see Supplementary Material 2 for species list). In this manuscript we only report results that relate to protein-coding genes of examined plastomes. We report de novo sequencing and assembling of complete cp-genomes of wild and cultured Allium species as well as outgroup species belonging to the same subfamily of Allioideae in Amaryllidaceae—Nothoscordum bivalve (L.) Britton. We have identified rapidly evolving plastome regions and genes under positive selection, and detected plastome traits that differentiate between species of three evolutionary lines of Allium genus adapted to certain contrasting habitat types: high-altitude vs. lowland habitats.
Plastomes were newly sequenced and assembled for the following species obtained alive from the outdoor collection of the Moscow State University Botanical Garden: A. fistulosum L., A. macleanii Baker, A. nutans L., A. obliquum L., A. pskemense B. Fedtsch., A. schoenoprasum L., A. victorialis L., A. zebdanense Boiss. & Noë. The specimens of A. semenovii Regelas well as A. platyspathum Schrenk were collected in the wild in 2016. The specimens of the abovementioned species from MSU Botanical Garden living collections and from wild natural habitats were deposited in the Moscow State University Herbarium, extracted DNA from Allium plants were deposited in DNA collection in MSU Biology Department. The plastomes of A. moly L and Nothoscordum bivalve (L.) Britton were sequenced using dry plant material obtained from Osnabrück Botanical Garden (Germany), kindly provided by Nikolai Friesen. All additional details, including taxonomic position of species used in the study, the source of specimens, GenBank/ENA accession numbers of plastome sequences, information about ecology and geographical location of wild collected specimens, accession number of vouchers are provided in Supplementary Table 2.
For cpDNA extraction from samples obtained from living specimens (Lomonosov MSU botanical garden collections),fresh leaves were cut and stored in the dark in a refrigerator at +4°C for 10 days. Chloroplasts were isolated from about 2 g (fresh weight) of leaves using protocol based on Shi et al. (2012) and Vieira et al. (2014) and described in detail in Logacheva et al. (2017). Then cpDNA was extracted using standard CTAB protocol (Doyle and Doyle, 1987). Quality of cpDNA was evaluated visually by gel-electrophoresis.
Another approach was used for herbarium samples of A. semenovii, A. platyspathum, A. moly, and N. bivalve. Fragments of dry leaves were used as material for total DNA extraction according to Doyle and Doyle (1987) with an additional stage of purification according to Krinitsina et al. (2015) because of high amount of impurities (presumably phenolic compounds). Quality of total DNA was evaluated using spectrophotometry (Nanophotometr-N-60, Implen). Quantification of nucleic acids was done using fluorimetry (Qubit 3.0, Thermo Fisher Scientific).
The libraries with insert sizes of 400–800 bp were constructed and then sequenced using high-throughput sequencing platform Illumina MiSeq (PE 2 × 250 bp or 2 × 300 bp). Paired-end libraries for each DNA sample were constructed at least twice using two different library preparation strategies to minimize protocol-associated biases and maximize assembly efficiency. First strategy included physical fragmentation by Covaris 220 followed by protocol with adapter ligation, using NEBNext® DNA Library Prep Master Mix Set for Illumina (E6040, NEB reagents) with single indexed primers from NEBNext® Multiplex Oligos for Illumina Kits (Index Primers Set 1–4), used according to the manufacturer’s recommendation. Another approach was DNA library preparation using a transposase-based method (Nextera), developed by Illumina. After tagmentation the libraries were amplified using NEB Q5® High-Fidelity DNA Polymerase (up to 12 cycles of amplification) and Nextera-compatible dual indexed primers.
The chloroplast genome assembly protocol included (1) quality trimming with Trimmomatic (Bolger et al., 2014), (2) filtering of reads using known chloroplast genome sequences of A. cepa (NC024813) and A. sativum (NC031829) by Bowtie2 mapper (Langmead and Salzberg, 2012), (3) producing of two contig sets for both filtered and non-filtered reads using de novo assemblers Velvet (Zerbino and Birney, 2008) and Spades (Bankevich et al., 2012). Assembled contigs were selected for the next assembly if they showed similarity to published Allium plastomes. The final de novo assembly was checked and fixed where necessary by PE reads mapping to the assembly in order to check for potential assembly artifacts using Bowtie2, VarScan (v.2.3.7) and SAMtools/BCFtools software packages (Li et al., 2009; Langmead and Salzberg, 2012; Koboldt et al., 2012). The coverage was as follows: A. semenovii ∼30×, A. macleanii ∼35×, A. fistulosum > 200×, A. platyspathum ∼35×, A. nutans > 300×, A. obliquum > 200×, A. pskemense ∼39×, A. schoenoprasum L. > 250×, A. victorialis ∼80×, A. zebdanense > 150×.
Plastomes of A. macleanii, A. nutans, A. obliquum, A. platyspathum, A. pskemense, A. schoenoprasum, A. victorialis., A. zebdanense were completely assembled using only the high-throughput sequencing approach. Some unassembled regions of A. semenovii plastome were sequenced by Sanger method. Primers for amplification were: Alsem 1 for: GTC CTC GGT AAC GAG ACA TAA; Alsem 1 rev: ACG TAG TCA ACT CCA TTC GT; Alsem 2 for: GTG CCC AAA ATG GTG TCA AT; Alsem 2 rev: ATC CAT GGT TTA TTC CTT ATC TCT; Alsem 3 for: GTA TGC CGT CTT CTG CTT G; Alsem 3 rev: AAG GGT TCT TTT AAA CTC TTT TGT T; Alsem 4 for: TGT TGG ACA ATA CTC GAC AC; Alsem 4 rev: GAC CAT AGA GGA GCC GTA TG; Alsem 5 for: GAG TGG AGC TAT ACC CAA TAG ATA; Alsem 5 rev: TAA GGT TAT CTC CCG CCA AT.
The Dual Organellar GenoMe Annotator (DOGMA) (Wyman et al., 2004) and GeSeq (Tillich et al., 2017) programs were used for preliminary gene annotation. From this initial annotation, putative start codons, stop codons, and intron positions were determined. Then putative start and stop codons together with intron positions were manually corrected based on comparisons with homologous genes of cp genomes of Lycoris squamigera (NC_040164.1), Narcissus poeticus voucher WSY: WSY0108940 (NC_039825.1), Lycoris radiata (NC_045077.1), Allium cepa (NC_024813.1), Agapanthus coddii voucher K:20081397 (NC_035971.1), Allium paradoxum (NC_039661), Allium herderianum (NC_042156.1), and Allium victorialis (NC_037240.1). All identified tRNAs were further verified by tRNAscan-SE 1.21 (Chan and Lowe, 2019). Complete nucleotide sequences of plastomes sequenced in this study were deposited in the GenBank database under accession numbers listed in Supplementary Table 1. After this results of annotations were manually checked to correct errors that appear as a result of the work of algorithms implemented in the annotation software.
To investigate the sets of protein coding genes that reflect the evolution of plastomes in the three main clades of the genus Allium (Friesen et al., 2006), we used all complete and partially assembled sequences and complete sequences of several other Allium species plastomes obtained from GenBank. All annotated sequences previously published in GenBank were re-annotated using in-house scripts and A. cepa (NC_024813) as basic reference sequences. A. praemixtum (NC_044412) and N. poeticus (NC_039825) were used for annotation of genes pseudogenized or absent in A. cepa. Then we excluded most cultivated species, besides taken as reference, to avoid possible bias due to unknown processes accompanying artificial selection of cultivated forms. The sequences of the protein coding genes were extracted, than extraction and translation were manually verified to correct possible errors that appear as a result of the work of algorithms implemented in the annotation software. Although a fairly large number of complete Allium plastome sequences are available online, taxonomic attribution of many species looks uncertain. We used the species that we sequenced and assembled during this project and added more plastomes from GenBank, choosing species that were important for the aims of our research, namely representatives of the first and the second evolutionary line (Friesen et al., 2006) with contrasting highest tolerated altitudes: A. paradoxum (M. Bieb.) G. Don (MH053150), A. ursinum L. (MH157875.1), A. prattii L. (NC_037432.1), two important agricultural species A. cepa L. (NC_024813), A. sativum L. (NC_031829), and two species with known intraspecific variability [A. obliquum (MG670111) and A. victorialis (MF687749)]. For the complete list of analyzed species see Supplementary Table 2.
Primary alignment was prepared with MAFFT v7.471 (Kazutaka and Standley, 2013). To identify pseudogenes, we aligned nucleotide sequences with MACSE and marked the first position of either frameshift, stop-codon or deletion spanning to the sequence end. Alignments were visualized with JalView v2.11.1.3 (Waterhouse et al., 2009). Indels-containing regions were inspected by eye for possible alignment errors. No additional alignments filtering was applied. Pseudogene heatmap was constructed with pandas (Reback et al., 2021), numpy (Harris et al., 2020), seaborn (Waskom et al., 2020), and matplotlib (Hunter, 2007) Python3 packages (Cock et al., 2009; Van Rossum and Drake, 2009).
We calculated GC content and proportion of gaps by window 100 bp in length using custom python script. For every window substitution count was estimated using a fixed tree topology (ParsimonyScorer class in Biopython v 1.78 (Talevich et al., 2012). Tree topology was the same as for HyPhy methods (explained below). Plots were constructed with ggplot2 R package (Wickham, 2016; R Core Team, 2018).
For selection and evolutionary rate analysis we used the concatenated sequences of 78 genes (see Supplementary Table 3). All individual gene sequences recognized as pseudogenes were replaced with gaps. Phylogenetic analysis was conducted with IQ-TREE v 2.0.6 (Chernomor et al., 2016; Minh et al., 2020). Evolutionary models and partitioning schemes were optimized using built-in methods (Kalyaanamoorthy et al., 2017) starting from individual partitions for every gene. The final scheme had seven partitions. Obtained tree was used for assessment of per-gene evolutionary rates and selective pressure detection. Tree was visualized with FigTree v1.4.4 (Rambaut and Drummond, 2012). We applied FUBAR v2.2 (Murrell et al., 2013), aBSREL 2.1 (Smith et al., 2015), and MEME 2.1.1 (Murrell et al., 2012) methods implemented in HyPhy framework v2.5.2 MP (Kosakovsky Pond et al., 2005). The latter was used to obtain Kn/Ks ratios for genes.
We have carried out analysis and identification of genes under positive or diversifying selection in Allium genus in its adaptive evolution to different solar irradiation and other abiotic conditions of its wide range in highlands and various lowlands in the temperate zone. A total of 49 species was analyzed, of which data for 13 species was obtained by our group. Two species sequenced by our group already had been stored in databases earlier (A. obliquum and A. victorialis), so we took 51 sequences in analysis total. We have chosen 8 species from the first evolutionary line, 7 from the second and 23 from the third. Cultivated species A. cepa was included in the analysis as a reference, A. sativum, A. fistulosum, A. tuberosum, A. chinense were excluded from the analysis. As far as species vertical limits are often species niche-limits (Hargreaves et al., 2014), we took the highest tolerable altitude of the species listed, using the data available from literature sources (see Supplementary Table 2) as well as the information presented on the herbarium labels for the corresponding species in the herbarium of the Moscow State University1. Our analysis consisted of a classical approach with Kn/Ks calculation, aBSREL (Smith et al., 2015), MEME (Murrell et al., 2012), and FUBAR (Murrell et al., 2013). We considered evidence of positive selection as sufficiently reliable only for those cases where it was confirmed by several methods.
To compare ratio of genes under selection among different gene ontologies we performed Fisher’s exact test for multiple samples. All genes, used for selection seeking, were divided into seven ontologies—ATP synthesis, Cytochrome complex and electron-transport chain, ndh genes, photosynthesis, translation, transcription and other. For all the ontologies were calculated ratio of sites under selection to sites without selection and compared with Fisher’s exact test for multiple samples with R package (Holm–Bonferroni method was used to counteract the problem of multiple comparison).
We have sequenced, assembled and annotated plastomes of A. zebdanense (MZ019480), A. moly (MZ019477), A. victorialis (NC_037240.1), A. macleanii (LT699703.1), A. nutans (LT799837.1), A. obliquum (MZ019478), A. schoenoprasum (LT699700.1), A. platyspathum (LT673892.1), A. semenovii (MZ019479), A. pskemense (MZ147623), A. fistulosum (LT674586.1). In addition, we have obtained a complete nucleotide sequence of the plastome of Nothoscordum bivalve. Together with genus Allium it belongs to the Allioideae subfamily, but to a different tribe Leucocoryneae. The plastome sequence of Nothoscordum bivalve was submitted as (MZ019481). The main parameters, i.e., total/LSC/SCC/IRs lengths and complete comparative annotations for most of assembled plastomes are represented in the (Supplementary Material 1). To expand the sampling, we took the plastome sequences of other wild Allium species published in GenBank. In total 41 plastome sequences of wild Allioideae species were analyzed. In addition our analysis also included representatives of two other Amaryllidaceae subfamilies, Agapanthoideae and Amaryllideae, data on which were found at Genbank. For the complete list of the studied species and descriptions of general ecology features of their natural habitats (see Supplementary Material 2).
To identify and correct possible annotation errors that arise as a result of the work of algorithms implemented in annotation software, we performed automatical reannotation of all previously published plastome sequences that were selected for comparative analysis in this work, using GeSeq. In addition to this we had determined start and stop codons of all protein coding genes using in-house scripts. The results of both procedures were then manually compared and finalized. This step allowed us to find some noteworthy inconsistencies with previous reports, in particular, regarding pseudogenization status of rps2, rps16, and ndhD reported by Xie et al. (2019). The authors claim that rps2 was lost in all Allium species (Allioideae), but we have found that pseudogenization occurred in only about a third of studied Allium species, while in the rest two thirds these genes proved to be in their true state, coding proteins without stop-codons (see Figure 1). Manual correction of automatic annotations has also influenced our conclusions regarding functionality of various genes like rps16 in A. platyspathum and A. nutans: we found that these genes in these species are true. The ndhD gene also proved to be in its true state in all investigated Allium species (excluding A. paradoxum), in contradiction to conclusions made by Xie et al. (2019).
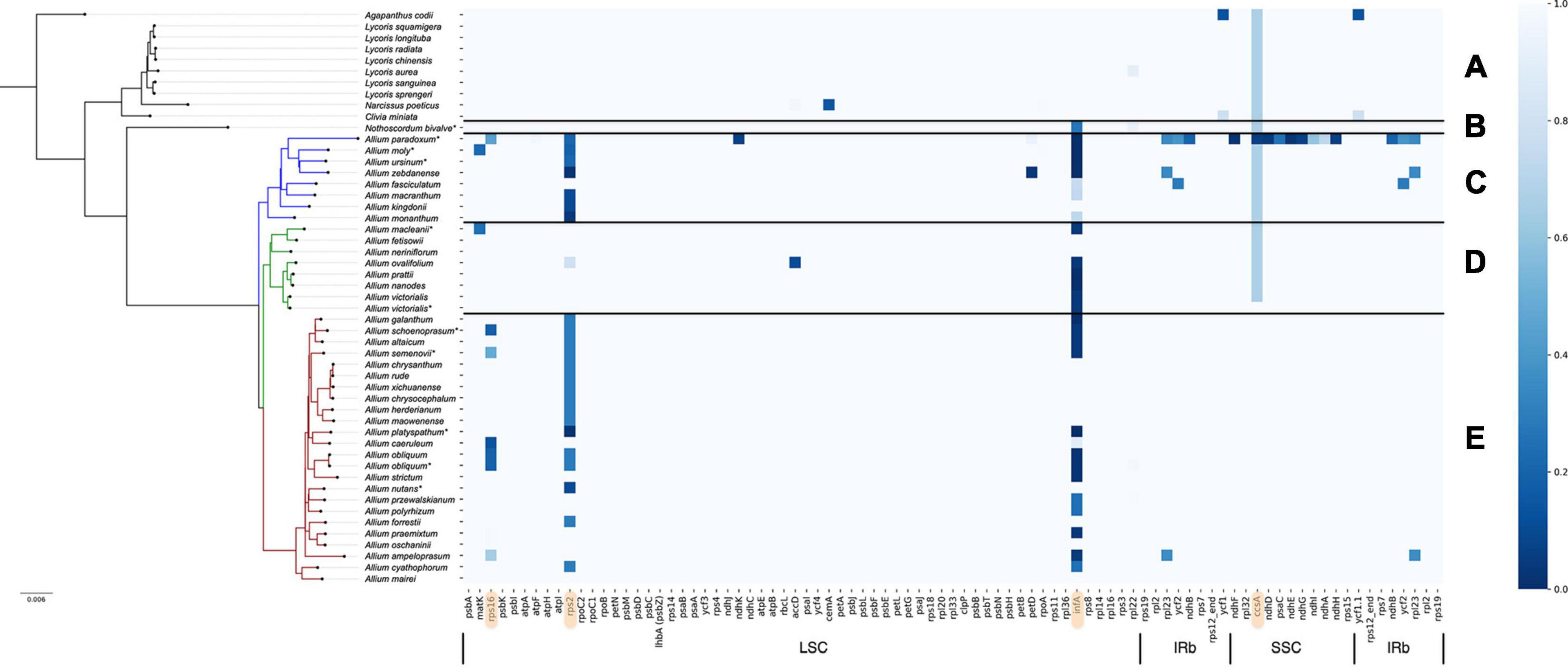
Figure 1. Pseudogenization or loss of protein-coding genes heatmap in the plastome of Allioideae (including all three evolution lines of Allium). Taxonomic position of species is denoted at the left side. Species sequenced by our group are marked with (*). (A) Shows Agapanthoideae and Amaryllidoideae subfamily species; (B) shows Nothoscordum bivalve as an outgroup for Allium genus; (C) shows the first evolutionary line Allium species; (D)—the second and (E)—the third. Genes are sorted in natural order in plastid. LSC, large single-copied region; SSC, small single-copied region; IRa and IRb, inverted repeats A and B. The darkest blue marks completely deleted genes; skyblue of varying shades marks pseudogenized genes (depending on the length of the preserved gene fragment); light blue marks true genes (translated without errors). Genes described below in detail are marked with peach color (rps16, rps2, infA, ccsA).
The set of plastomes obtained were used for analysis of deleted/pseudogenized genes patterns and their association with evolutionary lineages and with environmental adaptation of species. Plastomes of Allium species as well as other known Amaryllidaceae are generally similar in composition of their functional gene sets. The main differences were found in the functionality of the following genes: ccsA (responsible for heme attachment to cytochromes c), rps16 and rps2 (proteins of the small ribosomal subunit), infA (translation initiation factor I), see Figure 1. Pseudogenization of some other genes is presumably a sporadic event in the Allium genus, sometimes happening in all evolutionary lines: petD (encodes a subunit of the cytochrome b6/f complex), ycf1 (translocon on the inner plastid membrane), ycf2 (encodes a subunit of the 2-MD heteromeric AAA-ATPase complex which associated with the TIC complex (Kikuchi et al., 2018) and some other genes can undergo occasional defunctionalization.
Undoubtedly the sequence features of the rps2, infA, rps16, ccsA genes as well as their “normal” or “pseudo” state correlate with the genus evolutionary line to which species belong to.
We found that pseudogenization affects rps16 genes only in species of the third evolutionary line, namely A. schoenoprasum, A. obliquum, A. ampeloprasum, A. caeruleum, and A. platyspathum (see Figure 2). In plastomes of first and second evolutionary lines of Allium (with only one exception, A. paradoxum) as well as in all non-Allium Amaryllidaceae, rps16 gene is translated without errors.
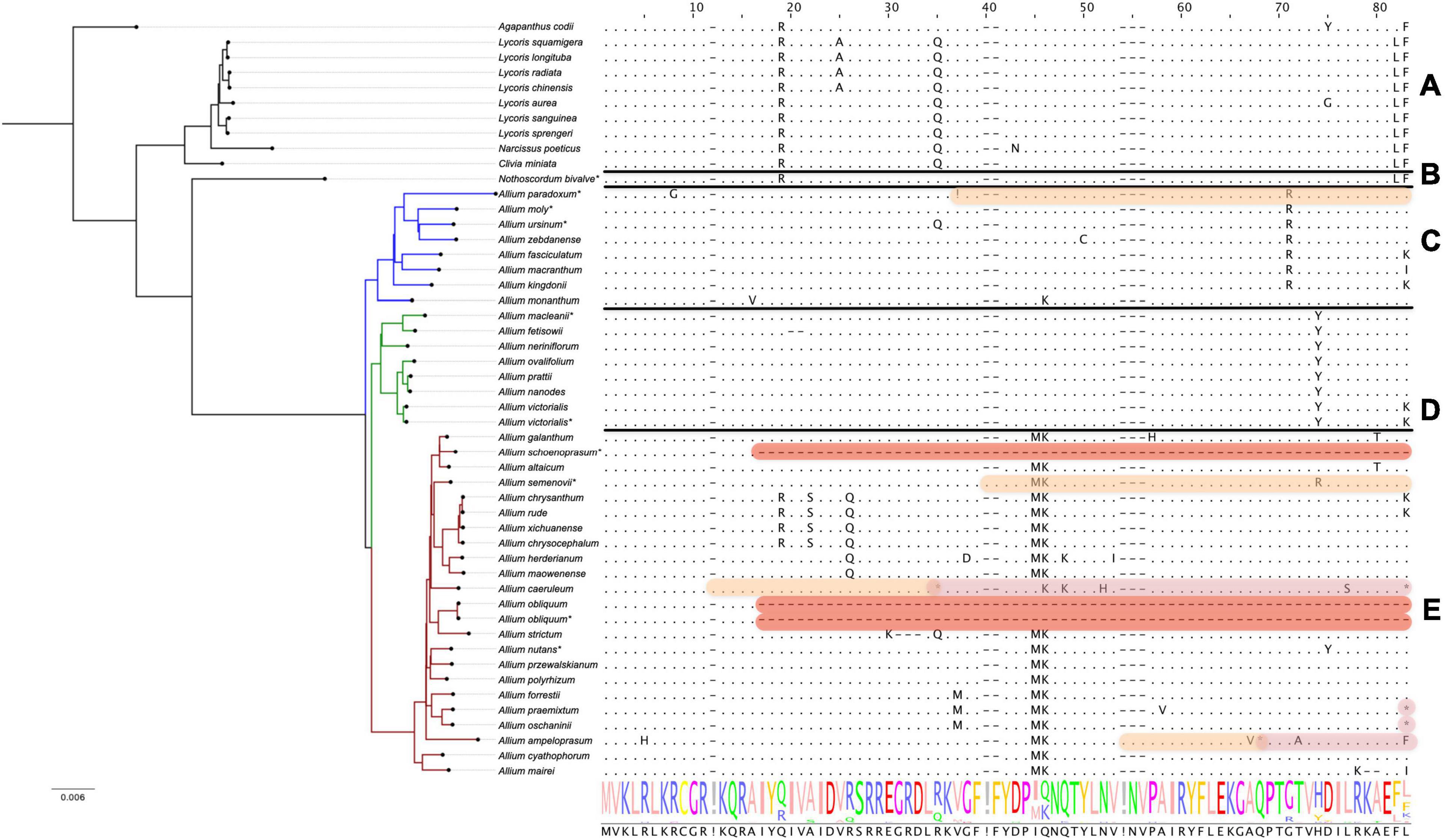
Figure 2. Protein sequence alignment of the plastid 30S ribosomal protein S16 encoded by rps16 genes. Àll species are sorted by phylogenetic tree: the first evolutionary line of Allium is blue, the second is green, the third is red and the remaining species are black. Species sequenced by our group are marked with *. (A) Shows Agapanthoideae and Amaryllidoideae subfamily species; (B) shows Nothoscordum bivalve as an outgroup for Allium genus; (C) shows the first evolutionary line Allium species; (D)—the second and (E)—the third. Non-conserved AA positions are highlighted as letters and conserved as dots. The whole AA sequence is shown as a normalized alignment logo below. Possible frameshifts are shown as! and stop codons as *. Damaged sequences are highlighted in peach color in case of frameshift, in purple in case of stop codon and in red in case of total AA loss. Colored sequences may be assumed to be a pseudogene. The gene is absent in A. platyspathum.
Defunctionalization of the rps2 gene (encoding plastid 30S ribosomal protein S2) was found to be characteristic of most Allium species (see Figure 3). There is a considerable amount of single nucleotide substitutions along with several indels in the sequence of the rps2 gene which leads to many non-synonymous AA substitutions and premature stop codons. Interestingly, at first glance, the species of the second evolutionary line seem to be less affected by this process, rps2 being pseudogenized only in one species, A. ovalifolium.

Figure 3. Protein sequence alignment of the plastid 30S ribosomal protein S2 encoded by rps2 genes. Àll species are sorted by phylogenetic tree: the first evolutionary line of Allium is blue, the second is green, the third is red and the remaining species are black. Species sequenced by our group are marked with *. (A) Shows Agapanthoideae and Amaryllidoideae subfamily species; (B) shows Nothoscordum bivalve as an outgroup for Allium genus; (C) shows the first evolutionary line Allium species; (D)—the second and (E)—the third. Non-conserved AA positions are highlighted as letters and conserved as dots. The whole AA sequence is shown as a normalized alignment logo below. Possible frameshifts are shown as ! and stop codons as *. Damaged sequences are highlighted in peach color in case of frameshift and in purple in case of stop codon. Colored sequences may be assumed to be a pseudogene.
The differences in the nucleotide sequences of the infA genes (encoding translation initiation factor IF-1) are undoubtedly correlated with the phylogeny of the Amaryllidaceae family. While Agapanthoideae and Amaryllidoideae species contain genes translated without errors, most Allioideae, including N. bivalve and most Allium species of the first, second and third evolutionary lines, have pseudogenized infA (see Figure 4). In some Allium species the infA genes are completely deleted (for example, in A. macleanii). In many species they contain stop codons at different distances from the start of translation (Figure 4). Pseudogenized infA genes, containing a stop codon at a short distance from translation start point, are found more often in plastomes of the species belonging to the first and second evolutionary lines. The observed patterns indicate that infA genes were pseudogenized multiple times after the separation of the Allioideae from other groups within Amaryllidaceae.
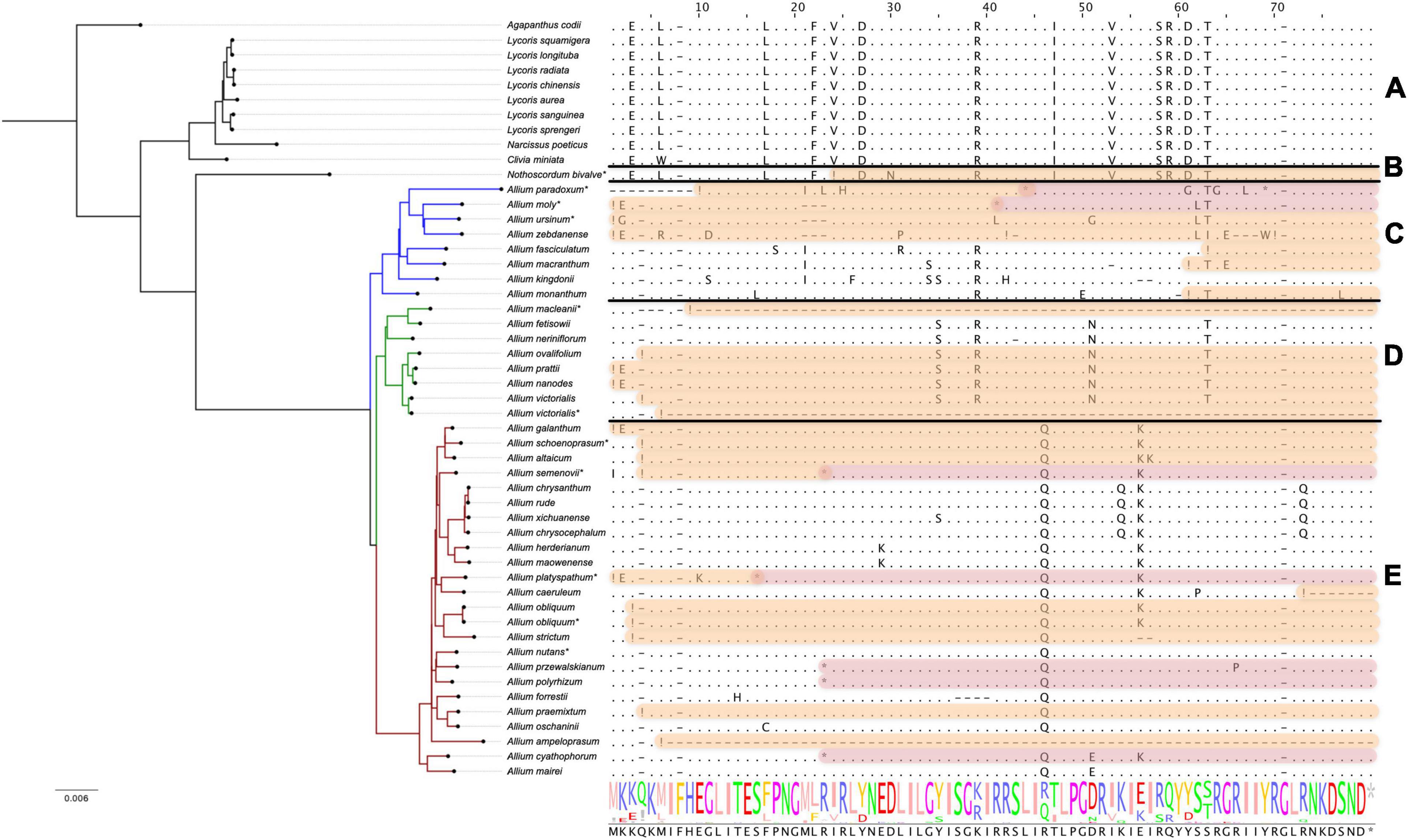
Figure 4. Protein sequence alignment of the translation initiation factor IF-1 encoded by infA genes. Àll species are sorted by phylogenetic tree: the first evolutionary line of Allium is blue, the second is green, the third is red and the remaining species are black. Species sequenced by our group are marked with *. (A) Shows Agapanthoideae and Amaryllidoideae subfamily species; (B) shows Nothoscordum bivalve as an outgroup for Allium genus; (C) shows the first evolutionary line Allium species; (D)—the second and (E)—the third. Non-conserved AA positions are highlighted as letters and conserved as dots. The whole AA sequence is shown as a normalized alignment logo below. Possible frameshifts are shown as ! and stop codons as *. Damaged sequences are highlighted in peach color in case of frameshift and in purple in case of stop codon. Colored sequences may be assumed to be a pseudogene.
Analysis of amino acid sequences that lack stop codons and frameshifts in the middle of the sequence has shown that in species of the second evolutionary line there occurs a radical amino acid substitution in Y34S position, while all Allium species have such substitutions at I46T. Amino acids of these positions participate in forming rRNA binding sites (nucleotide binding) (Hiratsuka et al., 1989).
The ccsA gene is undergoing pseudogenization within all non-Allium species as well as in all species of first and second evolution lines (see Figure 5). The plastome of A. victorialis (LT699702.1) sequenced in this work was the only exception to this rule, containing normally translated ccsA. Interestingly, the other specimen of A. victorialis (NC_037240.1) sequenced by Lee et al. (2017) has ccsA pseudogenized like other species of the second evolutionary line. We suppose that the plastome of A. victorialis (LT699702.1) could contain some inexplicable mistake in the ccsA gene and could be ignored when patterns of pseudogenization are discussed. Thus, the ccsA gene status (“normal” or “pseudo”) reflects the phylogenetic evolution of the Amaryllidaceae: Agapanthoideae and Amaryllidoideae species as well as basic clades of Allioideae form ccsA pseudogenes.
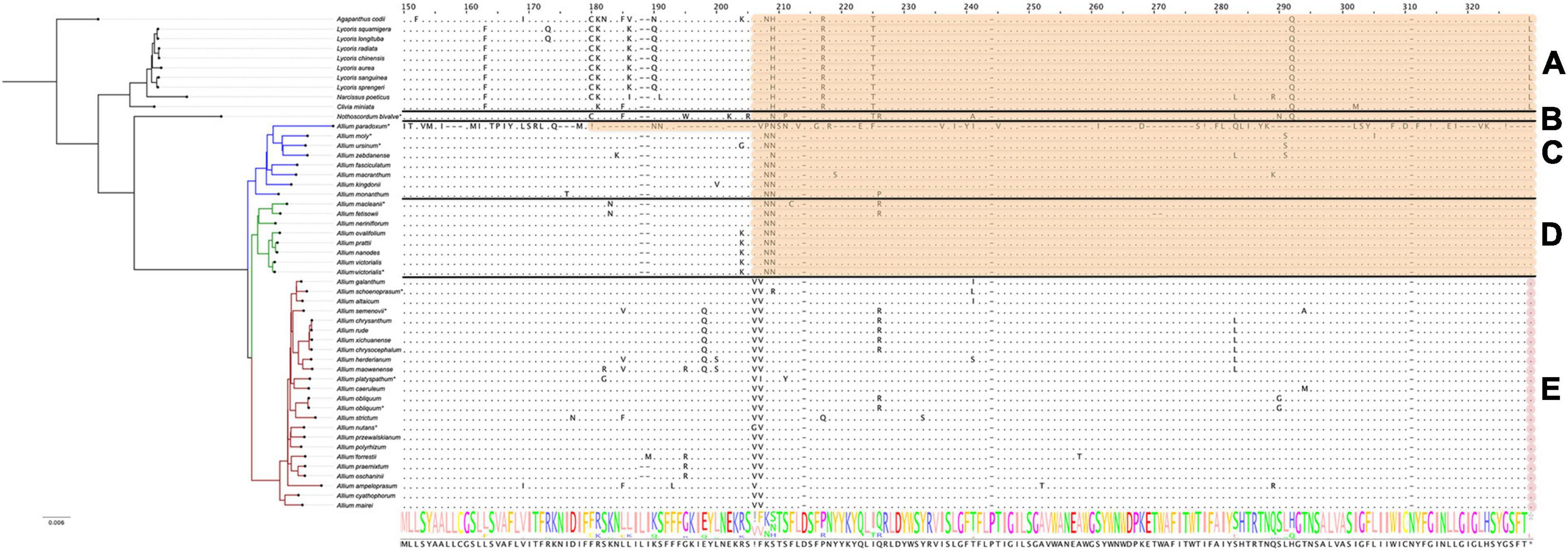
Figure 5. Protein sequence alignment of the cytochrome c biogenesis protein encoded by ccsA genes. Àll species are sorted by phylogenetic tree: the first evolutionary line of Allium is blue, the second is green, the third is red and the remaining species are black. Species sequenced by our group are marked with *. (A) Shows Agapanthoideae and Amaryllidoideae subfamily species; (B) shows Nothoscordum bivalve as an outgroup for Allium genus; (C) shows the first evolutionary line Allium species; (D)—the second and (E)—the third. Alignment here starts from the 150th AA position for better visualization. Non-conserved AA positions are highlighted as letters and conserved as dots. The whole AA sequence is shown as a normalized alignment logo below. Possible frameshifts are shown as ! and stop codons as *. Damaged sequences are highlighted in peach color in case of frameshift and in purple in case of stop codon. Colored sequences may be assumed to be a pseudogene.
Despite the fact that their products are obviously necessary for the normal plant system functioning, the reasons why the infA, rps16, rps2, and ccsA genes had lost their functionality remains unclear. It is possible that copies of these genes can be found in nuclear genomes. We checked the sequences of the assembled whole nuclear genome of A. sativum which is the only one available in NCBI presently (assembling accession number: GCA_014155895.1) for the presence of the sequences infA, rps16, rps2 but gained no positive results. Perhaps these genes can be found in nuclear genomes of other Allium species.
The evolution rate estimation is widely used in phylogenetic and evolutionary studies. Usually pseudogenes and genes with loose selective constraints have the highest rates, followed by positively selected genes. Methods for detecting positive or diversifying selection events are sensitive to evolutionary models, so we treated them as putatively selected only sites, discovered by both MEME and FUBAR methods of analysis (see Supplementary Table 3). The highest rate of evolution, 3,16, was detected in gene rpl32. No sites under positive selection are found by MEME and FUBAR in sequence of 59 a.a. length in protein 60S ribosome 32, coded by rpl32. We can therefore suppose a neutral model of evolution in most lineages, with selective constraints loosen in some species. Amino acid sequence of Allium paradoxum rpl32 protein has relatively low homology with sequences of the other Allium species and high number of gaps in alignment. In A. macranthum and A. fetisovii amino acid sequence of 60S ribosome 32 also differs from consensus alignment, so we can either suppose that this protein plays a role in adaptation or rather has lost its selective value in these species.
The next gene in the high evolutionary rate list is ycf1, encoding the long protein of 1,761 a.a. with partly known functions and a story of pseudogenization in various plant families. MEME gives 51 sites under positive selection, while only 4 of them coincide with FUBAR list. Selection was detected by AbsRel method in two lineages—A. mairei and A. oschaninii. We can suggest predominantly neutral evolution of ycf1 in most lineages, but in some cases we can suppose selective evolution as well. In A. macranthumwe can see long compensated frameshift, it is an evident trace of stabilizing selection.
The third gene in this list is psbZ, encoding a small protein of Photosystem II reaction center. MEME revealed only one site under selection and it does not match with FUBAR data. But the Kn/Ks ratio in psbZ is 1,26, one of the highest in all the examined genes. Natural selection obviously affects the sequence, but we cannot detect the particular site of its action. So, we would rather suggest neutral evolution after relaxation of selective constraints (Akashi et al., 2012). The last gene with evo rate >2 is the ndhF gene, coding NADH-plastoquinone_oxidoreductase_subunit_5. We detected it by MEME selection in 15 sites of 734 examined, 4 of them coinciding with FUBAR data. The aBSREL detected positive selection in the ndhF gene in three lineages—A. macranthum, A. forrestii, and A. neriniflorum. Kn/Ks ratio in the ndhF gene is 0,28, so we should stay away from the conclusion that selective mode of evolution is significant in all examined lineages, but we may say that in some of them natural selection can play a certain role.
matK has an evolutionary rate of 1,98, which is insignificantly lower than 2. MEME has found 13 sites from 521 under selection, and only 3 of them are the same as those found by FUBAR. aBSREL found only one lineage under selection—A. paradoxum, and in two species (A. moly and A. macleanii) matK was pseudogenized. matK is widely used as a phylogenetic marker and we agree here that it is reasonable in most cases, but some matK sites are still under positive or diversifying selection.
Two genes of small ribosomal proteins (rps15 and rps16) have close evo rates, rps15 evo rate is 1,68 and rps16 evo rate is 1,52. No selection was detected in rps15 by any method, so we would assume that its evolution is neutral and only negative selection affects it. In rps16 both MEME and FUBAR detect the same site under selection, so we are not able to argue there is no evidence of selection at all. In some species of the third evolutionary line rps16 is pseudogenized, but in other lineages it is functional and thus it can be presumably under selection in these lineages.
High percentage of gaps in the small single copy region is very interesting at first glance (Figure 6A), but can be reasonably explained by lineage-specific insertion in Nothoscordum and Narcissus. Species of genus Allium do not have this insertion, so they have many common gaps in alignment.
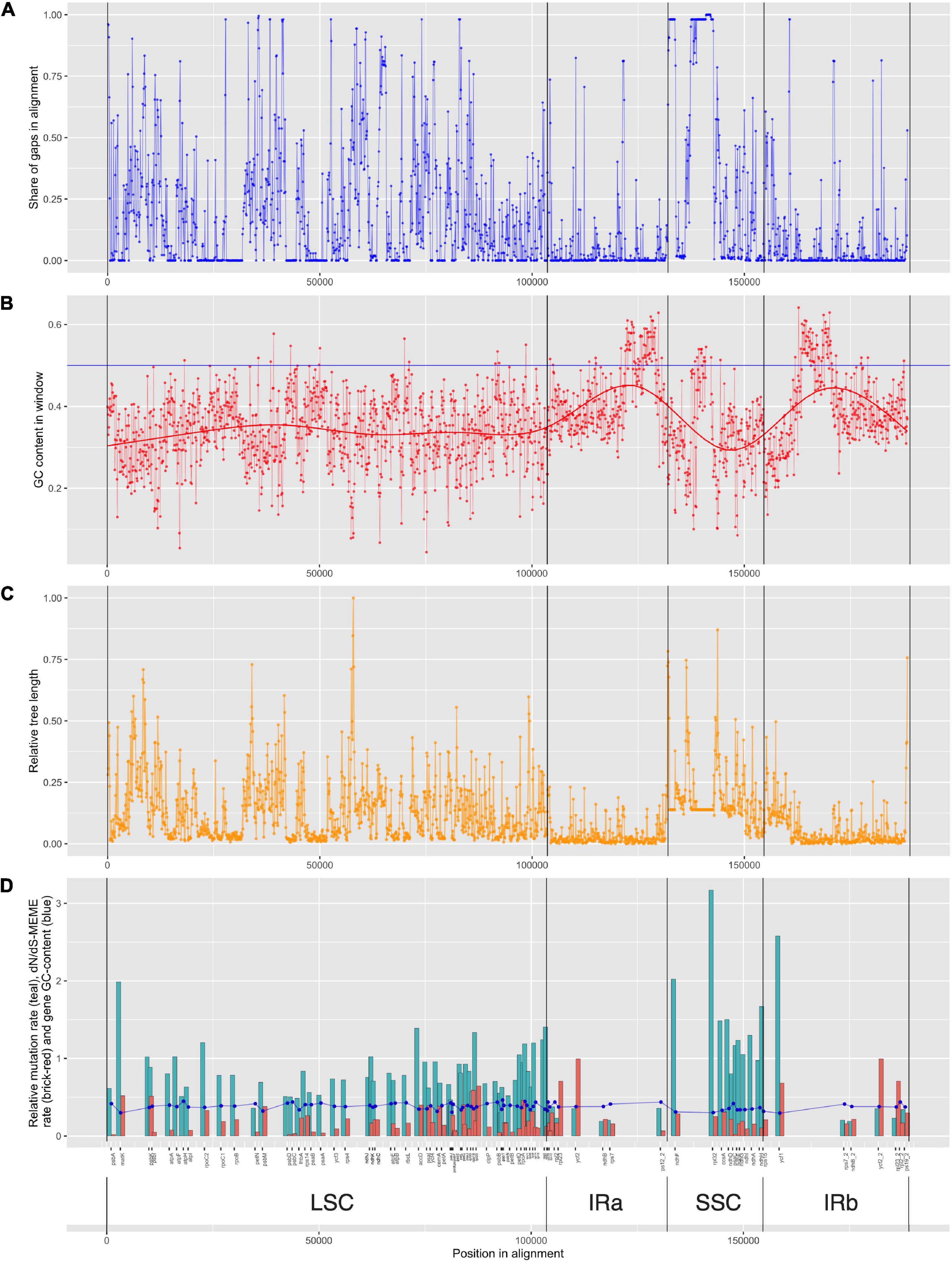
Figure 6. Information on alignment and gene features. Structural components of plastome genomes and genes’ positions are shown [LSC, large single-copied region; SSC, small single-copied region; IRa and IRb, inverted repeats (A,B)]. (A) Share of gaps in alignment in the window of 100 bp. Dots are connected with a line for visibility. (B) GC-content in the window of 100 bp. Dots are connected with a line for visibility. A bold line shows regression estimating of GC-content. (C) Relative tree length. Dots are connected with a line for visibility. (D) Relative mutation rate (teal), Kn/Ks-MEME rate (brick-red) bar-plot and gene GC-content (blue). Dots are connected with a line for visibility.
GC content in different genes varies from 0.29 to 0.45 (Supplementary Table 3 and Figure 6B). Genes with high evolutionary rates usually have lower GC content, e.g., ycf1 (0,29) or rpl32 (0,3), whereas genes with low evolutionary rates usually have higher GC content, like psbN (0,46) or atpH (0,44), but this is not a tight dependence. Genes with low evolutionary rate can also have low GC content, like psbL (0,30) (see Supplementary Table 3 and Figures 6B,C). The highest GC content is reported in repeats containing no protein-coding genes (see Figures 6B,D).
We found evidence of positive selection in the matK genes (its product takes part in the type II introns splicing), accD (the beta subunit of carboxyltransferase, which is part of the plastid acetyl-coA carboxylase), as well as in some ndh genes and petL (see Table 1). All these genes are involved in electron transport in photosystem II, in particular, in the cyclic transport of electrons. petL gene product is cytochrome b6-f complex subunit VI, a component of the cytochrome b6-f complex (Shi et al., 2016). Although only FUBAR results suggest some selection in petL, we found it worth mentioning due to its high relevance for cyclic electron transport. The ndh genes products are subunits of NADH dehydrogenase and are found to be under positive selection in many angiosperms (for example, Wang et al., 2018). Plants have mechanisms for enduring excess light irradiation using cyclic electron transfer (for example, it is known that mutant tobacco plants with a lost function of the ndhB gene demonstrate a greater sensitivity of photosynthesis to the width of stomata opening (Horváth et al., 2000). It is also known that the synthesis of the protein encoded by the ndhA gene occurs as a response to photooxidative stress (Martín et al., 1996, etc.). Photosystem II protein M gene psbM also has one confirmed site under positive selection. Taking all the above into account, we can assume that adaptive evolution of genes affecting cyclic electron transport around photosystem II, especially encoding the subunits of NADH-plastoquinone oxidoreductase, is supposedly caused by abiotic factors of high altitude regions, mostly by intense light and UV radiation.
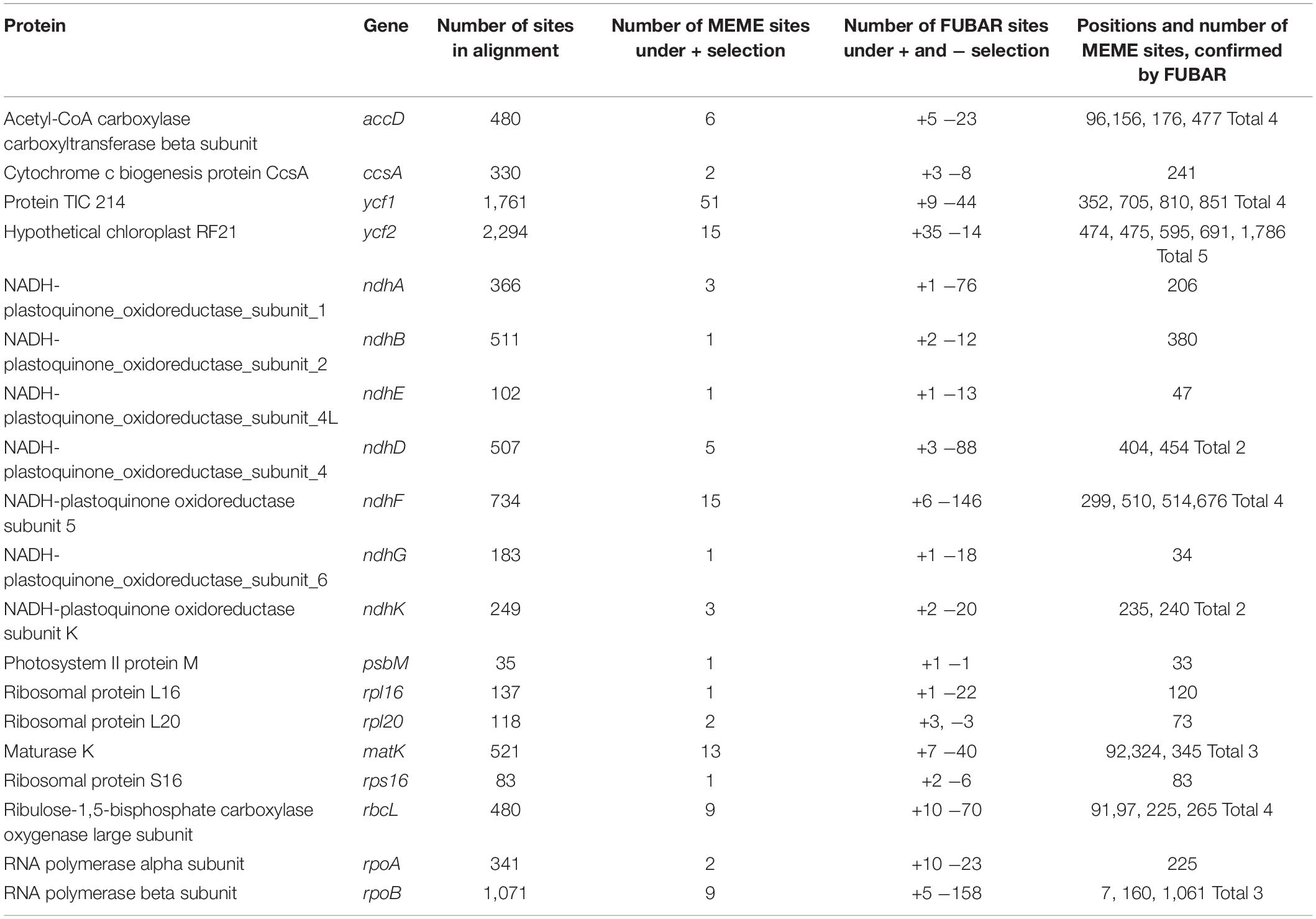
Table 1. Sites under positive of diversifying selection in protein coding genes.
More support of this assumption came from aBSrel analysis of selected branches. We analyzed the list of aBSrel findings of selection events and the maximal altitudes of habitats and found most species to be referred to as alpine species (occurring at altitudes of 3,000 m and higher), except for two rather lowland species—A. neriniflorum and A. strictum (Table 2). We did not find any links between selection events and taxonomic position of the species in the genus. In all branches besides A. paradoxumndh genes were discovered to be under selection, only in A. paradoxum signatures of selection were found in maturase K gene.
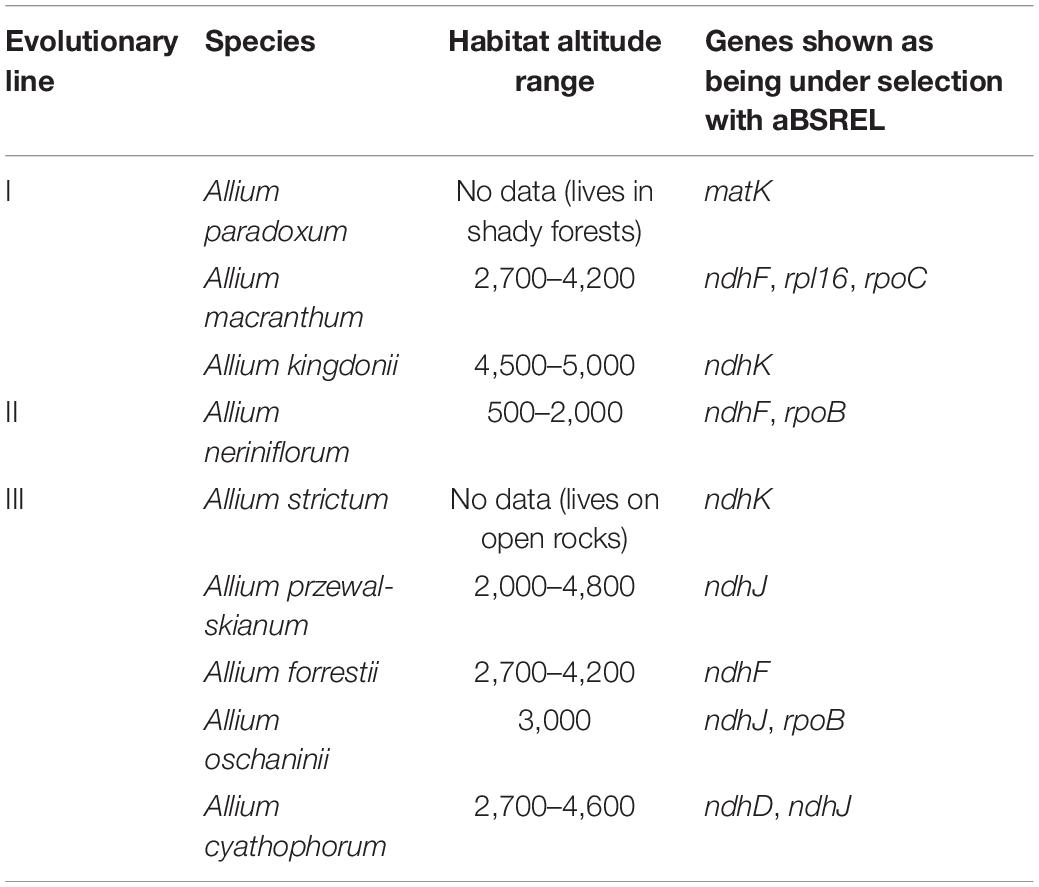
Table 2. Genes and branches with selection events.
Fisher’s exact text for multiple samples provided some more support for hypothesis of selection pressure on ndh genes. P-value 8.822e-06 allows us to reject H0, some annotations has more selected sites. The only differing annotation is “ndh genes,” it differs significantly from “photosynthesis” and marginally from “translation.” Other ontologies do not give significant signal (Supplementary Material 4).
According to our analysis, plastomes in the genus Allium evolve predominantly in neutral mode. It is important to remark that only A. paradoxum significantly differed from other Allium species in having large genomic rearrangements. First of all, major rearrangements occurred in the 4,825 bp inversion (in the region between the ndhE and rpl32 genes in SSC). A. paradoxum also demonstrates complete loss or pseudogenization of all its ndh (NADH dehydrogenase-like) genes. It was shown that A. paradoxum has the shortest known plastome of Allium species due to a large number of small deletions (145,819 bp). All the NADH-dehydrogenase complex genes (ndh genes) were found defunctionalized in the A. paradoxum plastome. This event may be supposedly associated with its adaptation to specific environmental conditions, rather uncommon among the species of Allium (shady humid forests). Some of A. paradoxum plastome genes have also lost their functionality in contrast to the rest of the studied species: rpl22 was deleted and rpl23 underwent pseudogenization (Omelchenko et al., 2020). We found that all other Allium species of the first evolutionary line had neither major rearrangements nor significant differences in the number of genes compared to Allium species of two other lines. The results comparing A. paradoxum and A. ursinum were published (Omelchenko et al., 2020).
Plants have evolved a suite of adaptive responses to cope with variable environmental conditions. Ranges of Allium species are wide not only regarding latitude and longitude, but also altitude. Numerous species occupy mountain territories, budding high altitude species from each evolutionary line (Friesen et al., 2006). That is why putative adaptations to specific high altitude environments were of our special interest. Natural areas of species habitats are different in climatic parameters in local areas. Considering temperature or humidity we can only speak of parameters that are more or less typical for the habitat of a particular species. But maximal altitudes as well as the total amount and spectrum of sunlight are detected more precisely and we can consider it as a limiting factor that could direct natural selection.
Some genes with discovered sites under positive selection—rpoA,rpoB and rbcL—code essential enzymes for chloroplast protein synthesis (rpoA and rpoB) and sugar synthesis (rbcL). RbcL gene evolves rapidly in many clades (see, for example, Sen et al., 2011; Galmés et al., 2014; Gao et al., 2019), and there is some data that its adaptive evolution may promote successful colonization of high altitude habitats (Zhao et al., 2019). The rbcL and rpoC2 genes were involved in adaptation of Cardamine species to high or low altitudes. Authors of the study speculated that amino acid residues found to be under positive selection in RUBISCO could possibly be involved in the modulation of RUBISCO aggregation/activation and enzymatic specificity (Hu et al., 2015). Also the rbcL is under selection pressure in shade-tolerant Oryza species (Gao et al., 2019).
Other genes of those that were found under positive diversifying selection in Allium genus in this work were also found under selection pressure in shade-tolerant Oryza species, namely accD (Gao et al., 2019) and in Cardamine species adapted to different altitudes, namely accD, ccsA, ycf1,rpl20 and matK (Hu et al., 2015). The matK gene also deserves a separate discussion. This gene is found under positive selection in our analysis of maturase K protein sequence in 3 sites from 521. The matK gene is under positive selection in many clades and its role in splicing of type II introns may also play a role in adaptation. Nevertheless, Kn/Ks ratio of matK is rather low (0,76) and we cannot be sure it is under selective pressure in the course of Allium adaptation to a wide range of environments, characteristic for this widespread genus.
Our analysis revealed that many genes from the ndh family are under positive selection—ndhA, ndhB, ndhD, ndhE, ndhF, ndhG, and ndhK. PetL is found only by FUBAR, but we are considering it here as well because its product is involved in the same biological process. All these genes play certain roles in electron transport across the PSII and especially in cyclic electron transport. PETL codes a component of the cytochrome b6-f complex (Shi et al., 2016) and genes from ndh family code subunits of NADH-dehydrogenase. We know that loss-of-function mutants of ndhB gene in tobacco demonstrate sensitivity of photosynthesis to moderate stomatal closure (Horváth et al., 2000) and ndhA protein is synthesized in response to photooxidative treatment (Martín et al., 1996). NdhK is a part of NADH-oxidizing subcomplex (Elortza et al., 1999) and is positively selected in some angiosperm clades (Wang et al., 2018), in particular in species adapted to contrasting high/low altitude habitats (Hu et al., 2015) and shade-tolerant and sun-loving species (Gao et al., 2019).
Cyclic electron transport across PSII is activated in response to intense light or when stomata are closed to prevent production of reactive oxygen compounds and photodamage of PSII and PSI. Under excess light and/or closed stomata (CO2 deficiency) plastoquinone pool regenerates and thereby a regeneration of NADP + occurs. NADPH cannot be expended in Calvin cycle in case of CO2 deficiency. Substrate for ferredoxin-NADP-reductase is deficient and excess of excited electrons can cause membrane oxidation, generation of active oxygen species and damage of photosystems. To prevent oxidative stress, plants start cyclic electron transport.
Genes from the ndh family are strongly overrepresented in the selected gene list in our study—we found sites under selection in 7 genes of 11 of this family. The most plausible explanation of this fact is their relevance in adaptation to conditions of high light intensity. Taking into account pseudogenization of ndh genes in A. paradoxum, we can accept this hypothesis as the first choice. Data from Fisher’s exact test provide moderate support to this hypothesis. Ndh is the only ontology that gives significant difference in ratio of selected to unselected sites and not in all comparisons. If we can speak of signatures of selection in this study, we can mention only genes from ndh family.
High altitudes have specific climate parameters, namely temperature extremes, strong winds, high solar radiation and lower atmospheric pressure (Rangwala and Miller, 2012). All these conditions affect leaf temperature, gas exchange constants, vapor pressure deficit, Michaelis-Menten constants for carboxylation and oxygenation and some other parameters affecting photosynthesis (Wang et al., 2017). Each extra 1,000 m in altitude makes photosynthesis more effective because of enhanced photosynthetic photon flux density and at the same time more problematic because of decreasing χ, which is a decreasing function of the leaf-to-air vapor pressure deficit (VPD) (Wang et al., 2017).
In mountain habitats with their fast circadian changes of light intensity and humidity and, consequently, with long periods of light phase of photosynthesis going on with closed stomata, cyclic electron transport is essential for preventing photodamage of PSII and PSI. In some plants high contents of phenolic chemicals play a role in preventing photodamage without switching to cyclic electron transport, and in A. ursinum high concentration of polyphenolic compounds was indeed reported (Lachowicz et al., 2017; Tóth et al., 2018).
Oxidative stress may affect not only electron transport, but RNA synthesis as well. In Mycobacterium tuberculosum mutations in rpoB gene can modulate bacterial survival in high oxygenic environments inside macrophages (O’Sullivan et al., 2005). In Escherichia coli cells, growing in aerobic conditions, the guanine base is oxidized to 8-oxo-7,8-dihydroguanine, which can pair with adenine as well as cytosine. So fidelity of RNA synthesis depends upon proper work of RNA polymerases and substitutions in its β and β’ subunit, coded by rpoB and rpoC genes are under selection in aerobic conditions (Inokuchi et al., 2013). Based on these facts we can hypothesize that highly oxygenic conditions in mountain habitats can cause selective pressure upon RNA polymerase function in chloroplasts, changing its protein sequence to establish stable function in conditions characterized by an excess of active oxygen, produced by overloaded Photosystem II.
So we can conclude that species of genus Allium appear to use various possible ways to prevent photodamage, but at high altitudes maintaining cyclic electron transport is most essential.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material. The custom scripts used in this study for figures preparation have been deposited in Github (https://github.com/nikitin-p/Allium_analysis).
VS, AK, and AS: conceptualization, ideas formulation, or evolution of overarching research goals and aims. IA, PN, MB, EK, and AES: software and bioinformatics analysis. VS, IA, AK, DO, ML, and AS: investigation, conducting a research and investigation process, specifically performing the experiments, or data/evidence collection. MA and SK: resources and provision of plants. VS, PN, and AS: data curation, management activities to annotate (produce metadata), scrub data, and maintain research data (including software code, where it is necessary for interpreting the data itself) for initial use and later reuse. VS, IA, PN, AK, and AS: writing—original draft preparation, creation and/or presentation of the published work, specifically writing the initial draft (including substantive translation). ML: writing—review and editing. PN: visualization and preparation of the figures. AS: project administration, management and coordination responsibility for the research activity planning and execution, funding acquisition, and acquisition of the financial support for the project. All authors contributed to the article and approved the submitted version.
This work was supported by RFBR Grant 18-04-01203.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Authors thank Dr. Sergei Lysenkov for useful discussion of statisitcal methods.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.674783/full#supplementary-material
Akashi, H., Osada, N., and Ohta, T. (2012). Weak selection and protein evolution. Genetics 192, 15–31. doi: 10.1534/genetics.112.140178
Bankevich, A., Nurk, S., Antipov, D. A., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing». J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Blazier, J. C., Guisinger, M. M., and Jansen, R. K. (2011). Recent loss of plastid-encoded ndh genes within Erodium (Geraniaceae). Plant Mol. Biol. 76, 263–272. doi: 10.1007/s11103-011-9753-5
Bolger, A. M., Lohse, M., and Usade, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Chan, P. P., and Lowe, T. M. (2019). tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol. Biol. 1962, 1–14. doi: 10.1007/978-1-4939-9173-0_1
Chase, M. W., Reveal, J. L., and Fay, M. F. (2009). A subfamilial classification for the expanded asparagalean families Amaryllidaceae, Asparagaceae and Xanthorrhoeaceae. Bot. J. Lin. Soc. 161, 132–136. doi: 10.1111/j.1095-8339.2009.00999.x
Chernomor, O., von Haeseler, A., and Minh, B. Q. (2016). Terrace aware data structure for phylogenomic inference from supermatrices. Syst. Biol. 65, 997–1008. doi: 10.1093/sysbio/syw037
Cock, P. J., Antao, T., Chang, J. T., Chapman, B. A., Cox, C. J., Dalke, A., et al. (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. doi: 10.1093/bioinformatics/btp163
Daniell, H., Lin, C. S., Yu, M., and Chang, W.-J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17:134. doi: 10.1186/s13059-016-1004-2
de Pamphilis, C., and Palmer, J. (1990). Loss of photosynthetic and chlororespiratory genes from the plastid genome of a parasitic flowering plant. Nature 348, 337–339. doi: 10.1038/348337a0
Dong, W. L., Wang, R. N., Zhang, N. Y., Fan, W. B., Fang, M. F., and Li, Z. H. (2018). Molecular evolution of chloroplast genomes of Orchid species: insights into phylogenetic relationship and adaptive evolution. Int. J. Mol. Sci. 19:716. doi: 10.3390/ijms19030716
Doyle, J. J., and Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phyt. Bull. 19, 11–15.
Elortza, F., Asturias, J. A., and Arizmendi, J. M. (1999). Chloroplast NADH dehydrogenase from Pisum sativum: characterization of its activity and cloning of ndhK gene. Plant Cell Physiol. 40, 149–154. doi: 10.1093/oxfordjournals.pcp.a029522
Filyushin, M. A., Beletsky, A. V., and Kochieva, E. Z. (2019). Characterization of the complete chloroplast genome of leek Allium porrum L. (Amaryllidaceae). Mitochondrial. DNA Part B. 4, 2602–2603. doi: 10.1080/23802359.2019.1640090
Filyushin, M. A., Beletsky, A. V., Mazur, A. M., and Kochieva, E. Z. (2016). The complete plastid genome sequence of garlic Allium sativum L. Mitochondrial. DNA Part B. 1, 831–832. doi: 10.1080/23802359.2016.1247669
Filyushin, M. A., Beletsky, A. V., Mazur, A. M., and Kochieva, E. Z. (2018). Characterization of the complete plastid genome of lop-sided onion Allium obliquum L. (Amaryllidaceae). Mitochondrial. DNA part B. 3, 393–394. doi: 10.1080/23802359.2018.1456369
Fonseca, L. H. M., and Lohmann, L. G. (2017). Plastome rearrangements in the Adenocalymma-Neojobertia“ clade (Bignonieae, Bignoniaceae) and its phylogenetic implications. Front. Plant Sci. 8:1875. doi: 10.3389/fpls.2017.01875
Friesen, N., Fritsch, R. M., and Blattner, F. R. (2006). Phylogeny and new intrageneric classification of Allium (Alliaceae) based on nuclear ribosomal DNA ITS sequences. Aliso Syst. Evol. Bot. 22, 372–395. doi: 10.5642/aliso.20062201.31
Fritsch, R. M. (2012). Geographic relations and morphological variation inside molecular clades of Central Asian Allium species of subg. Melanocrommyum (Amaryllidaceae)∗. Verh. Zool. Bot. Ges. Österreich 148/149, 245–263.
Fritsch, R. M., Blattner, F. R., and Gurushidze, M. (2010). New classification of Allium L. subg. Melanocrommyum (Webb & Berthel) Rouy (Alliaceae) based on molecular and morphological characters. Phyton 49, 145–220.
Galmés, J., Andralojc, P. J., Kapralov, M. V., Flexas, J., Keys, A. J., Molins, A., et al. (2014). Environmentally driven evolution of rubisco and improved photosynthesis and growth within the genus Limonium (Plumbaginaceae)». N. Phytol. 203, 989–999. doi: 10.1111/nph.12858
Gao, L. Z., Liu, Y. L., Zhang, D., Li, W., Gao, J., Liu, Y., et al. (2019). Evolution of Oryza chloroplast genomes promoted adaptation to diverse ecological habitats. Commun. Biol. 2:278. doi: 10.1038/s42003-019-0531-2
Govaerts, R., and Fritsch, R. (2021). World Checklist of [Allium]. Richmond: Facilitated by the Royal Botanic Gardens, Kew.
Green, B. R. (2011). Chloroplast genomes of photosynthetic eukaryotes: chloroplast genomes of photosynthetic eukaryotes. Plant J. 66, 34–44. doi: 10.1111/j.1365-313X.2011.04541.x
Guisinger, M. M., Kuehl, J. V., Boore, J. L., and Jansen, R. K. (2011). Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: rearrangements, repeats, and codon usage. Mol. Biol. Evol. 28, 583–600. doi: 10.1093/molbev/msq229
Haberhausen, G., and Zetsche, K. (1994). Functional loss of all ndh genes in an otherwise relatively unaltered plastid genome of the holoparasitic flowering plant Cuscuta reflexa. Plant Mol. Biol. 24, 217–222. doi: 10.1007/BF00040588
Haberle, R. C., Fourcade, H. M., Boore, J. L., and Jansen, R. K. (2008). Extensive rearrangements in the chloroplast genome of Trachelium caeruleum are associated with repeats and tRNA genes. J. Mol. Evol. 66, 350–361. doi: 10.1007/s00239-008-9086-4
Hanaoka, M., Kato, M., Anma, M., and Tanaka, K. (2012). SIG1, a Sigma Factor for the chloroplast RNA polymerase, differently associates with multiple DNA regions in the chloroplast chromosomes in vivo. Int. J. Mol. Sci. 13, 12182–12194. doi: 10.3390/ijms131012182
Hargreaves, A. L., Samis, K. E., and Eckert, C. G. (2014). Are species’ range limits simply niche limits writ large? a review of transplant experiments beyond the range. Am. Nat. 183, 157–173. doi: 10.1086/674525
Harris, C. R., Millman, K. J., van der Walt, S. J., Gommers, R., Virtanen, P., Cournapeau, D., et al. (2020). Array programming with numPy. Nature 585, 357–362. doi: 10.1038/s41586-020-2649-2
Hilu, K. W., Borsch, T., Müller, K., Soltis, D. E., Soltis, P. E., Savolainen, V., et al. (2003). Angiosperm phylogeny based on matK sequence information. Am. J. Bot. 90, 1758–1776. doi: 10.3732/ajb.90.12.1758
Hiratsuka, J., Shimada, H., Whittier, R., Ishibashi, T., Sakamoto, M., Mori, M., et al. (1989). The complete sequence of the rice (Oryza sativa) chloroplast genome: intermolecular recombination between distinct tRNA genes accounts for a major plastid DNA inversion during the evolution of the cereals. Mol. Gen. Genet. 217, 185–194. doi: 10.1007/BF02464880
Horváth, E. M., Peter, S. O., Joët, T., Rumeau, D., Cournac, L., Horváth, G. V., et al. (2000). Targeted inactivation of the plastid ndhB gene in tobacco results in an enhanced sensitivity of photosynthesis to moderate stomatal closure. Plant physiol. 123, 1337–1350. doi: 10.1104/pp.123.4.1337
Hsu, C. Y., Wu, C. S., and Chaw, S. M. (2016). Birth of four chimeric plastid gene clusters in Japanese umbrella pine. Genome Biol. Evol. 8, 1776–1784. doi: 10.1093/gbe/evw109
Hu, S., Sablok, G., Wang, B., Qu, D., Barbaro, E., Viola, R., et al. (2015). Plastome organization and evolution of chloroplast genes in Cardamine species adapted to contrasting habitats. BMC Genomics 16:306. doi: 10.1186/s12864-015-1498-0
Hunter, J. D. (2007). Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 9, 90–95. doi: 10.1109/mcse.2007.55
Huo, Y., Gao, L., Liu, B., Yang, Y., Kong, S., Sun, Y., et al. (2019). Complete chloroplast genome sequences of four Allium species: comparative and phylogenetic analyses. Sci. Rep. 9:12250. doi: 10.1038/s41598-019-48708-x
Inokuchi, H., Ito, R., Sekiguchi, T., and Sekiguchi, M. (2013). Search for proteins required for accurate gene expression under oxidative stress. J. Biol. Chem. 288, 32952–32962. doi: 10.1074/jbc.M113.507772
Jin, F. Y., Y, X., Xie, D. F., Li, H., Yu, Y., Zhou, S. D., et al. (2019). Comparative complete chloroplast genome analyses and contribution to the understanding of chloroplast phylogeny and adaptive evolution in subgenus nguinum. Russ. J. Genet. 55, 872–884. doi: 10.1134/S1022795419070081
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Kapralov, M. V., and Filatov, D. A. (2006). Molecular adaptation during adaptive radiation in the Hawaiian endemic genus Schiedea. PLoS One 1:e8. doi: 10.1371/journal.pone.0000008
Kazutaka, K., and Standley, D. M. (2013). MAFFT Multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kikuchi, S., Asakura, Y., Imai, M., Nakahira, Y., Kotani, Y., Hashiguchi, Y., et al. (2018). A Ycf2-FtsHi heteromeric AAA-ATPase complex is required for chloroplast protein import. Plant Cell 30, 2677–2703. doi: 10.1105/tpc.18.00357
Koboldt, D. C., Zhang, Q., Larson, D. E., Shen, D., McLellan, M. D., Lin, L., et al. (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. doi: 10.1101/gr.129684.111
Kosakovsky Pond, S. L., Frost, S. D. W., and Muse, S. V. (2005). HyPhy: hypothesis testing using phylogenies. Bioinformatics 21, 676–679. doi: 10.1093/bioinformatics/bti079
Krinitsina, A. A., Sizova, T. V., Zaika, M. A., Speranskaya, A. S., and Sukhorukov, A. P. (2015). A rapid and cost-effective method for DNA extraction from archival herbarium specimens. Biochemistry 80, 1478–1484. doi: 10.1134/s0006297915110097
Lachowicz, S., Kolniak-Ostek, J., Oszmiański, J., and Wiśniewski, R. (2017). Comparison of phenolic content and antioxidant capacity of bear garlic (Allium ursinum L.) in different maturity stages: phenolics and antioxidants in bear garlic. J. Food Process. Preserv. 41, 1–10. doi: 10.1111/jfpp.12921
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lee, J., Chon, J. K., Lim, J. S., Kim, E.-K., and Nah, G. (2017). Characterization of complete chloroplast genome of Allium victorialis and its application for barcode markers. Plant Breed. Biotechnol. 5, 221–227. doi: 10.9787/PBB.2017.5.3.221
Lee-Yaw, J. A., Kharouba, H. M., Bontrager, M., Mahony, C., Csergö, A. M., Noreen, A. M., et al. (2016). A synthesis of transplant experiments and ecological niche models suggests that range limits are often niche limits. Ecol. Lett. 19, 710–722. doi: 10.1111/ele.12604
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, Q.-Q., Zhou, S.-D., He, X.-J., Yu, Y., Zhang, Y.-C., and Wei, X.-Q. (2010). Phylogeny and biogeography of Allium (Amaryllidaceae: Allieae) based on nuclear ribosomal internal transcribed spacer and chloroplast rps16 sequences, focusing on the inclusion of species endemic to China. Ann. Bot. 106, 709–733. doi: 10.1093/aob/mcq177
Lin, C. S., Chen, J., Huang, Y. T., Chan, M.-T., Daniell, H., Chang, W.-J., et al. (2015). The location and translocation of Ndh genes of chloroplast origin in the Orchidaceae family. Sci. Rep. 5:9040. doi: 10.1038/srep09040
Lin, C. S., Chen, J. J. W., Chiu, C. C., Hsiao, H. C. W., Yang, C. J., Jin, X. H., et al. (2017). Concomitant loss of NDH complex-related genes within chloroplast and nuclear genomes in some Orchids. Plant J. Cell Mol. Biol. 90, 994–1006. doi: 10.1111/tpj.13525
Liu, L., Yusupov, Z., Suyunkulov, H., and Jiang, Z. (2020). The complete chloroplast genome of Allium ferganicum. Mitoch. DNA B Resour. 5, 2772–2773. doi: 10.1080/23802359.2020.1788454
Logacheva, M. D., Krinitsina, A. A., Belenikin, M. S., Khafizov, K., Konorov, E. A., Kuptsov, S. V., et al. (2017). Comparative analysis of inverted repeats of polypod fern (Polypodiales) plastomes reveals two hypervariable regions. BMC Plant Biol. 17:255. doi: 10.1186/s12870-017-1195-z
Macadlo, L. A., Ibrahim, I. M., and Puthiyaveetil, S. (2020). Sigma Factor 1 in chloroplast gene transcription and photosynthetic light acclimation. J. Exp. Bot. 71, 1029–1038. doi: 10.1093/jxb/erz464
Maragheh, F. P., Janus, D., Senderowicz, M., Haliloglu, K., and Kolano, B. (2019). Karyotype analysis of eight cultivated Allium species». J. Appl. Genet. 60, 1–11. doi: 10.1007/s13353-018-0474-1
Martín, M., Casano, L. M., and Sabater, B. (1996). Identification of the product of ndhA gene as a thylakoid protein synthesized in response to photooxidative treatment. Plant Cell Physiol. 37, 293–298. doi: 10.1093/oxfordjournals.pcp.a028945
Martín, M., and Sabater, B. (2010). Plastid Ndh genes in plant evolution. Plant Physiol. Biochem. 48, 636–645. doi: 10.1016/j.plaphy.2010.04.009
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Morton, B. R., and Clegg, M. T. (1993). A chloroplast DNA mutational hotspot and gene conversion in a noncoding region near rbcL in the Grass family (Poaceae). Curr. Genet. 24, 357–365. doi: 10.1007/BF00336789
Murrell, B., Moola, S., Mabona, A., Weighill, T., Sheward, D., Kosakovsky Pond, S. L., et al. (2013). FUBAR: a fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 30, 1196–1205. doi: 10.1093/molbev/mst030
Murrell, B., Wertheim, J. O., Moola, S., Weighill, T., Scheffler, K., and Kosakovsky Pond, S. L. (2012). Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 8:e1002764. doi: 10.1371/journal.pgen.1002764
Namgung, J., Do, H. D. K., Kim, C., Choi, H. J., and Kim, J. H. (2021). Complete chloroplast genomes shed light on phylogenetic relationships, divergence time, and biogeography of Allioideae (Amaryllidaceae). Sci. Rep. 11:3262. doi: 10.1038/s41598-021-82692-5
Ogihara, Y., Terachi, T., and Sasakuma, T. (1991). Molecular analysis of the hot spot region related to length mutations in wheat chloroplast DNAs. I. nucleotide divergence of genes and intergenic spacer regions located in the hot spot region. Genetics 129, 873–884. doi: 10.1093/genetics/129.3.873
Omelchenko, D. O., Krinitsina, A. A., Belenikin, M. S., Konorov, E. A., Kuptsov, S. V., Logacheva, M. D., et al. (2020). Complete plastome sequencing of Allium paradoxum reveals unusual rearrangements and the loss of the Ndh genes as compared to Allium ursinum and other onions. Gene 726:144154. doi: 10.1016/j.gene.2019.144154
O’Sullivan, D. M., McHugh, T. D., and Gillespie, S. H. (2005). Analysis of rpoB and pncA mutations in the published literature: an insight into the role of oxidative stress in Mycobacterium tuberculosis evolution? J. Antimicrob. Chemother. 55, 674–679. doi: 10.1093/jac/dki069
R Core Team, R. (2018). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Raman, G., and Park, S. (2015). Analysis of the complete chloroplast genome of a medicinal plant, Dianthus superbus var. longicalycinus, from a comparative genomics perspective. PLoS One 10:e0141329. doi: 10.1371/journal.pone.0141329
Rambaut, A., and Drummond, A. J. (2012). FigTree (version 1.4. 0). Edinburgh: Andrew Rambaut. Available online at: http://tree.bio.ed.ac.uk/software/figtree/
Rangwala, I., and Miller, J. R. (2012). Climate change in mountains: a review of elevation-dependent warming and its possible causes. Clim. Change 114, 527–547. doi: 10.1007/s10584-012-0419-3
Reback, J., McKinney, W., Jbrockmendel, Van Den Bossche, J., Augspurger, T., Cloud, P., et al. (2021). pandas-dev/pandas: pandas 1.2.3 (v1.2.3). Zenodo. doi: 10.5281/ZENODO.3509134
Sanchez-Puerta, M., Bachvaroff, T., and Delwiche, C. (2005). The complete plastid genome sequence of the haptophyte Emiliania huxleyi: a comparison to other plastid genomes. DNA Res. 12, 151–156. doi: 10.1093/dnares/12.2.151
Sassone, A. B., Arroyo-Leuenberger, S. C., and Giussani, L. M. (2014). Nueva circunscripción de la tribu Leucocoryneae (Amaryllidaceae. Allioideae) Darwinia. Nueva Serie 2, 197–206. doi: 10.14522/darwiniana/2014.22.584
Schwarz, E. N., Ruhlman, T. A., Sabir, J. S. M., Hajrah, N. H., Alharbi, N. S., Al−Malki, A. L., et al. (2015). Plastid genome sequences of Legumes reveal parallel inversions and multiple losses of Rps16 in Papilionoids: parallel inversions and Rps16 losses in Legumes. J. Syst. Evol. 53, 458–468. doi: 10.1111/jse.12179
Sen, L., Fares, M. A., Liang, B. Gao L., Wang B., Wang T., et al. (2011). Molecular evolution of rbcL in three gymnosperm families: identifying adaptive and coevolutionary patterns. Biol. Direct 6:29. doi: 10.1186/1745-6150-6-29
Shah, N. C. (2014). Status of cultivated & wild Allium species in India, A review. Scit. J. 1, 28–36.
Shi, C., Hu, N., Huang, H., Gao, J., Zhao, Y. J., and Gao, L. Z. (2012). An improved chloroplast DNA extraction procedure for whole plastid genome sequencing. PLoS One 7:e31468. doi: 10.1371/journal.pone.0031468
Shi, C., Wang, S., Xia, E. H., Jiang, J. J., Zeng, F. C., and Gao, L. Z. (2016). Full transcription of the chloroplast genome in photosynthetic eukaryotes. Sci. Rep. 6:30135. doi: 10.1038/srep30135
Silva, S. R., Diaz, Y. C., Penha, H. A., Pinheiro, D. G., Fernandes, C. C., Miranda, V. F., et al. (2016). The chloroplast genome of Utricularia reniformisSheds light on the evolution of the Ndh gene complex of terrestrial carnivorous plants from the Lentibulariaceae family. PloS One 11:e0165176. doi: 10.1371/journal.pone.0165176
Smith, M. D., Wertheim, J. O., Weaver, S., Murrell, B., Scheffler, K., and Kosakovsky Pond, S. L. (2015). Less is more: an adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol. Biol. Evol. 32, 1342–1353. doi: 10.1093/molbev/msv022
Talevich, E., Invergo, B. M., Cock, P. J., and Chapman, B. A. (2012). Bio.Phylo: a unified toolkit for processing, analyzing and visualizing phylogenetic trees in Biopython. BMC Bioinform. 13:209. doi: 10.1186/1471-2105-13-209
Tamura, M. N., Yamashita, J., Fuse, S., and Haraguchi, M. (2004). Molecular phylogeny of monocotyledons inferred from combined analysis of plastid matK and rbcL gene sequences. J. Plant Res. 117, 109–120. doi: 10.1007/s10265-003-0133-3
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq – versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Tóth, T., Kovarovič, J., Bystrická, J., Vollmannova, A., Musilová, J., and Lenková, M. (2018). The content of polyphenols and antioxidant activity in leaves and flowers of wild garlic (Allium ursinum L.). Acta Aliment. 47, 252–258. doi: 10.1556/066.2018.47.2.15
Vieira, L. N., Faoro, H., Fraga, H. P., Rogalski, M., de Souza, E. M., de Oliveira Pedrosa, F., et al. (2014). An improved protocol for intact chloroplasts and cpDNA isolation in conifers. PLoS One 9:e84792. doi: 10.1371/journal.pone.0084792
von Kohn, C., Kiełkowska, A., and Havey, M. J. (2013). Sequencing and annotation of the chloroplast DNAs and identification of polymorphisms distinguishing normal male-fertile and male-sterile cytoplasms of onion. Genome 56, 737–742. doi: 10.1139/gen-2013-0182
Vvedensky, A. I. (1935). Flora of USSR. V. 4. Genus 267, ed. L. Allium (Moscow; Leningrad: Ed.of USSR Academy of Science).
Wakasugi, T., Tsudzuki, J., Ito, S., Nakashima, K., Tsudzuki, T., and Sugiura, M. (1994). Loss of all Ndh genes as determined by sequencing the entire chloroplast genome of the black pine Pinus thunbergii. Proc. Natl. Acad. Sci. 91, 9794–9798. doi: 10.1073/pnas.91.21.9794
Wambugu, P. W., Brozynska, M., Furtado, A., Waters, D. L., and Henry, R. J. (2015). Relationships of wild and domesticated rices (Oryza AA genome species) based upon whole chloroplast genome sequences. Sci. Rep. 5:13957. doi: 10.1038/srep13957
Wang, H., Prentice, I. C., Davis, T. W., Keenan, T. F., Wright, I. J., and Peng, C. (2017). Photosynthetic responses to altitude: an explanation based on optimality principles. New Phytol. 213, 976–982. doi: 10.1111/nph.14332
Wang, X., Zhou, T., Bai, G., and Zhao, Y. (2018). Complete chloroplast genome sequence of Fagopyrum dibotrys: genome features, comparative analysis and phylogenetic relationships. Sci. Rep. 8:12379. doi: 10.1038/s41598-018-30398-6
Waskom, M., Botvinnik, O., Gelbart, M., Ostblom, J., Hobson, P., Lukauskas, S., et al. (2020). mwaskom/seaborn: v0.11.1. Zenodo. doi: 10.5281/ZENODO.592845
Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M., and Barton, G. J. (2009). Jalview version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. doi: 10.1093/bioinformatics/btp033
Wheeler, E. J., Mashayekhi, S., McNeal, D. W., Columbus, J. T., and Pires, J. C. (2013). Molecular systematics of Allium subgenus Amerallium (Amaryllidaceae) in North America. Am. J. Bot. 100, 701–711. doi: 10.3732/ajb.1200641
Wicke, S., Schneeweiss, G. M., dePamphilis, C. W., Müller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Xie, D. F., Tan, J. B., Yu, Y., Gui, L. J., Su, D. M., Zhou, S. D., et al. (2020). Insights into phylogeny, age and evolution of Allium (Amaryllidaceae) based on the whole plastome sequences. Ann. Bot. 125, 1039–1055. doi: 10.1093/aob/mcaa024
Xie, D. F., Yu, H. X., Price, M., Xie, C., Deng, Y. Q., Chen, J. P., et al. (2019). Phylogeny of Chinese Allium species in section daghestanica and adaptive evolution of Allium (Amaryllidaceae, Allioideae) species revealed by the chloroplast complete genome. Front. Plant Sci. 10:460. doi: 10.3389/fpls.2019.00460
Yan, C., Du, J., Gao, L., Li, Y., and Hou, X. (2019). The complete chloroplast genome sequence of watercress (Nasturtium officinale R. Br.): genome organization, adaptive evolution and phylogenetic relationships in Cardamineae. Gene 699, 24–36. doi: 10.1016/j.gene.2019.02.075
Yao, X., Tan, Y. H., Yang, J. B., Wang, Y., Corlett, R. T., and Manen, J. F. (2019). Exceptionally high rates of positive selection on the rbcL gene in the genus Ilex (Aquifoliaceae). BMC Evol. Biol. 19:192. doi: 10.1186/s12862-019-1521-1
Yusupov, Z., Deng, T., Volis, S., Khassanov, F., Makhmudjanov, D., and Tojibaev, K. (2020). Phylogenomics of Allium section Cepa (Amaryllidaceae) provides new insights on domestication of onion. Plant Divers. 43, 102–110. doi: 10.1016/j.pld.2020.07.008
Zeng, S., Zhou, T., Han, K., Yang, Y., Zhao, J., and Liu, Z. L. (2017). The complete chloroplast genome sequences of six Rehmannia species. Genes 8:103. doi: 10.3390/genes8030103
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn Graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Zhang, J., Yuan, H., Yang, Y., Fish, T., Lyi, S. M., Thannhauser, T. W., et al. (2016). Plastid ribosomal protein S5 is involved in photosynthesis, plant development, and cold stress tolerance in Arabidopsis. J. Exp. Bot. 67, 2731–2744. doi: 10.1093/jxb/erw106
Keywords: Allium, plastome, sequence, evolution, pseudogenization
Citation: Scobeyeva VA, Artyushin IV, Krinitsina AA, Nikitin PA, Antipin MI, Kuptsov SV, Belenikin MS, Omelchenko DO, Logacheva MD, Konorov EA, Samoilov AE and Speranskaya AS (2021) Gene Loss, Pseudogenization in Plastomes of Genus Allium (Amaryllidaceae), and Putative Selection for Adaptation to Environmental Conditions. Front. Genet. 12:674783. doi: 10.3389/fgene.2021.674783
Received: 02 March 2021; Accepted: 15 June 2021;
Published: 08 July 2021.
Edited by:
Yuriy L. Orlov, I.M. Sechenov First Moscow State Medical University, RussiaReviewed by:
Nikolai Friesen, University of Osnabrück, GermanyCopyright © 2021 Scobeyeva, Artyushin, Krinitsina, Nikitin, Antipin, Kuptsov, Belenikin, Omelchenko, Logacheva, Konorov, Samoilov and Speranskaya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Victoria A. Scobeyeva, c2tvYmVpLWtoYW51bUB5YW5kZXgucnU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.