
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 31 May 2021
Sec. Computational Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.668040
This article is part of the Research Topic Current State of Multi-omics Modeling and Simulations View all 5 articles
 Yi Xiang1
Yi Xiang1 Xiaohuan Zou2Huaqiu Shi1Xueming Xu1
Xiaohuan Zou2Huaqiu Shi1Xueming Xu1 Caixia Wu3Wenjuan Zhong1Jinfeng Wang1Wenting Zhou1Xiaoli Zeng1Miao He1Ying Wang3Li Huang1*†Xiangcai Wang1*†
Caixia Wu3Wenjuan Zhong1Jinfeng Wang1Wenting Zhou1Xiaoli Zeng1Miao He1Ying Wang3Li Huang1*†Xiangcai Wang1*†In the precision medicine of lung adenocarcinoma, the identification and prediction of tumor phenotypes for specific biomolecular events are still not studied in depth. Various earlier researches sheds light on the close correlation between genetic expression signatures and DNA copy number variations (CNVs), for which analysis of CNVs provides valuable information about molecular and phenotypic changes in tumorigenesis. In this study, we propose a comprehensive analysis combining genome-wide association analysis and an Elastic Net Regression predictive model, focus on predicting the levels of many gene expression signatures in lung adenocarcinoma, based upon DNA copy number features alone. Additionally, we predicted many other key phenotypes, including clinical features (pathological stage), gene mutations, and protein expressions. These Elastic Net prediction methods can also be applied to other gene sets, thereby facilitating their use as biomarkers in monitoring therapy.
The enormous genetic heterogeneity caused by multiple types of DNA aberrations is a key factor in tumorigenesis. Thus, the capacity to analyze tumor inhomogeneity is essential to elucidating cancer etiopathogenesis and to more accurately defining patient subgroups in precision medicine (Gao et al., 2016; Godek et al., 2016; Padmanabhan et al., 2017; Pommier et al., 2020). One limitation that currently limits us to evaluate the heterogeneity is the accurate description of tumor phenotypes. The establishment of various data integration and analysis platforms, e.g., the Cancer Genome Atlas (TCGA), has enabled us to take advantage of readily accessible large-scale genomic data from multiple platforms to better analyze the association between gene expression heterogeneity and key phenotypes in the process of tumorigenesis (Hoadley et al., 2018). In particular, numerous reports have gradually uncovered various gene expression signatures that can be used for delineating specific tumor phenotypes from invasion rates to characterization of tumorous immune microenvironment (Bild et al., 2006; Nevins and Potti, 2007). These messenger RNA expression signatures, besides protein expressions, genetic mutation loads, and clinical characteristics, offer a wide-ranging cancer molecular representation (Kumar et al., 2018; Chen Y. J. et al., 2020). Therefore, the application of genomic data from these data analysis platforms to illuminate the connection between genotypes and phenotypes is significant to identify the genomic changes that occur during tumorigenesis (Gatza et al., 2014; Watermann et al., 2020; Zengin and Önal-Süzek, 2020). Furthermore, the predictions of phenotypes that drive tumorigenesis according to DNA expression status would be valuable to the stratification of patients in precision medicine (George et al., 2019; Carrillo-Reixach et al., 2020; Tsimberidou et al., 2020), specifically in current clinical practice, due to the expensive expression profile and the routine acquisition of genetic mutation information.
Lung cancer is a kind of malignancies with highest morbidity and mortality in the world (Siegel et al., 2019). About 85% of lung cancers are Non-small cell lung cancer (NSCLC), with an only approximately 16% 5-year survival rate (Namani et al., 2019). Lung adenocarcinoma (LUAD) is the most common histological subtype of NSCLC (Zhu et al., 2019). Extensive research in recent years has led to great advances in exploring the oncogenesis and treatment strategies of LUAD, however it remains one of the most deadly and metastatic types of lung cancer (Denisenko et al., 2018). Patients with LUAD often do not respond well to conventional radiotherapy or chemotherapy, because of the delay diagnosis in the middle and advanced stages. Therefore, it is still urgent to elucidate the pathogenic and molecular mechanism of LUAD. The ubiquitous DNA copy number variations (CNVs) in the human genome, containing amplifications, losses, insertions and multisite mutations, plays a critical role in the occurrence and developmentof various tumor types (López et al., 2020; Staaf et al., 2013). Specifically, CNV in tumor tissues and cells can lead to irregular expressions of tumor-related genetic drivers as well as genomic and molecular phenotypic heterogeneity (Kim et al., 2020). Previous studies have shown that DNA CNVs are associated with higher risks and poorer prognosis of LUAD. It is increasingly recognized that therapy strategies for LUAD should focus on the relationship between mutation loads and epigenetic alterations. However, there is still a lot of room for exploration. TP53, EGFR and KRAS mutations, for example, are critical in the occurrence and development of lung cancers, but not all tumors are caused by the activation of these mutations alone and eliminated by the inhibition of these genes (Jänne et al., 2015; Kobayashi et al., 2005). Here, we used gene expression signatures downloaded from the TCGA platform, together with common clinical and molecular features, as the basis for constructing predictive models of lung adenocarcinoma phenotype. We adopted an comprehensive genomic method, containing genome-wide association analysis (Gatza et al., 2014; Watermann et al., 2020), and an ElasticNet-mediated regression approach (Zou and Hastie, 2005), to modeling complicated tumorous phenotypes based on DNA copy number variations (CNVs). By these findings, we aim to elucidate the significant correlations of CNVs with several genetic expression signatures and protein expression landscapes. In general, our proposed method can be used to link CNVs with multifaceted phenotypes, also to develop evaluation models for therapeutic significance using currently commonplace DNA-based clinical tools.
Gene mutation data, protein expression data, and clinical information of lung adenocarcinoma samples in TCGA were downloaded from Xena1 (Hoadley et al., 2018). For TCGA lung adenocarcinoma, we converted the gene expression data using the upper quartile scale and log2 transform, and then screened for genes expressed in more than 70% of the samples.
The collection, conversion and subsequent analysis of DNA copy number data were all based on GISTIC2.0 module. GISTIC2 gene-level CNVs information of human lung adenocarcinoma downloaded from TCGA GDAC FireBrowse (Mermel et al., 2011; Hoadley et al., 2018)2 are shown in Table 1, with no further processing, and the related clinicopathological characteristics are shown in Table 2. Using Ensembl 54 (hg18) genome build, gene-level copy number scores were derived through the extreme method as used in GISTIC2: Genes that fell completely within a circular binary segmentation (CBS)-identified copy number segment were assigned corresponding segment value. Genes that overlapped with multiple segments were assigned the greatest amplification or the least deletion value among the overlapped segments. Genes with no overlapping segments were excluded from further analyses (Xia et al., 2019).

Table 1. Sample information.
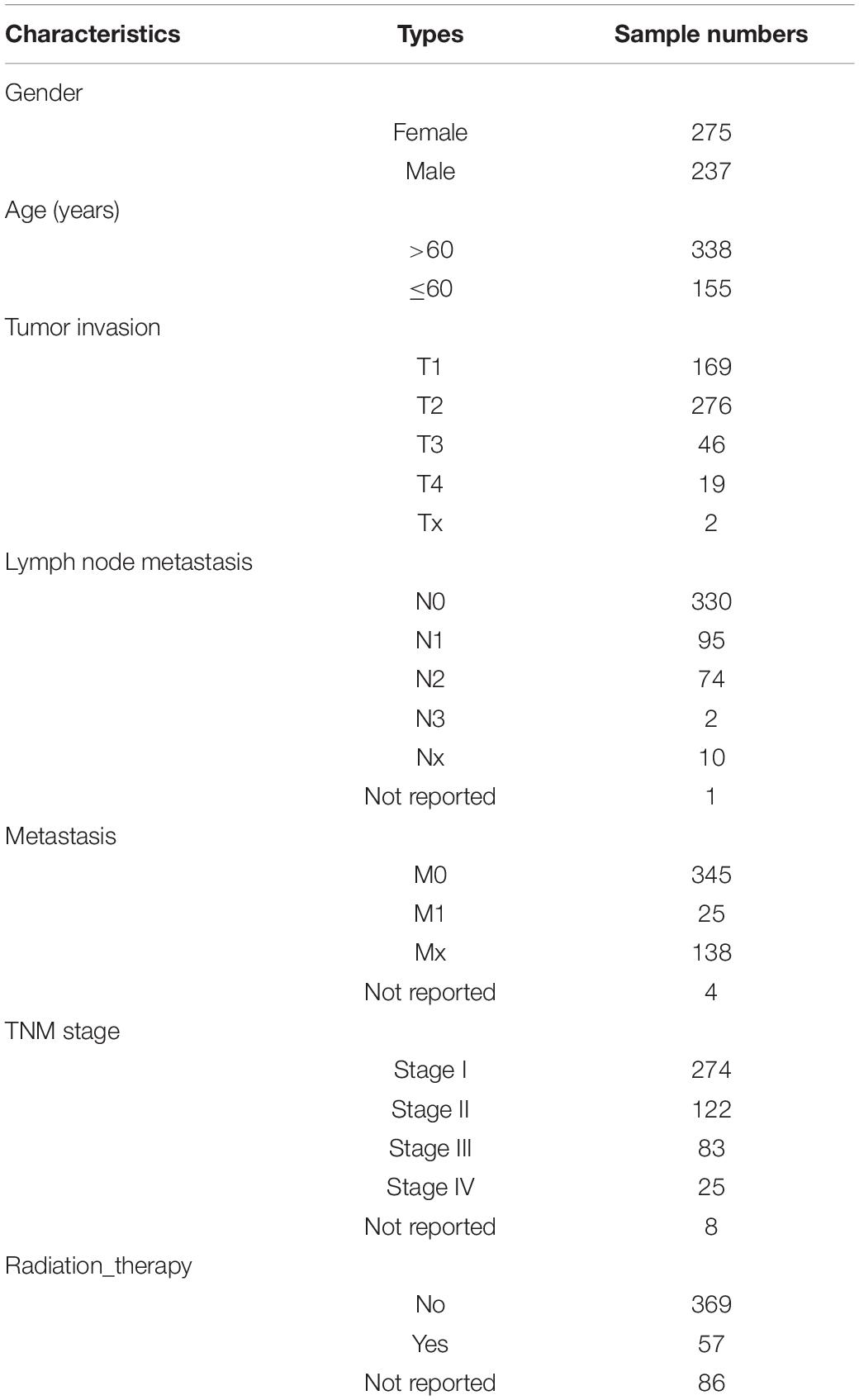
Table 2. Patient and clinicopathological characteristics.
Gene expression signature scores for 512 lung adenocarcinoma cases are composed of a panel of 531 previously published genetic expression signature, collected from a variety of earlier research, GSEA (Subramanian et al., 2005), and were partly summed up by Tanioka et al. (2018) that can be applied to completely describe tumor phenotypes. As for genetic signature scores, they were calculated in a manner consistent with their derivation. For 492 signatures with homogeneous expression across the genes, median expression value was used as signature score. The rest of the signatures were based on correlation to predetermined gene centroids or based on published algorithms. For correlation-based signatures, all predetermined training sets are available to download through our GitHub repository (see section Code Availability) (Xia et al., 2019). For each such signature, DWD was used to first merge gene expression matrix with corresponding training set and then Pearson/Spearman correlation/Euclidean distance was computed for each sample in the merged data. For several algorithm-based signatures, corresponding R code is provided to calculate each signature (see section Code Availability). All 531 signatures were applied to TCGA lung cancer data.
For identifying the association between CNVs and gene expression signatures, we employed two different statistical approach (Gatza et al., 2014; Watermann et al., 2020) on gene expression of TCGA lung adenocarcinoma cohorts and matched copy number data; the first involved calculating the Spearman correlation between the signature score and gene-level CNV score for each sample; the second statistical test involved performing a one-sided Fishers exact test for comparing the frequencies of CNV amplifications/deletions between higher score samples (top quartile) and lower score ones. The Benjamin-Hochberg (BH) method was applied to correct the q-value for each gene signatures for all analysis. The critical value of significance for the two statistical tests above (q-value) was set at 0.01.
The Elastic-Net regression method, a linear combination of L1/L2 regularization in the Ridge regression and LASSO (Least absolute shrinkage and selection operator) regression, was utilized for building the CNV-based tumor phenotype predictions (Zou and Hastie, 2005). First, gene-level CNV scores were converted into segment-level CNV scores, which were averaged as the mean CNV score for all genes in this DNA segment (Beroukhim et al., 2010; Zack et al., 2013; Cancer Genome Atlas Research Network, 2014; Qiu et al., 2018; Hua et al., 2020). To construct the model, the total sample was split 70%:30% for the training and the test sets, respectively, which were also stratified by clinical variates (i.e., overall survival, sex, TNM stage and histopathological stage). Only the training set was used to build the models. The fitted generalized linear models were used to determine the maximum and minimum observed values of λ for each α value in the training sets (CRAN Rpackage glmnet) (Candia and Tsang, 2019). Using the default parameters, two hundred turns of MonteCarlo cross-validation were performed for screening the tuning parameter (Bandalos, 1993), and the optimal parameter was confirmed as the most accurate classification method. The models with the optimal parameters were subsequently used in the validation sets, and the area under the receiver operating characteristic curve (AUC) was applied to assess the predictive value of models. The phenotypes with AUC values higher than 0.75 were considered as with high predictive performance.
Since the variables involved in the prediction in this study are continuous data, we divided the sample into two-halves (using the top quartile and bottom three quartiles) to perform model predictions. For clinical characteristics and gene mutation status, which have binary outcomes, the model was built directly for their prediction. For gene mutation prediction, we selected the top 20 genes with the highest mutation frequencies in lung adenocarcinoma. Clinical features included pathological stage (stage I-II vs. stage III-IV) and TNM stage.
SPSS software (version 23.0) or R software (version 4.0.3) were used for statistical analysis. Curve analysis of receiver operating characteristic (ROC) was utilized to evaluate the predictive performance of CNV-based Elastic Net regression models. The AUC, ranging from 0.5 (for an uninformative marker) to 1 (for a perfect predictive marker), was a measurement for how well patient survival can be predicted with the gene signatures.
First, we studied the potential correlations between multiple gene expression characteristics and gene CNVs. We used a group of 531 gene expression signatures reported previously to quantify various tumor phenotypes, including, but not limited to, tumor microenvironment characteristics and activated signaling pathways (Supplementary Table 1; Hoadley et al., 2018). We next applied a genome-wide association analysis to identify possible connections between each signature-based phenotype and DNA copy number data (Gatza et al., 2014; Watermann et al., 2020). Two different statistical methods were used for each signature to assess the correlation between DNA CNVs and the genetic signature for each genetic trait: The Spearman’s correlation was used for identifying negative/positive correlations between gene-level CNV and expression signature scores, and the Fisher’s exact test was performed to compare the differences in the probability of CNV gains/losses between the groups with higher expression signature scores (top 1/4) and low scores (other 3/4). B–H correction of q values were conducted for both sets of statistical methods (Benjamini and Hochberg, 1995). To further enhance accuracy, a given CNV was considered significantly associated with an expression signature only if the corrected q values for both statistical methods were < 0.01 (Figure 1A). Potential CNV drivers of a gene signature should increase the frequencies of CNV amplifications in samples with high signature scores and be positively correlated with its signature score. We used the methods above to analyze the relationship between each gene signature and CNV, and found that some gene signatures were significantly associated with CNVs, such as GSEA_Median_MYC_amplified_chr8q24, Pcorr_magnoid_PLOS.2012, and IMMUNE_Bindea_Cell _Th17_cells_Median_Immunity.2013 (Figures 1B–D and Supplementary Table 2). Taken together, this part of our study determined that the modeling methods could accurately find links between CNVs and specific genetic signatures, some had been confirmed in previous reports.
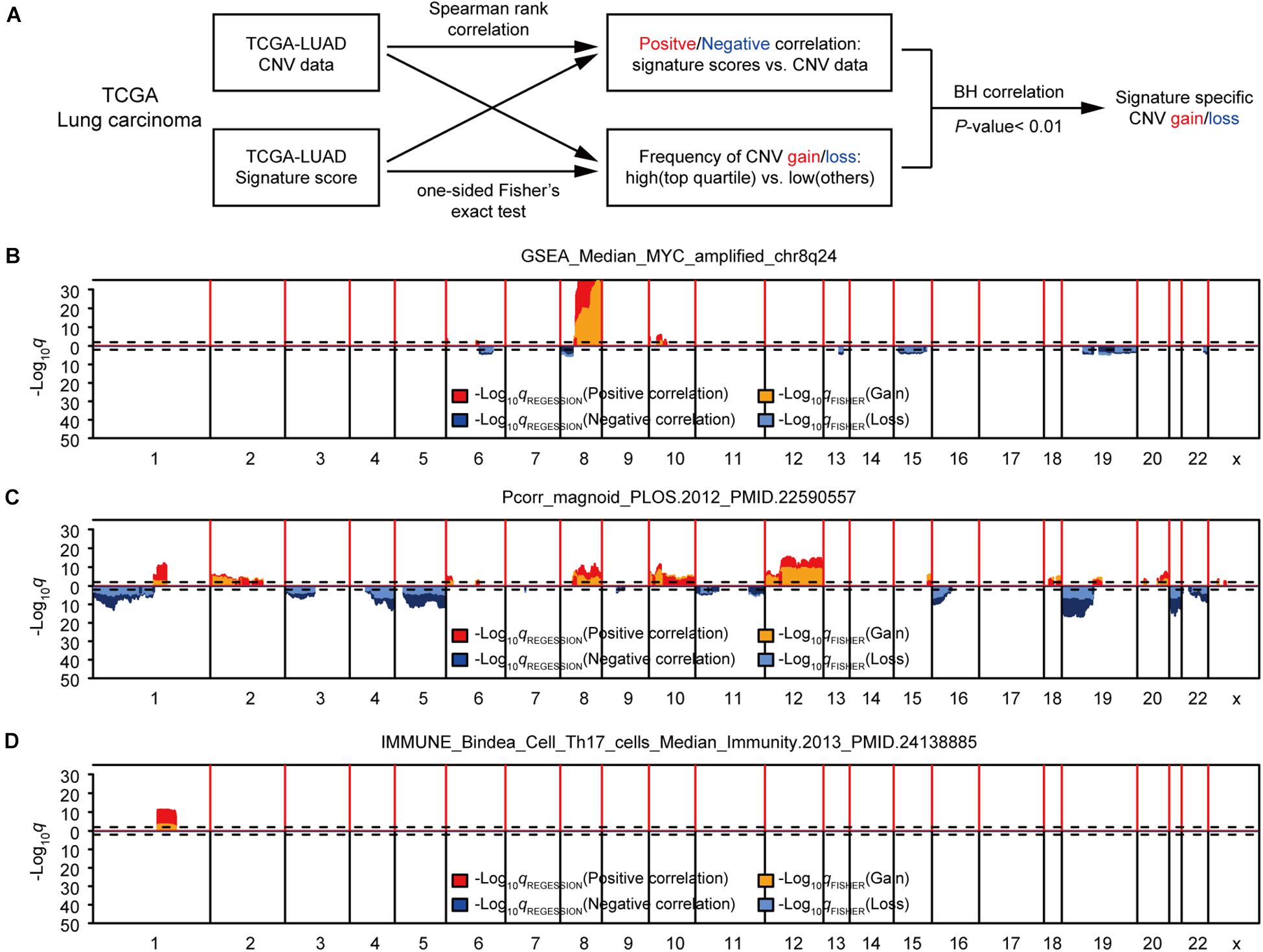
Figure 1. Identification of gene expression signature-specific CNVs in lung adenocarcinoma. (A) Schematic overview of the strategy used to identify CNVs associated with gene signatures. Gain/loss indicates DNA copy number gains or losses; Positive/Negative indicates positive or negative association. (B,D) Spearman rank correlation was used to identify genes that were positively (red) or negatively (dark blue) correlated with gene signatures, and Fisher’s exact test was used to compare the frequency of copy number gains (orange) or losses (light blue) in GSEA_Median_MYC_amplified_chr8q24 (B), Pcorr_magnoid_PLOS.2012 (C), and IMMUNE_Bindea_Cell_Th17_cells_Median_Immunity.2013 (D). Dashed lines indicate the significance threshold (q = 0.01). Only the q values for genes that were significant in both analyses were plotted. In each figure, chromosome boundaries are indicated by vertical black lines.
In view of the strong relationship, we next constructed models that can predict the levels of genetic expression signatures on the basis of DNA CNVs landscapes alone. For constructing the predictive models, a modeling method known as Elastic-Net was utilized, which can handle several potential co-linear variables that are present in regression models, and then screen out the most related elements for the final regression modeling (Zou and Hastie, 2005). Gene-level CNV scores were not used during model building, instead we applied the segment-level CNV scores of chromosomal regions proven being significant in multiple tumor types (Beroukhim et al., 2010; Zack et al., 2013; Qiu et al., 2018; Hua et al., 2020; Supplementary Table 3). The 512 cases of lung adenocarcinoma were divided 70% into the training group and 30% into the validation group. Models were constructed on the TCGA training set, and tested on the validation set, and model performance was assessed using AUC value (Figure 2A).
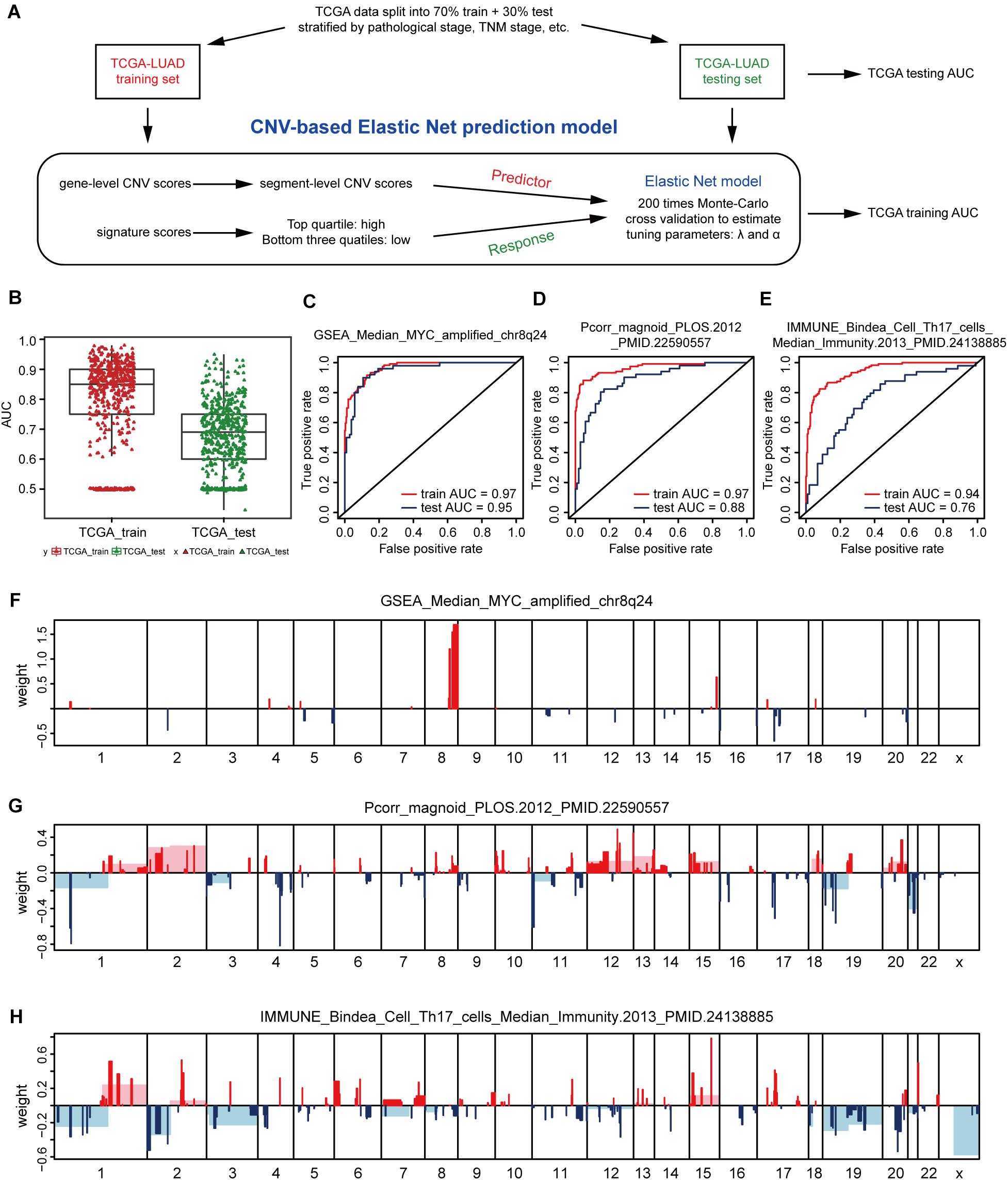
Figure 2. CNV-based gene signature prediction. (A) Schematic overview of the strategy used to build Elastic Net regression models for predicting gene expression signature levels. (B) AUC distributions of CNV-based prediction for the 531 gene signature scores of lung adenocarcinoma in the TCGA training and test set. (C–E) ROC curves and corresponding AUC values for three signatures in the training and test sets. GSEA_Median_MYC_amplified_chr8q24 (C), Pcorr_magnoid_PLOS.2012 (D), and IMMUNE_Bindea_Cell_Th17_cells_Median_Immunity.2013 (E). (F–H) CNV segments and whole chromosomal arms and their corresponding coefficients selected by the Elastic Net prediction models for the three signatures.
The AUC value of some genetic signatures indicated that the CNV-based Elastic Net prediction models can be good forecasters of certain gene signatures (Table 3 and Supplementary Table 4_AUC_signature). Among the 531 gene signatures, 142 in the test set had AUCs > 0.75 (Figure 2B), henceforth denoted as “highly predictable.” Of these 142 signatures, only 21 were DNA-based amplicon signatures that essentially measure specific CNA events and were therefore expected to produce high AUC values. For example, signature 11q13-amplicon had a fairly high AUC value in test set (AUC = 0.90). Notably, the three signatures highlighted in the previous correlation analysis, namely, GSEA_Median_MYC_amplified_chr8q24, Pcorr_magnoid_PLOS.2012, and IMMUNE_Bindea_Cell_Th17_cells_Median_Immunity.2013 were all highly predictable and their corresponding AUC were 0.95, 0.88, and 0.76, respectively (Figures 2C–E). The results of this part show the prediction efficiency of the Elastic Net model.
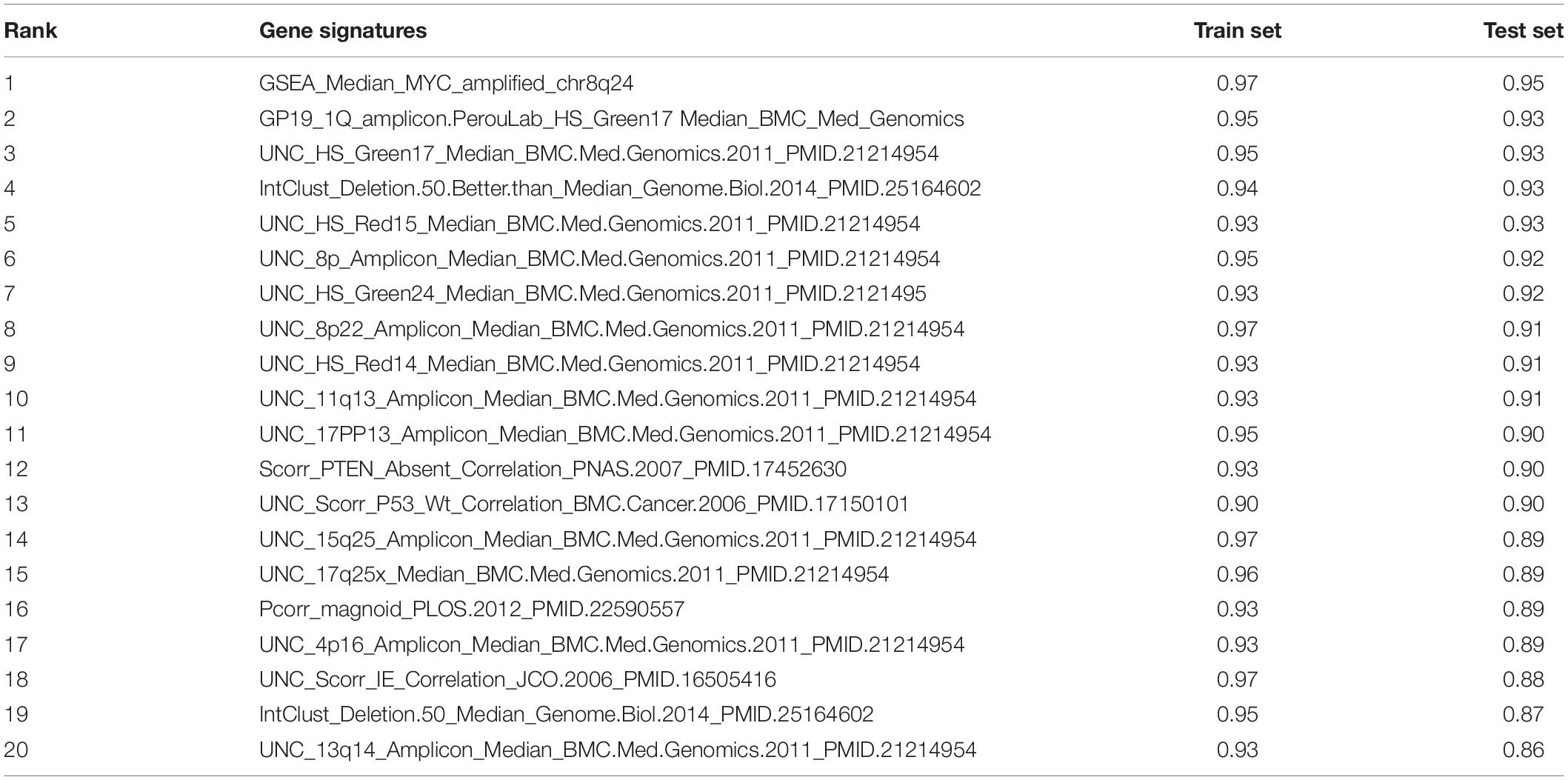
Table 3. Top 20 AUC value of gene signatures predicted by Elastic Net models.
For comparing these associated landscapes, the CNV regions and entire chromosome arms and the coefficients selected by Elastic Net models of the three features above are shown in Figures 2F–H (Supplementary Table 4). Remarkably, Pcorr_magnoid_PLOS.2012, and IMMUNE_Bindea_Cell_Th17_cells_Median_Immunity.2013 signatures had a large amount of correlation landscape with CNVs (Figures 2G,H). On the contrary, the correlation characteristics of GSEA_Median_MYC_amplified_chr8q24 signature had little intersection with those of the Elastic Net model (Figure 2F). In general, these consequences indicate that DNA CNV features can be used to predict gene expression signatures.
Next, the method of Elastic Net-mediated CNV-based prediction was used for constructing models of specific protein expressions. Reverse phase protein array (RPPA) information was used (Network, Cancer Genome Atlas Research, 2012; Cancer Genome Atlas Research Network, 2014) to measure the protein expression levels of TCGA lung adenocarcinoma samples (Figure 3A). A few studies have reported that DNA copy number has a significant impact on the expressions of some proteins (Geiger et al., 2010; Myhre et al., 2013; Liu et al., 2016). However, these studies only assessed the correlations with proteins encoded by individual genes. Here, Elastic Net predictive modeling was applied to account for genome-wide CNV changes in the prediction of protein expressions. By setting the AUC at > 0.75 as “high predictive value,” (Figure 3B) 8 of 131 proteins expression levels can be predicted with high accuracy (Table 4 and Supplementary Table 5), containing Claudin7 and CHK2 (Figures 3C,D). Notably, Beroukhim features 17p and 15q deletions were involved within the models, in agreement with the condition that Claudin7 negativity is common among patients with lung cancers (Figure 3E; Lu et al., 2015; Choi et al., 2016; Akizuki et al., 2017). Similarly, the result in Figure 3F was consistent with other previous reports about the lower expression of CHK2 in adenocarcinoma (Chen et al., 2012; Choi et al., 2012). Thus, our method can accurately predict these complex protein expressions based on a little fraction of genomic information.
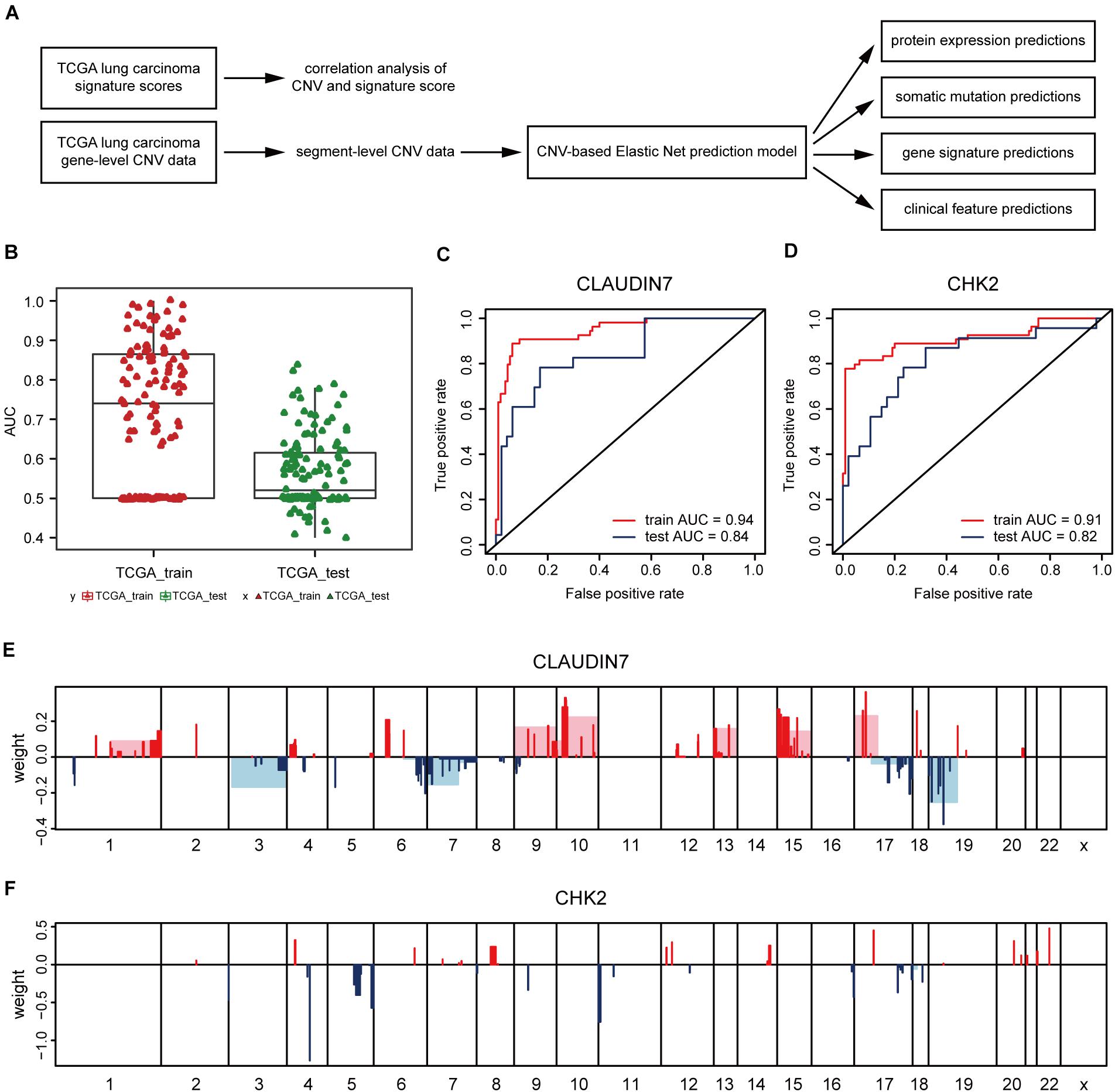
Figure 3. CNV-based protein expression prediction. (A) Flowchart showed the construction of CNV-based Elastic Net prediction models and the certification of its predictive value on gene expression, protein, mutation and clinical features. (B) AUC distributions of CNV-based expression level predictions for 131 proteins in the RPPA arrays. (C,D) ROC curves and corresponding AUC values of Claudin7 (C) and CHK2 (D) in the TCGA training and test sets. (E,F) Elastic Net-selected CNV segments and whole chromosomal arms and their coefficients for prediction models for the two proteins.
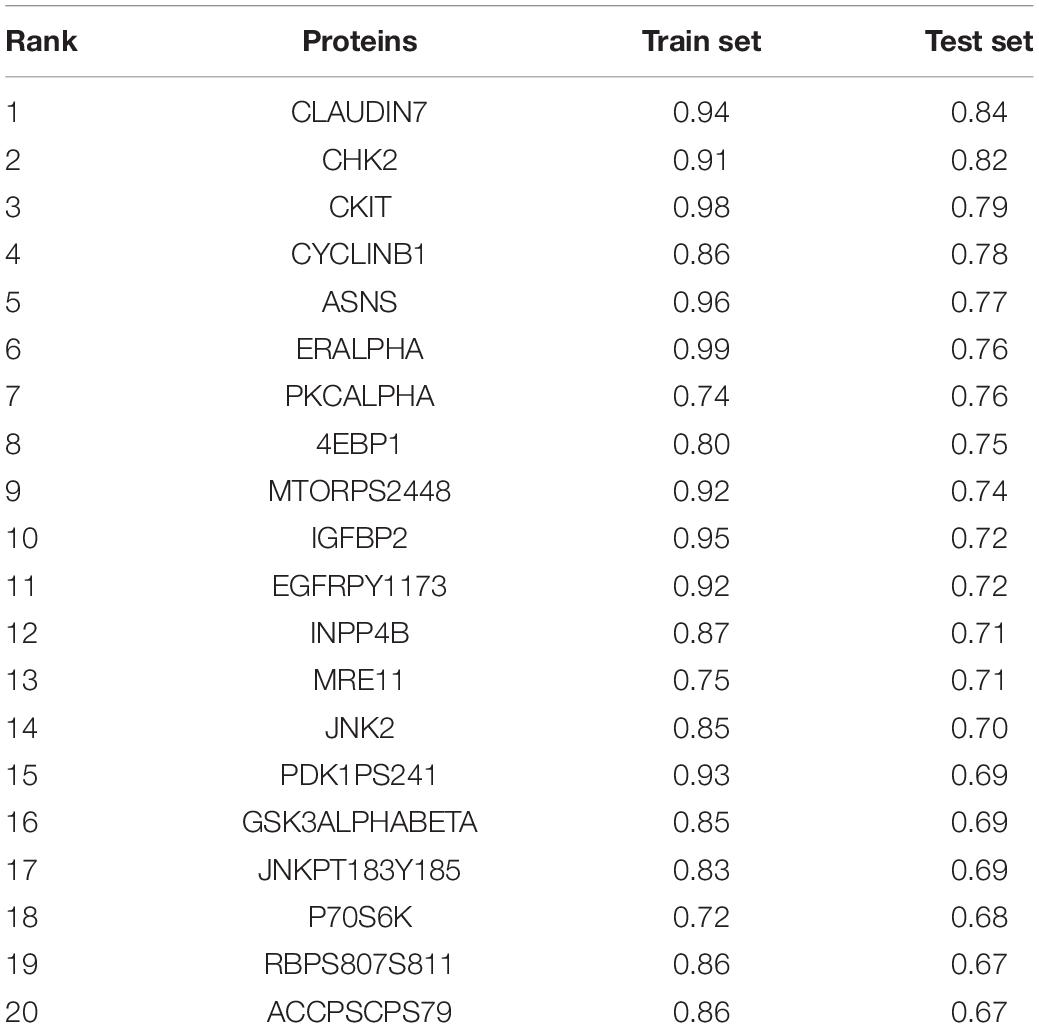
Table 4. Top 20 AUC value of protein expressions predicted by Elastic Net models.
Additionally, the capacity of CNV-based Elastic Net predictive models to foretell somatic mutation was tested. We first downloaded the mutation information of lung adenocarcinoma samples in TCGA database (Hofree et al., 2013), from which the top 20 genes with the highest mutation frequencies were selected (Figure 4A and Table 5). By setting the AUC threshold of validation set as 0.75, only the TP53 mutation was screened out (Figure 4B), corresponded with its widely recognized role as a cancer suppressor. And the differential expression of PIK3CA induced by Beroukhim 3q gain was speculated to take part in the tumorigenesis of lung adenocarcinoma when TP53 silencing (Yang et al., 2014; Hou et al., 2020; Wang F. et al., 2020; Figure 4C and Supplementary Table 6_TP53_weight). Moreover, when the AUC threshold was relaxed to 0.7, the TP53, RYR2, TTN, LRP1B and CSMD3 mutations satisfied the condition (Supplementary Table 6_AUC_snp), indicating their correlations with increased tumor mutation burden.
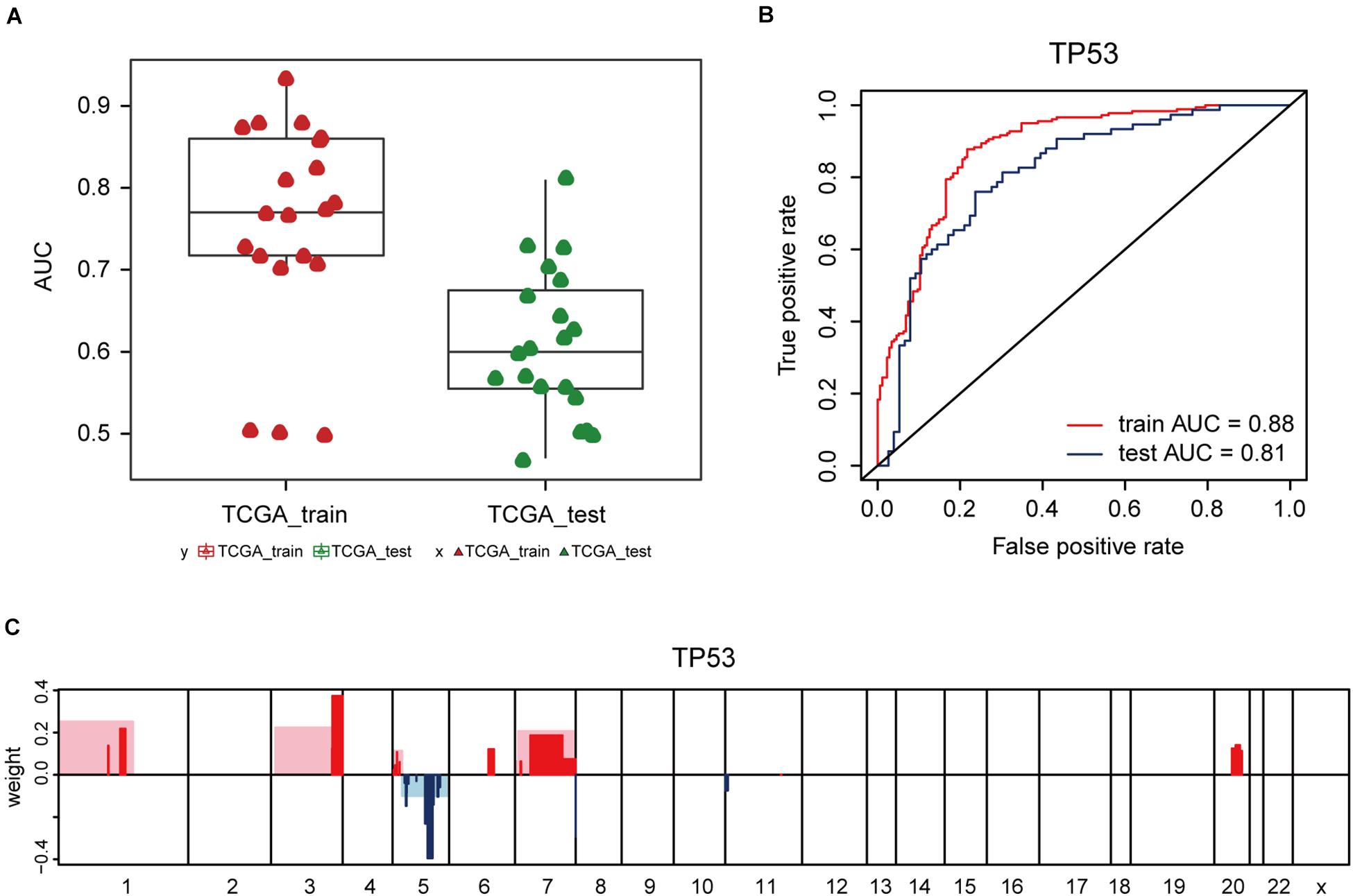
Figure 4. CNV-based genetic mutation prediction. (A) AUC distributions of CNV-based predictions for top 20 genes with the highest mutation frequencies selected from TCGA lung adenocarcinoma mutation data. (B) ROC curves and corresponding AUC values of TP53 mutation status in the TCGA training and test sets. (C) Elastic Net-selected CNV segments and whole chromosomal arms and their coefficients selected for prediction models for TP53 mutation.
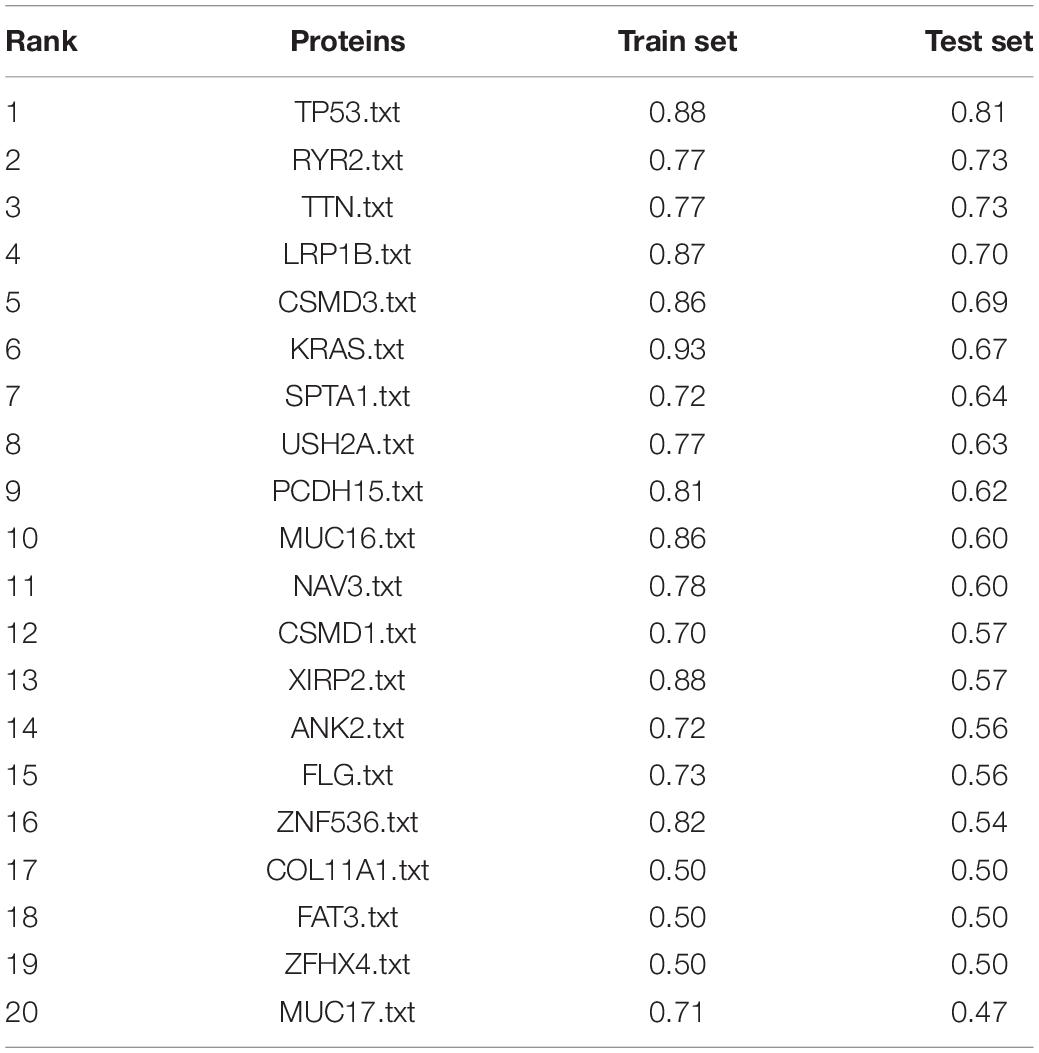
Table 5. Top 20 AUC value of somatic mutations predicted by Elastic Net models.
Furthermore, we explored whether CNV-based Elastic Net predictive regression models can be used to forecast clinical features. Here, pathological stage and TNM stage were selected to divide the sample into two halves (stage I –II, as one group; stage III –IV, as another). After grouping the samples for model building and prediction, the AUC of the prediction results was found to be 0.52 (Figure 5A). The model feature landscapes for TNM stage were shown in Figure 5B. For model building and prediction based on TNM stage, all AUCs were 0.5. Additionally, we acquired radiotherapy_information for TCGA lung adenocarcinoma samples, and applied the Elastic Net predictive model, but the prediction outcomes were poor, and all AUCs were 0.5.
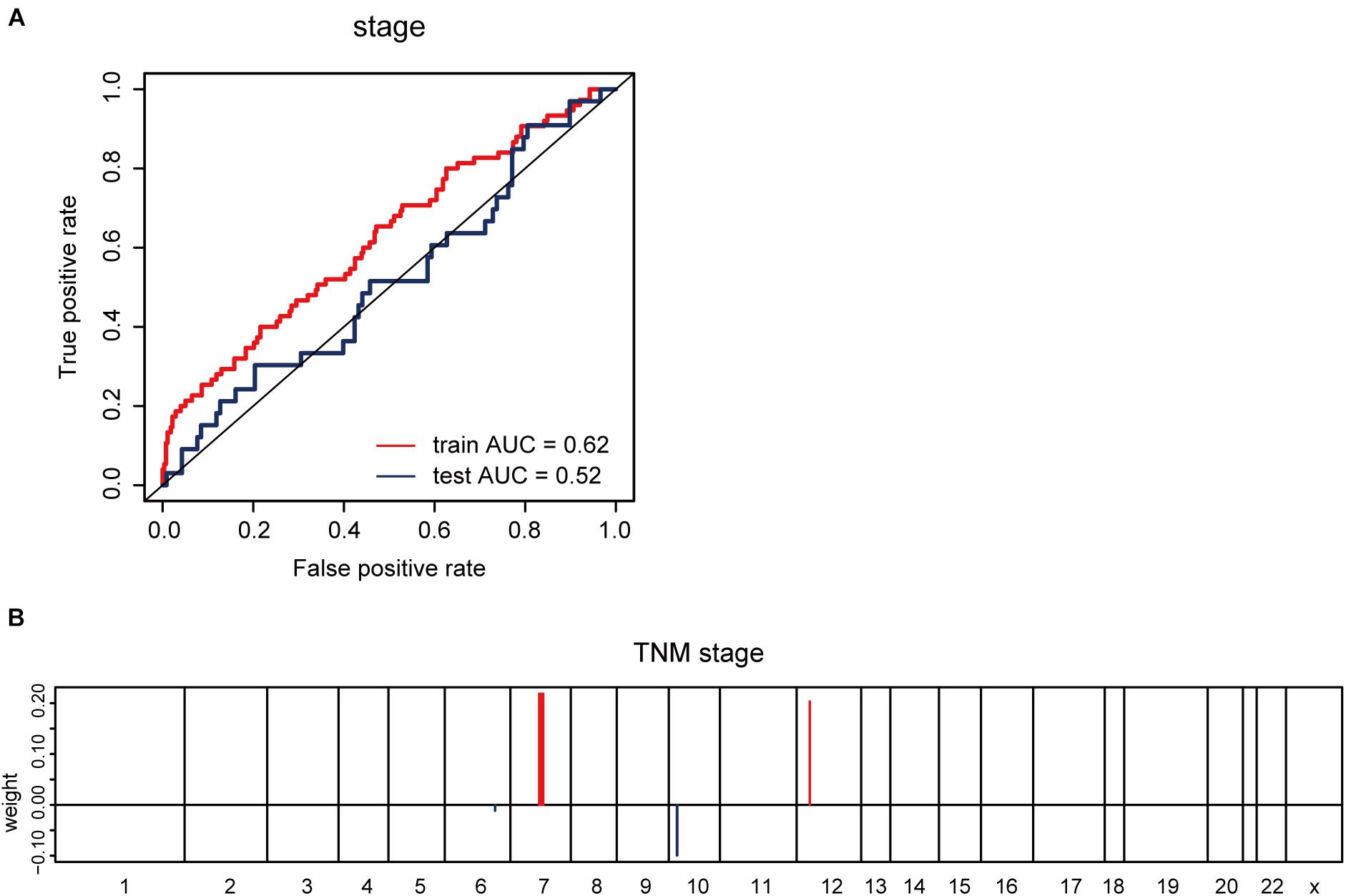
Figure 5. CNV-based prediction of clinical features. (A) ROC curves and corresponding AUC values for CNV-based prediction of clinical stage (stage I–II vs. stage III–IV) in the TCGA training and test sets. (B) Elastic Net-selected CNV segments and whole chromosomal arms and their coefficients for prediction model for pathological stage.
In this study, we applied chromosomal segment-level CNVs downloaded from TCGA database to perform Elastic Net predictive model building, and the resulting models were used for the forecasting of mutations, protein expression, clinical features, and gene signature scores in lung adenocarcinoma.
The ability to predict key tumor phenotypes is essential for elucidating the biocomplexity in multiple cancer types (Bailey et al., 2018; Ding et al., 2018). The standard treatment procedure for lung adenocarcinoma includes DNA-based gene expression profiling, whiles protein expression analysis (e.g., PDL1) is gaining prominence, which is mostly owing to immune therapy (Wang L. et al., 2020). Numerous previous studies have confirmed that lung cancer is likely to be driven by gene copy numbers in some degree due to the occurrence of numerous copy number events, many of which are known genetic drivers (Wilkerson et al., 2012; Campbell et al., 2016; Chen J. et al., 2020; Gillette et al., 2020). Therefore, we reasoned that when testing a copy number-driven tumor type, the diversity of DNA CNVs can be used to predict many key tumor phenotypes. To verify the above hypothesis, a wide-ranging manual archiving of arranged genetic expression signatures extracted from plenty of existing studies were utilized for analyzing tumorous phenotype and assess the predictability. Two methods were adopted to investigate the associations between every gene expression signature and DNA CNV; a genome-wide association analysis and Elastic Net regression modeling method. The correlation research was able to identify whether genes have positive or negative correlations with gene characteristics by evaluating the genes individually. The combination of the two methods synergistically produced a complete landscape of the connection between gene signature and CNV, including many known linkages, such as genetic signature for DNA gains and deletions, or some signaling pathway activities (e.g., TP53). Indeed, many of these signatures in the true validation group were highly predictable (AUC > 0.8). Combining the association landscape for each gene marker with its Elastic Net features has provided us with exact CNV region for deeper exploration of possible genetic drivers for multiple tumors. Besides, the following usage of Elastic Net predictive model method in many other phenotypes, such as the levels of proteins and mutations, has demonstrated the capacity to forecast various essential phenotypes of lung adenocarcinoma with high accuracy.
Nowadays, many similar studies are based on the expression levels of related genes to build the prediction models for molecular expression signatures, clinical indicators, disease progression and prognosis. However, our study starts from the perspective of DNA copy number data, intending to excavate the potential information contained in which, and predicting key tumor phenotypes, like mutation status or biomarker levels or complex expression phenotypes in a proposed copy number-driven tumor type, e.g., lung adenocarcinoma. In addition, instead of some simple methods such as Logistics regression and random forest used in some other conventional prediction model researches, the DNA CNVs-based prediction models in our study are built on the basis of Elastic Net regression. With the characteristics of both Ridge regression and LASSO regression, the Elastic Net algorithm can effectively achieve the screening of eigenvectors with group effect and compress the selected variables to avoid the model over-fitting, so as to maintain the simplicity and accuracy of the model at the same time.
This modeling strategy might have clinical benefit and offer an orthogonal method to addressing significant features like TP53 status, especially considering the increasing value of gene exons and genetic mutations goes with the diagnosis and treatment of tumors (Kinde et al., 2013; Garofalo et al., 2016; Uzilov et al., 2016; Valle et al., 2020). Our study indicates that a little fraction of genomic information plays a significant role in predicting many molecular phenotypes in lung cancer. This also provides possibility for the clinical use of Elastic Net model. For instance, a variety of cell cycle progression and apoptosis signatures, such as c-MYC signature assessed above, may play a predictive role in the efficacy of NEDD8 inhibitors targeting PI3K/c-MYC axis (Ochiiwa et al., 2021), or for MAPK inhibitors by targeting ABL1/2-mediated reactivation of MEK/ERK/c-MYC signaling (Tripathi et al., 2020). The Elastic Net prediction models of c-MYC signature were able to extract out the patients with high proliferation rates, which generally accompanied by MYC amplification, then the use of NEDD8 inhibitors is recommended for these patients. If followed by more extensive trial and verification, it might be possible to read out plenty of novel prediction markers for cancer diagnosis, efficacy, and prognosis from available genomic panel information, hence playing a better guiding role in tumor personalized medicine without increasing the expense.
The strengths of this present study are that the model has been validated in both the test and validation sets, and that some results have previously been shown to be associated with lung adenocarcinoma, such as GSEA_Median_MYC_amplified_chr8q24 and Pcorr_magnoid_PLOS.2012. Nevertheless, there are still many shortcomings in our study. For example, in the prediction of clinical features, we analyzed the pathological stage, TNM stage, and radiotherapy, but only the prediction outcome of the pathological stage in the validation set was > 0.5. No new sample sets with both CNV and expression profile data were found for further validation, so we used 70% TCGA data for testing, and another part of the data was used for validation.
Overall, our study exhibited the capability to construct CNV-based Elastic Net predictors for multiple key tumor phenotypes in patients with lung adenocarcinoma. Although most studies have focused on the search for genetic drivers of tumorigenesis, our results had the important connotation that DNA information can be used to predict significant complex tumor phenotypes, which could have applications in the clinical settings.
All code supporting the current study is deposited in GitHub (https://github.com/xyouli/DNA-based-predictors-of-non-genetic-cancer-phenotypes). All computational analyses are done using public R packages.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
The collection of human tissue samples or clinical data were not involved in this study. All data was downloaded from network open databases.
YX designed the study, wrote the manuscript, and took responsibility for the integrity of the data and the accuracy of the data analysis. YX and CW performed the database analysis from Xena and GSEA. YX, WZhon, and JW carried out the CNV identification. YX, HS, and XX did the CNV-based signature prediction. CW, WZhon, JW, WZhou, and XZe revised the manuscript. LH and XW planned and supervised all these works. All authors have read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.668040/full#supplementary-material
CNVs, copy number variations; CNV, copy number variation; LASSO, Least absolute shrinkage and selection operator; TCGA, the cancer genome atlas; DFS, disease-free survival; OS, overall survival; RPPA, reverse-phase protein arrays; HR, hazard ratios; CI, confidence interval; ROC, receiver operating characteristic; AUC, area under the curve.
Akizuki, R., Shimobaba, S., Matsunaga, T., Endo, S., and Ikari, A. (2017). Claudin-5, -7, and -18 Suppress Proliferation Mediated by Inhibition of Phosphorylation of Akt in Human Lung Squamous Cell Carcinoma. Biochim. Biophys. Acta. Mol. Cell. Res. 1864, 293–302. doi: 10.1016/j.bbamcr.2016.11.018
Bailey, M. H., Tokheim, C., Porta-Pardo, E., Sengupta, S., Bertrand, D., Weerasinghe, A., et al. (2018). Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 174, 1034–1035. doi: 10.1016/j.cell.2018.07.034
Bandalos, D. L. (1993). Factors Influencing Cross-Validation of Confirmatory Factor Analysis Models. Multivariate Behav. Res. 28, 351–374. doi: 10.1207/s15327906mbr2803_3
Benjamini, Y., and Hochberg, Y. (1995). Controlling the False Discovery Rate: a Practical and Powerful Approach to Multiple Testing. J. R. Statist. Soc. B 57, 289–300. doi: 10.1111/j.2517-6161.1995.tb02031.x
Beroukhim, R., Mermel, C. H., Porter, D., Wei, G., Raychaudhuri, S., Donovan, J., et al. (2010). The Landscape of Somatic Copy-Number Alteration across Human Cancers. Nature 463, 899–905. doi: 10.1038/nature08822
Bild, A. H., Yao, G., Chang, J. T., Wang, Q., Potti, A., Chasse, D., et al. (2006). Oncogenic Pathway Signatures in Human Cancers as a Guide to Targeted Therapies. Nature 439, 353–357. doi: 10.1038/nature04296
Campbell, J. D., Alexandrov, A., Kim, J., Wala, J., Berger, A. H., Pedamallu, C. S., et al. (2016). Distinct Patterns of Somatic Genome Alterations in Lung Adenocarcinomas and Squamous Cell Carcinomas. Nat. Gene. 48, 607–616. doi: 10.1038/ng.3564
Cancer Genome Atlas Research Network (2014). Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 511, 543–550. doi: 10.1038/nature13385
Candia, J., and Tsang, J. S. (2019). Enetxplorer: an R Package for the Quantitative Exploration of Elastic Net Families for Generalized Linear Models. BMC Bioinform. 20:189. doi: 10.1186/s12859-019-2778-5
Carrillo-Reixach, J., Torrens, L., Simon-Coma, M., Royo, L., Domingo-Sàbat, M., Abril-Fornaguera, J., et al. (2020). Epigenetic Footprint Enables Molecular Risk Stratification of Hepatoblastoma with Clinical Implications. J. Hepatol. 73, 328–341. doi: 10.1016/j.jhep.2020.03.025
Chen, J., Yang, H., Teo, A. S. M., Amer, L. B., Sherbaf, F. G., Tan, C. Q., et al. (2020). Genomic landscape of lung adenocarcinoma in East Asians. Nat. Genet. 52, 177–186. doi: 10.1038/s41588-019-0569-6
Chen, W., Liu, X., Qiao, T., and Yuan, S. (2012). Impact of Chk2-Small Interfering Rna on Cpg Odn7909-Enhanced Radiosensitivity in Lung Cancer A549 Cells. OncoTargets Ther. 5, 425–431. doi: 10.2147/ott.S38240
Chen, Y. J., Roumeliotis, T. I., Chang, Y. H., Chen, C. T., Han, C. L., Lin, M. H., et al. (2020). Proteogenomics of non-smoking lung cancer in East Asia delineates molecular signatures of pathogenesis and progression. Cell 182, 226–244.e17. doi: 10.1016/j.cell.2020.06.012
Choi, C. M., Yang, S. C., Jo, H. J., Song, S. Y., Jeon, Y. J., Jang, T. W., et al. (2012). Proteins Involved in DNA Damage Response Pathways and Survival of Stage I Non-Small-Cell Lung Cancer Patients”. Ann. Oncol. 23, 2088–2093. doi: 10.1093/annonc/mdr606
Choi, E. B., Yang, A. Y., Kim, S. C., Lee, J., Choi, J. K., Choi, C., et al. (2016). Parp1 Enhances Lung Adenocarcinoma Metastasis by Novel Mechanisms Independent of DNA Repair. Oncogene 35, 4569–4579. doi: 10.1038/onc.2016.3
Denisenko, T. V., Budkevich, I. N., and Zhivotovsky, B. (2018). Cell death-based treatment of lung adenocarcinoma. Cell Death Dis. 9:117. doi: 10.1038/s41419-017-0063-y
Ding, L., Bailey, M. H., Porta-Pardo, E., Thorsson, V., Colaprico, A., Bertrand, D., et al. (2018). Perspective on Oncogenic Processes at the End of the Beginning of Cancer Genomics. Cell 173, 305–320.e10. doi: 10.1016/j.cell.2018.03.033
Gao, R., Davis, A., McDonald, T. O., Sei, E., Shi, X., Wang, Y., et al. (2016). Punctuated Copy Number Evolution and Clonal Stasis in Triple-Negative Breast Cancer. Nat. Gene. 48, 1119–1130. doi: 10.1038/ng.3641
Garofalo, A., Sholl, L., Reardon, B., Taylor-Weiner, A., Amin-Mansour, A., Miao, D., et al. (2016). The Impact of Tumor Profiling Approaches and Genomic Data Strategies for Cancer Precision Medicine. Genome Med. 8:79. doi: 10.1186/s13073-016-0333-9
Gatza, M. L., Silva, G. O., Parker, J. S., Fan, C., and Perou, C. M. (2014). An Integrated Genomics Approach Identifies Drivers of Proliferation in Luminal-Subtype Human Breast Cancer. Nat. Gene. 46, 1051–1059. doi: 10.1038/ng.3073
Geiger, T., Cox, J., and Mann, M. (2010). Proteomic Changes Resulting from Gene Copy Number Variations in Cancer Cells. PLoS Genet. 6:e1001090. doi: 10.1371/journal.pgen.1001090
George, S. L., Izquierdo, E., Campbell, J., Koutroumanidou, E., Proszek, P., Jamal, S., et al. (2019). A Tailored Molecular Profiling Programme for Children with Cancer to Identify Clinically Actionable Genetic Alterations. Euro. J. Cancer 121, 224–235. doi: 10.1016/j.ejca.2019.07.027
Gillette, M. A., Satpathy, S., Cao, S., Dhanasekaran, S. M., Vasaikar, S. V., Krug, K., et al. (2020). Proteogenomic Characterization Reveals Therapeutic Vulnerabilities in Lung Adenocarcinoma. Cell 182, 200–225.e35. doi: 10.1016/j.cell.2020.06.013
Godek, K. M., Venere, M., Wu, Q., Mills, K. D., Hickey, W. F., Rich, J. N., et al. (2016). Chromosomal Instability Affects the Tumorigenicity of Glioblastoma Tumor-Initiating Cells. Cancer Discov. 6, 532–545. doi: 10.1158/2159-8290.Cd-15-1154
Hoadley, K. A., Yau, C., Hinoue, T., Wolf, D. M., Lazar, A. J., Drill, E., et al. (2018). Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 173, 291–304.e6. doi: 10.1016/j.cell.2018.03.022
Hofree, M., Shen, J. P., Carter, H., Gross, A., and Ideker, T. (2013). Network-Based Stratification of Tumor Mutations. Nat. Methods 10, 1108–1115. doi: 10.1038/nmeth.2651
Hou, J., Cao, X., Cheng, Y., and Wang, X. (2020). Roles of Tp53 Gene in the Development of Resistance to Pi3k Inhibitor Resistances in Crispr-Cas9-Edited Lung Adenocarcinoma Cells. Cell. Biol. Toxicol. 36, 481–492. doi: 10.1007/s10565-020-09523-7
Hua, X., Zhao, W., Pesatori, A. C., Consonni, D., Caporaso, N. E., Zhang, T., et al. (2020). Genetic and Epigenetic Intratumor Heterogeneity Impacts Prognosis of Lung Adenocarcinoma. Nat. Commun. 11:2459. doi: 10.1038/s41467-020-16295-5
Jänne, P. A., Yang, J. C., Kim, D. W., Planchard, D., Ohe, Y., Ramalingam, S. S., et al. (2015). Azd9291 in Egfr Inhibitor-Resistant Non-Small-Cell Lung Cancer. N. Engl. J. Med. 372, 1689–1699. doi: 10.1056/NEJMoa1411817
Kinde, I., Bettegowda, C., Wang, Y., Wu, J., Agrawal, N., IeShih, M., et al. (2013). Evaluation of DNA from the Papanicolaou Test to Detect Ovarian and Endometrial Cancers. Sci. Transl. Med. 5:167ra4. doi: 10.1126/scitranslmed.3004952
Kim, D., Lee, Y. S., Kim, D. H., and Bae, S. C. (2020). Lung cancer staging and associated genetic and epigenetic events. Mol. Cells 43, 1–9. doi: 10.14348/molcells.2020.2246
Kobayashi, S., Boggon, T. J., Dayaram, T., Jänne, P. A., Kocher, O., Meyerson, M., et al. (2005). Egfr mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 352, 786–792. doi: 10.1056/NEJMoa044238
Kumar, R., Raman, R., Kotapalli, V., Gowrishankar, S., Pyne, S., Pollack, J. R., et al. (2018). Ca/Nuclear Factor of Activated T Cells Signaling Is Enriched in Early-Onset Rectal Tumors Devoid of Canonical Wnt Activation”. J. Mol. Med. 96, 135–146. doi: 10.1007/s00109-017-1607-4
Liu, Y., Beyer, A., and Aebersold, R. (2016). On the Dependency of Cellular Protein Levels on Mrna Abundance”. Cell 165, 535–550. doi: 10.1016/j.cell.2016.03.014
López, S., Lim, E. L., Horswell, S., Haase, K., Huebner, A., Dietzen, M., et al. (2020). Interplay between whole-genome doubling and the accumulation of deleterious alterations in cancer evolution. Nat. Genet. 52, 283–293. doi: 10.1038/s41588-020-0584-7
Lu, Z., Kim, D. H., Fan, J., Lu, Q., Verbanac, K., Ding, L., et al. (2015). A Non-Tight Junction Function of Claudin-7-Interaction with Integrin Signaling in Suppressing Lung Cancer Cell Proliferation and Detachment. Mol. Cancer 14:120. doi: 10.1186/s12943-015-0387-0
Mermel, C. H., Schumacher, S. E., Hill, B., Meyerson, M. L., Beroukhim, R., and Getz, G. (2011). Gistic2.0 Facilitates Sensitive and Confident Localization of the Targets of Focal Somatic Copy-Number Alteration in Human Cancers. Genome Biol. 12:R41. doi: 10.1186/gb-2011-12-4-r41
Myhre, S., Lingjærde, O. C., Hennessy, B. T., Aure, M. R., Carey, M. S., Alsner, J., et al. (2013). Influence of DNA Copy Number and Mrna Levels on the Expression of Breast Cancer Related Proteins. Mol. Oncol. 7, 704–718. doi: 10.1016/j.molonc.2013.02.018
Namani, A., Zheng, Z., Wang, X. J., and Tang, X. (2019). Systematic identification of multi omics-based biomarkers in KEAP1 mutated TCGA lung adenocarcinoma. J. Cancer 10, 6813–6821. doi: 10.7150/jca.35489
Network, Cancer Genome Atlas Research. (2012). Comprehensive Genomic Characterization of Squamous Cell Lung Cancers. Nature 489, 519–525. doi: 10.1038/nature11404
Nevins, J. R., and Potti, A. (2007). Mining Gene Expression Profiles: expression Signatures as Cancer Phenotypes. Nat. Rev. Genet. 8, 601–609. doi: 10.1038/nrg2137
Ochiiwa, H., Ailiken, G., Yokoyama, M., Yamagata, K., Nagano, H., Yoshimura, C., et al. (2021). Tas4464, a Nedd8-Activating Enzyme Inhibitor, Activates Both Intrinsic and Extrinsic Apoptotic Pathways Via C-Myc-Mediated Regulation in Acute Myeloid Leukemia. Oncogene 40, 1217–1230. doi: 10.1038/s41388-020-01586-4
Padmanabhan, N., Ushijima, T., and Tan, P. (2017). How to Stomach an Epigenetic Insult: the Gastric Cancer Epigenome. Nat. Rev. Gastroenterol. Hepatol. 14, 467–478. doi: 10.1038/nrgastro.2017.53
Pommier, R. M., Sanlaville, A., Tonon, L., Kielbassa, J., Thomas, E., Ferrari, A., et al. (2020). Comprehensive Characterization of Claudin-Low Breast Tumors Reflects the Impact of the Cell-of-Origin on Cancer Evolution. Nat. Commun. 11:3431. doi: 10.1038/s41467-020-17249-7
Qiu, M., Xia, W., Chen, R., Wang, S., Xu, Y., Ma, Z., et al. (2018). The Circular Rna Circprkci Promotes Tumor Growth in Lung Adenocarcinoma. Cancer Res. 78, 2839–2851. doi: 10.1158/0008-5472.Can-17-2808
Siegel, R. L., Miller, K. D., and Jemal, A. (2019). Cancer Statistics, 2019. CA Cancer J. Clin. 69, 7–34. doi: 10.3322/caac.21551
Staaf, J., Isaksson, S., Karlsson, A., Jönsson, M., Johansson, L., Jönsson, P., et al. (2013). Landscape of somatic allelic imbalances and copy number alterations in human lung carcinoma. Int. J. Cancer 132, 2020–2031. doi: 10.1002/ijc.27879
Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., et al. (2005). Gene Set Enrichment Analysis: a Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. U. S. A. 102, 15545–15550. doi: 10.1073/pnas.0506580102
Tanioka, M., Fan, C., Parker, J. S., Hoadley, K. A., Hu, Z., Li, Y., et al. (2018). Integrated Analysis of Rna and DNA from the Phase III Trial Calgb 40601 Identifies Predictors of Response to Trastuzumab-Based Neoadjuvant Chemotherapy in Her2-Positive Breast Cancer. Clin. Cancer Res. 24, 5292–5304. doi: 10.1158/1078-0432.Ccr-17-3431
Tripathi, R., Liu, Z., Jain, A., Lyon, A., Meeks, C., Richards, D., et al. (2020). Combating Acquired Resistance to Mapk Inhibitors in Melanoma by Targeting Abl1/2-Mediated Reactivation of Mek/Erk/Myc Signaling. Nat. Commun. 11:5463. doi: 10.1038/s41467-020-19075-3
Tsimberidou, A. M., Fountzilas, E., Bleris, L., and Kurzrock, R. (2020). Transcriptomics and Solid Tumors: the Next Frontier in Precision Cancer Medicine. Semin. Cancer Biol. S1044-579X, 30196-6. doi: 10.1016/j.semcancer.2020.09.007
Uzilov, A. V., Ding, W., Fink, M. Y., Antipin, Y., Brohl, A. S., Davis, C., et al. (2016). Development and Clinical Application of an Integrative Genomic Approach to Personalized Cancer Therapy. Genome Med. 8:62.
Valle, B. L., Rodriguez-Torres, S., Kuhn, E., Díaz-Montes, T., Parrilla-Castellar, E., Lawson, F. P., et al. (2020). Hist1h2bb and Methylation and Somatic Mutations as Precision Medicine Biomarkers for Diagnosis and Prognosis of High-Grade Serous Ovarian Cancer. Cancer Prev. Res. 13, 783–794. doi: 10.1158/1940-6207.Capr-19-0412
Wang, F., Zhao, N., Gao, G., Deng, H. B., Wang, Z. H., Deng, L. L., et al. (2020). Prognostic Value of Tp53 Co-Mutation Status Combined with Egfr Mutation in Patients with Lung Adenocarcinoma. J. Cancer Res. Clin. Oncol. 146, 2851–2859. doi: 10.1007/s00432-020-03340-5
Wang, L., Luo, X., Cheng, C., Amos, C. I, Cai, G., and Xiao, F. (2020). A gene expression-based immune signature for lung adenocarcinoma prognosis. Cancer Immunol. Immunother. 69, 1881–1890. doi: 10.1007/s00262-020-02595-8
Watermann, C., Pasternack, H., Idel, C., Ribbat-Idel, J., Brägelmann, J., Kuppler, P., et al. (2020). Recurrent Hnscc Harbor an Immunosuppressive Tumor Immune Microenvironment Suggesting Successful Tumor Immune Evasion. Clin. Cancer Res. 27:0197. doi: 10.1158/1078-0432.Ccr-20-0197
Wilkerson, M. D., Yin, X., Walter, V., Zhao, N., Cabanski, C. R., Hayward, M. C., et al. (2012). Differential Pathogenesis of Lung Adenocarcinoma Subtypes Involving Sequence Mutations, Copy Number, Chromosomal Instability, and Methylation. PLoS One 7:e36530. doi: 10.1371/journal.pone.0036530
Xia, Y., Fan, C., Hoadley, K. A., Parker, J. S., and Perou, C. M. (2019). Genetic determinants of the molecular portraits of epithelial cancers. Nat. Commun. 10:5666. doi: 10.1038/s41467-019-13588-2
Yang, Y., Ahn, Y. H., Chen, Y., Tan, X., Guo, L., Gibbons, D. L., et al. (2014). Zeb1 Sensitizes Lung Adenocarcinoma to Metastasis Suppression by Pi3k Antagonism. J. Clin. Investig. 124, 2696–2708. doi: 10.1172/jci72171
Zack, T. I., Schumacher, S. E., Carter, S. L., Cherniack, A. D., Saksena, G., Tabak, B., et al. (2013). Pan-Cancer Patterns of Somatic Copy Number Alteration. Nat. Genet. 45, 1134–1140. doi: 10.1038/ng.2760
Zengin, T., and Önal-Süzek, T. (2020). Analysis of Genomic and Transcriptomic Variations as Prognostic Signature for Lung Adenocarcinoma. BMC Bioinform. 21:368. doi: 10.1186/s12859-020-03691-3
Zhu, X., Chen, L., Liu, L., and Niu, X. (2019). Emt-mediated acquired Egfr-Tki resistance in Nsclc: mechanisms and strategies. Front. Oncol. 9:1044. doi: 10.3389/fonc.2019.01044
Keywords: Elastic Net, DNA copy number, lung adenocarcinoma, gene expression signature, predictive model
Citation: Xiang Y, Zou X, Shi H, Xu X, Wu C, Zhong W, Wang J, Zhou W, Zeng X, He M, Wang Y, Huang L and Wang X (2021) Elastic Net Models Based on DNA Copy Number Variations Predicts Clinical Features, Expression Signatures, and Mutations in Lung Adenocarcinoma. Front. Genet. 12:668040. doi: 10.3389/fgene.2021.668040
Received: 16 February 2021; Accepted: 26 April 2021;
Published: 31 May 2021.
Edited by:
Yun Xiao, Harbin Medical University, ChinaReviewed by:
Qiuyan Guo, First Affiliated Hospital of Harbin Medical University, ChinaCopyright © 2021 Xiang, Zou, Shi, Xu, Wu, Zhong, Wang, Zhou, Zeng, He, Wang, Huang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Huang, aGxlbGxlbkBnbXUuZWR1LmNu; Xiangcai Wang, d2FuZ3hpYW5nY2FpQGNzY28uYWMuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.