 Jerzy K. Kulski
Jerzy K. Kulski Shingo Suzuki
Shingo Suzuki Takashi Shiina
Takashi Shiina
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 28 May 2021
Sec. Human and Medical Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.665899
This article is part of the Research Topic Population Genomic Architecture: Conserved Polymorphic Sequences (CPSs), Not Linkage Disequilibrium View all 9 articles
The major histocompatibility complex (MHC) on chromosome 6p21 is one of the most single-nucleotide polymorphism (SNP)-dense regions of the human genome and a prime model for the study and understanding of conserved sequence polymorphisms and structural diversity of ancestral haplotypes/conserved extended haplotypes. This study aimed to follow up on a previous analysis of the MHC class I region by using the same set of 95 MHC haplotype sequences downloaded from a publicly available BioProject database at the National Center for Biotechnology Information to identify and characterize the polymorphic human leukocyte antigen (HLA)-class II genes, the MTCO3P1 pseudogene alleles, the indels of transposable elements as haplotypic lineage markers, and SNP-density crossover (XO) loci at haplotype junctions in DNA sequence alignments of different haplotypes across the extended class II region (∼1 Mb) from the telomeric PRRT1 gene in class III to the COL11A2 gene at the centromeric end of class II. We identified 42 haplotypic indels (20 Alu, 7 SVA, 13 LTR or MERs, and 2 indels composed of a mosaic of different transposable elements) linked to particular HLA-class II alleles. Comparative sequence analyses of 136 haplotype pairs revealed 98 unique XO sites between SNP-poor and SNP-rich genomic segments with considerable haplotype shuffling located in the proximity of putative recombination hotspots. The majority of XO sites occurred across various regions including in the vicinity of MTCO3P1 between HLA-DQB1 and HLA-DQB3, between HLA-DQB2 and HLA-DOB, between DOB and TAP2, and between HLA-DOA and HLA-DPA1, where most XOs were within a HERVK22 sequence. We also determined the genomic positions of the PRDM9-recombination suppression sequence motif ATCCATG/CATGGAT and the PRDM9 recombination activation partial binding motif CCTCCCCT/AGGGGAG in the class II region of the human reference genome (NC_ 000006) relative to published meiotic recombination positions. Both the recombination and anti-recombination PRDM9 binding motifs were widely distributed throughout the class II genomic regions with 50% or more found within repeat elements; the anti-recombination motifs were found mostly in L1 fragmented repeats. This study shows substantial haplotype shuffling between different polymorphic blocks and confirms the presence of numerous putative ancestral recombination sites across the class II region between various HLA class II genes.
Haplotypes are combinations of alleles at different loci of phased DNA segregating together in multigenerational families (Bodmer et al., 1986; Lloyd et al., 2016; Alper and Larsen, 2017) essentially as DNA sequences that are identical by descent (IBD) via recent shared ancestry (Druet and Farnir, 2011; Browning and Browning, 2012; Thompson, 2013; Zhou et al., 2020b). The word haplotype (single, from haploid) was first introduced by Ruggero Ceppellini in 1966/67 to describe immunoglobin allotypes as corresponding “to the product of a single gene dose” and was appropriated almost immediately by immunogeneticists to describe the linked alleles in the highly polymorphic, multilocus human major histocompatibility complex (MHC) super locus on chromosome 6 (Bodmer, 2019) that consists of three distinct genomic regions, classes I, II and III with clusters of human leukocyte antigen (HLA) genes involved in the regulation of the innate and adaptive immune system, autoimmunity, and transplantation (Shiina et al., 2004, 2009; Vandiedonck and Knight, 2009; Trowsdale, 2011). During the past 30 years, the study of human MHC population haplotypes for transplantation and disease has developed into a formidable field of segregated haplotype blocks analyzed by congruence (Baschal et al., 2012) and conserved polymorphic sequences (CPSs) of ancestral haplotypes (AH), and conserved extended haplotypes (CEHs) (Awdeh et al., 1983; Dawkins et al., 1983, 1999; Contu et al., 1989; Degli-Esposti et al., 1992; Yunis et al., 2003; Alper et al., 2006; Aly et al., 2006; Bilbao et al., 2006; Smith et al., 2006; Lam et al., 2013; Gambino et al., 2018). After the transition into the third millennium and the publication of the analysis of the first human genomic sequence (International Human Genome Sequencing Consortium, 2001; Venter et al., 2001), haplotype studies began to spread in earnest from the continuous analysis of the MHC super locus (Jeffreys et al., 2001; Ahmad et al., 2003; Kauppi et al., 2003; Miretti et al., 2005; Blomhoff et al., 2006) to other regions of the human genome (Daly et al., 2001; Gabriel et al., 2002; Jeffreys et al., 2004; Kauppi et al., 2004; Conrad et al., 2006; The International HapMap Consortium, 2007; Baschal et al., 2012; Browning and Browning, 2020; Nait Saada et al., 2020) and across to other species (Guryev et al., 2006; Kauppi et al., 2007; Villa-Angulo et al., 2009; Ando et al., 2019; Lan et al., 2019). Genomic haplotype blocks are now more commonly described in terms of haplotype estimations using the less structurally precise population linkage disequilibrium (LD) statistics and inferred LD-allelic block analyses (Al Bkhetan et al., 2019; Park, 2019) instead of the more accurately deduced pedigree-defined segments/blocks (Alper and Larsen, 2017). The LD-phased DNA sequences are useful but can generate false information that might be misleading in disease association studies (Slatkin, 2008; Tewhey et al., 2011; Alper and Larsen, 2017; Choi et al., 2018; Al Bkhetan et al., 2019). IBD segmental mapping of recent ancestry between individuals in families and populations based on sequence similarity, genotypes, and single-nucleotide polymorphism (SNP) profiles is a newly developed and tested imputation used either with or without LD analysis for inferred haplotype detection (Browning and Browning, 2012; Thompson, 2013; Zhou et al., 2020b).
Major histocompatibility complex disease association studies are most commonly performed at the level of correlations with genotypes, alleles, SNPs (Trowsdale, 2011; Ferreira et al., 2012; Kennedy et al., 2017), microsatellites (Oka et al., 2003; Tamiya et al., 2005; Charfi et al., 2020), and retrotransposon insertion polymorphisms (Dunn et al., 2006; Wang et al., 2017). However, the customary genome-wide association studies (GWAS) of the MHC genomic region are limited severely by the biological complexity of the diseases under investigation and the statistical unreliability and substantial irreproducibility of many analyses that should be examined by also using haplotype genomic structure and haplotypic disease markers (Dawkins et al., 1983; Alper and Larsen, 2017; Kennedy et al., 2017; Lokki and Paakkanen, 2019) including for autoimmune disorders such as systemic sclerosis (Arnett et al., 2010), Graves’ disease (Macel et al., 2001), selective immunoglobulin A deficiency (Ferreira et al., 2012), Parkinson disease (Wissemann et al., 2013), type 1 diabetes (T1D), and celiac disease (Farina et al., 2019).
The main barrier to expanding large-scale haplotype studies at the genomic sequence level in different worldwide populations has been the difficulty of accurately and reliably obtaining long stretches of phased DNA within the MHC and other genomic regions to perform comparative haplomics (O’Neill, 2009). Although next-generation sequencing methods can generate phased DNA and haplotypes (Huang et al., 2017; Choi et al., 2018; Suzuki et al., 2018), much of this is still experimental and relatively too expensive and complicated for most research laboratories to incorporate easily into their current sequencing and genotyping protocols and analytical pipelines. The use of homozygous cell lines is one approach to overcoming the uncertainty of using diploid DNA and the current technical problems of generating phased DNA (Dorak et al., 2006; Horton et al., 2008; Norman et al., 2017). These phased MHC genomic sequences provide representative haplotype panels for better informed large population studies, mapping heterozygous sequence reads (Traherne, 2008; Lam et al., 2013, 2015) and disease associations (Alper and Larsen, 2017; Lokki and Paakkanen, 2019). Although Norman et al. (2017) produced an important database for 95 MHC homozygous cell lines of assembled MHC genomic sequences, their own DNA sequence analyses were limited to describing the multilocus alleles and haplotypes of the HLA classical class I and class II genes, MUC22 and the structural diversity of C4 duplications.
Major histocompatibility complex haplotype diversity is driven largely by segmental shuffling and meiotic recombination (Traherne et al., 2006), and this exchange between genomic segments or blocks can be identified by high and low SNP-density XOs at the junctions of different haplotypic blocks (Larsen et al., 2014; Kulski et al., 2021). The analysis of haplotype segmental exchange provides an important insight into IBD due to recent common ancestry for at least 3,400 generations (Traherne et al., 2006), the evolutionary history of ancestral recombinations, and the mechanisms that are involved in generating IBD segment, haplotype, and SNP diversity (Zhou et al., 2020a,b). Therefore, SNP-density XOs between neighboring haplotype blocks are a potential qualitative and quantitative measure of segmental exchanges in the MHC (Traherne et al., 2006; Lam et al., 2013, 2015; Larsen et al., 2014; Kulski et al., 2021) as well as for inferred IBD segments in at least 11 other regions of the human genome (Browning and Browning, 2020; Nait Saada et al., 2020).
Many repeat elements and transposable elements (TEs) that make up > 50% of the human DNA content have contributed to various diseases (Ayarpadikannan and Kim, 2014; Payer et al., 2017; Payer and Burns, 2019), gene regulation and recombination (Moolhuijzen et al., 2010; Myers et al., 2010; Altemose et al., 2017; Chuong et al., 2017) as well as to the duplicated segmental organization of the human and other primate MHC genomic structures (Kulski et al., 1997, 1999a,b, 2000a,b, 2004; Anzai et al., 2003). Because of their mobility, hypermutability, and potential participation in recombination, TEs are integral to molecular drive (Dover, 1993) and together with point mutations, gene conversion (Adamek et al., 2015), and balancing selection (van Oosterhout, 2009), have contributed to generating haplotypic polymorphisms in the MHC class I and class II regions (Andersson et al., 1998; Shi et al., 2014; Kulski et al., 2021). The role of TEs in recombination events is evidenced in part by the structural biallelic Alus, short interspersed nuclear element–VNTR–Alus (SVAs), long terminal repeats (LTRs), and human endogenous retroviruses (HERVs) located either near or within putative recombination hotspots throughout the human genome (Katzourakis et al., 2007; Konkel and Batzer, 2010; Burns and Boeke, 2012; Ayarpadikannan and Kim, 2014; Thomas et al., 2018; Wallace et al., 2018) and the MHC class I and class II genomic regions (Kulski et al., 2011, 2021). In a study of expression quantitative trait loci within the genomic sequences of lymphoblastoid cell lines, Spirito et al. (2019) found that the chromosomal location 6p21.32, which includes the extended MHC class II region from TNXB to DAXX, was one of the two most enriched genomic regions where structurally polymorphic TEs influenced gene expression.
As part of our previous studies on the importance of TEs as evolutionary and haplotypic markers both in population and comparative sequence analyses, we reported on their role in haplotype shuffling and their linkages to HLA class I alleles in the MHC class I region (Kulski et al., 2021). In this study, we have extended our analysis of haplotype shuffling and the linkages between TE and HLA gene alleles within the MHC class II region of the Norman et al. (2017) sequences to identify and characterize (1) the particular haplotypic linkages between the HLA class II genic and intergenic structurally polymorphic TEs and (2) ancestral SNP-density XO loci in DNA sequence alignments of different haplotype blocks or segments across the ∼1 Mb-extended MHC class II genomic region from the telomeric PRRT1 gene to the centromeric COL11A2 gene. We identified a variety of structural bi-allelic TEs that may be useful as lineage markers and confirmed the presence of numerous regions of haplotype exchanges between low and high SNP density XOs at putative ancestral recombination sites that are consistent with and extend the observations of other investigators who have mapped recombination hotspots in the HLA class II region.
We identified 41 structural bi-allelic TE haplotypic markers and confirmed the presence of numerous regions of haplotype exchanges between low and high SNP density XOs at putative ancestral recombination sites that are widely distributed across the ∼1 Mb-extended MHC class II genomic region from the telomeric PRRT1 gene to the centromeric COL11A2 gene.
The main sequences and methods used in this study were previously described by Kulski et al. (2021). Essentially, the haplotype data of 95 MHC genomic sequences sequenced and assembled from HLA-homozygous cell lines by Norman et al. (2017) at the National Center for Biotechnology Information (NCBI) BioProject with the accession number PRJEB67631 were downloaded as Fasta files and used for the analyses described later. The other MHC genomic sequences used in haplotype analyses were the GRChr38.p13 (GCF_000001405.39) of the chromosome 6 reference NC_ 000006.12 at the NCBI2, Ensembl3, University of California, Santa Cruz4 browsers and databases, eight human reference haplotypes described by Horton et al. (2008), one chimpanzee sequence of the MTCO3P1 pseudogene (AC275796.1), four gorilla MTCO3P1 sequences (AC270181.1, CT025711.1, CT025621.2, AC270177.1), and one orangutan MTCO3P1 sequence (AC206450.4). All of the Fasta sequences downloaded from the public archives were submitted to the RepeatMasker webserver5 for output files of annotated members of the interspersed repetitive DNA families, their locations in the sequence, and their relative similarity or identity in comparison with reference sequences of short interspersed retrotransposable elements (SINEs), long interspersed retrotransposable elements (LINEs), LTRs, HERVs, DNA elements, small RNA, and simple repeats using the Dfam database (3.0) for the repeat sequence comparisons (Hubley et al., 2016).
Norman et al. (2017) provided the alleles for HLA-DRB1, -DRB2, -DRB3, -DRB4, and -DRB5 -DQA1, -DQB1, -DPA1, and -DPB1 for all the 95 cell line sequences shown in Supplementary Table 1. We extracted the sequences and assigned the haplotyped alleles to another seven loci, HLA-DQA2, -DQB2, -DOB, -DOA, -DPB2, -DPA3, and the 660-bp pseudogene MTCO3P1 in 90 of the 95 cell line sequences by comparing them to the HLA allele sequences in IPD-IMGT/HLA (6 Release 3.42.0) and those in GenBank7 using the DNA sequence assembly software Sequencher ver.5.0 (Gencode8). The new HLA class II alleles are reported here without providing any further information about the novel nucleotide or amino acid differences (Supplementary Table 2). A laboratory identifier number (ID_1 to ID_95) was added to each of the Norman et al. (2017) sequences (Supplementary Tables 1, 2) for ease of identification in comparative sequence analysis. The TE dimorphisms (absence or presence) were easily recognized in each of the RepeatMasker outputs because of their periodic positions within or close proximity of other TE elements and short tandem repeats (STRs). Comparative sequence alignments between two or more sequences to evaluate SNP densities and determine SNP-density XO regions between SNP-poor regions of < 10 SNPs per 100 kb and SNP-rich regions of > 50 SNPs per 100 kb were performed with the web-based MultiPipMaker alignment program9 by uploading the Fasta sequence files, a RepeatMasker output file and using the MultiPipMaker setting for single coverage as described by Schwartz et al. (2000) to generate the optimal sequence alignment. SNPs in the alignments were counted twice manually and averaged. Obvious assembly errors, polynucleotides, simple microsatellite repeats, and indels were not counted as SNPs. Also, a series of many adjoining SNPs (e.g., > 5 SNPs in a string of 50 nucleotides) or SNPs within 50 bp of obvious sequencing errors with runs of unspecified nucleotides (Ns) and/or inconsistent long strings of deletions were not counted. The length of sequence alignments usually ranged between 50 and 500 kb depending on (1) the segments targeted for the analysis and the ease of SNP manual counting in the pdf outputs of the nucleotide alignments and/or (2) the length of the percentage identity plot output for reproduction as a convenient and readable image. The targeted sequences were selected and trimmed from the Fasta files previously downloaded from the NCBI BioProject, accession number PRJEB6763. The software program Genetyx ver.20 (GENETYX Co., Tokyo, Japan) was used with the Selector function set to select and trim to obtain the required Fasta file sequences with the genomic sequence target positions guided by those listed in the RepeatMasker output text file. SNP-density plots of selected haplotype sequence alignments were drawn using Microsoft Excel for Mac 2019 from inputs of sequence alignments created by the online MultiPipMaker.
The “find” option of the Preview v11 software (Apple Inc.) was used to search for the PRDM9 binding motifs CCTCCCCT/AGGGGAGG and ATCCATG/CATGGAT in MultiPipMaker pdf outputs of the centromeric end of MHC class III and the entire MHC class II region to the COL11A2 gene (Figure 1) in the trimmed human genomic reference sequence GRChr38.p13 (NC_000006.12). No text wrapping or different formats or layouts of the same sequence were applied in the search.
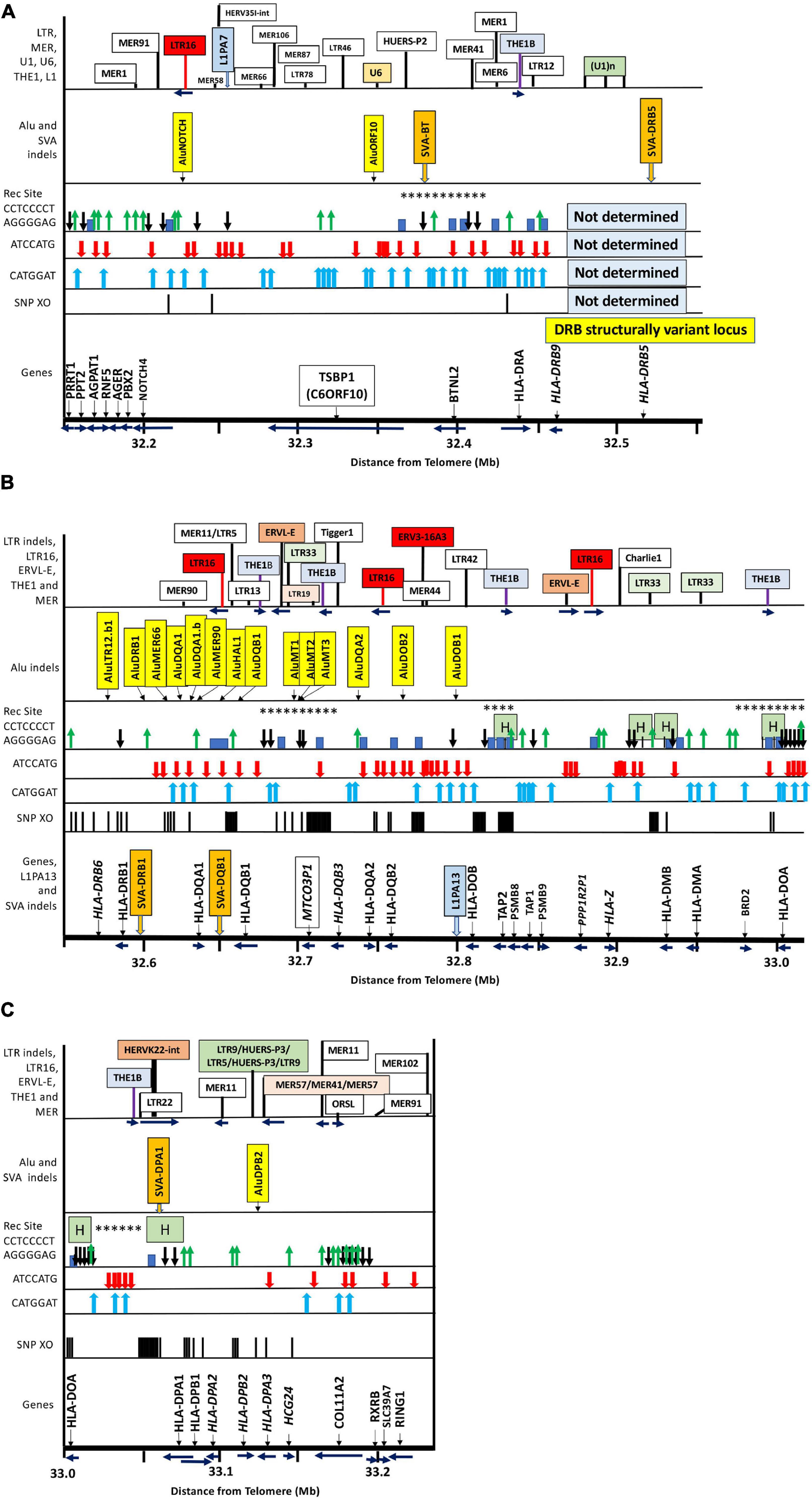
Figure 1. Locations of gene markers (pseudogenes in italics), SNP-density crossover points (haplotype shuffling), recombination sites (Rec Site), PRDM9 partial binding and suppression motifs, Alu and SVA indels, and particular repeat elements used as location tags within ∼1 Mb of MHC class III/class II genomic sequence from PRRT1 to RING1 and the nucleotide position 32.150 to 33.22 Mb distance from telomere on chromosome 6 (sequence NC_000006 at NCBI, UCSC, ENSEMBL): (A) The MHC class III/II boundary from 32.15 to 32.55 Mb including the Class III genes PRRT1 to BTNL2 and the Class II genes, HLA-DRA and HLA-DRB9 and HLA-DRB5– within the DRB structural variant locus; (B) the MHC class II region from 32.55 to 33.01 Mb with the location of the duplicated HLA class II genes from HLA-DRB6 to HLA-DOA; (C) the MHC class II region from 33 to 33.22 Mb with the location of the duplicated HLA class II genes from HLA-DOA to HLA-DPA3 with the extended centromeric region containing the COL11A2, RXB, SLC3A7 and RING1 genes. Each Figure A to C contains labeled boxes showing the following comparative items: Genomic position of SNP-density crossover points (SNP XO) indicated by vertical black lines. The genomic position of the PRDM9-suppression sequence motifs ATCCATG and CATGGAT indicated by red and blue vertical arrows, respectively. The ‘Rec Site’ boxes represent the putative regions of ancestral meiotic recombinations and gene conversions as indicated by the PRDM9 partial binding motif CCTCCCCT (black vertical arrow with head down) and its complimentary sequence AGGGGAG (green vertical arrow with head up). The blue blocks are putative recombination sites identified by Lam et al. (2013). The H green boxes are ‘hotspots’ identified by Jeffreys et al. (2001), Kauppi et al. (2005), Kong et al. (2010) and Pratto et al. (2014), and highlighted in the NCBI browser (Table 7). The asterix (∗) are the meiotic recombination positions identified in sperm studies by Cullen et al. (1997, 2002). The ‘Alu indels’ boxes show the location of the dimorphic Alu listed in Table 3, and the dimorphic SVA are shown in the ‘Alu indels’ boxes for (A) and (C) and in the ‘Genes’ box for (B). The top boxes of ‘LTR indels’ show the genomic position of selected TE as location tags for orientation and because some of them such as MER1 and MER11 harbor PRDM9 motifs or because some such as LTR16, LTR19, LTR33 and THE sequences have a possible role in recombination initiation and/or suppression.
The T-Coffee multiple sequence alignment tool (Notredame et al., 2000) at EMBL-EBI10 was used to submit multiple sequences of MTCO3P1 in the Fasta format (Supplementary Table 3) with an input maximum file size of 1 Mb resulting in the following outputs of an alignment file in the CLUSTALW (1.83), a simple guide tree or phylogram (dnd format), a neighbor-joining phylogenetic tree without distance corrections (ph format), and a percentage identity matrix (pim format) created by Clustal2.1.
Figure 1 shows a summary map of the locations of MHC class II gene markers (Genes), SNP-density XO sites, published recombination sites (Rec Site), ATCCATG and CATGGAT PRDM9 recognition motifs, and dimorphic TE that we identified and analyzed in the extended MHC class II region from the telomeric PRRT1 gene in the class III region to the centromeric COL11A2 gene in the class II region on the short arm of chromosome 6, GRCh38.p12 Primary Assembly NC_000006.12; NCBI, UCSC, or ENSEMBL browsers on the Web. The PRDM9 recognition motifs are only for the genomic reference sequence NC_000006.12, and the variations between haplotypes are not shown. Table 1 shows a summary of the types of TE repeats identified by RepeatMasker in the MHC class III genomic region from PRRT1 to DRB5 (400 kb) and the MHC class II region from DRB1 to the COL11A2 gene (650 kb). Overall, there were ∼1206 TE within 1.050 Mb of a genomic sequence, 474 SINES (353 Alus, 121 MIRs), 383 LINES (260 L1, 110 L2, and 13 L3/CR1), 184 LTR elements, 148 DNA elements, and 17 unclassified elements at 51.3% of the 1,050-kb genomic content. Of the % content of the different family types of TEs, there are relatively fewer SINEs and LINEs and more HERVs and DNA elements in the MHC class II than the class III region. Most of these TEs are inherited from the hominoids (great apes) and fixed in the extended MHC class II region of humans.
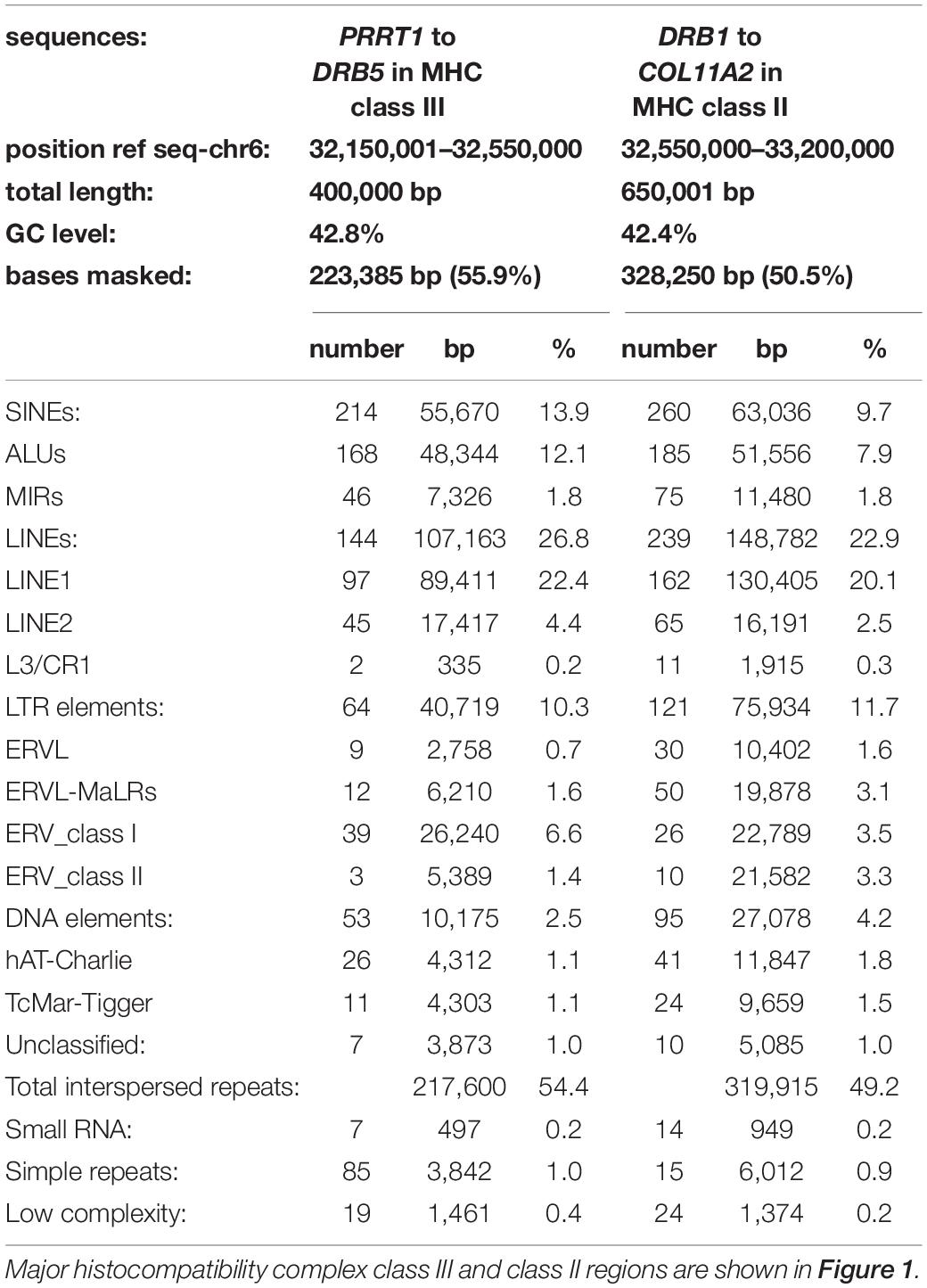
Table 1. Summary of TE repeat sequence families in the extended MHC class II region.
Supplementary Table 1 shows a comparative analysis of HLA class II gene alleles at 14 loci, including the pseudogene MTCO3P1 from HLA-DR to HLA-DP and haplotype (allele) shuffling between genomic sequences (Hap ID) obtained from 95 homozygous cell lines (Norman et al., 2017). There are two to eight identical long-range haplotypes from DR to DOA loci that share the same combinations of alleles. For example, there are eight cell lines with the 8-locus haplotype DRB1∗04/DQA1∗03/DQB1∗03/MTCO3P1∗08/DQA2∗01/DQB2∗ 01/DOB∗01/DOA∗01. Most of the other haplotypes have an obvious transition between gene alleles at least at one of the 10 loci between HLA-DRB1 and HLA-DPA3. Table 2 shows 26 distinct DRB1/DQA1/DQB1/MTCO3P1/DQA2/DQB2 haplotype lineages in 87 of the Norman et al. (2017) sequences. The two most common haplotypes were 10 DRB1∗03/DQA1∗05/DQB1∗02/MTCO3P1∗01/DQA2∗01/DQB2 ∗01 and eight DRB1∗04/DQA1∗03/DQB1∗03/MTCO3P1∗08/DQA2∗01/DQB2∗01. Various allelic transitions have occurred in a location between the HLA-DQB1 and MTCO3P1 loci. For example, there are four different haplotype linkages between the HLA-DQB1 alleles and MTCO3P1∗03 and seven between HLA-DRB1 and MTCO3P1∗03.
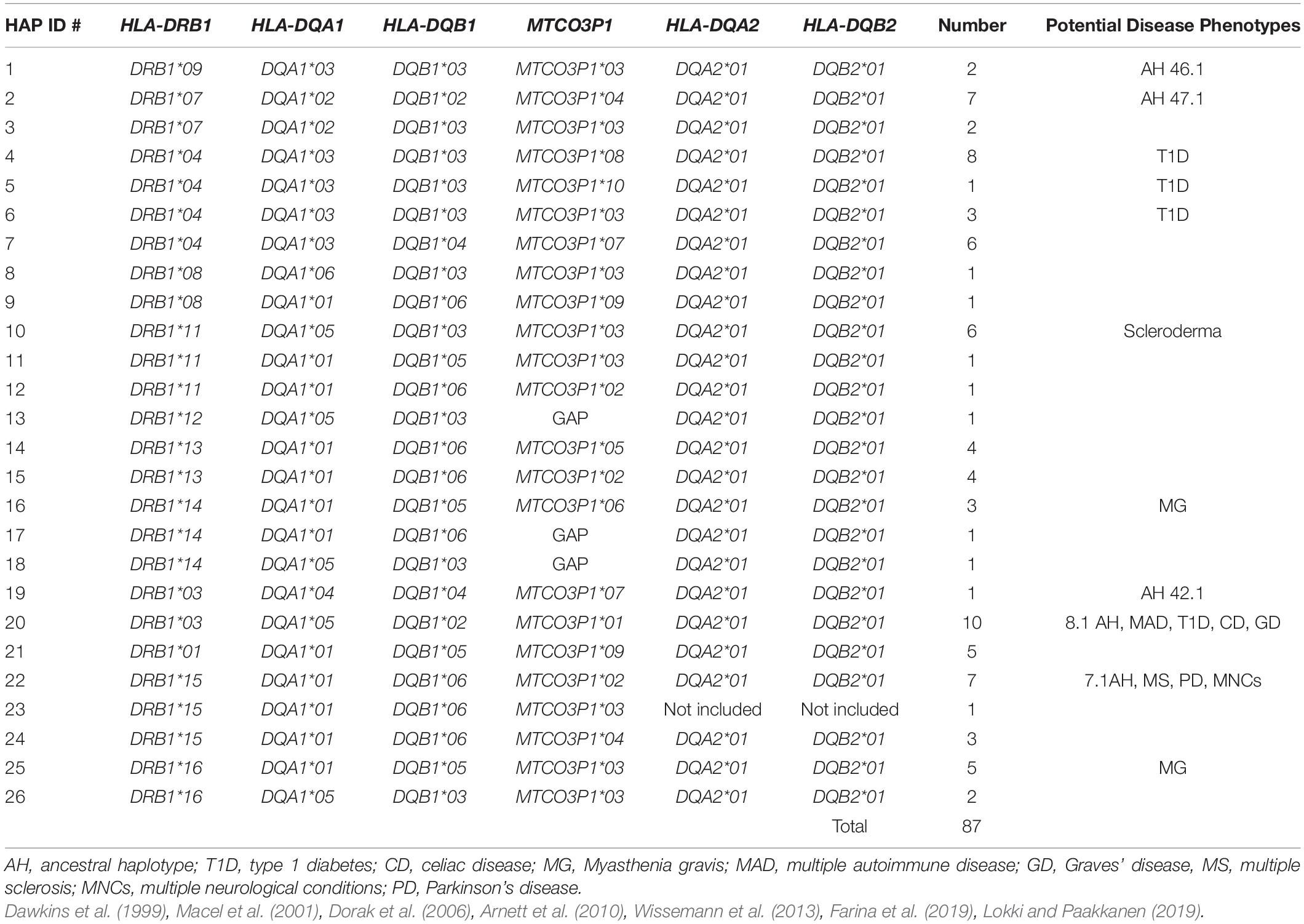
Table 2. Twenty-six distinct DRB1-DQA1-DQB1-MTCO3P1-DQA2-DQB2 haplotype lineages in 87 of the Norman et al. (2017) sequences.
The alleles for the 660-bp MTCO3P1 pseudogene were determined in 84 of the Norman et al. (2017) sequences because there are at least 19 SNP-density XOs near its locus (Figure 1). Ten alleles (Supplementary Table 3) were determined for 84 MTCO3P1 sequences, and these were all haplotypic (Supplementary Table 2). MTCO3P1 haplospecificities were observed between MTCO3P1∗01 and DRB1∗03/DQA1∗05/DQB1∗02; MTCO3P1∗06 and DRB1∗14/DQA1∗01:04/DQB1∗05:03; and MTCO3P1∗09 and DRB1∗01/DQA1∗01/DQB1∗05. The haplotype DRB1∗13/DQA1∗01/DQB1∗06 was linked with either MTCO3P1∗02 or MTCO3P1∗05. On the other hand, MTCO3P1∗03 in 22 sequences was linked with DRB1∗04, DRB1∗07, DRB1∗08, DRB1∗09, DRB1∗11, DRB1∗15, and DRB1∗16. The linkage of MTCO3P1∗03 with seven different HLA-DRB1 allelic lineages suggests that this particular MTCO3P1 allele was transmitted via many ancestral recombination events via haplotype shuffling and is probably the oldest of the 10 alleles. Some of the other MTCO3P1 alleles were also linked with multiple HLA-DRB1 alleles: MTCO3P1∗02 with DRB1∗13 and DRB1∗15; MTCO3P1∗04 with DRB1∗07 and DRB1∗15, MTCO3P1∗07 with DRB1∗03, DRB1∗04 and DRB1∗08; and MTCO3P1∗09 with DRB1∗01 and DRB1∗08 (Table 2).
Figure 2 shows a phylogenetic tree of the 10 human MTCO3P1 allele sequences aligned and compared with the MTCO3P1 sequences of a chimpanzee, four gorillas, and an orangutan. Two human MTCO3P1 sequence clusters separated from the ape sequences, suggesting that the MTCO3P1 alleles are human-specific. One cluster of four human MTCO3P1 alleles 01, 02, 05, and 06 that are mostly associated with the DRB2 (DR52 haplotype) lineage separated from the MTCO3P1∗04 allele branch that was linked with the DRB6/DRB7/DRB8 lineages. This separation suggests that the DRB2 (DR52 haplotype) lineage had branched from the DRB6 (DR1/DR51 haplotype) and DRB7/DRB8 (DR53 haplotype) lineages as previously proposed by Andersson (1998). The other cluster of five human MTCO3P1 alleles has MTCO3P1∗09 linked with the DRB6 (DR1 haplotype) and the HLA-DRB1∗08 lineage; MTCO3P1∗08 and MTCO3P1∗10 linked to the DRB7/DRB8 (DR53 haplotype) lineage; and MTCO3P1∗03 and MTCO3P1∗07 linked with various other DR loci. This suggests that numerous recombination events had occurred after the proposed initial evolutionary separation between the DR53 haplotype (DRB7/DRB8) and haplotype lineages of DR52 (DRB2), DR51 (DRB5 and DRB6), DR1 (DRB6), and DR8 (DRB1∗08 allelic lineage) more than 65 million years ago (Andersson, 1998; Andersson et al., 1998).

Figure 2. Phylogenetic tree of the 10 human MTCO3P1 allele sequences that were aligned by clustalW and compared to the MTCO3P1 sequences of a chimpanzee, four gorillas, and an orangutan.
Eighty-nine of the 95 human MHC haplotypes were examined for the presence of dimorphic TE represented by Alus, SVAs, LTRs, and HERVs in RepeatMasker outputs of the interspersed repetitive DNA families; their locations in the sequence and their relative similarity were compared with reference sequences of SINEs, LINEs, LTRs, HERVs, DNA elements, small RNA, and simple repeats. The five class II polymorphic Alu insertions, AluORF10, AluDRB1, AluDQA1, AluDQA2, and AluDPB2, were easily identified within the RepeatMasker outputs on the basis of their location and flanking sequences as previously described (Kulski et al., 2010). Of the 44 TE indels (absent or present) examined in the present analysis (Table 3), two were monomorphic (SVA-BT, LTR16) and appear to have been fixed in the human genome at least before the divergence of humans and chimpanzees (data not shown). One of the indels, TE-ID#44 that is located in the region between HLA-DQB1 and MTCO3P1, is a 7-kb mosaic composed of at least 10 other TE family members, including LTR9, AluSx, HUERS-P3, MER51, MER58, MER61, MER63, and LTR33 (Table 4). The MLT1E, MSTC, and AluSx TE sequences within the 7-kb insertion were deleted separately from some of the other ID #44 insertion sequences. Moreover, the 7-kb insertion (with the LTR9 and LTR33 sequences) was linked haplotypically to all 22 MTCO3P1∗03, six HLA-DQB1∗0502, 15 of 16 HLA-DQB1∗03:01, and one of five HLA-DQB1∗06 sequences (Supplementary Table 4). Although any combination of these particular TEs could be used as genetic markers for these haplotypes, caution is required in constructing primers and probes because the MLT1E, MSTC, and AluSx TE sequences are duplicated in a region between HLA-DQB2 and HLA-DOB (Table 4), and a relatively full-length LTR9-AluSx-HUERS-P3 sequence is inserted between exons 2 and 3 within the HLA-DPB2 pseudogene sequence (Supplementary Figure 1).
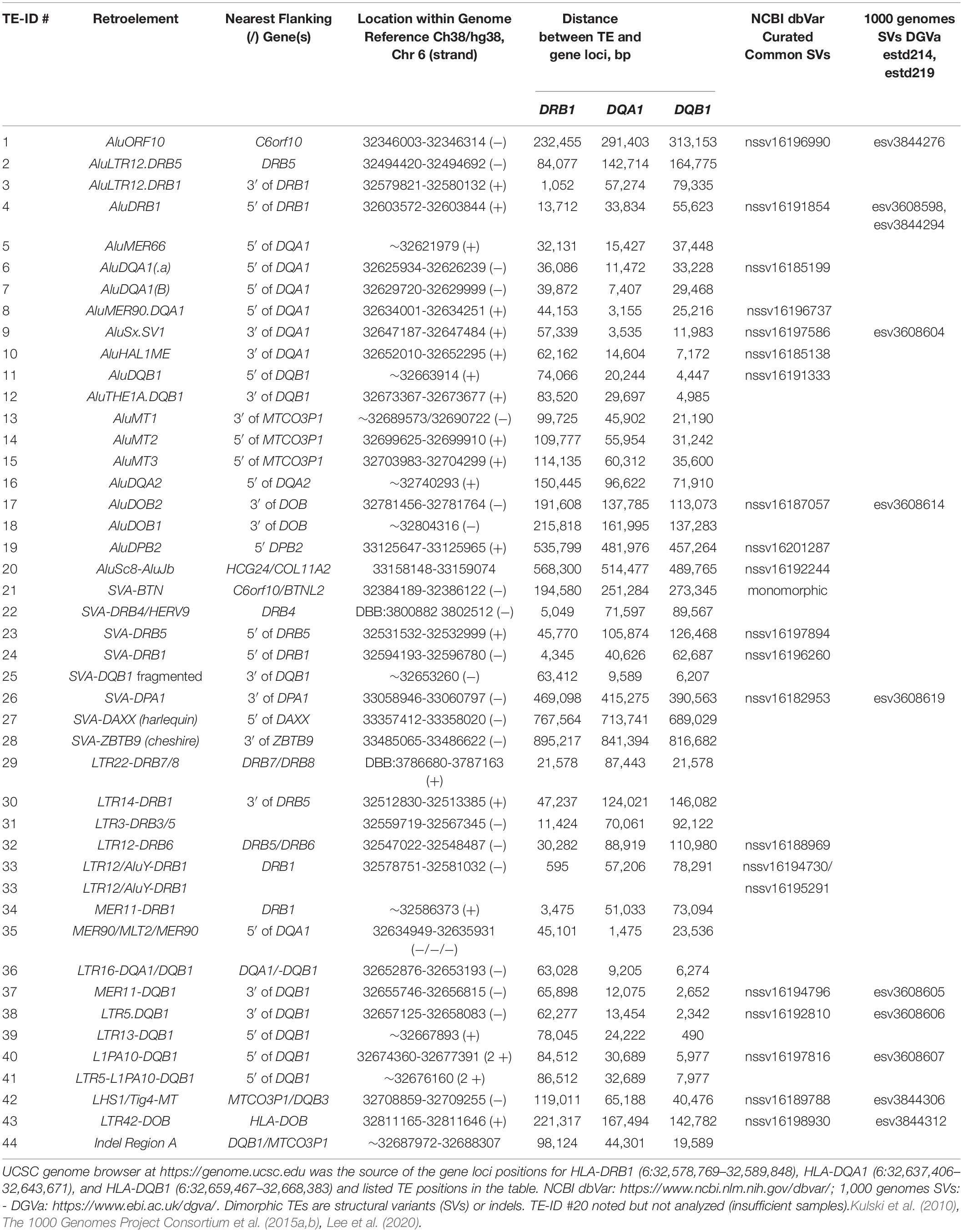
Table 3. Dimorphic TE (indels, absent or present) analyzed in this study.
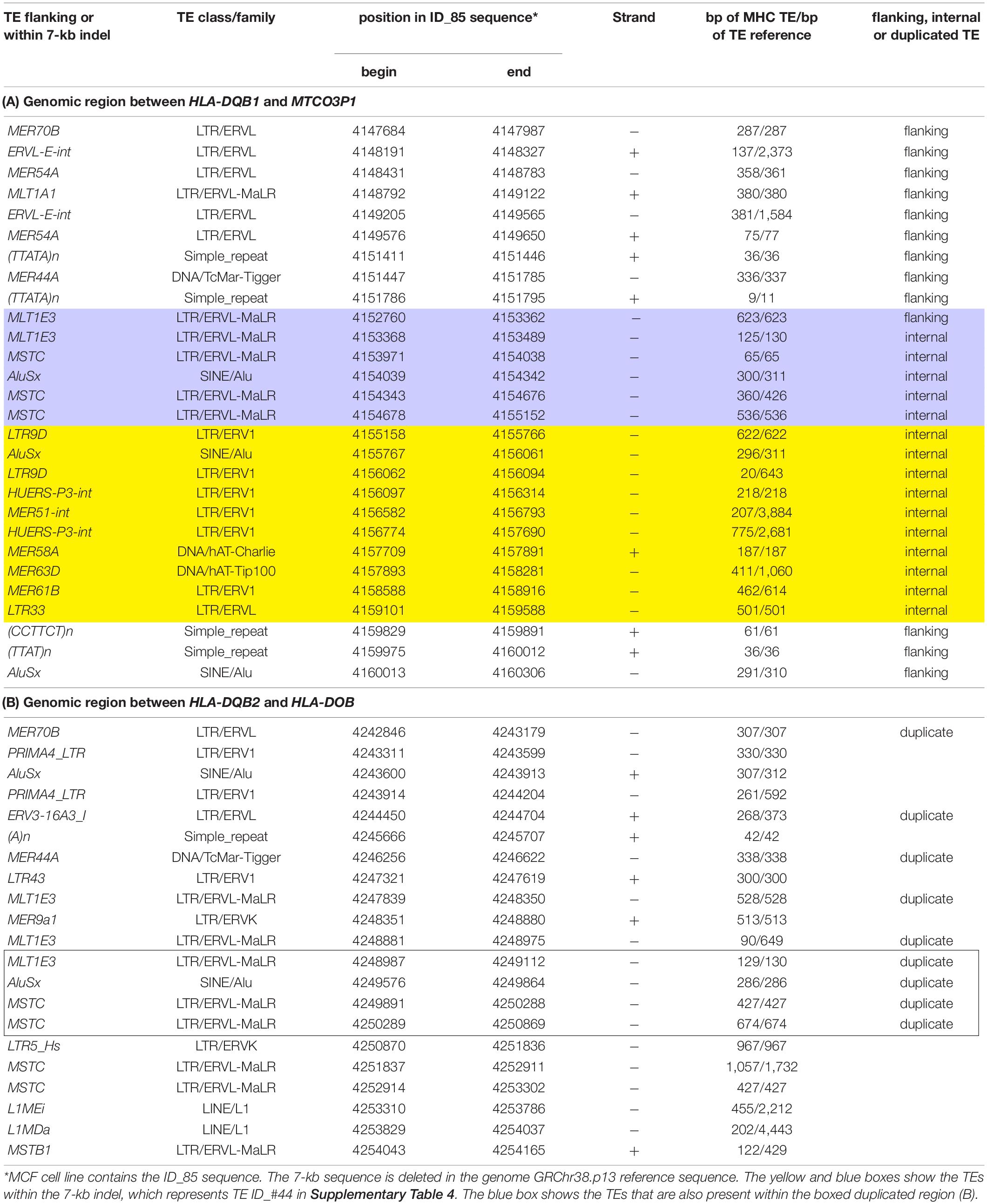
Table 4. A list of TEs flanking or within (A) a 7-kb indel (boxed) located between HLA-DQB1 and the MTCO3P1 pseudogene compared with the TEs in (B) a partially duplicated region located between HLA-DQB2 and HLA-DOB.
Supplementary Table 4 shows the dimorphic TE linkages with each other and the HLA class II loci, including the MTCO3P1 pseudogene alleles in the 89 haplotype sequences. Supplementary Table 5 provides a summary of 148 percentage linkage counts between some of the dimorphic TE, the HLA class II gene alleles, and the MTCO3P1 alleles. Many of these dimorphic TE insertions are haplotypic, but four of the dimorphic Alu insertions appear to be haplospecific: AluMER66∗2 and HLA-DRB1∗01:01:01, AluHAL∗2 and DQA1∗01, AluDQB1∗2 and HLA-DQB1∗02, and the AluMT1∗2 insertion within the DRB1∗07/DQA1∗02/DQB1∗02/MTCO3P1∗04 haplotype. In addition, 10 of the 17 AluDQB1 insertions are also linked to all 10 sequences with the 8.1 Ancestral haplotype (DRB1∗03:01:01:01/DQA1∗05:01:01:02//DQB1∗02:01:01/MTCO 3P1∗01).
The number of different MHC class II haplotypes for the Norman et al. (2017) DNA sequences can vary markedly depending on what linkage markers, alleles, and genomic distances are used for the haplotype counts. For example, we counted 29 distinct haplotypes using 14 loci covering a distance of 128,187 bp from the HLA-DRB1 gene to the MTCO3P1 pseudogene for 89 sequences, including nine Alu and two LTR indels (LTR13 and LTR33) and the allelic lineages of HLA-DRB1, HLA-DQA1, HLA-DQB1, and MTCO3P1. On the other hand, the number increased to 53 different haplotypes simply by extending the distance of the sequence coverage a further 100,000 bp toward HLA-DOB with the addition of another five loci, four for Alu indels and one for the LTR42 indel located near the HLA-DOB gene (Supplementary Table 6). In comparison, we counted 26 distinct six-loci DRB1/DQA1/DQB1/MTCO3P1/DQA2/DQB2 haplotype/allelic lineages in 87 sequences (Table 2). Table 5 shows the six AluDOB2/AluDOB1/LTR42.DOB haplotypes in 84 sequences, and Table 6 provides 11 HLA-DPB2/AluDPB2/HLA-DPA3 haplotypes and the number for each combination in 72 sequences.
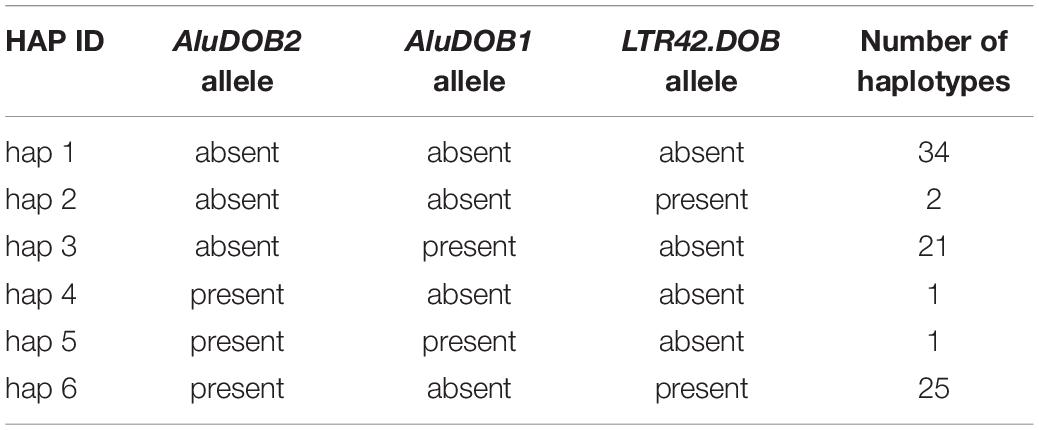
Table 5. AluDOB2/AluDOB1/LTR42.DOB haplotypes.
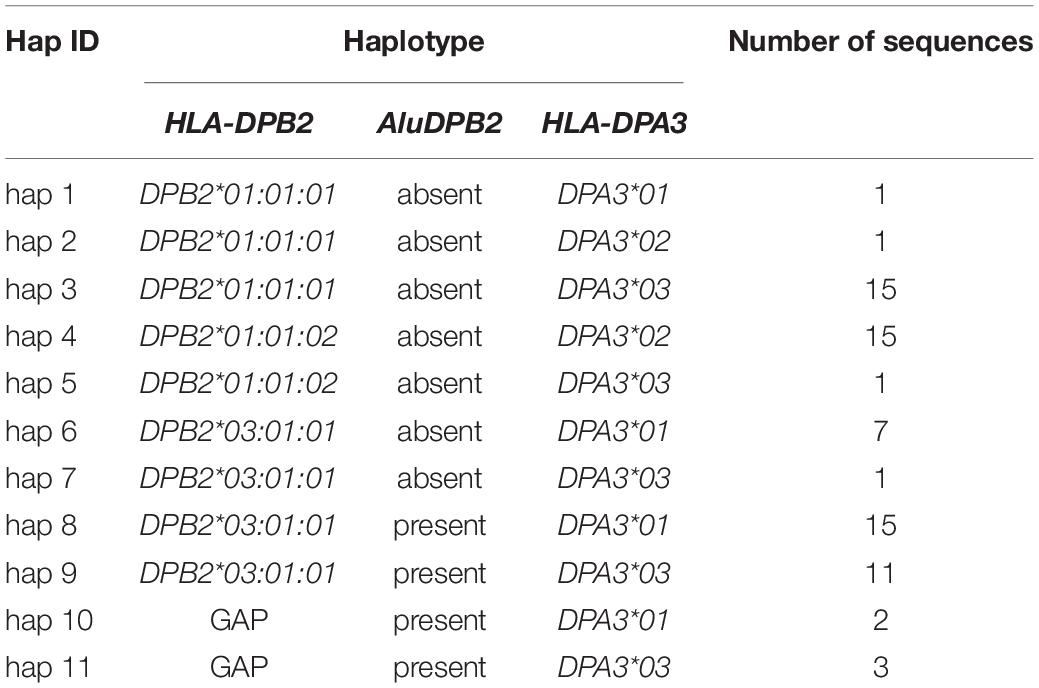
Table 6. HLA-DPB2/AluDPB2/HLA-DPA3 haplotypes.
Because there were numerous gaps and various sequence rearrangements in the DR haplotype region between HLA-DRA and HLA-DRB1, even within the same haplotypes, we could not identify with any confidence the correct positions of the TE indels relative to each other and the HLA-DR genes. Instead, we simply counted the numbers for the presence of nine particular TE indels for each haplotype sequence (Supplementary Table 7). The TE counts in 89 sequences were for U1 (300 counts), LTR12 (212), HERV9-LTR12 (139), LTR43 (42), LTR5 (41), MER77 (26), LTR22 (24), LTR14 (21), and LTR14-HERVK14 (8). The TE associated with particular DR haplotypes based on the eight haplotype sequences of Horton et al. (2008) is shown in Supplementary Table 8.
Supplementary Tables 1, 2, 4 show numerous gene allele XOs between various shuffling haplotypes across the MHC class II region from the HLA-DRB1 locus to the HLA-DP locus. Sequence alignment comparisons were performed between various MHC class II genomic sequences to locate the site of the last identifiable SNP at the junction between a SNP-rich and a SNP-poor block. Table 7 presents a summary of the number of sequence comparisons and the number of SNP-density XO sites detected. Supplementary Table 9 lists the 171 XOs at 98 unique SNP-density XO nucleotide positions identified from HLA-DRB6 to HLA-DPB1 in 121 paired-sequence alignments of different haplotypes. The 98 SNP-density XO positions relative to HLA class II genes and previously identified recombination sites are shown in Figure 1. Most XOs occurred within or between various TE elements, but some were also within HLA-DRB1, -DQB2, -DOB, -DMB, -DOA, and -DPA3 gene sequences. In most cases, the XOs were identified in locations between the different gene loci. Fourteen of 98 unique SNP XO sites were identified in the region between the pseudogenes MTCO3P1 and HLA-DQB3, and five XOs were in locations between HLA-DQB1 and MTCO3P1. Thus, 19 XOs were in the vicinity of MTCO3P1, compared with 11 XOs in the vicinity of TAP2 and PSMB8, 7 XOs between HLA-DQB2 and HLA-DOB, and 3 XOs between DOB and TAP2. Also, 13 XOs were found in locations between HLA-DOA and HLA-DPA1 mostly within the HERVK22 sequence and one within the SVA-DPA1 indel. Of the 121 sequence alignments with XOs, 81 had a single XO, 31 had 2 XO sites, 8 had 3 XO sites, and 1 (6_ PGF v 73_SPL) had 4 XO sites.

Table 7. Number of haplotype pair comparisons and SNP-density XO sites detected.
The longest SNP-free alignments in the 121 comparisons between different haplotype pairs were from:
(1) HLA-DRB1 to HLA-DOA (439–466 kb) for the sequence comparisons between 9-WT47 and 13-SLE005 (DRB1∗13:02/DQA1∗01:02/DQB1∗06:04/MTCO3P1 ∗05), and 64-YAR and 48-ISH3 (DRB1∗04/DQA1∗03:01/DQB1∗03:02/MTCO3P1∗08),
(2) HLA-DRB1 to 3’HLA-DPA1 (460 kb) between 59-AZH and 38-CALEGORO (DRB1∗16:01/DQA1∗01;02/DQB1∗05:02/MTCO3P1∗05,
(3) HLA-DRB1 to telomeric (3’) of HLA-DOA (408 kb) between 19-LO541265 and 16-PF04015 (DRB1∗03:01/DQA1∗05:01/DQB1∗02:01/MTCO3P1∗01).
No SNP-density XOs were detected in four paired sequence alignments between the same haplotypes: 51_QBL v 90_LD2B; 62_AKIBA v 93_KAWASAKI; 37_DBB v 58_BEI; and 15_QBL v 26_DUCAF (Supplementary Tables 4, 9).
SNP-density plots across the entire class II region for the same haplotype pair and five different haplotypes are shown in Figure 3. Two different SNP-rich haplotypes (ID_1 v ID_48) produced a typical SNP density profile of four major peaks and troughs decreasing in height and intensity from the HLA-DRB1 gene over 600 kb to the COL11A2 gene. The two highest SNP-density peaks were in the region of the HLA-DRB1 gene and the DQA1/DQB1 gene cluster and then smaller peaks in the regions of MTCO3P to DQA2 and separately in the HLA-DP cluster. By comparison, the SNP density was very low in a 191.5-kb genomic region between HLA-DOB and HLA-DOA that included the TAP2/PSMB8/TAP1/PSMB9 genes, HLA-DMB, HLA-DMA, and BRD2. On the other hand, the SNP plot between the same haplotype sequences (ID_51 v ID_90) produced no peaks or troughs because there were essentially no SNPs (SNP-poor) to be counted over 600 kb of sequence. Figure 3B shows a few narrow peaks labeled “A” that are assembly and alignment errors, inversions, and/or long runs of unspecified nucleotides. The other four SNP density plots (C) to (F) in Figure 3 show discernible SNP-density XO points at the junctions of SNP-poor and SNP-rich segments in the alignments between different haplotypes (Supplementary Table 4); three XOs in (C) and (D), two XO in (E), and one XO in (F).
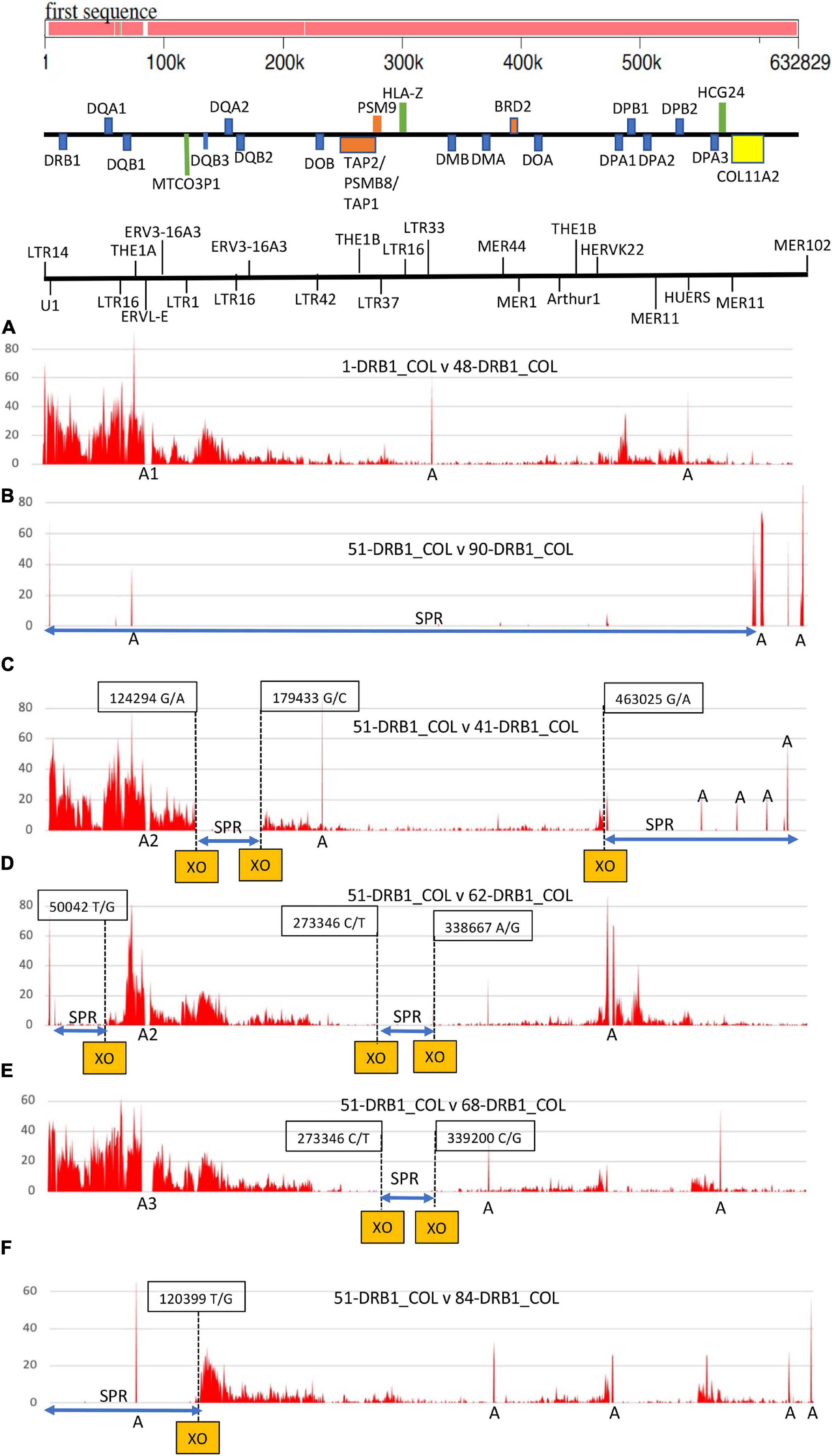
Figure 3. Single-nucleotide polymorphism (SNP) density plots of six pairs of MHC class II haplotypes from HLA-DRB1 to COL11A2. The genomic region and distances for the haplotype sequences are indicated at the top of the Figure with a gene map and an abbreviated TE map highlighting U1 and a few LTR, MER and HERV elements. The six SNP plots are comparisons between (A) 1_ DRB1∗01:01/DQA1∗01:01/DQB1∗05:01:01/MTCO3P1∗09 versus 48_ DRB1∗04:06/DPA1∗02:02/MTCO3P1∗08; and then 51_DRB1∗15:01:01:01/DQA1∗01:02/DQB1∗06:02/MTCO3P1∗02 versus (B) 90_ DRB1∗15:01/DQA1∗01:02/DQB1∗06:02/MTCO3P1∗02, (C) 41_ DRB1∗04:01/DQA1∗03:01/DQB1∗03:02/MTCO3P1∗08, (D) 62_ DRB1∗15:02/DQA1∗01:03/DQB1∗06:01/MTCO3P1∗04, (E) 68_ DRB1∗11:03/DPA1∗01:03/DPB1∗04:02/MTCO3P1∗03, and (F) 84_ DRB1∗15:01:01/DQA1∗01:02:01/DQB1∗06:02/MTCO3P1∗02. The Y-axis presents the number of SNPs per 500 nucleotides (window size). The X-axis shows the SNP positions (SNPs/500 nucleotides) in a block of 632829 nucleotides from HLA-DRB1 to COL11A2 (A–F). The letter ‘A’ along the X-axis marks regions of sequence gaps, poor assembly, inversions or long runs of unspecified nucleotides. ‘A1’ is the position of a MER11/LTR5 indel (TE ID #37 and #38) between 76999 and 79305; ‘A2’ is the position of a 4562-bp LTR5/L1PA10 indel (TE ID #41); and A3 is the position of a 9324-bp indel at 77368-86691 that includes the presence or absence of MER4 and L1PA10 together with the THE1A-AluY-THE1A and LTR5 (see TE IDs #12, #40 and #41 in Table 3 and Supplementary Table 3). The dashed vertical lines mark the SNP-density crossover (XO) points between haplotype pairs. The boxed number is the XO sequence position for sequence ID_51.
Myers et al. (2005, 2010) identified a consensus PRDM9 binding motif CCTCCC[CT]AGCCA[CT] associated with recombination hotspots and genomic instability in humans, whereas Altemose et al. (2017) found an ATCCATG motif that might inhibit recombination and that they considered was one of the most common non-PRDM9 recombination-influencing motifs. Table 8 shows the meiotic recombination sites and genomic positions within the MHC class II region annotated by the NCBI Genome Data Viewer11. Most of these sites have the nucleotide motif with similarity to the predicted 13-mer PRDM9 binding motif CCNCCNTNNCCNC (16 nt).
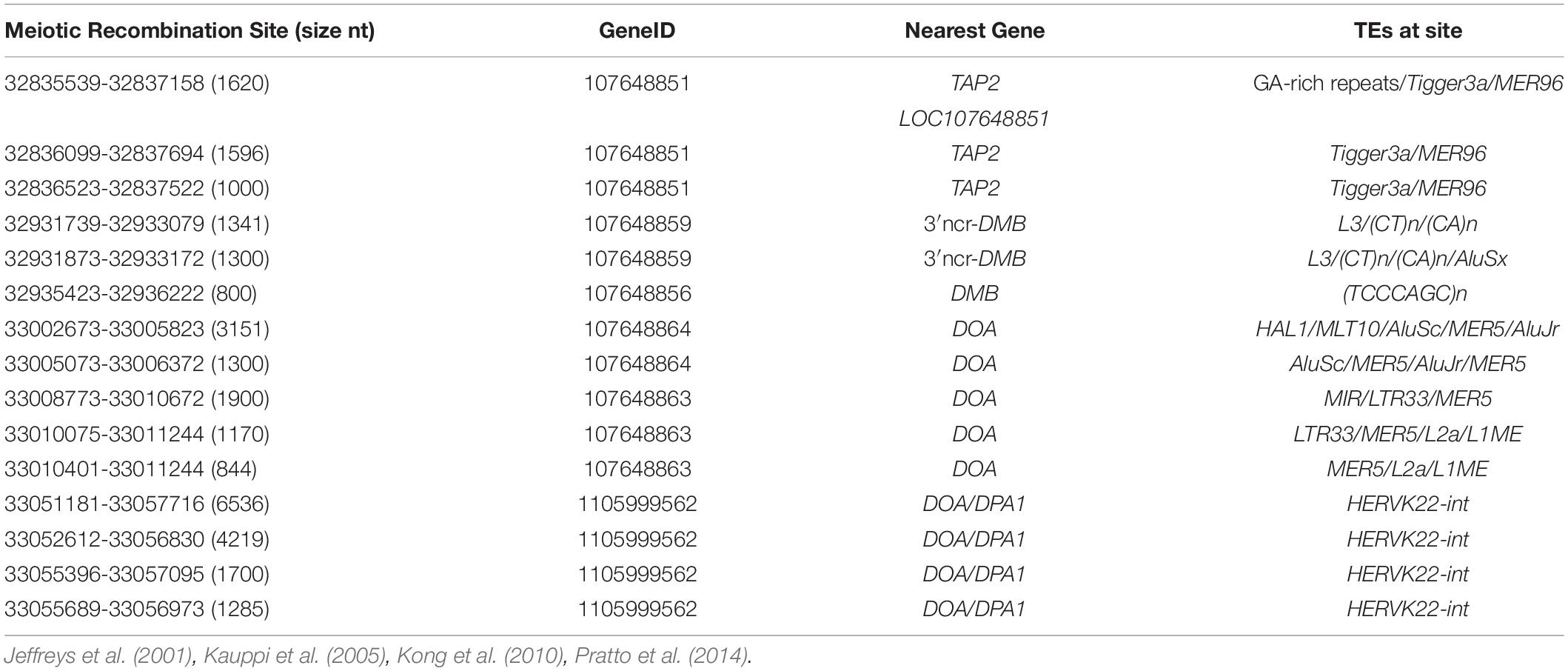
Table 8. Meiotic recombination sites within the MHC class II region annotated by the NCBI Genome Data Viewer.
We searched for CCTCCCCT and ATCCATG and their reverse complementary sequences AGGGGAGG and CATGGAT, respectively, to identify their distribution in the centromeric end of MHC class III and the entire class II region from PRRT1 to COL11A2 in the human genomic reference sequence GRChr38.p13 (NC_ 000006.12), which is the DRB1∗15:01/DQA1∗01:02/DQB1∗06:02/DPB1∗04:01 haplotype or 7.1AH represented by the MHC-PGF homozygous cell line (Supplementary Table 1) previously described by Horton et al. (2008). We detected 204 copies of the four motifs with 50 to 68.7% of them within different repeat elements (Table 9); the highest percentages were for CCTCCCCT within simple repeats (16.1%), AGGGGAGG within MIR (11.4%), ATCCATG within L1 (43.3%), and CATGGAT within L1 (33.9%). The MIR element was also near many of these motifs, as were several different TEs from the TcMar-Tigger, hAT-Charlie ERVL-MaLR, and ERV1 repeat families such as Charlie, Tigger, MER20, MER5, and THE elements as well as the ancient LTR16 and ERVL-E-int of the ERVL family (Supplementary Table 10).
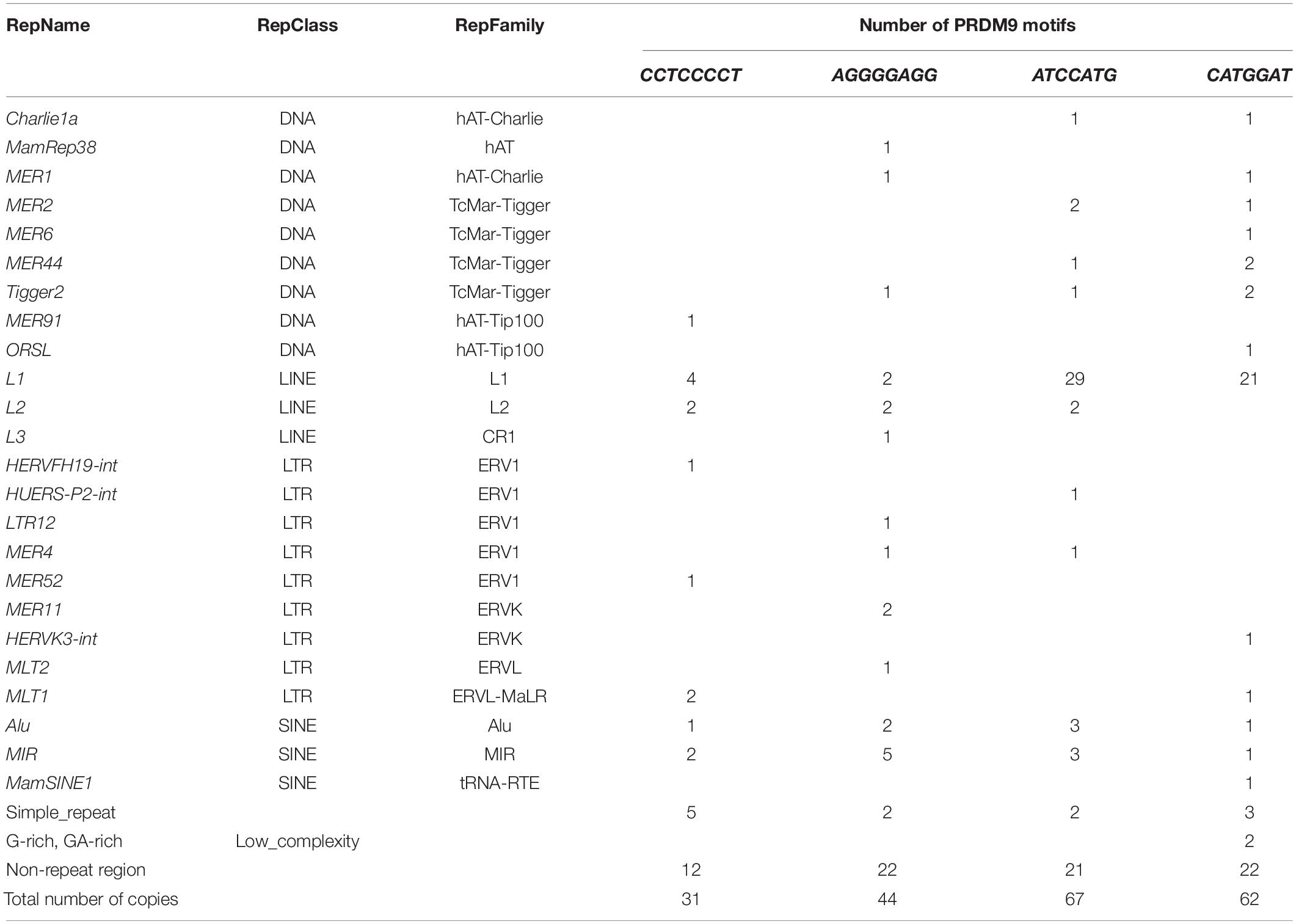
Table 9. Number of PRDM9 motifs located in the repeat elements distributed from RNF5 to RING1 in the extended MHC II genomic region.
The locations of the PRDM9 recombination motifs are shown in Figure 1 relative to the positions of recombination hotspots, the SNP-density XOs, the Alu and SVA indels, LTR, MER, L1, and other TE location tags, the centromeric MHC class III genes from PRRT1 to BTNL2, and the HLA class II genes, MTCO3P1 and COL11A2 in the MHC class II region. Of the 31 CCTCCCCT and 44 AGGGGAGG 8mers, 20 and 28 of them, respectively, were in the class II region between the HLA-DRB1 gene and the COL11A2 gene. Of these, 17 CCTCCCCT and 12 AGGGGAGG were in the previously identified recombination hotspots near MTCO3P1, within or bordering the TAP2/PSMB8/TAP1/PSMB9 region, on either side of HLA-DMB and -DMA, within and flanking -DOA, and within the HLA-DP gene cluster. Six copies of CCTCCCCT and seven copies of AGGGGAGG were within the COL11A2 gene sequence, with only one copy each of the PRDM9 suppressive motifs, ATCCATG and CATGGAT. Many of these motifs are also found near the SNP-density XO nucleotides.
Haplotype shuffling (SNP-density XOs) at the MHC haplotype boundaries has received relatively little attention (Smith et al., 2006; Traherne et al., 2006; Lam et al., 2013; Larsen et al., 2014; Kulski et al., 2021) when compared with the much greater focus on genotyping SNPs and applying LD statistical analysis to estimate haplotypes (Ahmad et al., 2003; Miretti et al., 2005; Blomhoff et al., 2006; de Bakker et al., 2006; Baschal et al., 2012; Lam et al., 2015). However, several historical studies show that statistically inferred haplotype sequences often miss the importance of CPSs of the CEH (Alper et al., 2006) and AH (Dawkins et al., 1999) in matching donors and recipients for transplantations and for identifying the haplotypes involved in autoimmune diseases (Dawkins et al., 1983) such as T1D (Alper and Larsen, 2017). The present study has taken advantage of the Norman et al. (2017) phased haplotype sequences to examine SNP-density XO points to measure haplotype shuffling in the class II region. The MHC haplotype boundaries or junctions are potential “hotspots” in genome-wide association disease studies. Previously, we investigated the occurrence of TE indels and haplotype exchanges in class I genomic region (Kulski et al., 2021) and now broadened our analysis to TE indels and haplotype switching at the junctions between SNP-rich and SNP-poor blocks in the class II region, covering 620 kb of genomic sequence from the HLA-DRB1 gene to the COL11A2 gene (Figure 1). Haplotype shuffling at more than 50 sequence locations was indicated by various genomic markers, including the HLA-class II alleles, MTCO3P1 alleles, and 42 of 44 TE markers listed in Table 3. The HLA-class II alleles for HLA-DRB1, -DRB2, -DRB3, and -DRB4 and -DRB5, -DQA1, -DQB1, -DPA1, and -DPB1 were determined by Norman et al. (2017), but to better assess the structure of the haplotype changes, we also included the alleles for HLA-DQA2, -DQB2, -DOB, -DOA, -DPB2, and -DPA3 and the pseudogene MTCO3P1. In general, the total number of alleles for each of these HLA-class II gene loci are regularly updated and presented by the IPD-IMGT/HLA Database (Robinson et al., 2019) and show that the greatest SNP diversity occurs in the 82-kb genomic region from HLA-DRB1 (2,838 alleles) to HLA-DQB1 (1,930 alleles) and to a lesser extent in the 268-kb genomic region from HLA-DQA2 (38 alleles) to HLA-DOA (12 alleles). HLA-DPB1 at the centromeric end of the MHC class II region has generated 1,654 alleles, whereas the neighboring HLA-DPA1 gene is less diverse with 216 alleles.
The haplotype estimations and population frequencies of five haplotypic Alu indels, AluORF10, AluDRB1, AluDQA1, AluDQA2, and AluDPB2 (Table 3), were investigated previously in Caucasians, Japanese (Kulski et al., 2010), Chinese Han (Shi et al., 2014), and 12 other Chinese ethnic populations (Cun et al., in preparation). The population frequencies for three of these Alu and five others were also reported by The 1000 Genomes Project Consortium et al. (2015a, b) using data from 2,504 unrelated individuals from 26 populations around the world. AluDQA1 and AluDRB1 belong to the AluY subgroup, and AluDQA2, AluDPB2, and AluORF10 are within the youngest AluYa5 or AluYb8 subgroup (Kulski et al., 2010). Whereas AluDQA1 appears to be the oldest of the five Alu indels based on its subfamily sequence and for having the highest frequency in different populations and for its association with most of the different HLA-DR supertypes (Supplementary Table 5), the frequency of the AluDQA2 insertion was higher in the Caucasians than in the Chinese or Japanese populations, which supports the hypothesis that it originated in Caucasians (Kulski et al., 2010; Shi et al., 2014). Moreover, five of the 10 AluDQA2 insertions were linked to four of 17 AluDQB1 insertions and to five of the 10 8.1 Ancestral haplotypes HLA-A1-B8-C7-DRB3-DQ2 (Supplementary Table 4), which is a common European haplotype (Aly et al., 2006; Smith et al., 2006; Gambino et al., 2018). The AluDRB1 indel has a wide frequency range from 0.10 to 0.455 and a strong percentage association with only HLA-DRB1∗15 and -DRB1∗16 in most populations studied so far. These results confirm that the AluDRB1 insertion probably originated in an ancestral HLA-DRB1 allele as a progenitor of the DR51 supertypes (Kulski et al., 2010), which contained HLA-DRB1∗15 or -DRB1∗16 (Andersson, 1998; Suzuki et al., 2018). The AluDPB2 insertion also has a wide frequency range from 0.278 to 0.574 in 15 populations but with low- to high-level percentage associations with many different HLA-DRB1 alleles (Supplementary Table 5). This is not surprising because the AluDPB2 locus is 536 kb from the HLA-DRB1 locus, with the likelihood of numerous ancient recombination events occurring in between the two loci. In contrast, the AluORF10 had a strong association with HLA-DRB1∗15, mostly in Caucasians (89.1%) and a strong association with HLA-DRB1∗16 in eight East Asian populations (Cun et al., in preparation). It is evident from this and previous studies that the closer the dimorphic Alu is to the HLA-DRB1 locus, the stronger the haplotypic linkage/association, hitchhiking, and recombination resistance (Kulski et al., 2010; 2011). For example, the AluDRB1 that is most strongly associated with HLA-DRB1∗15 and HLA-DRB1∗16 is located within 14 kb of the HLA-DRB1 locus, whereas AluORF10 and AluDP2, which are 233 and 536 kb, respectively, from the DRB1 locus, are associated with many more different DRB1 alleles. Thus, these five genotyped and haplotyped dimorphic Alu elements are genomic markers that provide evolutionary and “identical by descent” lineage evidence about the common ancestral state, diversity, and genomic rearrangements of the MHC class II region.
The 660-bp MTCO3P1 pseudogene (NCBI gene ID 404026) was haplotyped as a non-HLA class II allelic marker because of high-frequency haplotype exchanges in the vicinity of its locus between the genomic loci of HLA-DQB1 and HLA-DQA2. MTCO3P1 has a high sequence identity with cytochrome c oxidase III (NCBI gene ID 4514) in the mitochondrial DNA, and there are numerous other MTCO3 pseudogene loci distributed throughout the human genome (chromosomes 2, 3, 4, 7, 9, and 16 and Supplementary Figure 2). We identified 10 MTCO3P1 sequence variants (Supplementary Table 3) in 84 sequences, with gaps or insufficient sequence information available in the remaining 11 sequences. MTCO3P1∗06, MTCO3P1∗08, and MTCO3P1∗10 appear to be haplospecific, whereas the other seven variants are haplotypic with MTCO3P1∗03 linked to nine different HLA-DRB1 haplotypes (Table 2) and to the 7-kb #44 indel composed of at least 10 other TE family members (Table 4 and Supplementary Table 4). Most of the haplotype shuffling was detected in the genomic region between MTCO3P1 and HLA-DQB3 in 31 paired sequence comparisons and between the HLA-DQB1 and MTCO3P1 loci in 11 cases (Figure 1 and Supplementary Table 9). Although we identified 10 alleles for MTCO3P1, there are at least 102 sequence variants archived at the National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site—Genomics database12 that possibly could be linked to many more MHC class II haplotypes. The MTCO3P1 genomic sequence might be involved haplotypically with Alzheimer’s disease and Alzheimer’s disease-related neuropathologies (National Institute on Aging Genetics of Alzheimer’s Disease Data Storage Site—Genomics database). In this regard, the GWAS survey by Chesmore et al. (2018) revealed that MTCO3P1 was the third most pleiotropic sequence in the human genome with 32 phenotype associations after the top-ranking ABO gene on chromosome 9 with 39 phenotype associations—a gene that forms the basis of the ABO blood group diversity. The HLA-DRB1, -DQB1, and -DQA1 genes also were among the 10 top-ranking pleiotropic genes in the Chesmore et al. (2018) study and, together with the MTCO3P1 variants, might be associated with systemic lupus erythematosus, T1D, immunoglobulin A nephropathy, Crohn’s disease, multiple sclerosis, narcolepsy, and systemic sclerosis among various other disease phenotypes. Some of the MTCO3P1 alleles together with dimorphic haplotypic TE markers such as the LTR13-DQB1, AluMT2, AluMT3, and the 7-kb #44 indel might be useful genomic markers for subdividing MHC class II haplotype disease associations into different categories, such as the HLA-DRB1∗03/DQA1∗03/DQB1∗03 haplotypes associated with TID and/or autoimmune Addison’s disease (Table 2, Pani et al., 2002; Gambelunghe et al., 2005). The LTR13-DQB1 indel was linked to six different DRB1-MTCO3P1 haplotypes (Supplementary Table 4), some of which might be useful for predicting the onset of T1D and/or Addison’s disease in multiple populations (Valdes et al., 2012; Vadva et al., 2019). In addition, there are four TcMar-Tigger DNA elements (Smit and Riggs, 1996) located between MTCO3P1 and HLA-DQB3, including a 2,411-bp Tigger1 sequence adjoining the HLA-DQB3 pseudogene locus in all of the haplotypes examined in this study. Tigger repeat sequences can generate microRNAs for the regulation of gene expression at the post-translational level (Qin et al., 2015), and they have been found overrepresented in cell-free DNA extruded from cultured human bone osteosarcoma cells (Bronkhorst et al., 2018).
The identification of dimorphic TEs near the junctions of duplicated genes (Kulski et al., 2021) and at ectopic and meiotic recombination sites (Myers et al., 2010; Altemose et al., 2017; Kent et al., 2017) as well as at SNP-density XO sites (Figure 1) further emphasizes their role in contributing to genomic diversity. The two highest SNP-density levels between different MHC class II haplotypes were seen in the genomic region between HLA-DRB1 and HLA-DOB (Figure 3), which also contained 13 of the 15 Alu indels, two of the five SVA indels, two MER11 indels, two LTR5 indels, and a single indel location each for LTR13, LTR33, and LTR42 (Figure 1 and Table 3) and 44 of the 98 SNP-density XO points (Supplementary Table 9) in the MHC class II region. This connection between TE indels and SNP-density XOs confirms our previous finding that the dimorphic Alu and SVA are located close to or within putative recombination hotspots throughout the MHC classes I, II, and III genomic regions (Kulski et al., 2011, 2021), suggesting that they might be involved in DNA repair in response to genomic stress and damage (Kulski et al., 2000a). Although the expression of most Alu elements in the human genome is silenced by methylation, they are transcriptionally active in germ cells during early development and in response to cellular and genomic stress and damage as a result of heat shock (Schmid, 1996), viral infections (Chu et al., 1998; Tucker and Glaunsinger, 2017), autoimmune diseases (Yüksel et al., 2016; Wu et al., 2019), and cancer pathogenesis (Moolhuijzen et al., 2010; Kaczkowski et al., 2016). Many Alu elements of the AluJ, AluS, and AluY subfamilies are transcriptionally active with highly expressed self-cleaving ribozyme activity during T-cell activation and thermal and endoplasmic reticulum stress (Hernandez et al., 2020). Furthermore, Wang et al. (2017) identified three TE indels, Alu-5072, Alu-5075, and SVA-282, in the class II region as potential enhancers for HLA-DRB5, HLA-DQB1-AS1, and HLA-DPB2 associated with GWAS phenotypes of lymphoma, Hodgkin lymphoma, and chronic hepatitis B infection, respectively. Alu-5057 is probably the AluDRB1 indel at the 5′ end of HLA-DRB1, whereas SVA-282 is likely the SVA-DPA1 indel at the 3′ end of HLA-DPA1 (Figure 1). Thus, the question remains whether the other 19 Alu and six SVA indels identified in the extended class II region in this study (Table 3) also have enhancer functions as reported by Wang et al. (2017) for Alu-5072, Alu-5075, and SVA-282. On the basis of these findings, the transcriptional activity and role of Alu and SVA in the human MHC during epigenetic regulation need to be investigated further and better defined.
There are ∼121 LTR sequences interspersed between the DRB1 and COL11A2 gene loci, but few have been investigated specifically as genetic risk markers in disease association studies. DQ-LTR13 was associated with DRB1∗0401 T1D susceptibility (Bieda et al., 2002; Krach et al., 2003) and autoimmune Addison’s disease due to an LD with DQB1∗0302 and DRB1∗0403 (Pani et al., 2002; Gambelunghe et al., 2005). HERV LTRs found in the class II region such as MER11, MER41, MER44, LTR5, LTR9, and LTR12 are known to regulate the transcription of neighboring genes outside the MHC genomic region (Bi et al., 1997; Bièche et al., 2003; Buzdin et al., 2006; Chuong et al., 2017; Daskalakis et al., 2018). The solitary 482-bp LTR42 indel that is located 1-kb telomeric of the 3′ end of HLA-DOB coding gene is positively linked to the AluDOB2 insertion and negatively linked to the AluDOB1 insertion (Table 5) and might act to regulate the transcriptional and/or translational activity of either HLA-DQB2 or HLA-DOB. There are only two HERVs in the class II region centromeric of HLA-DRB1 that are greater than 5-kb in length; the LTR12 (526 bp)/HERVK22 (6,805 bp) and LTR9 (666 bp)/HUERS-P3-int (5589 bp)/LTR9 (666 bp) sequences. The HERVK22 sequence is located near the SVA-DPA1 indel at the 3’ end of the HLA-DPA1 gene, and it is a putative ancestral meiotic recombination hotspot with SNP-density XOs (haplotype shuffling) occurring within its sequence (Supplementary Table 9). The HUERS sequence is fragmented and interrupted by a 1-kb LTR5 insertion in all of the 90 haplotype sequences that were examined, and it is located within the HLA-DPB2 pseudogene and less than 1 kb from the AluDPB2 indel (Supplementary Figure 1). There are also HERVs (HERVK3, HERV9, ERVLE, HERVIP10, and HERVK14) located in the HLA-DR super haplotype region (Supplementary Table 7, Andersson et al., 1998; Doxiadis et al., 2008; Horton et al., 2008) and in the class I region (Kulski et al., 1999a, 2021). These and other ERVs provide promoter and enhancer exaptation and non-coding transcripts of viral accessory proteins as regulatory units (Buzdin et al., 2006, 2017; Daskalakis et al., 2018) that might have a pathogenic role in several autoimmune diseases such as rheumatoid arthritis and systemic lupus erythematosus by providing epitopes, superantigens, and/or hypomethylation motifs (Moyes et al., 2007; Balada et al., 2010; Tugnet et al., 2013; Trela et al., 2016). The diseases associated with MHC haplotypes (Dawkins et al., 1983; Lokki and Paakkanen, 2019) are still poorly defined, and current knowledge about genomic disease associations remains largely at the level of SNPs and alleles in GWAS. In this regard, structurally polymorphic TEs are potential haplotype disease markers to associate particular MHC haplotypes and disease traits.
In previous studies, the major recombination hotspots in the MHC class II region were identified between HLA-DQB1 and HLA-DQA2 (near MTCO3P1); within intron 2 of TAP2; between TAP2 and HLA-DMB; between BRD2 and HLA-DOA; between HLA-DOA and HLA-DPA1; and between HLA-DPB1 and RING1 (Jeffreys et al., 2001; Cullen et al., 2002; Miretti et al., 2005; Table 8). Our comparative sequence analysis of 121 different haplotype pairs revealed 98 unique XO sites between SNP-poor and SNP-rich genomic segments with considerable haplotype shuffling in the proximity of the putative recombination hotspots (Figure 1). The majority of SNP-density XO sites occurred across various regions, including within HLA-DRB1; between the HLA-DRB1 and HLA-DQA1 loci; between HLA-DQA1 and HLA-DQB1; in the vicinity of MTCO3P1 between HLA-DQB1 and HLA-DQB3; between HLA-DQB2 and HLA-DOB; between DOB and TAP2; within TAP2; and between HLA-DOA and HLA-DPA1 where eight XO sites were found within a HERVK22 sequence. The SNP XO sites were located mostly outside of TEs, but 43 of the 98 unique XO-SNPs were found within TE sequences, including within different Alu subfamilies, L1 subfamilies, MER20, LTR1, LTR5, LTR19, LTR33, LTR41, Tigger3b, MLT1H, MER3, SVA-DPA1, and HERVK22-int (Supplementary Table 9). We also determined the genomic positions of the PRDM9-recombination suppression sequence motif ATCCATG/CATGGAT and the PRDM9 recombination activation partial binding motif CCTCCCCT/AGGGGAG in the class II region of the human reference genome (NC_ 000006) relative to published meiotic recombination positions (Figure 1).
A possible limitation in our comparative analysis between the SNP XO sites and recombination sites (Figure 1) is that there are large differences between our data and those of others, such as with methodologies, amounts of data analyzed, and the genotypes or haplotypes studied. However, our comparative analysis generally supports and extends the previous observations of the centromeric class II region by Larsen et al. (2014) for 158 common European haplotypes, and we concur with them that the SNP density XOs or haplotype break point locations and frequencies vary significantly between different CEH/AHs. Also, the exact historical sequence recombination leading to the breakage or shuffling of a dominant haplotype based solely on SNP transitions between high and low (or absent) densities is difficult to localize precisely or with confidence. We did not have sufficient sequences with the same haplotypes to demonstrate that some dominant sequences break down gradually in many locations, whereas some others break down at specific locations (Larsen et al., 2014). Instead, we found that SNP-density XOs were mostly at specific locations, as shown in Figure 3 and listed in Supplementary Table 9. Most of the XO sites that we observed depended on which haplotypes were compared, and the differences between various haplotypes are probably due to different CEH/AH expansion timelines and recombination events. Taken together, our study and that of Larsen et al. (2014) show substantial haplotype shuffling between different polymorphic blocks and suggest the presence of numerous putative ancestral recombination sites across the class II region located between various HLA class II genes.
Of 125 haplotype-pairwise-sequence comparisons in this study, 121 showed evidence of haplotype shuffling at least once within the 620-kb genomic region between the telomeric HLA-DRB1 and the centromeric COL11A2 gene (Supplementary Table 9). Much of this exchange occurred between or in close vicinity to the PRDM9-mediated recombination activation motif CCTCCCCT/AGGGGAG and/or recombination suppression motif ATCCATG/CATGGAT (Figure 1) that is recognized by the PRDM9-mediated recombination machinery to alter chromatin structure and PRDM9 initiated meiotic recombination (Myers et al., 2010; Altemose et al., 2017; Parvanov et al., 2017). Our analysis of the PRDM9 motifs was limited mainly to the reference genome GRChr38.p13 for this study, but we found some site differences between MHC haplotypes that still need to be evaluated in greater detail (data not shown). Both the recombination and anti-recombination motifs are widely distributed throughout the class I and class II genomic regions (Kulski et al., 2021), with 50% or more found within repeat elements. The anti-recombination motifs mostly involved the L1 fragmented repeats, whereas the recombination activation partial binding motifs were distributed more widely between L1, MIR, and simple repeats (Table 9). In addition, many have accumulated within regions of previously identified ancestral meiotic recombination sites (Table 8) and in proximity to regions of pseudogene conversions and near the loci of the dimorphic Alu and SVA insertions. Multiple recombinations in the MHC might be relatively rare events (Cullen et al., 1997, 2002; Smith et al., 2006), but we found multiple SNP-density XOs within the class II region in 40 of 121 (33%) pairwise sequence analyses of different haplotypes (Table 7) including zero to three XOs in the comparison between haplotype ID_51 and three other haplotypes (ID_41, _62, and _68) in Figure 3. The present study on haplotype shuffling and TE indels within the class II region extends our previous study within the class I region using the same Norman et al. (2017) haplotype sequences. The important class III region that harbors many immune regulatory genes was not examined in detail here, although we found evidence of haplotype exchanges in the genomic regions between MICB and TNF, C4/C2 duplication region (data not shown), and between NOTCH4 and TSBP1 (Figure 1), similar to the SNP-density XO regions described by Lam et al. (2013) in the proximity of the meiotic hotspots reported by Cullen et al. (1997, 2002).
Comparative genomic analysis of haplotype diversity and shuffling advances our understanding of the evolutionary history of human genomic architecture and the variability of population structures and ancestry. Our previous analysis of haplotype shuffling in the MHC class I region (Kulski et al., 2021) and the class II region in this study further highlights the metameric design of the MHC genomic region characterized by the presence of duplicated HLA-genes, LTR16/HERV16 duplications, and various repeating TEs embedded within distinct subgenomic segments Kulski et al., 1999b, 2000a,b, 2002, 2004, 2021), polymorphic frozen blocks (Dawkins et al., 1999), and conserved regions of framework genes (Amadou, 1999). These segments and blocks have evolved with considerable haplotype shuffling of CPSs by meiotic recombination, unequal recombinations, and gene conversions often involving TEs. In this context, it appears that the location of many of the dimorphic TEs within the MHC is the remains of DNA repair “sutures” or “scars” in response to genomic stress and damage and/or meiotic recombinations and unequal XO events (Schmid, 1996; Kulski et al., 2000a; Hernandez et al., 2020). The length of class II genomic region from HLA-DRA1 to -DPB2 (∼689 kb) is ∼3 × shorter than the class I region from HLA-F to MICB (∼1.8 Mb), but it seems to have a greater frequency of multiple haplotype shuffling events at the junctions between SNP-poor and SNP-rich blocks (Kulski et al., 2021, and this study). This may be due in part to differences in CPSs, TEs, genomic structure, and evolutionary age of class I and class II regions; the primate class II allelic lineages appear to be much older than those of the class I allelic lineages (Andersson, 1998; Kulski et al., 2000b, 2004; Tìšickı and Vinkler, 2015). Also, the mean recombination rate is ∼4 × greater in the class II region than in the class I region (Radwan et al., 2020). The information gathered so far for putative ancestral recombination sites and haplotype shuffling between different polymorphic blocks in this and previous studies is still largely based on the availability of a limited number of MHC genomic sequences representing approximately 50–80 MHC ancestral haplotypes, a very small fraction of the thousands of different haplotypes that are distributed in worldwide populations (Goodin et al., 2018). Many more MHC genomic haplotype sequences need to be added to those already generated by Horton et al. (2008), Lam et al. (2015), and Norman et al. (2017) for better informed comparative genomics, population studies, disease associations, and genetic manipulations such as knock-in and knock-out in regulatory and functional studies. The advancement of new sequencing strategies (Cheng et al., 2021; Ebert et al., 2021; Human Genome Structural Variation Consortium et al., 2021) might allow these types of analyses to be extended to many more fully phased HLA haplotypes in the future. Such work will lead to a better understanding of the role of TEs and the different DNA processes involved in the evolution of the human MHC haplotypes, IBD segments, and their association with autoimmune disorders and other disease traits.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
JK carried out the analyses of the repeat elements, haplotype sequence comparisons, SNP-density XOs, and interpretation of the data and wrote the manuscript. SS and TS analyzed and interpreted parts of the data and provided the alleles for the classical and non-classical HLA class II genes, HLA pseudogenes, and the MTCO3P1 pseudogene and provided the SNP density plots. All authors checked the final version of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by MEXT KAKENHI (16H06502) and by the Practical Research Project for Allergic Diseases and Immunology (Research on Technology of Medical Transplantation) from the Japan Agency for Medical Research and Development (20ek0510032s0101).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.665899/full#supplementary-material
AH, ancestral haplotype; CEH, conserved extended haplotype; CPS, conserved polymorphic sequence; CR1, chicken repeat 1; EMBL-EBI, European Molecular Biology Laboratory - The European Bioinformatics Institute; ERV, endogenous retrovirus; GWAS, genome-wide association study; HERVs, human endogenous retrovirus; HLA, Human Leukocyte Antigen; IBD, identical by descent; indel, insertion-deletion; LD, linkage disequilibrium; LINES, long interspersed retrotransposable elements; LTR, long terminal repeat; MER, medium reiterated repeat; MHC, major histocompatibility complex; MIR, mammalian-wide interspersed repeat; NCBI, National Center for Biotechnology Information; Rec Site, recombination site; SINES, short interspersed retrotransposable elements; SNP, single nucleotide polymorphism; STR, short tandem repeat; SVA, short interspersed nuclear element–VNTR–Alu; T1D, type 1 diabetes; TE, transposable element; UCSC, University of California, Santa Cruz; XO, crossover.
Adamek, M., Klages, C., Bauer, M., Kudlek, E., Drechsler, A., Leuser, B., et al. (2015). Seven novel HLA alleles reflect different mechanisms involved in the evolution of HLA diversity: description of the new alleles and review of the literature. Hum. Immunol. 76, 30–35. doi: 10.1016/j.humimm.2014.12.007
Ahmad, T., Neville, M., Marshall, S. E., Armuzzi, A., Mulcahy-Hawes, K., Crawshaw, J., et al. (2003). Haplotype-specific linkage disequilibrium patterns define the genetic topography of the human MHC. Hum. Mol. Genet. 12, 647–656. doi: 10.1093/hmg/ddg066
Al Bkhetan, Z., Zobel, J., Kowalczyk, A., Verspoor, K., and Goudey, B. (2019). Exploring effective approaches for haplotype block phasing. BMC Bioinformatics 20:540. doi: 10.1186/s12859-019-3095-8
Alper, C. A., and Larsen, C. E. (2017). “Pedigree-Defined haplotypes and their applications to genetic studies,” in Haplotyping Methods in Molecular Biology, eds I. Tiemann-Boege and A. Betancourt, (New York, NY: Springer New York), 113–127. doi: 10.1007/978-1-4939-6750-6_6
Alper, C. A., Larsen, C. E., Dubey, D. P., Awdeh, Z. L., Fici, D. A., and Yunis, E. J. (2006). The haplotype structure of the human major histocompatibility complex. Hum. Immunol. 67, 73–84. doi: 10.1016/j.humimm.2005.11.006
Altemose, N., Noor, N., Bitoun, E., Tumian, A., Imbeault, M., Chapman, J. R., et al. (2017). A map of human PRDM9 binding provides evidence for novel behaviors of PRDM9 and other zinc-finger proteins in meiosis. ELife 6:e28383. doi: 10.7554/eLife.28383
Aly, T. A., Eller, E., Ide, A., Gowan, K., Babu, S. R., Erlich, H. A., et al. (2006). Multi-SNP analysis of MHC region: remarkable conservation of HLA-A1-B8-DR3 haplotype. Diabetes 55, 1265–1269. doi: 10.2337/db05-1276
Amadou, C. (1999). Evolution of the Mhc class I region: the framework hypothesis. Immunogenetics 49, 362–367. doi: 10.1007/s002510050507
Andersson, G. (1998). Evolution of the human HLA-DR region. Front. Biosci. 3:d739–d745. doi: 10.2741/A317
Andersson, G., Svensson, A.-C., Setterblad, N., and Rask, L. (1998). Retroelements in the human MHC class II region. Trends Genet. 14, 109–114. doi: 10.1016/S0168-9525(97)01359-0
Ando, A., Imaeda, N., Matsubara, T., Takasu, M., Miyamoto, A., Oshima, S., et al. (2019). Genetic association between swine leukocyte antigen class II haplotypes and reproduction traits in microminipigs. Cells 8:783. doi: 10.3390/cells8080783
Anzai, T., Shiina, T., Kimura, N., Yanagiya, K., Kohara, S., Shigenari, A., et al. (2003). Comparative sequencing of human and chimpanzee MHC class I regions unveils insertions/deletions as the major path to genomic divergence. Proc. Natl. Acad. Sci. U.S.A. 100, 7708–7713. doi: 10.1073/pnas.1230533100
Arnett, F. C., Gourh, P., Shete, S., Ahn, C. W., Honey, R. E., Agarwal, S. K., et al. (2010). Major histocompatibility complex (MHC) class II alleles, haplotypes and epitopes which confer susceptibility or protection in systemic sclerosis: analyses in 1300 Caucasian, African-American and Hispanic cases and 1000 controls. Ann. Rheum. Dis. 69, 822–827. doi: 10.1136/ard.2009.111906
Awdeh, Z. L., Raum, D., Yunis, E. J., and Alper, C. A. (1983). Extended HLA/complement allele haplotypes: evidence for T/t-like complex in man. Proc. Natl. Acad. Sci. U.S.A. 80, 259–263. doi: 10.1073/pnas.80.1.259
Ayarpadikannan, S., and Kim, H.-S. (2014). The impact of transposable elements in genome evolution and genetic instability and their implications in various diseases. Genomics Inform. 12:98. doi: 10.5808/GI.2014.12.3.98
Balada, E., Vilardell-Tarrés, M., and Ordi-Ros, J. (2010). Implication of human endogenous retroviruses in the development of autoimmune diseases. Int. Rev. Immunol. 29, 351–370. doi: 10.3109/08830185.2010.485333
Baschal, E. E., Jasinski, J. M., Boyle, T. A., Fain, P. R., Eisenbarth, G. S., and Siebert, J. C. (2012). Congruence as a measurement of extended haplotype structure across the genome. J. Transl. Med. 10, 32. doi: 10.1186/1479-5876-10-32
Bi, S., Gavrilova, O., Gong, D.-W., Mason, M. M., and Reitman, M. (1997). Identification of a placental enhancer for the human leptin gene. J. Biol. Chem. 272, 30583–30588. doi: 10.1074/jbc.272.48.30583
Bièche, I., Laurent, A., Laurendeau, I., Duret, L., Giovangrandi, Y., Frendo, J.-L., et al. (2003). Placenta-Specific INSL4 expression is mediated by a human endogenous retrovirus element. Biol. Reprod. 68, 1422–1429. doi: 10.1095/biolreprod.102.010322
Bieda, K., Pani, M. A., van der Auwera, B., Seidl, C., Tönjes, R. R., Gorus, F., et al. (2002). A retroviral long terminal repeat adjacent to the HLA DQB1 gene (DQ-LTR13) modifies Type I diabetes susceptibility on high risk DQ haplotypes. Diabetologia 45, 443–447. doi: 10.1007/s00125-001-0753-x
Bilbao, J. R., Calvo, B., Aransay, A. M., Martin-Pagola, A., Perez de Nanclares, G., Aly, T. A., et al. (2006). Conserved extended haplotypes discriminate HLA-DR3-homozygous Basque patients with type 1 diabetes mellitus and celiac disease. Genes Immun. 7, 550–554. doi: 10.1038/sj.gene.6364328
Blomhoff, A., Olsson, M., Johansson, S., Akselsen, H. E., Pociot, F., Nerup, J., et al. (2006). Linkage disequilibrium and haplotype blocks in the MHC vary in an HLA haplotype specific manner assessed mainly by DRB1∗03 and DRB1∗04 haplotypes. Genes Immun. 7, 130–140. doi: 10.1038/sj.gene.6364272
Bodmer, W. (2019). Ruggero Ceppellini: a perspective on his contributions to genetics and immunology. Front. Immunol. 10:4. doi: 10.3389/fimmu.2019.01280
Bodmer, W., Trowsdale, J., Young, J., and Bodmer, J. (1986). Gene clusters and the evolution of the major histocompatibility system. Philos. Trans. R. Soc. Lond. B Biol. Sci. 312, 303–315. doi: 10.1098/rstb.1986.0009
Bronkhorst, A. J., Wentzel, J. F., Ungerer, V., Peters, D. L., Aucamp, J., de Villiers, E. P., et al. (2018). Sequence analysis of cell-free DNA derived from cultured human bone osteosarcoma (143B) cells. Tumor Biol. 40:101042831880119. doi: 10.1177/1010428318801190
Browning, S. R., and Browning, B. L. (2012). Identity by descent between distant relatives: detection and applications. Annu. Rev. Genet. 46, 617–633. doi: 10.1146/annurev-genet-110711-155534
Browning, S. R., and Browning, B. L. (2020). Probabilistic estimation of identity by descent segment endpoints and detection of recent selection. Am. J. Hum. Genet. 107, 895–910. doi: 10.1016/j.ajhg.2020.09.010
Burns, K. H., and Boeke, J. D. (2012). Human transposon tectonics. Cell 149, 740–752. doi: 10.1016/j.cell.2012.04.019
Buzdin, A. A., Prassolov, V., and Garazha, A. V. (2017). Friends-Enemies: endogenous retroviruses are major transcriptional regulators of human DNA. Front. Chem. 5:35. doi: 10.3389/fchem.2017.00035
Buzdin, A., Kovalskaya-Alexandrova, E., Gogvadze, E., and Sverdlov, E. (2006). At Least 50% of human-specific HERV-K (HML-2) long terminal repeats serve in vivo as active promoters for host nonrepetitive DNA transcription. J. Virol. 80, 10752–10762. doi: 10.1128/JVI.00871-06
Charfi, A., Mahfoudh, N., Kamoun, A., Frikha, F., Dammak, C., Gaddour, L., et al. (2020). Association of HLA alleles with primary sjögren syndrome in the south tunisian population. Med. Princ. Pract. 29, 32–38. doi: 10.1159/000501896
Cheng, H., Concepcion, G. T., Feng, X., Zhang, H., and Li, H. (2021). Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175. doi: 10.1038/s41592-020-01056-5
Chesmore, K., Bartlett, J., and Williams, S. M. (2018). The ubiquity of pleiotropy in human disease. Hum. Genet. 137, 39–44. doi: 10.1007/s00439-017-1854-z
Choi, Y., Chan, A. P., Kirkness, E., Telenti, A., and Schork, N. J. (2018). Comparison of phasing strategies for whole human genomes. PLoS Genet. 14:e1007308. doi: 10.1371/journal.pgen.1007308
Chu, W.-M., Ballard, R., Carpick, B. W., Williams, B. R. G., and Schmid, C. W. (1998). Potential Alu function: regulation of the activity of double-stranded RNA-activated kinase PKR. Mol. Cell. Biol. 18, 58–68. doi: 10.1128/MCB.18.1.58
Chuong, E. B., Elde, N. C., and Feschotte, C. (2017). Regulatory activities of transposable elements: from conflicts to benefits. Nat. Rev. Genet. 18, 71–86. doi: 10.1038/nrg.2016.139
Conrad, D. F., Jakobsson, M., Coop, G., Wen, X., Wall, J. D., Rosenberg, N. A., et al. (2006). A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nat. Genet. 38, 1251–1260. doi: 10.1038/ng1911
Contu, L., Carcassi, C., and Dausset, J. (1989). The “Sardinian” HLA-A30,B18,DR3,DQw2 haplotype constantly lacks the21-OHA andC4B genes. Is it an ancestral haplotype without duplication? Immunogenetics 30, 13–17. doi: 10.1007/BF02421464
Cullen, M., Noble, J., Erlich, H., Thorpe, K., Beck, S., Klitz, W., et al. (1997). Characterization of Recombination in the HLA Class 11 Region. Am. J. Hum. Genet. 60, 397–407.
Cullen, M., Perfetto, S. P., Klitz, W., Nelson, G., and Carrington, M. (2002). High-Resolution patterns of meiotic recombination across the human major histocompatibility complex. Am. J. Hum. Genet. 71, 759–776. doi: 10.1086/342973
Daly, M. J., Rioux, J. D., Schaffner, S. F., Hudson, T. J., and Lander, E. S. (2001). High-resolution haplotype structure in the human genome. Nat. Genet. 29, 229–232. doi: 10.1038/ng1001-229
Daskalakis, M., Brocks, D., Sheng, Y.-H., Islam, M. S., Ressnerova, A., Assenov, Y., et al. (2018). Reactivation of endogenous retroviral elements via treatment with DNMT- and HDAC-inhibitors. Cell Cycle 17, 811–822. doi: 10.1080/15384101.2018.1442623
Dawkins, R. L., Christiansen, F. T., Kay, P. H., Garlepp, M., Mccluskey, J., Hollingsworth, P. N., et al. (1983). Disease associations with complotypes, supratypes and haplotypes. Immunol. Rev. 70, 5–22. doi: 10.1111/j.1600-065X.1983.tb00707.x
Dawkins, R., Leelayuwat, C., Gaudieri, S., Tay, G., Hui, J., Cattley, S., et al. (1999). Genomics of the major histocompatibility complex: haplotypes, duplication, retroviruses and disease. Immunol. Rev. 167, 275–304. doi: 10.1111/j.1600-065X.1999.tb01399.x
de Bakker, P. I. W., McVean, G., Sabeti, P. C., Miretti, M. M., Green, T., Marchini, J., et al. (2006). A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat. Genet. 38, 1166–1172. doi: 10.1038/ng1885
Degli-Esposti, M. A., Leaver, A. L., Christiansen, F. T., Witt, C. S., Abraham, L. J., and Dawkins, R. L. (1992). Ancestral haplotypes: conserved population MHC haplotypes. Hum. Immunol. 34, 242–252. doi: 10.1016/0198-8859(92)90023-G
Dorak, M. T., Shao, W., Machulla, H. K. G., Lobashevsky, E. S., Tang, J., Park, M. H., et al. (2006). Conserved extended haplotypes of the major histocompatibility complex: further characterization. Genes Immun. 7, 450–467. doi: 10.1038/sj.gene.6364315
Dover, G. A. (1993). Evolution of genetic redundancy for advanced players. Curr. Opin. Genet. Dev. 3, 902–910. doi: 10.1016/0959-437X(93)90012-E
Doxiadis, G. G. M., de Groot, N., and Bontrop, R. E. (2008). Impact of endogenous intronic retroviruses on major histocompatibility complex class II diversity and stability. J. Virol. 82, 6667–6677. doi: 10.1128/JVI.00097-08
Druet, T., and Farnir, F. P. (2011). Modeling of identity-by-descent processes along a chromosome between haplotypes and their genotyped ancestors. Genetics 188, 409–419. doi: 10.1534/genetics.111.127720
Dunn, D. S., Inoko, H., and Kulski, J. K. (2006). The association between non-melanoma skin cancer and a young dimorphic Alu element within the major histocompatibility complex class I genomic region. Tissue Antigens 68, 127–134. doi: 10.1111/j.1399-0039.2006.00631.x
Ebert, P., Audano, P. A., Zhu, Q., Rodriguez-Martin, B., Porubsky, D., Bonder, M. J., et al. (2021). Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 372:eabf7117. doi: 10.1126/science.abf7117
Farina, F., Picascia, S., Pisapia, L., Barba, P., Vitale, S., Franzese, A., et al. (2019). HLA-DQA1 and HLA-DQB1 alleles, conferring susceptibility to celiac disease and type 1 diabetes, are more expressed than non-predisposing alleles and are coordinately regulated. Cells 8:751. doi: 10.3390/cells8070751
Ferreira, R. C., Pan-Hammarström, Q., Graham, R. R., Fontán, G., Lee, A. T., Ortmann, W., et al. (2012). High-Density SNP mapping of the HLA region identifies multiple independent susceptibility loci associated with selective IgA deficiency. PLoS Genet. 8:e1002476. doi: 10.1371/journal.pgen.1002476
Gabriel, S. B., Schaffner, S. F., Nguyen, H., Moore, J. M., Roy, J., Blumenstiel, B., et al. (2002). The structure of haplotype blocks in the human genome. Science 296, 2225–2229. doi: 10.1126/science.1069424
Gambelunghe, G., Kockum, I., Bini, V., Giorgi, G. D., Celi, F., Betterle, C., et al. (2005). Retrovirus-like long-terminal repeat DQ-LTR13 and genetic susceptibility to Type 1 diabetes and autoimmune Addison’s disease. Diabetes 54, 900–905. doi: 10.2337/diabetes.54.3.900
Gambino, C. M., Aiello, A., Accardi, G., Caruso, C., and Candore, G. (2018). Autoimmune diseases and 8.1 ancestral haplotype: an update. HLA 92, 137–143. doi: 10.1111/tan.13305
Goodin, D. S., Khankhanian, P., Gourraud, P.-A., and Vince, N. (2018). Highly conserved extended haplotypes of the major histocompatibility complex and their relationship to multiple sclerosis susceptibility. PLoS One 13:e0190043. doi: 10.1371/journal.pone.0190043
Guryev, V., Smits, B. M. G., van de Belt, J., Verheul, M., Hubner, N., and Cuppen, E. (2006). Haplotype block structure is conserved across mammals. PLoS Genet. 2:e121. doi: 10.1371/journal.pgen.0020121
Hernandez, A. J., Zovoilis, A., Cifuentes-Rojas, C., Han, L., Bujisic, B., and Lee, J. T. (2020). B2 and ALU retrotransposons are self-cleaving ribozymes whose activity is enhanced by EZH2. Proc. Natl. Acad. Sci. U.S.A. 117, 415–425. doi: 10.1073/pnas.1917190117
Horton, R., Gibson, R., Coggill, P., Miretti, M., Allcock, R. J., Almeida, J., et al. (2008). Variation analysis and gene annotation of eight MHC haplotypes: the MHC haplotype project. Immunogenetics 60, 1–18. doi: 10.1007/s00251-007-0262-2
Huang, M., Tu, J., and Lu, Z. (2017). Recent advances in experimental whole genome haplotyping methods. Int. J. Mol. Sci. 18:1944. doi: 10.3390/ijms18091944
Hubley, R., Finn, R. D., Clements, J., Eddy, S. R., Jones, T. A., Bao, W., et al. (2016). The Dfam database of repetitive DNA families. Nucleic Acids Res. 44, D81–D89. doi: 10.1093/nar/gkv1272
Human Genome Structural Variation Consortium, Porubsky, D., Ebert, P., Audano, P. A., Vollger, M. R., Harvey, W. T., et al. (2021). Fully phased human genome assembly without parental data using single-cell strand sequencing and long reads. Nat. Biotechnol. 39, 302–308. doi: 10.1038/s41587-020-0719-5
International Human Genome Sequencing Consortium, (2001). Initial sequencing and analysis of the human genome. Nature 409, 860–921. doi: 10.1038/35057062
Jeffreys, A. J., Holloway, J. K., Kauppi, L., May, C. A., Neumann, R., Slingsby, M. T., et al. (2004). Meiotic recombination hot spots and human DNA diversity. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 359, 141–152. doi: 10.1098/rstb.2003.1372
Jeffreys, A. J., Kauppi, L., and Neumann, R. (2001). Intensely punctate meiotic recombination in the class II region of the major histocompatibility complex. Nat. Genet. 29, 217–222. doi: 10.1038/ng1001-217
Kaczkowski, B., Tanaka, Y., Kawaji, H., Sandelin, A., Andersson, R., Itoh, M., et al. (2016). Transcriptome analysis of recurrently deregulated genes across multiple cancers identifies new pan-cancer biomarkers. Cancer Res. 76, 216–226. doi: 10.1158/0008-5472.CAN-15-0484
Katzourakis, A., Pereira, V., and Tristem, M. (2007). Effects of recombination rate on human endogenous retrovirus fixation and persistence. J. Virol. 81, 10712–10717. doi: 10.1128/JVI.00410-07
Kauppi, L., Jasin, M., and Keeney, S. (2007). Meiotic crossover hotspots contained in haplotype block boundaries of the mouse genome. Proc. Natl. Acad. Sci. U.S.A. 104, 13396–13401. doi: 10.1073/pnas.0701965104
Kauppi, L., Jeffreys, A. J., and Keeney, S. (2004). Where the crossovers are: recombination distributions in mammals. Nat. Rev. Genet. 5, 413–424. doi: 10.1038/nrg1346
Kauppi, L., Sajantila, A., and Jeffreys, A. (2003). Recombination hotspots rather than population history dominate linkage disequilibrium in the MHC class II region. Hum. Mol. Genet. 12, 33–40. doi: 10.1093/hmg/ddg008
Kauppi, L., Stumpf, M. P. H., and Jeffreys, A. J. (2005). Localized breakdown in linkage disequilibrium does not always predict sperm crossover hot spots in the human MHC class II region. Genomics 86, 13–24. doi: 10.1016/j.ygeno.2005.03.011
Kennedy, A. E., Ozbek, U., and Dorak, M. T. (2017). What has GWAS done for HLA and disease associations? Int. J. Immunogenet. 44, 195–211. doi: 10.1111/iji.12332
Kent, T. V., Uzunoviæ, J., and Wright, S. I. (2017). Coevolution between transposable elements and recombination. Philos. Trans. R. Soc. B Biol. Sci. 372, 20160458. doi: 10.1098/rstb.2016.0458
Kong, A., Thorleifsson, G., Gudbjartsson, D. F., Masson, G., Sigurdsson, A., Jonasdottir, A., et al. (2010). Fine-scale recombination rate differences between sexes, populations and individuals. Nature 467, 1099–1103. doi: 10.1038/nature09525
Konkel, M. K., and Batzer, M. A. (2010). A mobile threat to genome stability: the impact of non-LTR retrotransposons upon the human genome. Semin. Cancer Biol. 20, 211–221. doi: 10.1016/j.semcancer.2010.03.001
Krach, K., Pani, M. A., Seidl, C., Autreve, J. V., der Auwera, B. J. V., Gorus, F. K., et al. (2003). DQ-LTR13 modifies Type 1 diabetes (IDDM) susceptibility on high risk DQ haplotypes: reply to the comments of Pascual et al. Diabetologia 46, 870–871. doi: 10.1007/s00125-003-1114-8
Kulski, J. K., Anzai, T., Shiina, T., and Inoko, H. (2004). Rhesus macaque class i duplicon structures, organization, and evolution within the alpha block of the major histocompatibility complex. Mol. Biol. Evol. 21, 2079–2091. doi: 10.1093/molbev/msh216
Kulski, J. K., Gaudieri, S., and Dawkins, R. L. (2000a). “Transposable elements and the metamerismatic evolution of the HLA class I region,” in Major Histocompatibility Complex, ed. M. Kasahara, (Tokyo: Springer Japan), 158–177. doi: 10.1007/978-4-431-65868-9_11
Kulski, J. K., Gaudieri, S., and Dawkins, R. L. (2000b). Using Alu J elements as molecular clocks to trace the evolutionary relationships between duplicated HLA class I genomic segments. J. Mol. Evol. 50, 510–519. doi: 10.1007/s002390010054
Kulski, J. K., Gaudieri, S., Bellgard, M., Balmer, L., Giles, K., Inoko, H., et al. (1997). The evolution of MHC diversity by segmental duplication and transposition of retroelements. J. Mol. Evol. 45, 599–609.
Kulski, J. K., Gaudieri, S., Inoko, H., and Dawkins, R. L. (1999a). Comparison between two human endogenous retrovirus (HERV)-Rich regions within the major histocompatibility complex. J. Mol. Evol. 48, 675–683. doi: 10.1007/PL00006511
Kulski, J. K., Gaudieri, S., Martin, A., and Dawkins, R. L. (1999b). Coevolution of PERB11 (MIC) and HLA class I genes with HERV-16 and retroelements by extended genomic duplication. J. Mol. Evol. 49, 84–97. doi: 10.1007/PL00006537
Kulski, J. K., Shigenari, A., and Inoko, H. (2011). Genetic variation and hitchhiking between structurally polymorphic Alu insertions and HLA-A, -B, and -C alleles and other retroelements within the MHC class I region. Tissue Antigens 78, 359–377. doi: 10.1111/j.1399-0039.2011.01776.x
Kulski, J. K., Shigenari, A., Shiina, T., and Inoko, H. (2010). Polymorphic major histocompatibility complex class II Alu insertions at five loci and their association with HLA-DRB1 and -DQB1 in Japanese and Caucasians. Tissue Antigens 76, 35–47. doi: 10.1111/j.1399-0039.2010.01465.x
Kulski, J. K., Shiina, T., Anzai, T., Kohara, S., and Inoko, H. (2002). Comparative genomic analysis of the MHC: the evolution of class I duplication blocks, diversity and complexity from shark to man. Immunol. Rev. 190, 95–122. doi: 10.1034/j.1600-065X.2002.19008.x
Kulski, J. K., Suzuki, S., and Shiina, T. (2021). SNP-Density crossover maps of polymorphic transposable elements and HLA genes within MHC class i haplotype blocks and junction. Front. Genet. 11:594318. doi: 10.3389/fgene.2020.594318
Lam, T. H., Shen, M., Chia, J.-M., Chan, S. H., and Ren, E. C. (2013). Population-specific recombination sites within the human MHC region. Heredity 111, 131–138. doi: 10.1038/hdy.2013.27
Lam, T., Tay, M., Wang, B., Xiao, Z., and Ren, E. (2015). Intrahaplotypic variants differentiate complex linkage disequilibrium within human MHC haplotypes. Sci. Rep. 5:16972. doi: 10.1038/srep16972
Lan, H., Zhou, T., Wan, Q.-H., and Fang, S.-G. (2019). Genetic diversity and differentiation at structurally varying MHC haplotypes and microsatellites in bottlenecked populations of endangered crested Ibis. Cells 8:377. doi: 10.3390/cells8040377
Larsen, C. E., Alford, D. R., Trautwein, M. R., Jalloh, Y. K., Tarnacki, J. L., Kunnenkeri, S. K., et al. (2014). Dominant sequences of human major histocompatibility complex conserved extended haplotypes from HLA-DQA2 to DAXX. PLoS Genet. 10:e1004637. doi: 10.1371/journal.pgen.1004637
Lee, Y., Lee, J., Kim, J., and Kim, Y.-J. (2020). Insertion variants missing in the human reference genome are widespread among human populations. BMC Biol. 18:167. doi: 10.1186/s12915-020-00894-1
Lloyd, S. S., Steele, E. J., and Dawkins, R. L. (2016). “Analysis of haplotype sequences,” in Next Generation Sequencing - Advances, Applications and Challenges, ed. J. K. Kulski, (Rijeka: InTechOpen). doi: 10.5772/61794
Lokki, M., and Paakkanen, R. (2019). The complexity and diversity of major histocompatibility complex challenge disease association studies. HLA 93, 3–15. doi: 10.1038/srep30381
Macel, L. M. Z., Rodrigues, S. S., Dibbern, R. S., Navarro, P. A. A., and Donadi, E. A. (2001). Association of the HLA-DRB1∗0301 and HLA-DQA1∗0501 Alleles with Graves’ disease in a population representing the gene contribution from several ethnic backgrounds. Thyroid 11, 31–35. doi: 10.1089/10507250150500630
Miretti, M. M., Walsh, E. C., Ke, X., Delgado, M., Griffiths, M., Hunt, S., et al. (2005). A high-resolution linkage-disequilibrium map of the human major histocompatibility complex and first generation of tag single-nucleotide polymorphisms. Am. J. Hum. Genet. 76, 634–646. doi: 10.1086/429393
Moolhuijzen, P., Kulski, J. K., Dunn, D. S., Schibeci, D., Barrero, R., Gojobori, T., et al. (2010). The transcript repeat element: the human Alu sequence as a component of gene networks influencing cancer. Funct. Integr. Genomics 10, 307–319. doi: 10.1007/s10142-010-0168-1
Moyes, D., Griffiths, D. J., and Venables, P. J. (2007). Insertional polymorphisms: a new lease of life for endogenous retroviruses in human disease. Trends Genet. 23, 326–333. doi: 10.1016/j.tig.2007.05.004
Myers, S., Bottolo, L., McVean, G., and Donnelly, P. (2005). A fine-scale map of recombination rates and hotspots across the human genome. Science 310, 321–324. doi: 10.1126/science.1117196
Myers, S., Bowden, R., Tumian, A., Bontrop, R. E., Freeman, C., MacFie, T. S., et al. (2010). Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science 327, 876–879. doi: 10.1126/science.1182363
Nait Saada, J., Kalantzis, G., Shyr, D., Cooper, F., Robinson, M., Gusev, A., et al. (2020). Identity-by-descent detection across 487,409 British samples reveals fine scale population structure and ultra-rare variant associations. Nat Commun. 11:6130.
Norman, P. J., Norberg, S. J., Guethlein, L. A., Nemat-Gorgani, N., Royce, T., Wroblewski, E. E., et al. (2017). Sequences of 95 human MHC haplotypes reveal extreme coding variation in genes other than highly polymorphic HLA class I and II. Genome Res. 27, 813–823. doi: 10.1101/gr.213538.116
Notredame, C., Higgins, D. G., and Heringa, J. (2000). T-coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217. doi: 10.1006/jmbi.2000.4042
Oka, A., Hayashi, H., Tomizawa, M., Okamoto, K., Suyun, L., Hui, J., et al. (2003). Localization of a non-melanoma skin cancer susceptibility region within the major histocompatibility complex by association analysis using microsatellite markers. Tissue Antigens 61, 203–210. doi: 10.1034/j.1399-0039.2003.00007.x
Pani, M. A., Seidl, C., Bieda, K., Seissler, J., Krause, M., Seifried, E., et al. (2002). Preliminary evidence that an endogenous retroviral long-terminal repeat (LTR13) at the HLA-DQB1 gene locus confers susceptibility to Addison’s disease. Clin. Endocrinol. (Oxf.) 56, 773–777. doi: 10.1046/j.1365-2265.2002.t01-1-01548.x
Park, L. (2019). Population-specific long-range linkage disequilibrium in the human genome and its influence on identifying common disease variants. Sci. Rep. 9:11380. doi: 10.1038/s41598-019-47832-y
Parvanov, E. D., Tian, H., Billings, T., Saxl, R. L., Spruce, C., Aithal, R., et al. (2017). PRDM9 interactions with other proteins provide a link between recombination hotspots and the chromosomal axis in meiosis. Mol. Biol. Cell 28, 488–499. doi: 10.1091/mbc.e16-09-0686
Payer, L. M., and Burns, K. H. (2019). Transposable elements in human genetic disease. Nat. Rev. Genet. 20, 760–772. doi: 10.1038/s41576-019-0165-8
Payer, L. M., Steranka, J. P., Yang, W. R., Kryatova, M., Medabalimi, S., Ardeljan, D., et al. (2017). Structural variants caused by Alu insertions are associated with risks for many human diseases. Proc. Natl. Acad. Sci. U.S.A. 114, E3984–E3992. doi: 10.1073/pnas.1704117114
Pratto, F., Brick, K., Khil, P., Smagulova, F., Petukhova, G. V., and Camerini-Otero, R. D. (2014). Recombination initiation maps of individual human genomes. Science 346, 1256442–1256442. doi: 10.1126/science.1256442
Qin, S., Jin, P., Zhou, X., Chen, L., and Ma, F. (2015). The role of transposable elements in the origin and evolution of MicroRNAs in human. PLoS One 10:e0131365. doi: 10.1371/journal.pone.0131365
Radwan, J., Babik, W., Kaufman, J., Lenz, T. L., and Winternitz, J. (2020). Advances in the evolutionary understanding of MHC polymorphism. Trends Genet. 36, 298–311. doi: 10.1016/j.tig.2020.01.008
Robinson, J., Barker, D. J., Georgiou, X., Cooper, M. A., Flicek, P., and Marsh, S. G. E. (2019). IPD-IMGT/HLA database. Nucleic Acids Res. 48:gkz950. doi: 10.1093/nar/gkz950
Schmid, C. (1996). Alu structure, origin, evolution, significance, and function of one-tenth of human DNA. Prog Nucleic Acids Res. Mol. Biol. 53, 283–319. doi: 10.1016/S0079-6603(08)60148-8
Schwartz, S., Zhang, Z., Frazer, K. A., Smit, A., Riemer, C., Bouck, J., et al. (2000). PipMaker—A web server for aligning two genomic DNA sequences. Genome Res. 10, 577–586. doi: 10.1101/gr.10.4.577
Shi, L., Kulski, J. K., Zhang, H., Dong, Z., Cao, D., Zhou, J., et al. (2014). Association and differentiation of MHC class I and II polymorphic Alu insertions and HLA-A, -B, -C and -DRB1 alleles in the Chinese Han population. Mol. Genet. Genomics 289, 93–101. doi: 10.1007/s00438-013-0792-2
Shiina, T., Hosomichi, K., Inoko, H., and Kulski, J. K. (2009). The HLA genomic loci map: expression, interaction, diversity and disease. J. Hum. Genet. 54, 15–39. doi: 10.1038/jhg.2008.5
Shiina, T., Inoko, H., and Kulski, J. K. (2004). An update of the HLA genomic region, locus information and disease associations: 2004. Tissue Antigens 64, 631–649. doi: 10.1111/j.1399-0039.2004.00327.x
Slatkin, M. (2008). Linkage disequilibrium — understanding the evolutionary past and mapping the medical future. Nat. Rev. Genet. 9, 477–485. doi: 10.1038/nrg2361
Smit, A. F. A., and Riggs, A. D. (1996). Tiggers and other DNA transposon fossils in the human genome. Proc. Natl. Acad. Sci. U.S.A. 93, 1443–1448. doi: 10.1073/pnas.93.4.1443
Smith, W. P., Vu, Q., Li, S. S., Hansen, J. A., Zhao, L. P., and Geraghty, D. E. (2006). Toward understanding MHC disease associations: partial resequencing of 46 distinct HLA haplotypes. Genomics 87, 561–571. doi: 10.1016/j.ygeno.2005.11.020
Spirito, G., Mangoni, D., Sanges, R., and Gustincich, S. (2019). Impact of polymorphic transposable elements on transcription in lymphoblastoid cell lines from public data. BMC Bioinformatics 20(Suppl. 9):495. doi: 10.1186/s12859-019-3113-x
Suzuki, S., Ranade, S., Osaki, K., Ito, S., Shigenari, A., Ohnuki, Y., et al. (2018). Reference grade characterization of polymorphisms in full-length HLA class I and II genes with short-read sequencing on the ION PGM system and long-reads generated by single molecule, real-time sequencing on the PacBio platform. Front. Immunol. 9:2294. doi: 10.3389/fimmu.2018.02294
Tamiya, G., Shinya, M., Imanishi, T., Ikuta, T., Makino, S., Okamoto, K., et al. (2005). Whole genome association study of rheumatoid arthritis using 27 039 microsatellites. Hum. Mol. Genet. 14, 2305–2321. doi: 10.1093/hmg/ddi234
Tìšickı, M., and Vinkler, M. (2015). Trans-Species polymorphism in immune genes: general pattern or MHC-Restricted phenomenon? J. Immunol. Res. 2015:838035. doi: 10.1155/2015/838035
Tewhey, R., Bansal, V., Torkamani, A., Topol, E. J., and Schork, N. J. (2011). The importance of phase information for human genomics. Nat. Rev. Genet. 12, 215–223. doi: 10.1038/nrg2950
The 1000 Genomes Project Consortium, Auton, A., and Abecasis, G. R. (2015a). A global reference for human genetic variation. Nature 526, 68–74. doi: 10.1038/nature15393.
The 1000 Genomes Project Consortium, Sudmant, P. H., Rausch, T., Gardner, E. J., Handsaker, R. E., Abyzov, A., et al. (2015b). An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81. doi: 10.1038/nature15394
The International HapMap Consortium, (2007). A second generation human haplotype map of over 3.1 million SNPs. Nature 449, 851–861. doi: 10.1038/nature06258
Thomas, J., Perron, H., and Feschotte, C. (2018). Variation in proviral content among human genomes mediated by LTR recombination. Mob. DNA 9:36. doi: 10.1186/s13100-018-0142-3
Thompson, E. A. (2013). Identity by descent: variation in meiosis, across genomes, and in populations. Genetics 194, 301–326. doi: 10.1534/genetics.112.148825
Traherne, J. A. (2008). Human MHC architecture and evolution: implications for disease association studies. Int. J. Immunogenet. 35, 179–192. doi: 10.1111/j.1744-313X.2008.00765.x
Traherne, J. A., Horton, R., Roberts, A. N., Miretti, M. M., Hurles, M. E., Stewart, C. A., et al. (2006). Genetic analysis of completely sequenced disease-associated MHC haplotypes identifies shuffling of segments in recent human history. PLoS Genet. 2:e9. doi: 10.1371/journal.pgen.0020009
Trela, M., Nelson, P. N., and Rylance, P. B. (2016). The role of molecular mimicry and other factors in the association of Human Endogenous Retroviruses and autoimmunity. APMIS 124, 88–104. doi: 10.1111/apm.12487
Trowsdale, J. (2011). The MHC, disease and selection. Immunol. Lett. 137, 1–8. doi: 10.1016/j.imlet.2011.01.002
Tucker, J. M., and Glaunsinger, B. A. (2017). Host noncoding retrotransposons induced by DNA viruses: a SINE of infection? J. Virol. 91:e00982-17. doi: 10.1128/JVI.00982-17
Tugnet, N., Rylance, P., Roden, D., Trela, M., and Nelson, P. (2013). Human endogenous retroviruses (HERVs) and autoimmune rheumatic disease: is there a link? Open Rheumatol. J. 7, 13–21. doi: 10.2174/1874312901307010013
Vadva, Z., Larsen, C. E., Propp, B. E., Trautwein, M. R., Alford, D. R., and Alper, C. A. (2019). A New pedigree-based SNP haplotype method for genomic polymorphism and genetic studies. Cells 8:835. doi: 10.3390/cells8080835
Valdes, A. M., Erlich, H. A., Carlson, J., Varney, M., Moonsamy, P. V., and Noble, J. A. (2012). Use of class I and class II HLA loci for predicting age at onset of type 1 diabetes in multiple populations. Diabetologia 55, 2394–2401. doi: 10.1007/s00125-012-2608-z
van Oosterhout, C. (2009). A new theory of MHC evolution: beyond selection on the immune genes. Proc. R. Soc. B Biol. Sci. 276, 657–665. doi: 10.1098/rspb.2008.1299
Vandiedonck, C., and Knight, J. C. (2009). The human Major Histocompatibility Complex as a paradigm in genomics research. Brief. Funct. Genomic. Proteomic. 8, 379–394. doi: 10.1093/bfgp/elp010
Venter, J. C., Adams, M. D., Myers, E. W., Li, P. W., Mural, R. J., Sutton, G. G., et al. (2001). The sequence of the human genome. Science 291, 1304–1351. doi: 10.1126/science.1058040
Villa-Angulo, R., Matukumalli, L. K., Gill, C. A., Choi, J., Van Tassell, C. P., and Grefenstette, J. J. (2009). High-resolution haplotype block structure in the cattle genome. BMC Genet. 10:19. doi: 10.1186/1471-2156-10-19
Wallace, A. D., Wendt, G. A., Barcellos, L. F., de Smith, A. J., Walsh, K. M., Metayer, C., et al. (2018). To ERV is human: a phenotype-wide scan linking polymorphic human endogenous retrovirus-K insertions to complex phenotypes. Front. Genet. 9:298. doi: 10.3389/fgene.2018.00298
Wang, L., Norris, E. T., and Jordan, I. K. (2017). Human retrotransposon insertion polymorphisms are associated with health and disease via gene regulatory phenotypes. Front. Microbiol. 8:1418. doi: 10.3389/fmicb.2017.01418
Wissemann, W. T., Hill-Burns, E. M., Zabetian, C. P., Factor, S. A., Patsopoulos, N., Hoglund, B., et al. (2013). Association of Parkinson disease with structural and regulatory variants in the HLA region. Am. J. Hum. Genet. 93, 984–993. doi: 10.1016/j.ajhg.2013.10.009
Wu, H., Chen, Y., Zhu, H., Zhao, M., and Lu, Q. (2019). The pathogenic role of dysregulated epigenetic modifications in autoimmune diseases. Front. Immunol. 10:2305. doi: 10.3389/fimmu.2019.02305
Yüksel, ş, Kucukazman, S. O., KarataŞ, G. S., Ozturk, M. A., Prombhul, S., and Hirankarn, N. (2016). Methylation status of Alu and LINE-1 interspersed repetitive sequences in Behcet’s disease patients. BioMed Res. Int. 2016, 1–9. doi: 10.1155/2016/1393089
Yunis, E. J., Larsen, C. E., Fernandez-Viña, M., Awdeh, Z. L., Romero, T., Hansen, J. A., et al. (2003). Inheritable variable sizes of DNA stretches in the human MHC: conserved extended haplotypes and their fragments or blocks. Tissue Antigens 62, 1–20. doi: 10.1034/j.1399-0039.2003.00098.x
Zhou, Y., Browning, B. L., and Browning, S. R. (2020a). Population-specific recombination maps from segments of identity by descent. Am. J. Hum. Genet. 107, 137–148.
Keywords: major histocompatibility complex, haplotypes, DNA sequences, Retroelements, single-nucleotide polymorphism-density crossovers, polymorphisms, indels, shuffling
Citation: Kulski JK, Suzuki S and Shiina T (2021) Haplotype Shuffling and Dimorphic Transposable Elements in the Human Extended Major Histocompatibility Complex Class II Region. Front. Genet. 12:665899. doi: 10.3389/fgene.2021.665899
Received: 09 February 2021; Accepted: 12 April 2021;
Published: 28 May 2021.
Edited by:
Ramcés Falfán-Valencia, Instituto Nacional de Enfermedades Respiratorias-México (INER), MexicoReviewed by:
Martin Maiers, National Marrow Donor Program, United StatesCopyright © 2021 Kulski, Suzuki and Shiina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jerzy K. Kulski, a3Vsc2tpQG1lLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.