
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Genet., 29 September 2021
Sec. Toxicogenomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.664717
This article is part of the Research TopicGene-environment interactions: impact of inorganic and organic toxicants on redox status, DNA stability and epigenetic profileView all 5 articles
 Victor D. Martinez1,2,3,4*
Victor D. Martinez1,2,3,4* Wan L. Lam4,5
Wan L. Lam4,5Inorganic arsenic is a well-established human carcinogen, able to induce genetic and epigenetic alterations. More than 200 million people worldwide are exposed to arsenic concentrations in drinking water exceeding the recommended WHO threshold (10μg/l). Additionally, chronic exposure to levels below this threshold is known to result in long-term health effects in humans. The arsenic-related health effects in humans are associated with its biotransformation process, whereby the resulting metabolites can induce molecular damage that accumulates over time. The effects derived from these alterations include genomic instability associated with oxidative damage, alteration of gene expression (including coding and non-coding RNAs), global and localized epigenetic reprogramming, and histone posttranslational modifications. These alterations directly affect molecular pathways involved in the onset and progression of many conditions that can arise even decades after the exposure occurs. Importantly, arsenic metabolites generated during its biotransformation can also pass through the placental barrier, resulting in fetal exposure to this carcinogen at similar levels to those of the mother. As such, more immediate effects of the arsenic-induced molecular damage can be observed as detrimental effects on fetal development, pregnancy, and birth outcomes. In this review, we focus on the genetic and epigenetic damage associated with exposure to low levels of arsenic, particularly those affecting early developmental stages. We also present how these alterations occurring during early life can impact the development of certain diseases in adult life.
Exposure to arsenic in drinking water has health effects in humans, including disorders of the skin, alterations in the vascular/respiratory systems, and cancer (World Health Organization, 2004; Podgorski and Berg, 2020). The WHO has recommended that the levels of arsenic in drinking water should not exceed 10μg/l (Gorchev and Ozolins, 1984). Worldwide, ~220 million people are potentially exposed to high arsenic concentrations (Podgorski and Berg, 2020).
Genetic and epigenetic alterations have been associated with exposure to arsenic (Figure 1). Damage at the genetic level is associated with the generation of reactive oxygen species (ROS), while epigenetic effects are linked with arsenic metabolism. Gestational (in utero) exposure to arsenic has been also associated with health effects arising in pre- and perinatal stages as well as during childhood, as well as an increased risk of developing diseases during adulthood (Naujokas et al., 2013; Quansah et al., 2015; Howard, 2018; Navas-Acien et al., 2019; Salmeri et al., 2020). An increasing number of publications illustrates the recent interest in the association between prenatal exposure and health effects (Supplementary Figure 1).
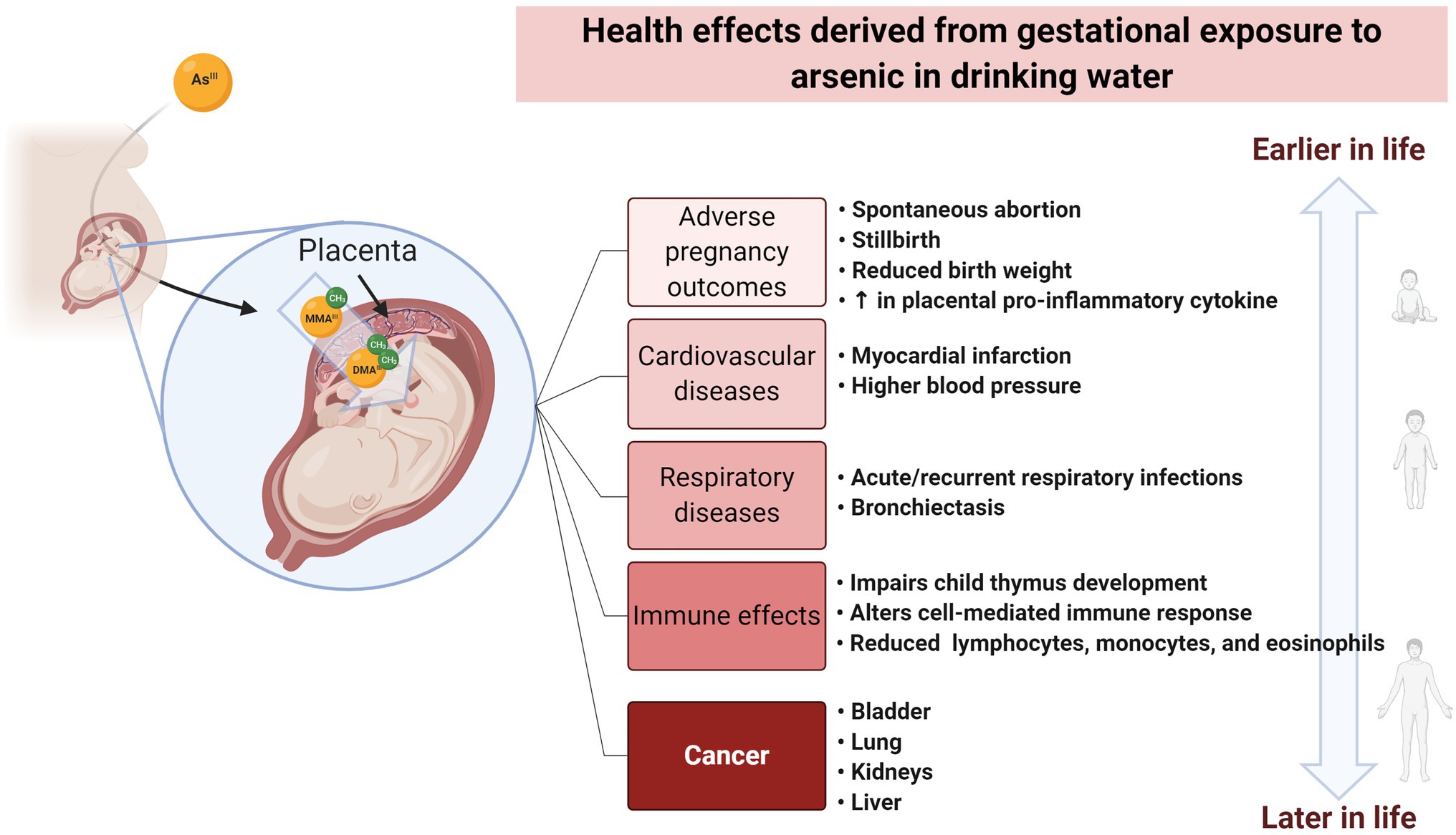
Figure 1. Health effects derived from gestational exposure to arsenic in drinking water. Arsenic ingested by the mother can cross the placental barrier. Gestational exposure to arsenic has been associated with effects at pre-and perinatal stages, as well as during childhood. These effects include adverse pregnancy outcomes and alterations to the cardiovascular, respiratory, and immune system. Moreover, in utero exposure has been correlated with an increased risk of developing cancer and other types of diseases during the adult life. Created with BioRender.com.
Arsenic is found in its inorganic forms: arsenite (As[III], oxidation state+3) and arsenate (As[V], oxidation state+5). Arsenate is the main form present in drinking water and is readily absorbed by the gastrointestinal tract (Figure 2). The metabolization occurs in the liver, where As[V] is reduced to As[III], in a reaction catalyzed by the purine nucleoside phosphorylase (PNP) and glutathione-S-transferase omega. Subsequently, additional redox reactions involving As[V] and As[III] occur (Drobna et al., 2009; Minatel et al., 2018).
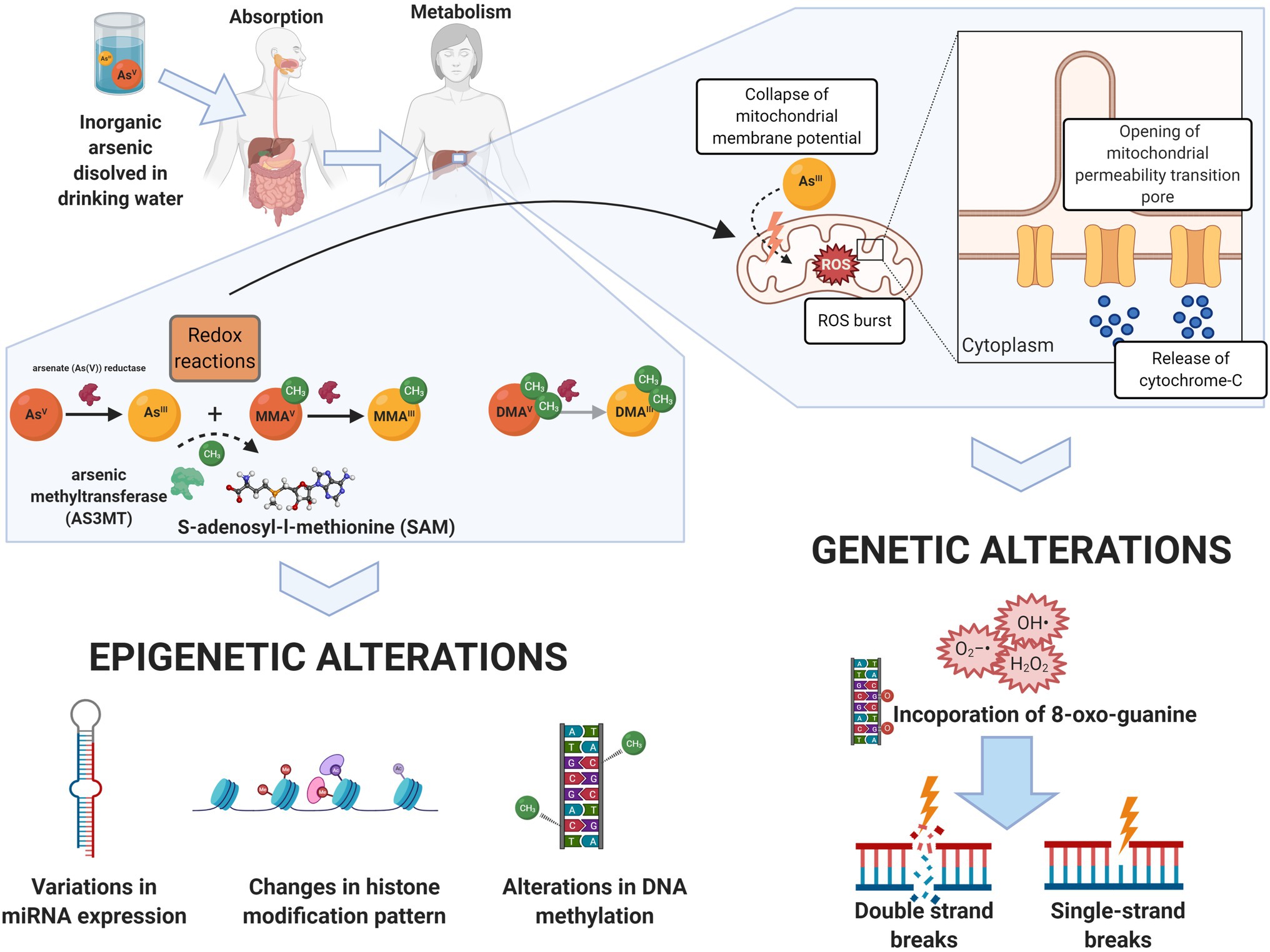
Figure 2. Arsenic biotransformation as mechanisms for molecular damage. Arsenic dissolved in drinking water (mainly as arsenate, in its oxidation state +5, As[V]) is readily absorbed by the gastrointestinal tract. Biotransformation of arsenic occurs in the liver, where arsenite (As[III]) and As[V] goes through a series of redox reactions, resulting in the generation of mono and dimethylated arsenic intermediaries: Monomethylarsonous acid (MMA[III]), monomethylarsonic acid (MMA[V]), dimethylarsinous acid (DMA[III]), and dimethylarsinic acid (DMA[V]). S-adenosylmethionine (SAM) is the methyl donor during arsenic biotransformation to both oxidation state III and oxidation state V arsenic species. The transfer of a methyl group from SAM to trivalent arsenicals is catalyzed by the arsenic6 (+3 oxidation state) methyltransferase (AS3MT), producing methylated and dimethylated arsenicals. Epigenetic alterations, including global and local DNA methylation changes, alteration of miRNA expression, and changes in the histone modification pattern, are associated with this process. On the other hand, the redox reactions involved in arsenic biotransformation lead to ROS-mediated disruption of the mitochondrial electron transport chain, which subsequently results in DNA damage such as double- and single-strand breaks, and specific mutational patterns. Created with BioRender.com.
Sequential addition of methyl groups to the intermediaries generates mono- and dimethylated arsenicals (+3/+5). The arsenic [+3] methyltransferase (AS3MT) catalyzes the transfer of a methyl group from S-adenosylmethionine (SAM) to trivalent arsenicals. This reaction generates monomethylarsonous acid (MMA[III]), monomethylarsonic acid (MMA[V]), dimethylarsinous acid (DMA[III]), and dimethylarsinic acid (DMA[V]; Dopp et al., 2004; Antonelli et al., 2014; see Figure 2). This process facilitates its excretion through urine; however, prolonged exposure (even at low levels) might convert this detoxification process into one of the possible mechanisms inducing genetic and epigenetic alterations linked to arsenic exposure.
Polymorphisms in genes involved in the arsenic biotransformation pathway are considered a major cause of inter-individual variations in the susceptibility to arsenic-related health conditions (reviewed in Paul et al., 2015; Minatel et al., 2018). For example, single nucleotide variants (SNVs) in AS3MT, PNP, and GSTO(1/2) are significantly associated with arsenic-induced dermatological lesions (De Chaudhuri et al., 2008; Paul et al., 2015). It has been proposed that these SNVs could modify physicochemical features of these enzymes particularly in regions close to the arsenic binding site (De Chaudhuri et al., 2008).
Genetic and epigenetic mechanisms are involved in arsenic-induced health effects (summarized in Figure 2 and Table 1).
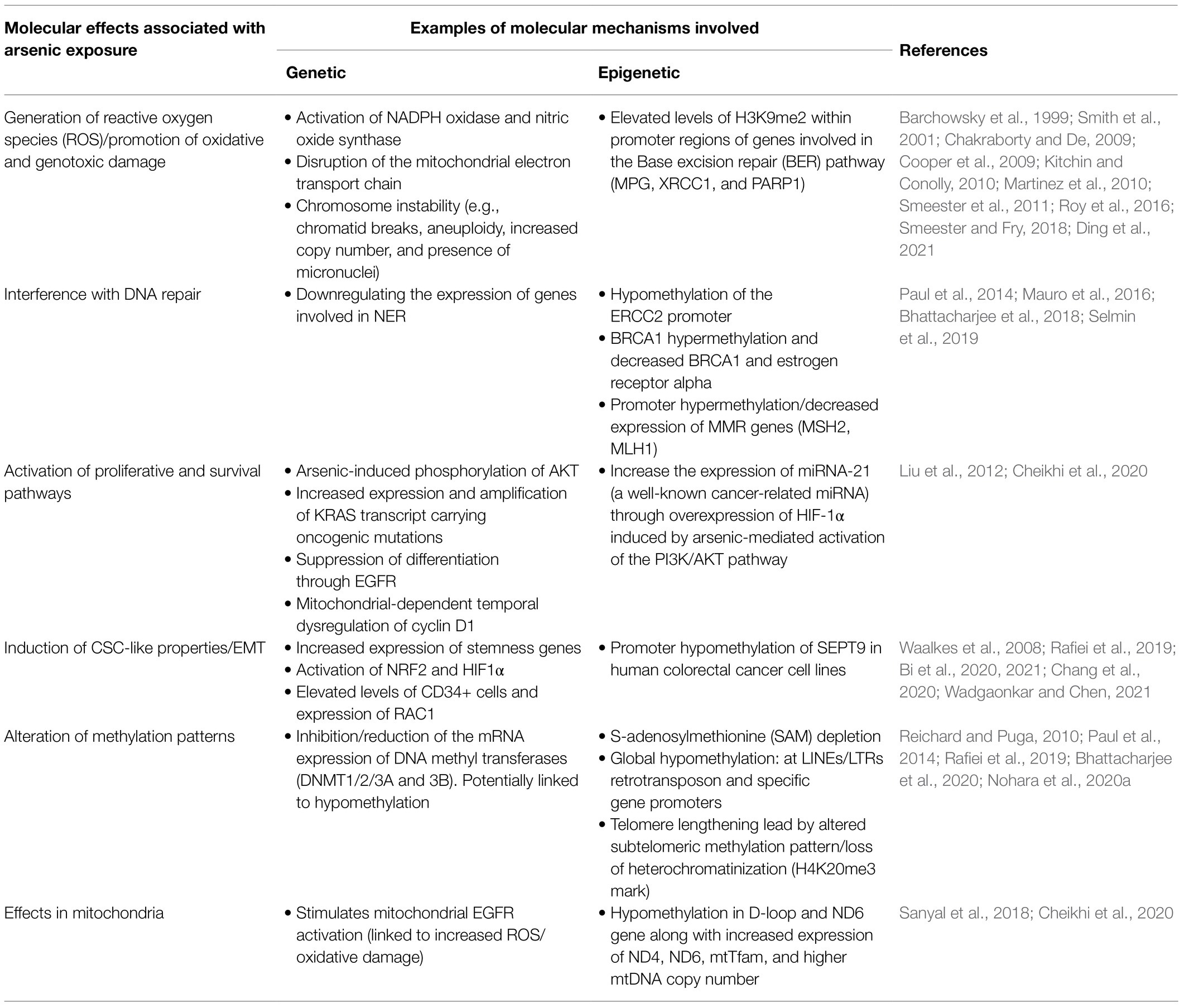
Table 1. Genetic and epigenetic molecular changes associated with arsenic exposure.
Genetic mechanisms associated with arsenic exposure include the promotion of oxidative and genotoxic damage and a decrease in DNA repair (Kitchin and Conolly, 2010; Smeester et al., 2011; Minatel et al., 2018; Smeester and Fry, 2018). At the epigenetic level, arsenic has been shown to induce DNA hypo- and hypermethylation, disrupt the expression of microRNAs (miRNAs) and alter the histone modification pattern (Smeester et al., 2011; Chervona et al., 2012; see Figure 2 or Table 1). Some of these mechanisms have been observed as a response to gestational exposure to arsenic (Pilsner et al., 2012; Nohara et al., 2020b).
Arsenic can act as a co-mutagen with other chemicals and UV light, leading to increased oxidative stress and subsequent DNA damage (Hartwig et al., 1997; Rossman et al., 2004). Additionally, It has been shown that arsenic is associated with a distinctive mutational spectrum in lung squamous cell carcinomas arising in never smokers (Martinez et al., 2013). The toxicity of the trivalent forms of arsenic is higher than the pentavalent species, owing to higher affinity for sulfhydryl groups and cysteine-binding capacity, resulting in loss of protein activity, impaired recruitment, and loss of the protein–protein/protein-DNA interaction capacity (Shen et al., 2013; Moe et al., 2016; Hirano, 2020).
The cycling between As[III] and As[V] generates oxygen-derived radicals, such as superoxide anion (O2•−), hydroxyl radical (•OH), hydrogen peroxide (H2O2), singlet oxygen (1O2), and peroxyl radicals (Halliwell and Whiteman, 2004; see Figure 2). One of the main mechanisms of arsenic-induced ROS generation involves the disruption of the mitochondrial electron transport chain via ROS accumulation and/or by inducing permeability transition linked to the high affinity of arsenic for sulfhydryl groups, leading to apoptosis (Pulido and Parrish, 2003; Liu et al., 2005). MMA[III] can also induce the formation of ROS, promoting the formation of DNA adducts, double-strand breaks, and specific mutational patterns (Wang et al., 2001; Kligerman et al., 2010; Naranmandura et al., 2011). DMA[III] seems to be more readily taken up by cells and capable of inducing the formation of micronuclei due to the generation of free radicals (Kenyon and Hughes, 2001; Zamora et al., 2014). The levels of mutagenic oxidative DNA lesions (8-oxo-7,8-dihydro-2'-deoxyguanosine and 8-nitroguanine), DNA strand breaks, and micronucleus frequency in cord blood have been shown to be associated with arsenic exposure in utero (Navasumrit et al., 2019).
Arsenic interferes with DNA repair by downregulating the expression of genes involved in the nucleotide excision repair process (Walter et al., 2007; Nollen et al., 2009; Osmond et al., 2010; Minatel et al., 2018). Arsenic also disrupts proliferative and survival pathways (Wang et al., 2014a; Person et al., 2015), such as PI3K/Akt signaling, through arsenic-induced phosphorylation of AKT (Gao et al., 2004; Liu et al., 2012, 2020). Likewise, it has been shown that arsenic can induce cancer stem cell (CSC)-like properties. For example, arsenic promotes a metabolic shift from mitochondrial TCA cycle to glycolysis through activation of NRF2 and HIF1α, a defining characteristic of arsenic-induced CSCs (Bi et al., 2020; Wadgaonkar and Chen, 2021). Arsenic can also preserve a germinative state in cultured human epidermal cells and alters signal transduction related to proliferative potential (Patterson et al., 2005). Fetal arsenic exposure can also increase the effects of skin carcinogens (topical 12-O-tetradecanoyl phorbol-13-acetate, TPA) in mice by elevating the levels of CD34+ cells and expression of RAC1 (involved in self-renewal stimulation; Waalkes et al., 2008). Bronchial epithelial cells exposed to arsenic, acquired CSC-like features such as asymmetric division, self-renewal, and increased expression of stemness genes (Chang et al., 2020; Bi et al., 2021).
Arsenic-mediated malignant transformation has been linked to disruption of the KRAS gene. Arsenite-mediated transformation of human prostate epithelia has been linked to increased expression and amplification of KRAS transcript carrying oncogenic mutations (G12S and A59T; Merrick et al., 2019). Additionally, long-term arsenite exposure can cause activation of human endogenous retroviruses related to KRAS gene fusion and oncogenic amplification, which has been associated with arsenic-mediated epigenetic effects resulting in de-repression of retroviral sequences (Merrick et al., 2020).
Prolonged exposure to arsenic can lead to the depletion of SAM. The depletion is likely due to arsenite methylation competing with many cellular processes that require methyl groups provided by SAM, including DNA, RNA, and histone methylation (Simeonova and Luster, 2000; Reichard et al., 2007; Ouyang et al., 2020). Thus, the arsenic-induced depletion of the cellular pool of methyl groups can have an impact on epigenetic reprogramming and thus modify disease susceptibility.
Exposure to arsenic has been linked to both hypo- and hypermethylation affecting gene expression in humans (Bailey and Fry, 2014). Global hypomethylation can be explained by the competition for the pool of SAM-provided methyl groups and by the arsenic-mediated inhibition of DNA methyltransferase (DNMT) enzymes (Reichard and Puga, 2010). In addition to global hypomethylation, arsenic reduces methylation at gene promoters and specific regions of the DNA. Long-term (20days) exposure to low levels (1 and 0.1μM) of inorganic arsenic induces promoter hypomethylation of SEPT9 in human colorectal cancer cell lines, which is correlated with epithelial-mesenchymal transition (Rafiei et al., 2019). Similarly, arsenic induces hypomethylation of the ERCC2 promoter both in vivo and in vitro, leading to ERCC2 overexpression, inhibition of Cdk-activating kinase complex release, and decrease of p53 phosphorylation (serine 392; Paul et al., 2014). The disruption of ERCC2 and the downstream effects might contribute to increased DNA damage observed among arsenic exposed individuals. On the other hand, arsenic can also decrease methylation levels in specific regions of the DNA harboring non-coding sequences. Long interspersed nuclear element-1 (LINE-1). LINE-1 methylation levels are lower among individuals living in arsenic-endemic areas compared to those in non-endemic areas, particularly among female individuals (Hossain et al., 2017). The mechanisms governing hypomethylation of specific DNA sequences linked to arsenic exposure are not yet fully elucidated. It has been proposed that gestational arsenic exposure can increase hypomethylated cytosines, with accumulation in the promoter regions of the active full-length L1MdA subfamily of LINEs, potentially enhancing retrotransposition and cryptic promoter activity of 5' long-terminal repeats for coding genes and non-coding RNAs (Nohara et al., 2020a). LINE-1 hypomethylation has been associated with cardiovascular disease (CVD; Muka et al., 2016), which is also a hallmark of arsenic-induced chronic conditions. Arsenic also induces global hypomethylation (Alu/LINE-1 methylation status) in children chronically exposed to arsenic (even at low levels), resulting in genotoxic stress (Alegría-Torres et al., 2016; Bandyopadhyay et al., 2016).
The mechanisms underlying arsenic-induced gene-level hypermethylation are still unknown. A positive correlation has been described between the concentration of arsenic in the mother’s urine and global DNA hypomethylation in an infant’s cord blood sample (Pilsner et al., 2012). The effects of prenatal exposure to arsenic on the fetal epigenome have been also observed in a sex-dependent manner. In utero exposure to arsenic (measured through maternal urinary arsenic concentration) induced the expression of a completely different set of genes involved in epigenetic pathways in male vs. female fetal placentas (Winterbottom et al., 2019). On the other hand, in utero and early life exposure has been associated with increased DNA methylation in the promoter region of extracellular matrix remodeling gene MMP9, accompanied by a reduction of protein levels (Chicoine et al., 2002; Gonzalez-Cortes et al., 2017). Similarly, gestational exposure to arsenic (before the 25th week) has been associated with increased methylation in repetitive sequences, as well as with higher methylation in promoter regions of tumor suppressor genes (CDKN2A and TP53) in umbilical cord leukocytes (Kile et al., 2012; Cardenas et al., 2015).
Recent studies indicate that exposure to arsenic can affect the methylation status of human mitochondrial DNA (mtDNA). Arsenic is able to induce hypomethylation of the D-loop region (critical for mtDNA replication and transcription) and of the ND6 gene, along with increased expression of ND4, ND6, mtTfam and higher mtDNA copy number in individuals exposed to arsenic in drinking water (Sanyal et al., 2018). Arsenic-induced mitochondrial damage has been also associated with increased ROS/oxidative damage following exposure. For example, exposure to low levels of arsenic stimulates mitochondrial EGFR activation as an upstream mechanism for arsenic dysregulation of reserve cells, increasing mtROS, and proliferative signaling in Murine C2C12 myoblasts (Cheikhi et al., 2020). Other cellular events associated with disruption of the EGFR includes is the suppression of differentiation through EGFR and mitochondrial-dependent temporal dysregulation of cyclin D1 (Cheikhi et al., 2020), which might explain arsenic-mediated impairment of differentiation in different cell types (Macoch et al., 2013). Mechanisms related to low-level exposure differ from those linked to high-level exposure, where mitochondrial ATP generation is inhibited, resulting in cell death (Cheikhi et al., 2020). In children, low to moderate level arsenic exposures (<100ppb) cause strength and motor deficits (Parvez et al., 2011), possibly as the result of targeting of muscle and progenitor cell mitochondria, with disruption of muscle metabolism, maintenance, and regeneration (Ambrosio et al., 2014).
Experimental models have contributed to elucidate the correlation between the epigenetic modifications induced by gestational exposure to arsenic and disease development. The offspring of pregnant mice transplacentally exposed to arsenic in drinking water showed a sex-dependent increase in tumor incidence after 74 (males) and 90weeks (females) compared with non-exposed mice (Waalkes et al., 2003). Promoter hypomethylation of genes involved in estrogen signaling with the concurrent increase in mRNA expression has been observed in hepatic tumors in adult mice exposed to arsenic in utero (Waalkes et al., 2004). Changes in the histone modification pattern, including an increase in H3K4 trimethylation in the promoter region of Fatty acid binding protein 4 (Fabp4) and genome-wide hypo-acetylation at H3K9, have been observed in brain samples of the offsprings of mice prenatally exposed to arsenic, compared to non-exposed mice (Nohara et al., 2012; Cronican et al., 2013). Exposure to arsenic can increase the expression of miRNA-21 (a well-known cancer-related miRNA) through overexpression of HIF-1α induced by arsenic-mediated activation of the PI3K/AKT pathway (Liu et al., 2020). The expression of other miRNAs involved in apoptosis (miR-143), cell proliferation/migration (miR-200b and miR-222), and apoptosis (miR-27a), is also disrupted by exposure to inorganic arsenic (Wang et al., 2014b, 2016; Ngalame et al., 2016; Zhang et al., 2016). Moreover, in utero arsenic exposure (measured in cord blood) strengthens the negative association between miR-1290 and birthweight in a cohort of mother-infant pairs enrolled in a prospective birth cohort in Bangladesh (Rahman et al., 2018).
Fetal and early postnatal stages of development are particularly sensitive to the impact of environmental exposure to arsenic. Such exposures may compromise early developmental processes and predispose the fetus to adverse health risks later in life (Barker, 2007; Grandjean et al., 2015). Furthermore, changes to the phenotype in the following generations can be observed even in the absence of direct environmental exposures (Skinner, 2011; Nilsson et al., 2018).
The intrauterine environment to which the fetus is exposed is regulated by the placenta, a fetomaternal organ that regulates critical aspects of embryonic development and pregnancy, including immune responses, gas and nutrient transfer, endocrine function, and protection from environmental exposures (Carter, 2015; Wilson and Robinson, 2018). Arsenic compounds can cross through the placental barrier, so exposure in pregnant women is significant for fetal development (Ramsey et al., 2013). The concentration of arsenic found in the human placenta correlates with both maternal and infant levels, as well as with the levels in the household drinking water (Punshon et al., 2015).
Increasing evidence suggests that gestational exposure to arsenic affects fetal development and induces disease phenotypes that develop only later in life (Nohara et al., 2020b). In Northern Chile, the levels of arsenic in drinking water increased from about 90 to 870ppb in 1958 (Romero et al., 2003; Thomas, 2013). In the 1970s, remediation measures reduced levels to approximately 110ppb, generating two cohorts of individuals exposed to significantly different levels of arsenic (Smith et al., 2012). The adult mortality for individual types of cancer (urinary bladder, respiratory tract, kidney, and liver), and all cancer types combined was significantly higher in the cohort exposed to the highest levels of arsenic in drinking water (born between 1958 and 1970; Smith et al., 2012). A study analyzing childhood cancer mortality for the same region found an increased risk in childhood (ages 10–19) of liver cancer mortality (Liaw et al., 2008).
Arsenic-related health effects resulting from gestational or early life exposure to arsenic in drinking water have been also observed in CVDs and other conditions. For example, early-life (3–8years old) exposure to inorganic arsenic was significantly associated with higher values of blood pressure and left ventricular mass (a predictor of adverse cardiovascular events and premature death) and with lower ejection fraction (indicating impaired contraction) in Mexican children exposed to arsenic concentrations in drinking water between 3 and 398ppb (Osorio-Yáñez et al., 2015). Additionally, early life exposure appears to have gender-specific effects among the pediatric population. In a rural population in Bangladesh, young girls (12–18years old) were at increased risk of mortality from all causes (Rahman et al., 2013). Furthermore, in utero exposure has been also associated with increased susceptibility to impaired thymic and lung function (Dauphiné et al., 2011; Ahmed et al., 2012), body size (Saha et al., 2012), and motor function (Parvez et al., 2011). A similar situation was observed for bronchiectasis and other chronic obstructive pulmonary disease, acute myocardial infarction, chronic renal disease, and all non-cancer causes of death (Thomas, 2013; see Figure 1).
The role of arsenic in the development of other significantly associated conditions, such as CVD, are also closely related to its biotransformation. Increase in ROS generation can result in disturbance of endothelial function (inhibition of endothelial nitric oxide synthase) leading to lipid peroxidation, which is one of the most commonly proposed mechanisms in arsenic-induced CVD (Barchowsky et al., 1999; Liao et al., 2006; Sidhu et al., 2015). Moreover, exposure to inorganic arsenic through drinking water induces pathological hypertrophy of the heart in male rats by activating the calcineurin-nuclear factor of activated T cells (NFAT) signaling pathway (Kabir et al., 2021).
Several studies show that arsenic exposure at different stages of life has detrimental effects on human health, both in the short term (prenatal, perinatal, and early childhood), as well as later in life (increased risk of developing diseases). Effects during fetal development and early life stages are defined by the dynamics of the transit of arsenic across the placental barrier. Factors such as concentration, length of exposure (mother), and effects of the genotype might influence significantly the health effects associated with gestational exposure. Additionally, the effects during fetal development should consider that the organs responsible for arsenic metabolism are not fully developed yet (full maturity of the liver takes up to 2years after birth; Beath, 2003). Therefore, the metabolism of arsenic during the gestational/postnatal period could present some differences compared to the established model in a fully-functional liver.
The precise mechanisms underlying the long-term arsenic-related effects are yet to be elucidated. Arsenic-related epigenetic alterations seem to play a significant role in these effects, suggesting that imprinting could be involved (Smeester et al., 2011; Bailey et al., 2013; Sanders et al., 2014). Moreover, arsenic-induced epigenetic alterations of imprinted genes (such as Igf2 and H19) generated during prenatal exposure can have deleterious effects (spermatotoxicity) in male offsprings of subsequent generations (Yin et al., 2021). However, how these alterations could impact fetal development and drive long-term tumorigenesis is still unknown.
One possible explanation for the arsenic-induced long-term effects is the cumulative molecular damage, as a result of chronic exposure. While some of the early molecular alterations induced by arsenic could be repaired, others (particularly epigenetic) could persist and progressively generate a favorable scenario for disease-prone alterations. The alterations identified in the adult population might already reflect some degree of molecular damage accumulation. In contrast, alterations identified during early stages of development could represent the first indications of arsenic-induced molecular damage. Thus, the identification of specific genetic/epigenetic alterations linked to gestational arsenic exposure would contribute to monitoring those individuals at higher risk of developing health effects during childhood, as well as later in life.
Cohorts of exposed human populations have been pivotal in identifying some of the health outcomes linked to early arsenic exposure; however, it is difficult to use these cohorts to identify the series of events occurring over time, particularly at a molecular level. Identifying the sequence of molecular events involved in arsenic-induced molecular damage remains as one of the critical gaps in our understanding of the modes of action of this environmental compound. Clues to decipher early events in arsenic-induced disease could be obtained from longitudinal in vitro models, where exposure at physiologically-relevant doses could be mimicked. Such models could potentially generate the preliminary information that could be validated in in vivo models.
Importantly, the combined use of new technologies will help to elucidate an extended network of biological effects associated with arsenic exposure. So far, molecular events associated with arsenic exposure have been analyzed individually. The incorporation of different omics technologies in the analysis of these longitudinal in vitro cohorts will help to elucidate how changes at DNA and RNA level are acting concertedly. For example, the use of next generation sequencing technologies will help to interpret methylation and other modification in DNA and how these changes can influence changes in the levels of messenger RNAs, as well as of long and short non-coding RNAs. Ultimately, integrating these molecular events will contribute to the development of integrated genetic and epigenetic-based signatures as biomarkers of the cumulative effects of arsenic exposure that could lead to early detection of arsenic-related diseases.
VM is the principal and corresponding author, and compiled and analyzed the literature on arsenic-related health effects. VM and WL designed the mini-review article. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the Canadian Institutes of Health Research (FDN-143345) and National Institutes of Health (R01HD089713), and funds from the BC Cancer Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank Kim M. Lonergan for critical reading of the manuscript and insightful comments.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.664717/full#supplementary-material
Ahmed, S., Ahsan, K. B., Kippler, M., Mily, A., Wagatsuma, Y., Hoque, A. M. W., et al. (2012). In utero arsenic exposure is associated with impaired thymic function in newborns possibly via oxidative stress and apoptosis. Toxicol. Sci. 129, 305–314. doi: 10.1093/toxsci/kfs202
Alegría-Torres, J. A., Carrizales-Yánez, L., Díaz-Barriga, F., Rosso-Camacho, F., Motta, V., Tarantini, L., et al. (2016). DNA methylation changes in Mexican children exposed to arsenic from two historic mining areas in San Luis potosí. Environ. Mol. Mutagen. 57, 717–723. doi: 10.1002/em.22062
Ambrosio, F., Brown, E., Stolz, D., Ferrari, R., Goodpaster, B., Deasy, B., et al. (2014). Arsenic induces sustained impairment of skeletal muscle and muscle progenitor cell ultrastructure and bioenergetics. Free Radic. Biol. Med. 74, 64–73. doi: 10.1016/j.freeradbiomed.2014.06.012
Antonelli, R., Shao, K., Thomas, D. J., Sams, R., and Cowden, J. (2014). AS3MT, GSTO, and PNP polymorphisms: impact on arsenic methylation and implications for disease susceptibility. Environ. Res. 132, 156–167. doi: 10.1016/j.envres.2014.03.012
Bailey, K. A., and Fry, R. C. (2014). Arsenic-associated changes to the epigenome: what are the functional consequences? Curr. Environ. Health Rep. 1, 22–34. doi: 10.1007/s40572-013-0002-8
Bailey, K. A., Wu, M. C., Ward, W. O., Smeester, L., Rager, J. E., García-Vargas, G., et al. (2013). Arsenic and the epigenome: interindividual differences in arsenic metabolism related to distinct patterns of DNA methylation. J. Biochem. Mol. Toxicol. 27, 106–115. doi: 10.1002/jbt.21462
Bandyopadhyay, A. K., Paul, S., Adak, S., and Giri, A. K. (2016). Reduced LINE-1 methylation is associated with arsenic-induced genotoxic stress in children. Biometals 29, 731–741. doi: 10.1007/s10534-016-9950-4
Barchowsky, A., Klei, L. R., Dudek, E. J., Swartz, H. M., and James, P. E. (1999). Stimulation of reactive oxygen, but not reactive nitrogen species, in vascular endothelial cells exposed to low levels of arsenite. Free Radic. Biol. Med. 27, 1405–1412. doi: 10.1016/S0891-5849(99)00186-0
Barker, D. J. P. (2007). The origins of the developmental origins theory. J. Intern. Med. 261, 412–417. doi: 10.1111/j.1365-2796.2007.01809.x
Beath, S. V. (2003). Hepatic function and physiology in the newborn. Semin. Neonatol. 8, 337–346. doi: 10.1016/S1084-2756(03)00066-6
Bhattacharjee, P., Das, A., Giri, A. K., and Bhattacharjee, P. (2020). Epigenetic regulations in alternative telomere lengthening: understanding the mechanistic insight in arsenic-induced skin cancer patients. Sci. Total Environ. 704:135388. doi: 10.1016/j.scitotenv.2019.135388
Bhattacharjee, P., Sanyal, T., Bhattacharjee, S., and Bhattacharjee, P. (2018). Epigenetic alteration of mismatch repair genes in the population chronically exposed to arsenic in West Bengal, India. Environ. Res. 163, 289–296. doi: 10.1016/j.envres.2018.01.002
Bi, Z., Zhang, Q., Fu, Y., Seno, A., Wadgaonkar, P., Qiu, Y., et al. (2021). Cooperation between NRF2-mediated transcription and MDIG-dependent epigenetic modifications in arsenic-induced carcinogenesis and cancer stem cells. Semin. Cancer Biol. doi: 10.1016/j.semcancer.2021.03.030 [Epub ahead of print]
Bi, Z., Zhang, Q., Fu, Y., Wadgaonkar, P., Zhang, W., Almutairy, B., et al. (2020). Nrf2 and HIF1α converge to arsenic-induced metabolic reprogramming and the formation of the cancer stem-like cells. Theranostics 10, 4134–4149. doi: 10.7150/thno.42903
Cardenas, A., Koestler, D. C., Houseman, E. A., Jackson, B. P., Kile, M. L., Karagas, M. R., et al. (2015). Differential DNA methylation in umbilical cord blood of infants exposed to mercury and arsenic in utero. Epigenetics 10, 508–515. doi: 10.1080/15592294.2015.1046026
Carter, A. M. (2015). Placental gas exchange and the oxygen supply to the fetus. Compr. Physiol. 5, 1381–1403. doi: 10.1002/cphy.c140073
Chakraborty, T., and De, M. (2009). Clastogenic effects of inorganic arsenic salts on human chromosomes in vitro. Drug Chem. Toxicol. 32, 169–173. doi: 10.1080/01480540802594509
Chang, Q., Bi, Z., Fu, Y., Rice, M. K. A., Zhang, Q., Wadgaonkar, P., et al. (2020). Characterization of arsenic-induced cancer stem-like cells. Methods Mol. Biol. 2117, 293–303. doi: 10.1007/978-1-0716-0301-7_19
Cheikhi, A., Anguiano, T., Lasak, J., Qian, B., Sahu, A., Mimiya, H., et al. (2020). Arsenic stimulates myoblast mitochondrial epidermal growth factor receptor to impair myogenesis. Toxicol. Sci. 176, 162–174. doi: 10.1093/toxsci/kfaa031
Chervona, Y., Hall, M. N., Arita, A., Wu, F., Sun, H., Tseng, H.-C., et al. (2012). Associations between arsenic exposure and global posttranslational histone modifications among adults in Bangladesh. Cancer Epidemiol. Biomark. Prev. 21, 2252–2260. doi: 10.1158/1055-9965.EPI-12-0833
Chicoine, E., Estève, P.-O., Robledo, O., Van Themsche, C., Potworowski, E. F., and St-Pierre, Y. (2002). Evidence for the role of promoter methylation in the regulation of MMP-9 gene expression. Biochem. Biophys. Res. Commun. 297, 765–772. doi: 10.1016/S0006-291X(02)02283-0
Cooper, K. L., Liu, K. J., and Hudson, L. G. (2009). Enhanced ROS production and redox signaling with combined arsenite and UVA exposure: contribution of NADPH oxidase. Free Radic. Biol. Med. 47, 381–388. doi: 10.1016/j.freeradbiomed.2009.04.034
Cronican, A. A., Fitz, N. F., Carter, A., Saleem, M., Shiva, S., Barchowsky, A., et al. (2013). Genome-wide alteration of histone H3K9 acetylation pattern in mouse offspring prenatally exposed to arsenic. PLoS One 8:e53478. doi: 10.1371/journal.pone.0053478
Dauphiné, D. C., Ferreccio, C., Guntur, S., Yuan, Y., Hammond, S. K., Balmes, J., et al. (2011). Lung function in adults following in utero and childhood exposure to arsenic in drinking water: preliminary findings. Int. Arch. Occup. Environ. Health 84, 591–600. doi: 10.1007/s00420-010-0591-6
De Chaudhuri, S., Ghosh, P., Sarma, N., Majumdar, P., Sau, T. J., Basu, S., et al. (2008). Genetic variants associated with arsenic susceptibility: study of purine nucleoside phosphorylase, arsenic (+3) methyltransferase, and glutathione S-transferase omega genes. Environ. Health Perspect. 116, 501–505. doi: 10.1289/ehp.10581
Ding, X., Zhang, A., Li, C., Ma, L., Tang, S., Wang, Q., et al. (2021). The role of H3K9me2-regulated base excision repair genes in the repair of DNA damage induced by arsenic in HaCaT cells and the effects of Ginkgo biloba extract intervention. Environ. Toxicol. 36, 850–860. doi: 10.1002/tox.23088
Dopp, E., Hartmann, L. M., Florea, A.-M., von Recklinghausen, U., Pieper, R., Shokouhi, B., et al. (2004). Uptake of inorganic and organic derivatives of arsenic associated with induced cytotoxic and genotoxic effects in Chinese hamster ovary (CHO) cells. Toxicol. Appl. Pharmacol. 201, 156–165. doi: 10.1016/j.taap.2004.05.017
Drobna, Z., Styblo, M., and Thomas, D. J. (2009). An overview of arsenic metabolism and toxicity. Curr. Protoc. Toxicol. 42, 4.31.1–4.31.6. doi: 10.1002/0471140856.tx0431s42
Gao, N., Shen, L., Zhang, Z., Leonard, S. S., He, H., Zhang, X.-G., et al. (2004). Arsenite induces HIF-1alpha and VEGF through PI3K, Akt and reactive oxygen species in DU145 human prostate carcinoma cells. Mol. Cell. Biochem. 255, 33–45. doi: 10.1023/B:MCBI.0000007259.65742.16
Gonzalez-Cortes, T., Recio-Vega, R., Lantz, R. C., and Chau, B. T. (2017). DNA methylation of extracellular matrix remodeling genes in children exposed to arsenic. Toxicol. Appl. Pharmacol. 329, 140–147. doi: 10.1016/j.taap.2017.06.001
Gorchev, H. G., and Ozolins, G. (1984). WHO guidelines for drinking-water quality. WHO Chron. 38, 104–108.
Grandjean, P., Barouki, R., Bellinger, D. C., Casteleyn, L., Chadwick, L. H., Cordier, S., et al. (2015). Life-long implications of developmental exposure to environmental stressors: new perspectives. Endocrinology 156, 3408–3415. doi: 10.1210/en.2015-1350
Halliwell, B., and Whiteman, M. (2004). Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? Br. J. Pharmacol. 142, 231–255. doi: 10.1038/sj.bjp.0705776
Hartwig, A., Groblinghoff, U. D., Beyersmann, D., Natarajan, A. T., Filon, R., and Mullenders, L. H. (1997). Interaction of arsenic(III) with nucleotide excision repair in UV-irradiated human fibroblasts. Carcinogenesis 18, 399–405. doi: 10.1093/carcin/18.2.399
Hirano, S. (2020). Biotransformation of arsenic and toxicological implication of arsenic metabolites. Arch. Toxicol. 94, 2587–2601. doi: 10.1007/s00204-020-02772-9
Hossain, K., Suzuki, T., Hasibuzzaman, M. M., Islam, M. S., Rahman, A., Paul, S. K., et al. (2017). Chronic exposure to arsenic, LINE-1 hypomethylation, and blood pressure: a cross-sectional study in Bangladesh. Environ. Health 16:20. doi: 10.1186/s12940-017-0231-7
Howard, S. G. (2018). Developmental exposure to endocrine disrupting chemicals and type 1 diabetes mellitus. Front. Endocrinol. 9:513. doi: 10.3389/fendo.2018.00513
Kabir, R., Sinha, P., Mishra, S., Ebenebe, O. V., Taube, N., Oeing, C. U., et al. (2021). Inorganic arsenic induces sex-dependent pathological hypertrophy in the heart. Am. J. Physiol. Heart Circ. Physiol. 320, H1321–H1336. doi: 10.1152/ajpheart.00435.2020
Kenyon, E. M., and Hughes, M. F. (2001). A concise review of the toxicity and carcinogenicity of dimethylarsinic acid. Toxicology 160, 227–236. doi: 10.1016/S0300-483X(00)00458-3
Kile, M. L., Baccarelli, A., Hoffman, E., Tarantini, L., Quamruzzaman, Q., Rahman, M., et al. (2012). Prenatal arsenic exposure and DNA methylation in maternal and umbilical cord blood leukocytes. Environ. Health Perspect. 120, 1061–1066. doi: 10.1289/ehp.1104173
Kitchin, K. T., and Conolly, R. (2010). Arsenic-induced carcinogenesis--oxidative stress as a possible mode of action and future research needs for more biologically based risk assessment. Chem. Res. Toxicol. 23, 327–335. doi: 10.1021/tx900343d
Kligerman, A. D., Malik, S. I., and Campbell, J. A. (2010). Cytogenetic insights into DNA damage and repair of lesions induced by a monomethylated trivalent arsenical. Mutat. Res. 695, 2–8. doi: 10.1016/j.mrgentox.2009.09.007
Liao, Y.-H., Hwang, L.-C., Kao, J.-S., Yiin, S.-J., Lin, S.-F., Lin, C.-H., et al. (2006). Lipid peroxidation in workers exposed to aluminium, gallium, indium, arsenic, and antimony in the optoelectronic industry. J. Occup. Environ. Med. 48, 789–793. doi: 10.1097/01.jom.0000229782.71756.8e
Liaw, J., Marshall, G., Yuan, Y., Ferreccio, C., Steinmaus, C., and Smith, A. H. (2008). Increased childhood liver cancer mortality and arsenic in drinking water in northern Chile. Cancer Epidemiol. Biomark. Prev. 17, 1982–1987. doi: 10.1158/1055-9965.EPI-07-2816
Liu, J., Chen, B., Lu, Y., Guan, Y., and Chen, F. (2012). JNK-dependent Stat3 phosphorylation contributes to Akt activation in response to arsenic exposure. Toxicol. Sci. 129, 363–371. doi: 10.1093/toxsci/kfs199
Liu, S.-X., Davidson, M. M., Tang, X., Walker, W. F., Athar, M., Ivanov, V., et al. (2005). Mitochondrial damage mediates genotoxicity of arsenic in mammalian cells. Cancer Res. 65, 3236–3242. doi: 10.1158/0008-5472.CAN-05-0424
Liu, J., Niu, Q., Hu, Y., Ran, S., and Li, S. (2020). The mechanism of trivalent inorganic arsenic on HIF-1α: a systematic review and meta-analysis. Biol. Trace Elem. Res. 198, 449–463. doi: 10.1007/s12011-020-02087-x
Macoch, M., Morzadec, C., Fardel, O., and Vernhet, L. (2013). Inorganic arsenic impairs differentiation and functions of human dendritic cells. Toxicol. Appl. Pharmacol. 266, 204–213. doi: 10.1016/j.taap.2012.11.008
Martinez, V. D., Buys, T. P. H., Adonis, M., Benítez, H., Gallegos, I., Lam, S., et al. (2010). Arsenic-related DNA copy-number alterations in lung squamous cell carcinomas. Br. J. Cancer 103, 1277–1283. doi: 10.1038/sj.bjc.6605879
Martinez, V. D., Thu, K. L., Vucic, E. A., Hubaux, R., Adonis, M., Gil, L., et al. (2013). Whole-genome sequencing analysis identifies a distinctive mutational spectrum in an arsenic-related lung tumor. J. Thorac. Oncol. 8, 1451–1455. doi: 10.1097/JTO.0b013e3182a4dd8e
Mauro, M., Caradonna, F., and Klein, C. B. (2016). Dysregulation of DNA methylation induced by past arsenic treatment causes persistent genomic instability in mammalian cells. Environ. Mol. Mutagen. 57, 137–150. doi: 10.1002/em.21987
Merrick, B. A., Phadke, D. P., Bostrom, M. A., Shah, R. R., Wright, G. M., Wang, X., et al. (2019). Arsenite malignantly transforms human prostate epithelial cells in vitro by gene amplification of mutated KRAS. PLoS One 14:e0215504. doi: 10.1371/journal.pone.0215504
Merrick, B. A., Phadke, D. P., Bostrom, M. A., Shah, R. R., Wright, G. M., Wang, X., et al. (2020). KRAS-retroviral fusion transcripts and gene amplification in arsenic-transformed, human prostate CAsE-PE cancer cells. Toxicol. Appl. Pharmacol. 397:115017. doi: 10.1016/j.taap.2020.115017
Minatel, B. C., Sage, A. P., Anderson, C., Hubaux, R., Marshall, E. A., Lam, W. L., et al. (2018). Environmental arsenic exposure: from genetic susceptibility to pathogenesis. Environ. Int. 112, 183–197. doi: 10.1016/j.envint.2017.12.017
Moe, B., Peng, H., Lu, X., Chen, B., Chen, L. W. L., Gabos, S., et al. (2016). Comparative cytotoxicity of fourteen trivalent and pentavalent arsenic species determined using real-time cell sensing. J. Environ. Sci. (China) 49, 113–124. doi: 10.1016/j.jes.2016.10.004
Muka, T., Koromani, F., Portilla, E., O’Connor, A., Bramer, W. M., Troup, J., et al. (2016). The role of epigenetic modifications in cardiovascular disease: a systematic review. Int. J. Cardiol. 212, 174–183. doi: 10.1016/j.ijcard.2016.03.062
Naranmandura, H., Xu, S., Sawata, T., Hao, W. H., Liu, H., Bu, N., et al. (2011). Mitochondria are the main target organelle for trivalent monomethylarsonous acid (MMA(III))-induced cytotoxicity. Chem. Res. Toxicol. 24, 1094–1103. doi: 10.1021/tx200156k
Naujokas, M. F., Anderson, B., Ahsan, H., Aposhian, H. V., Graziano, J. H., Thompson, C., et al. (2013). The broad scope of health effects from chronic arsenic exposure: update on a worldwide public health problem. Environ. Health Perspect. 121, 295–302. doi: 10.1289/ehp.1205875
Navas-Acien, A., Spratlen, M. J., Abuawad, A., LoIacono, N. J., Bozack, A. K., and Gamble, M. V. (2019). Early-life arsenic exposure, nutritional status, and adult diabetes risk. Curr. Diab. Rep. 19:147. doi: 10.1007/s11892-019-1272-9
Navasumrit, P., Chaisatra, K., Promvijit, J., Parnlob, V., Waraprasit, S., Chompoobut, C., et al. (2019). Exposure to arsenic in utero is associated with various types of DNA damage and micronuclei in newborns: a birth cohort study. Environ. Health 18:51. doi: 10.1186/s12940-019-0481-7
Ngalame, N. N. O., Makia, N. L., Waalkes, M. P., and Tokar, E. J. (2016). Mitigation of arsenic-induced acquired cancer phenotype in prostate cancer stem cells by miR-143 restoration. Toxicol. Appl. Pharmacol. 312, 11–18. doi: 10.1016/j.taap.2015.12.013
Nilsson, E. E., Sadler-Riggleman, I., and Skinner, M. K. (2018). Environmentally induced epigenetic transgenerational inheritance of disease. Environ. Epigenet. 4:dvy016. doi: 10.1093/eep/dvy016
Nohara, K., Nakabayashi, K., Okamura, K., Suzuki, T., Suzuki, S., and Hata, K. (2020a). Gestational arsenic exposure induces site-specific DNA hypomethylation in active retrotransposon subfamilies in offspring sperm in mice. Epigenetics Chromatin 13:53. doi: 10.1186/s13072-020-00375-3
Nohara, K., Suzuki, T., and Okamura, K. (2020b). Gestational arsenic exposure and paternal intergenerational epigenetic inheritance. Toxicol. Appl. Pharmacol. 409:115319. doi: 10.1016/j.taap.2020.115319
Nohara, K., Tateishi, Y., Suzuki, T., Okamura, K., Murai, H., Takumi, S., et al. (2012). Late-onset increases in oxidative stress and other tumorigenic activities and tumors with a Ha-ras mutation in the liver of adult male C3H mice gestationally exposed to arsenic. Toxicol. Sci. 129, 293–304. doi: 10.1093/toxsci/kfs203
Nollen, M., Ebert, F., Moser, J., Mullenders, L. H. F., Hartwig, A., and Schwerdtle, T. (2009). Impact of arsenic on nucleotide excision repair: XPC function, protein level, and gene expression. Mol. Nutr. Food Res. 53, 572–582. doi: 10.1002/mnfr.200800480
Osmond, M. J., Kunz, B. A., and Snow, E. T. (2010). Age and exposure to arsenic alter base excision repair transcript levels in mice. Mutagenesis 25, 517–522. doi: 10.1093/mutage/geq037
Osorio-Yáñez, C., Ayllon-Vergara, J. C., Arreola-Mendoza, L., Aguilar-Madrid, G., Hernández-Castellanos, E., Sánchez-Peña, L. C., et al. (2015). Blood pressure, left ventricular geometry, and systolic function in children exposed to inorganic arsenic. Environ. Health Perspect. 123, 629–635. doi: 10.1289/ehp.1307327
Ouyang, Y., Wu, Q., Li, J., Sun, S., and Sun, S. (2020). S-adenosylmethionine: a metabolite critical to the regulation of autophagy. Cell Prolif. 53:e12891. doi: 10.1111/cpr.12891
Parvez, F., Wasserman, G. A., Factor-Litvak, P., Liu, X., Slavkovich, V., Siddique, A. B., et al. (2011). Arsenic exposure and motor function among children in Bangladesh. Environ. Health Perspect. 119, 1665–1670. doi: 10.1289/ehp.1103548
Patterson, T. J., Reznikova, T. V., Phillips, M. A., and Rice, R. H. (2005). Arsenite maintains germinative state in cultured human epidermal cells. Toxicol. Appl. Pharmacol. 207, 69–77. doi: 10.1016/j.taap.2004.11.020
Paul, S., Banerjee, N., Chatterjee, A., Sau, T. J., Das, J. K., Mishra, P. K., et al. (2014). Arsenic-induced promoter hypomethylation and over-expression of ERCC2 reduces DNA repair capacity in humans by non-disjunction of the ERCC2-Cdk7 complex. Metallomics 6, 864–873. doi: 10.1039/c3mt00328k
Paul, S., Majumdar, S., and Giri, A. K. (2015). Genetic susceptibility to arsenic-induced skin lesions and health effects: a review. Genes Environ. 37:23. doi: 10.1186/s41021-015-0023-7
Person, R. J., Ngalame, N. N. O., Makia, N. L., Bell, M. W., Waalkes, M. P., and Tokar, E. J. (2015). Chronic inorganic arsenic exposure in vitro induces a cancer cell phenotype in human peripheral lung epithelial cells. Toxicol. Appl. Pharmacol. 286, 36–43. doi: 10.1016/j.taap.2015.03.014
Pilsner, J. R., Hall, M. N., Liu, X., Ilievski, V., Slavkovich, V., Levy, D., et al. (2012). Influence of prenatal arsenic exposure and newborn sex on global methylation of cord blood DNA. PLoS One 7:e37147. doi: 10.1371/journal.pone.0037147
Podgorski, J., and Berg, M. (2020). Global threat of arsenic in groundwater. Science 368, 845–850. doi: 10.1126/science.aba1510
Pulido, M. D., and Parrish, A. R. (2003). Metal-induced apoptosis: mechanisms. Mutat. Res. 533, 227–241. doi: 10.1016/j.mrfmmm.2003.07.015
Punshon, T., Davis, M. A., Marsit, C. J., Theiler, S. K., Baker, E. R., Jackson, B. P., et al. (2015). Placental arsenic concentrations in relation to both maternal and infant biomarkers of exposure in a US cohort. J. Expo. Sci. Environ. Epidemiol. 25, 599–603. doi: 10.1038/jes.2015.16
Quansah, R., Armah, F. A., Essumang, D. K., Luginaah, I., Clarke, E., Marfoh, K., et al. (2015). Association of arsenic with adverse pregnancy outcomes/infant mortality: a systematic review and meta-analysis. Environ. Health Perspect. 123, 412–421. doi: 10.1289/ehp.1307894
Rafiei, G., Shirkoohi, R., Saffari, M., Salehipour, P., and Modarressi, M. H. (2019). The impact of long-term exposure to low levels of inorganic arsenic on the hypomethylation of SEPT9 promoter in epithelial-mesenchymal transformed colorectal cancer cell lines. Int. J. Mol. Cell. Med. 8, 130–138. doi: 10.22088/IJMCM.BUMS.8.2.130
Rahman, M. L., Liang, L., Valeri, L., Su, L., Zhu, Z., Gao, S., et al. (2018). Regulation of birthweight by placenta-derived miRNAs: evidence from an arsenic-exposed birth cohort in Bangladesh. Epigenetics 13, 573–590. doi: 10.1080/15592294.2018.1481704
Rahman, M., Sohel, N., Yunus, M., Chowdhury, M. E., Hore, S. K., Zaman, K., et al. (2013). Increased childhood mortality and arsenic in drinking water in Matlab, Bangladesh: a population-based cohort study. PLoS One 8:e55014. doi: 10.1371/journal.pone.0055014
Ramsey, K. A., Larcombe, A. N., Sly, P. D., and Zosky, G. R. (2013). In utero exposure to low dose arsenic via drinking water impairs early life lung mechanics in mice. BMC Pharmacol. Toxicol. 14:13. doi: 10.1186/2050-6511-14-13
Reichard, J. F., and Puga, A. (2010). Effects of arsenic exposure on DNA methylation and epigenetic gene regulation. Epigenomics 2, 87–104. doi: 10.2217/epi.09.45
Reichard, J. F., Schnekenburger, M., and Puga, A. (2007). Long term low-dose arsenic exposure induces loss of DNA methylation. Biochem. Biophys. Res. Commun. 352, 188–192. doi: 10.1016/j.bbrc.2006.11.001
Romero, L., Alonso, H., Campano, P., Fanfani, L., Cidu, R., Dadea, C., et al. (2003). Arsenic enrichment in waters and sediments of the Rio Loa (Second Region, Chile). Appl. Geochem. 18, 1399–1416. doi: 10.1016/S0883-2927(03)00059-3
Rossman, T. G., Uddin, A. N., and Burns, F. J. (2004). Evidence that arsenite acts as a cocarcinogen in skin cancer. Toxicol. Appl. Pharmacol. 198, 394–404. doi: 10.1016/j.taap.2003.10.016
Roy, P., Mukherjee, A., and Giri, S. (2016). Evaluation of genetic damage in tobacco and arsenic exposed population of Southern Assam, India using buccal cytome assay and comet assay. Ecotoxicol. Environ. Saf. 124, 169–176. doi: 10.1016/j.ecoenv.2015.10.019
Saha, K. K., Engström, A., Hamadani, J. D., Tofail, F., Rasmussen, K. M., and Vahter, M. (2012). Pre- and postnatal arsenic exposure and body size to 2 years of age: a cohort study in rural Bangladesh. Environ. Health Perspect. 120, 1208–1214. doi: 10.1289/ehp.1003378
Salmeri, N., Villanacci, R., Ottolina, J., Bartiromo, L., Cavoretto, P., Dolci, C., et al. (2020). Maternal arsenic exposure and gestational diabetes: a systematic review and meta-analysis. Nutrients 12:3094. doi: 10.3390/nu12103094
Sanders, A. P., Smeester, L., Rojas, D., DeBussycher, T., Wu, M. C., Wright, F. A., et al. (2014). Cadmium exposure and the epigenome: exposure-associated patterns of DNA methylation in leukocytes from mother-baby pairs. Epigenetics 9, 212–221. doi: 10.4161/epi.26798
Sanyal, T., Bhattacharjee, P., Bhattacharjee, S., and Bhattacharjee, P. (2018). Hypomethylation of mitochondrial D-loop and ND6 with increased mitochondrial DNA copy number in the arsenic-exposed population. Toxicology 408, 54–61. doi: 10.1016/j.tox.2018.06.012
Selmin, O. I., Donovan, M. G., Skovan, B., Paine-Murieta, G. D., and Romagnolo, D. F. (2019). Arsenic-induced BRCA1 CpG promoter methylation is associated with the downregulation of ERα and resistance to tamoxifen in MCF7 breast cancer cells and mouse mammary tumor xenografts. Int. J. Oncol. 54, 869–878. doi: 10.3892/ijo.2019.4687
Shen, S., Li, X.-F., Cullen, W. R., Weinfeld, M., and Le, X. C. (2013). Arsenic binding to proteins. Chem. Rev. 113, 7769–7792. doi: 10.1021/cr300015c
Sidhu, M. S., Desai, K. P., Lynch, H. N., Rhomberg, L. R., Beck, B. D., and Venditti, F. J. (2015). Mechanisms of action for arsenic in cardiovascular toxicity and implications for risk assessment. Toxicology 331, 78–99. doi: 10.1016/j.tox.2015.02.008
Simeonova, P. P., and Luster, M. I. (2000). Mechanisms of arsenic carcinogenicity: genetic or epigenetic mechanisms? J. Environ. Pathol. Toxicol. Oncol. 19, 281–286.
Skinner, M. K. (2011). Environmental epigenetic transgenerational inheritance and somatic epigenetic mitotic stability. Epigenetics 6, 838–842. doi: 10.4161/epi.6.7.16537
Smeester, L., and Fry, R. C. (2018). Long-term health effects and underlying biological mechanisms of developmental exposure to arsenic. Curr. Environ. Health Rep. 5, 134–144. doi: 10.1007/s40572-018-0184-1
Smeester, L., Rager, J. E., Bailey, K. A., Guan, X., Smith, N., García-Vargas, G., et al. (2011). Epigenetic changes in individuals with arsenicosis. Chem. Res. Toxicol. 24, 165–167. doi: 10.1021/tx1004419
Smith, K. R., Klei, L. R., and Barchowsky, A. (2001). Arsenite stimulates plasma membrane NADPH oxidase in vascular endothelial cells. Am. J. Phys. Lung Cell. Mol. Phys. 280, L442–L449. doi: 10.1152/ajplung.2001.280.3.L442
Smith, A. H., Marshall, G., Liaw, J., Yuan, Y., Ferreccio, C., and Steinmaus, C. (2012). Mortality in young adults following in utero and childhood exposure to arsenic in drinking water. Environ. Health Perspect. 120, 1527–1531. doi: 10.1289/ehp.1104867
Thomas, D. J. (2013). The die is cast: arsenic exposure in early life and disease susceptibility. Chem. Res. Toxicol. 26, 1778–1781. doi: 10.1021/tx4003337
Waalkes, M. P., Liu, J., Chen, H., Xie, Y., Achanzar, W. E., Zhou, Y.-S., et al. (2004). Estrogen signaling in livers of male mice with hepatocellular carcinoma induced by exposure to arsenic in utero. J. Natl. Cancer Inst. 96, 466–474. doi: 10.1093/jnci/djh070
Waalkes, M. P., Liu, J., Germolec, D. R., Trempus, C. S., Cannon, R. E., Tokar, E. J., et al. (2008). Arsenic exposure in utero exacerbates skin cancer response in adulthood with contemporaneous distortion of tumor stem cell dynamics. Cancer Res. 68, 8278–8285. doi: 10.1158/0008-5472.CAN-08-2099
Waalkes, M. P., Ward, J. M., Liu, J., and Diwan, B. A. (2003). Transplacental carcinogenicity of inorganic arsenic in the drinking water: induction of hepatic, ovarian, pulmonary, and adrenal tumors in mice. Toxicol. Appl. Pharmacol. 186, 7–17. doi: 10.1016/S0041-008X(02)00022-4
Wadgaonkar, P., and Chen, F. (2021). Connections between endoplasmic reticulum stress-associated unfolded protein response, mitochondria, and autophagy in arsenic-induced carcinogenesis. Semin. Cancer Biol. doi: 10.1016/j.semcancer.2021.04.004 [Epub head of print]
Walter, I., Schwerdtle, T., Thuy, C., Parsons, J. L., Dianov, G. L., and Hartwig, A. (2007). Impact of arsenite and its methylated metabolites on PARP-1 activity, PARP-1 gene expression and poly(ADP-ribosyl)ation in cultured human cells. DNA Repair (Amst) 6, 61–70. doi: 10.1016/j.dnarep.2006.08.008
Wang, W., Cheng, S., and Zhang, D. (2014a). Association of inorganic arsenic exposure with liver cancer mortality: a meta-analysis. Environ. Res. 135, 120–125. doi: 10.1016/j.envres.2014.08.034
Wang, M., Ge, X., Zheng, J., Li, D., Liu, X., Wang, L., et al. (2016). Role and mechanism of miR-222 in arsenic-transformed cells for inducing tumor growth. Oncotarget 7, 17805–17814. doi: 10.18632/oncotarget.7525
Wang, T. S., Hsu, T. Y., Chung, C. H., Wang, A. S., Bau, D. T., and Jan, K. Y. (2001). Arsenite induces oxidative DNA adducts and DNA-protein cross-links in mammalian cells. Free Radic. Biol. Med. 31, 321–330. doi: 10.1016/S0891-5849(01)00581-0
Wang, Z., Humphries, B., Xiao, H., Jiang, Y., and Yang, C. (2014b). MicroRNA-200b suppresses arsenic-transformed cell migration by targeting protein kinase Cα and Wnt5b-protein kinase Cα positive feedback loop and inhibiting Rac1 activation. J. Biol. Chem. 289, 18373–18386. doi: 10.1074/jbc.M114.554246
Wilson, S. L., and Robinson, W. P. (2018). Utility of DNA methylation to assess placental health. Placenta 64(Suppl. 1), S23–S28. doi: 10.1016/j.placenta.2017.12.013
Winterbottom, E. F., Moroishi, Y., Halchenko, Y., Armstrong, D. A., Beach, P. J., Nguyen, Q. P., et al. (2019). Prenatal arsenic exposure alters the placental expression of multiple epigenetic regulators in a sex-dependent manner. Environ. Health 18:18. doi: 10.1186/s12940-019-0455-9
World Health Organization (2004). IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, Volume 84 - Some Drinking-Water Disinfectants and Contaminants, Including Arsenic: This Publication Represents The Views and Expert Opinions of an IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, which met in Lyon, October 15–22, 2002. Lyon: IARC.
Yin, G., Xia, L., Hou, Y., Li, Y., Cao, D., Liu, Y., et al. (2021). Transgenerational male reproductive effect of prenatal arsenic exposure: abnormal spermatogenesis with Igf2/H19 epigenetic alteration in CD1 mouse. Int. J. Environ. Health Res. 7, 1–13. doi: 10.1080/09603123.2020.1870668
Zamora, P. L., Rockenbauer, A., and Villamena, F. A. (2014). Radical model of arsenic(III) toxicity: theoretical and EPR spin trapping studies. Chem. Res. Toxicol. 27, 765–774. doi: 10.1021/tx4004227
Keywords: arsenic, DNA methylation, in utero exposure, fetal development, genetics, epigenetics
Citation: Martinez VD and Lam WL (2021) Health Effects Associated With Pre- and Perinatal Exposure to Arsenic. Front. Genet. 12:664717. doi: 10.3389/fgene.2021.664717
Edited by:
António Sebastião Rodrigues, Universidade NOVA de Lisboa, PortugalReviewed by:
Erik Tokar, National Toxicology Program Division (NIEHS), United StatesCopyright © 2021 Martinez and Lam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Victor D. Martinez, dmljdG9yLm1hcnRpbmV6QGRhbC5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.