 Xiao Yang
Xiao Yang Xiucai Ye
Xiucai Ye Xuehong Li3*
Xuehong Li3* Lesong Wei
Lesong Wei
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
METHODS article
Front. Genet., 31 March 2021
Sec. Computational Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.663572
This article is part of the Research TopicArtificial Intelligence in Bioinformatics and Drug Repurposing: Methods and ApplicationsView all 13 articles
Motivation: DNA N4-methylcytosine (4mC) and N6-methyladenine (6mA) are two important DNA modifications and play crucial roles in a variety of biological processes. Accurate identification of the modifications is essential to better understand their biological functions and mechanisms. However, existing methods to identify 4mA or 6mC sites are all single tasks, which demonstrates that they can identify only a certain modification in one species. Therefore, it is desirable to develop a novel computational method to identify the modification sites in multiple species simultaneously.
Results: In this study, we proposed a computational method, called iDNA-MT, to identify 4mC sites and 6mA sites in multiple species, respectively. The proposed iDNA-MT mainly employed multi-task learning coupled with the bidirectional gated recurrent units (BGRU) to capture the sharing information among different species directly from DNA primary sequences. Experimental comparative results on two benchmark datasets, containing different species respectively, show that either for identifying 4mA or for 6mC site in multiple species, the proposed iDNA-MT outperforms other state-of-the-art single-task methods. The promising results have demonstrated that iDNA-MT has great potential to be a powerful and practically useful tool to accurately identify DNA modifications.
DNA modifications have been identified in multiple species. DNA modification plays an irreplaceable role in many basic biological functions (Fu and He, 2012; Shen and Zou, 2020). It refers to add methyl or hydroxymethyl groups to the nucleotides of DNA molecules. In particular, it is essential in the normal development of organisms such as aging, carcinogenesis, and X chromosome inactivation. Due to its importance, DNA methylation is one of the most widely studied epigenetic modifications (Bergman and Cedar, 2013; Smith and Meissner, 2013). Currently, four out of the DNA modifications, such as N4-methylcytosine (4mC), N6-methyladenine (6mA), 5-methylcytosine (5mC), and 5-hydroxymethylcytosine (5hmC), have been extensively studied (Cheng and Baldi, 2006; Guohua et al., 2017; He et al., 2019; Luo et al., 2020; Zuo et al., 2020c).
Schweizer (2008) proposed that 4mC has the effect of protecting the host DNA from degradation by restriction enzymes and belongs to restriction-modification (RM) systems. Timinskas et al. (1995) proposed 4mC can methylate the 4th amino group of cytosine in DNA under the catalysis of N-4 cytosine-specific DNA methyltransferase (DNMT). Iyer et al. (2011) proposed 4mC can distinguish the self and foreign DNA of prokaryotes and repair DNA replication errors. 5hmC arises from the oxidation of 5-methylcytosine (5mC) by Fe2 + and 2-oxoglutarate-dependent 10–11 translocation (TET) family proteins (Hu et al., 2019). Thomson and Meehan (2016) proposed 5hmC can be used as an identifier of cell type or disease state. It is an intermediate product produced during the 5mC demethylation process. Szulwach et al. (2011) proposed 5hmC is critical in neurodevelopment and diseases (Tang et al., 2018; Zhang Y. et al., 2019). 6mA is a non-canonical DNA base modification present at low levels and maybe a carrier of heritable epigenetic information in eukaryotes (Greer et al., 2015; Mondo et al., 2017) and is found in the genomes of certain protists and fungi and might exist in other eukaryotes (Wion and Casadesús, 2006). The role of 6mA is very extensive. For example, it protects against restriction enzymes in bacteria (Heyn and Esteller, 2015) and unravels the DNA double helix structure during the cell cycle (Fang et al., 2012), which is catalyzed by two classes of DNA adenine methyltransferases (Wion and Casadesús, 2006; Zhang L. et al., 2019).
Numerous studies have shown that 5hmC, 6mA, and 4mC, and others are widely present in the genome, and significant progress has been made (Wu et al., 2016; Ao et al., 2019; Hu et al., 2019; Zhu et al., 2019; Zou et al., 2019; Cai et al., 2020; Fu et al., 2020; Hong et al., 2020). However, methylation-related technologies-the short-read sequencing and long-read have major disadvantages. For example, short-read technology can convert unmethylated cytosine to uracil. However, it has intrinsic disadvantages, such as low positioning efficiency and low accuracy. Long-read sequencing can be used to identify DNA modifications. There is a problem that it does not have a high signal-to-noise ratio for DNA modification. In nature, 5hmC, 6mA, and 4mC content are low, and the requirements for detection technology are relatively high. Therefore, we perform predictive calculations in advance, which can improve the efficiency of the experiment, to reduce the cost of the experiment, and provide guidance information for subsequent implementations.
Recently, there have been many machine learning methods to predict DNA methylation sites (Basith et al., 2019; Chen and Zou, 2019; Dou et al., 2020; Lv et al., 2020b). For instance, Ni et al. (2019) proposed DeepSignal, a deep learning approach to detect DNA methylation states from Nanopore sequencing reads. Besides, Liu et al. (2016) designed a two-way neural network with long short-term memory, called DeepMod. It can also identify DNA methylation sites in E. coli and Homo sapiens. Chen et al. (2019) developed a computational method called i6mA-Pred, to identify 6mA sites targeted to the rice genome, in which the optimal nucleotide chemical properties obtained by the using feature selection technique were used to encode the DNA sequences. Similarly, Yu and Dai (2019) created SNNRice6mA based on deep learning to identify 6mA in rice.
Kong and Zhang (2019) proposed a new machine learning-based method, namely i6mA-DNCP, which proved that there is also 6mA sites also in the rice genome. In i6mA-DNCP, dinucleotide composition and dinucleotide-based DNA properties were first employed to represent DNA sequences. Chen et al. (2017) developed iDNA4mC, the first webserver to identify 4mC sites, in which DNA sequences are encoded with both nucleotide chemical properties and nucleotide frequency. Later on, Wei et al. (2019b) developed a new predictor named 4mcPred-IFL to identify 4mC sites, in which they proposed an iterative feature representation algorithm that enables learning informative features from several sequential models in a supervised iterative mode. Basith et al. (2019) developed a novel computational predictor, called the Sequence-based DNA N6-methyladenine predictor (SDM6A), which is a two-layer ensemble approach for identifying 6mA sites in the rice genome. Manavalan et al. (2019a) designed the first method for identifying 4mC sites in the mouse genome, called 4mCpred-EL. Similarly, Hasan et al. (2020) invented a method to identify the 4mC sites, called i4mC-ROSE in the Fragaria vesca and Rosa genome. However, the training data of the above methods are all derived from specific species. And when extended to other species, it may produce a low true-positive rate with a high false-positive rate. Therefore, there is urgent to develop a generic DNA modification site predictor that can be used in different species. In other biological and medical fields, machine learning-based computational methods have been widely used, including microRNAs and cancer association prediction (Yuming et al., 2015; Jiang et al., 2018; Ding et al., 2020a; Wang et al., 2021), function prediction of proteins (Ding et al., 2019d, 2020b; Wang Y. et al., 2019; Wang H. et al., 2019; Tao et al., 2020; Zou et al., 2020b; Yang et al., 2021), drugs complex network analysis (Ding et al., 2017, 2019a,b,c, 2020c; Guo et al., 2020b) and dry weight assessment of hemodialysis patients (Guo et al., 2020a).
In this study, we developed a new deep learning-based multi-task method, called iDNA-MT, for identifying 4mC site and 6mA site in multiple species, respectively. This method combines both the bidirectional gated recurrent units (BGRU) and multi-task learning to learn sharing information hiding in different species for better characterizing a DNA sequence. Afterward, the sharing features are fed into the corresponding fully connected layers, specifically designed for a certain task, to identify the modification site. Several experiments were carried out to investigate the performance of the proposed iDNA-MT. Experimental results on two benchmark datasets showed that iDNA-MT achieved significantly better performance than state-of-the-art single-task methods for identifying 4mC site and 6mA site, respectively. In addition, our model can provide a powerful tool for identifying 4mC sites and 6mA sites in multiple species, respectively, and facilitate our knowledge of their biological functions.
For a fair comparison, we employed the same benchmark datasets derived from Lv et al. (2020a). Four species of 4mC site data and four species of 6mA site data were selected. The 4mC site data contains four species (C. equisetifolia, F. vesca, S. cerevisiae, and Tolypocladium) that were collected from the MDR database (Liu et al., 2016) and MethSMRT database (Pohao et al., 2017). The 6mA site data for four species (Tolypocladium, C. elegans, C. equisetifolia, and R. chinensis) were extracted from the MethSMRT database (Pohao et al., 2017), MethSMRT database (Pohao et al., 2017), and MDR database (Liu et al., 2016). The benchmark data is divided into two parts. One part is used as a training dataset, and the other one is a testing dataset. The function of the training dataset is to train and evaluate the predictive model, while the purpose of the testing dataset is to test the performance of the model. The number of positive and negative samples is the same in the training dataset and testing dataset. A summary of the different species datasets used for benchmarking is displayed in Table 1.

Table 1. Summary of benchmark datasets used in this study.
In this section, we introduce the network architecture of our model iDNA-MT, as illustrated in Figure 1. This network architecture consists of three main components: (i) sequence processing module, (ii) sharing module, and (iii) task-specific output module. To make DNA sequences recognized easily by the neural network, the sequence processing module is designed to encode the original DNA sequences into matrices by one-hot encoding (Quang and Xie, 2016). Next, the encoded matrix is passed through a bidirectional GRU to extract different levels of dependency relationships between subsequences, and then a max-pooling layer is employed to automatically measure which feature plays a key role in NDA methylation site identification in each unit of the GRU. Finally, the features learned from the max-pooling layer are sent to the task-specific output module to identify 6mA sites in four species, respectively. The task-specific output module contains four parts and each part consists of fully connected layers that are designed in terms of the size of the training set of each species. The model is implemented using Pytorch. Below each module of our model is described in detail.
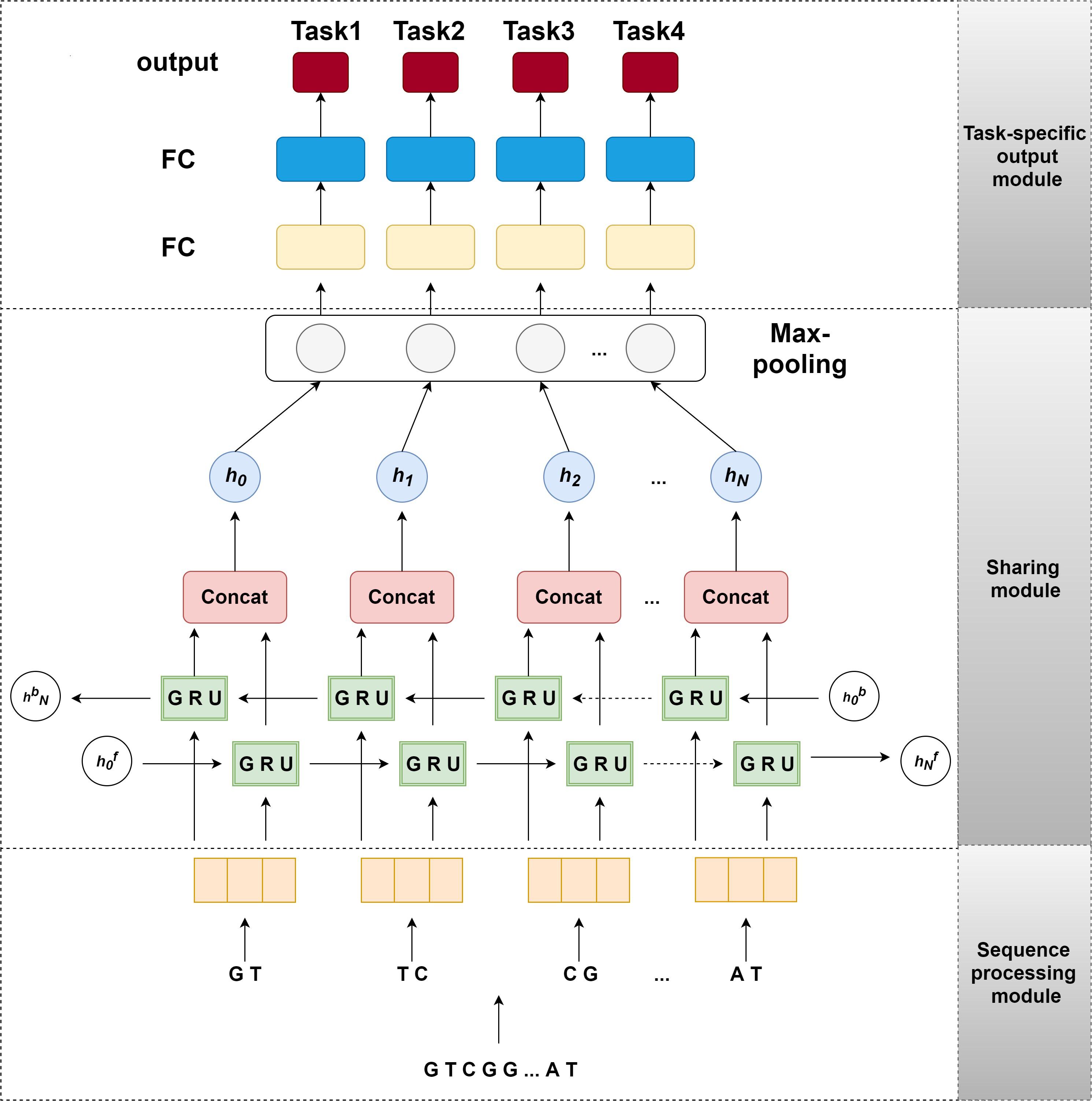
Figure 1. The structure of iDNA-MT. The sequence processing module uses 2-gram to split an original DNA sequence into overlapping subsequences and converts them into feature vectors by one-hot encoding. Next, the feature vectors of subsequences are sent into sharing module, containing a BGRU and a max-pooling layer, to capture the sharing information among different species. Finally, the output of sharing module is fed into the task-specific output module to predict the modification site (i.e., 4mA or 6mC) of a certain species.
DNA modification identification is the task to separate the DNA sequences into related classes of DNA modifications, while text categorization is the problem of assigning text documents to predefined categories. To apply text categorization techniques to DNA sequences, we first employed n-gram nucleobases to define “words” in DNA sequences (Dong et al., 2006; Dao et al., 2020; Wang et al., 2020; Zhang et al., 2020). The n-grams are the set of all possible subsequences of nucleobases. Then, we split the DNA sequences into overlapping n-gram nucleobases. The number of possible it is 4n, since there are four types of nucleobases (Yang et al., 2020). In this study, to avoid low-frequency words in the encoding, the n-gram number n is set to 2. For example, we split a DNA sequence into overlapping 2-gram nucleobase sequences as follows: GTTGT…CTT→ “GT,” “TT,” “TG,” “GT,”…, “CT,” “TT.”
For a given DNA sequence P of length L, it can be expressed as follows:
where Ri is the i -th word. These words are first randomly initialized embedded by one-hot embedding, which is referred to as “word embeddings.” Here, we defined the sequence of word embeddings as:
where xi ∈ ℝd is the d-dimensional embedding of the i -th word. In the proposed method, such a sequence is fed into the bidirectional GRU to extract dependency information.
GRU is one of the widely used deep learning techniques, which is designed to specifically address the problems of learning long-distance correlations in a sequence (Cho et al., 2014). Bidirectional GRU is the most important part of the sharing module, which is employed to automatically extract long-terms and short-term dependency relationships in DNA sequences. The structure of the basic unit of GRU is shown in Figure 2. The unit receives two input vectors: the embedding vector of the subsequence and the hidden state of the previous time step. The special thing about them is that they can be trained to keep information from long ago. Based on the two inputs, two gates, namely, reset gate and update gate, coordinate with each other to capture short-term and long-term dependencies in sequences. The reset gate is used to control how much of the previous information to forget. Likewise, the update gate helps the model to determine how much of the past information, from previous time steps, needs to be passed along to the future.
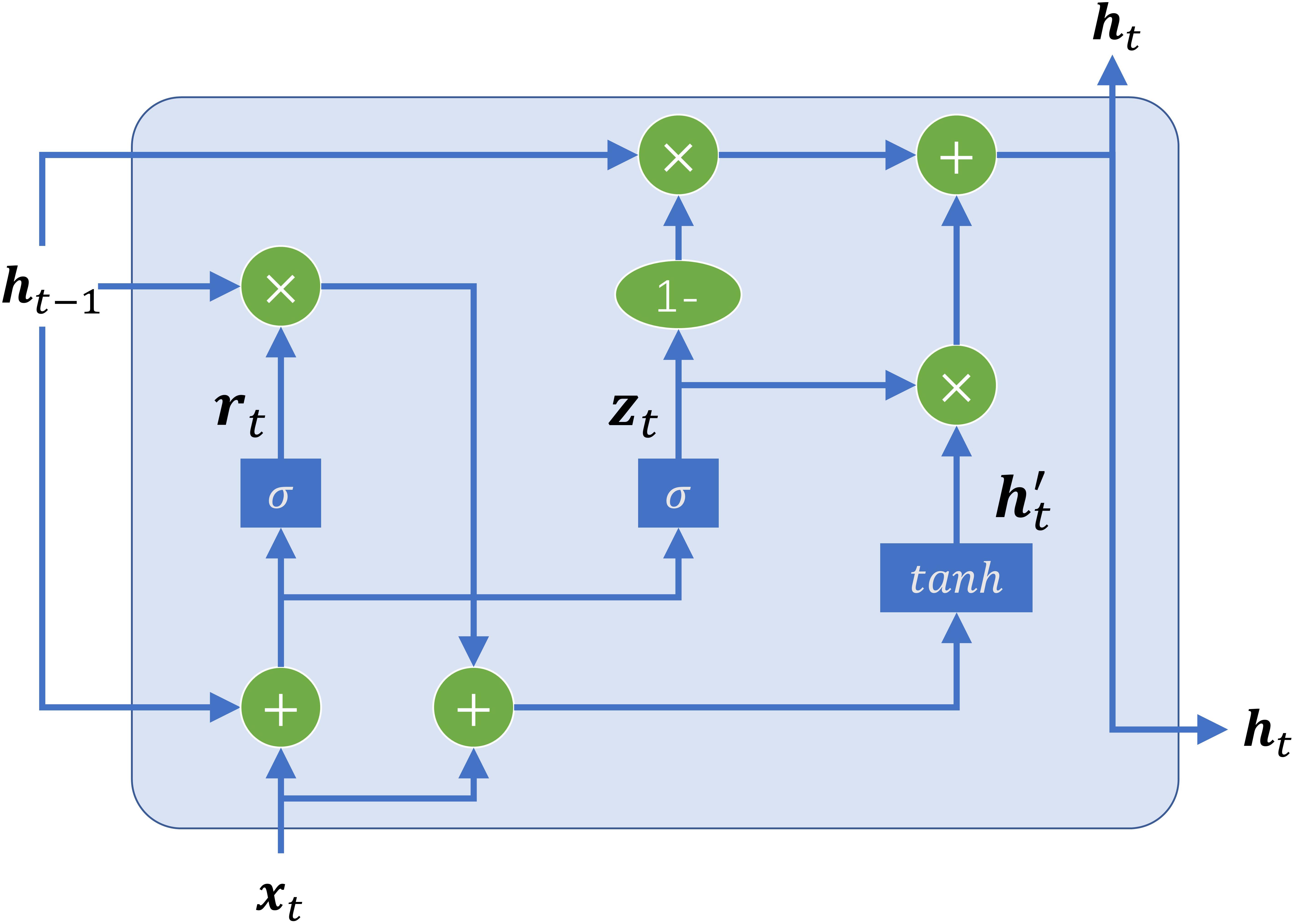
Figure 2. The structure of GRU cell. There are two gates, including a reset gate (denoted as rt) and an update gate (denoted as zt), to control the information flowing in and out of the cell. The reset gate control how much of the previous information to store. The update gate control how much of the past information needs to be passed along to the next time step. x is the embedding matrix of the input subsequence, h is the output of the GRU cell, and t denotes the t -th time step.
For a given time step t, there are four components composite the GRU-based recurrent neural network: a reset gate rt with corresponding weight matrices Wr, Ur; an update gate zt with corresponding weight matrices Wz, Uz; a candidate hidden state with corresponding weight matrices W,U; and a new hidden state ht. The equations of GRU are the following:
where xt denotes the input of the current time step, σ denotes the logistic sigmoid function to transform input values to the interval (0,1), ht−1 denotes the output of the last time step, ⊙ denotes element-wise multiplication, and tanh is a non-linear activation function to ensure the values in the candidate hidden state remain in the interval (−1,1). Hence, the new hidden state ht holds information for the current step and previous steps and passes it down to the network.
However, a standard GRU network process a sequence in temporal order, resulting in that the outputs only contain the forward sequence information. To fully extract the information of a sequence, it is significant to capture not only the forward information but also the backward information at each time step. Therefore, we attempt to add another GRU network that captures the backward sequence information by processing a DNA sequence in the opposite temporal order. Combine it with the standard GRU network to form a bidirectional GRU, which can exploit information both from the past and the future.
To better capture the dependency information of subsequences with large time step distances, in this study, we combined the forward and backward hidden vectors generated by bidirectional GRU in each step. Therefore, the i -th subsequence can be expressed as the following vector:
where h is the hidden vector, and denote the hidden vectors generated by the forward GRU and the backward GRU, respectively.
The feature vector h of each subsequence, generated by bidirectional GRU, is fed into a max-pooling layer to capture the most significant feature in identifying the DNA modification to represent this subsequence. Then, all the most significant features of subsequences are concatenated into a vector to represent a DNA sequence, which is shown in the following equation:
where i is the i -th subsequence, n is the number of subsequences in a DNA sequence, and the y is regarded as the feature vector of a target sequence. The max-pooling layer attempts to find the most important dependencies in subsequences.
This module consists of four sets of fully connected layers corresponding to each task, respectively. In each fully connected layer with a relu activation function, its output is calculated by the following equation:
where is the output of the previous layer of j -th task, is the current layer output of j -th task, is the weight matrix, and is the bias vector. In each layer, the “Batch Normalization” technique was used to improve generalization performance (Cheng and Baldi, 2006). Finally, a softmax layer is added on the top of final output fj to perform the final prediction. Note that the parameters of different set of the fully connected layer are designed differently in terms of the amount of data of the corresponding task.
The task-specific features y, generated by the sharing module, are ultimately sent into one set of fully connected layers in terms of it belonging to which task. For classification tasks, we used binary cross-entropy loss function as the objective:
where N denotes the number of training samples, yi denotes the label (i.e., 1 or 0) of sample i, pi denotes the probability that sample i is predicted to be positive. Our global loss function is the linear combination of loss function for all tasks:
where αk is the weight for task k.
It is worth noting that the samples for training each task can come from completely different datasets. Following the study (Liu et al., 2016), the training is carried out in a stochastic manner by looping over the tasks:
1. Select a task randomly.
2. Select a training sample from this task randomly.
3. Update the parameters for this task by taking a gradient step in terms of this sample.
4. Go to 1.
To evaluate the performance of our model, four commonly used metrics are employed to evaluate the performance of the model (Zou et al., 2016; Jin et al., 2019, 2020; Manayalan et al., 2019; Manavalan et al., 2019b; Hong et al., 2020; Lv et al., 2020b; Qiang et al., 2020; Su et al., 2020a,b,c, 2019a,b; Wei et al., 2020, 2014, 2019a, 2018a,b; Zhao et al., 2020; Zou et al., 2020a), including sensitivity (SN), specificity (SP), overall accuracy (ACC), and Matthew’s correlation coefficient (MCC), respectively. They are formulated as:
where TP, TN, FP, and FN represent the numbers of true positives, true negatives, false positives, and false negatives, respectively. SN and SP are used to evaluate positive and negative predictive ability. MCC and ACC were used to evaluate the overall prediction performance. Besides, the ROC curve (receiver operating characteristic curve) can be used to visualize the performance of the classifier. In addition, we calculate the area under the ROC curve (AUC) to evaluate the prediction performance of the model. The range of AUC is 0.5–1. The higher the AUC score, the better the prediction performance of the model.
To evaluate the performance of our model iDNA-MT for identifying 4mC and 6mA site in multiple species, we compared it with seven state-of-the-art models based on random forest (RF), which were all single-task learning methods and used different feature descriptors to identify 4mC and 6mA site in each species, respectively, including K-tuple nucleotide frequency component (KNFC), nucleotide chemical property and nucleotide frequency (NCPNF), and mono-nucleotide binary encoding (MNBE), and their four combinations (Lv et al., 2020a).
The experimental results of different methods are listed in Figure 3. From Figure 3, we can observe that for 4mC site identification, our proposed iDNA-MT significantly outperforms all the other competing methods in three species (C. equisetifolia, Tolypocladium, and S. cerevisiae) in terms of five metrics (SN, SP, ACC, MCC, and AUC), while the model using MNBE achieves the best performance amongst all methods. For 6mA site identification, iDNA-MT exhibits better performance than any other RF-based models in each species. These results indicate that using both BGRU and multi-task learning can extract more effective and discriminative features to represent DNA sequences for identifying 4mA site and 6mC site and be generalized well on different species. There are two main reasons for the outstanding performance of our model. First, compared with the RF-based methods that use handcrafted features to train models, which need prior knowledge, iDNA-MT can automatically capture effective features by data driving. Second, the proposed iDNA-MT employs the BGRU to learn long-distance dependency information of DNA subsequences, and then introduce the multi-task learning technique to capture the shared information hidden in data from different species to improve the performance of each task, to improve the accuracy for identifying 4mC and 6mA site in multiply species, respectively. Therefore, iDNA-MT can achieve better performance than other state-of-the-art single-task learning methods. Note that the detailed comparison results of iDNA-MT and seven state-of-the-art methods can be found in Supplementary Table S1.
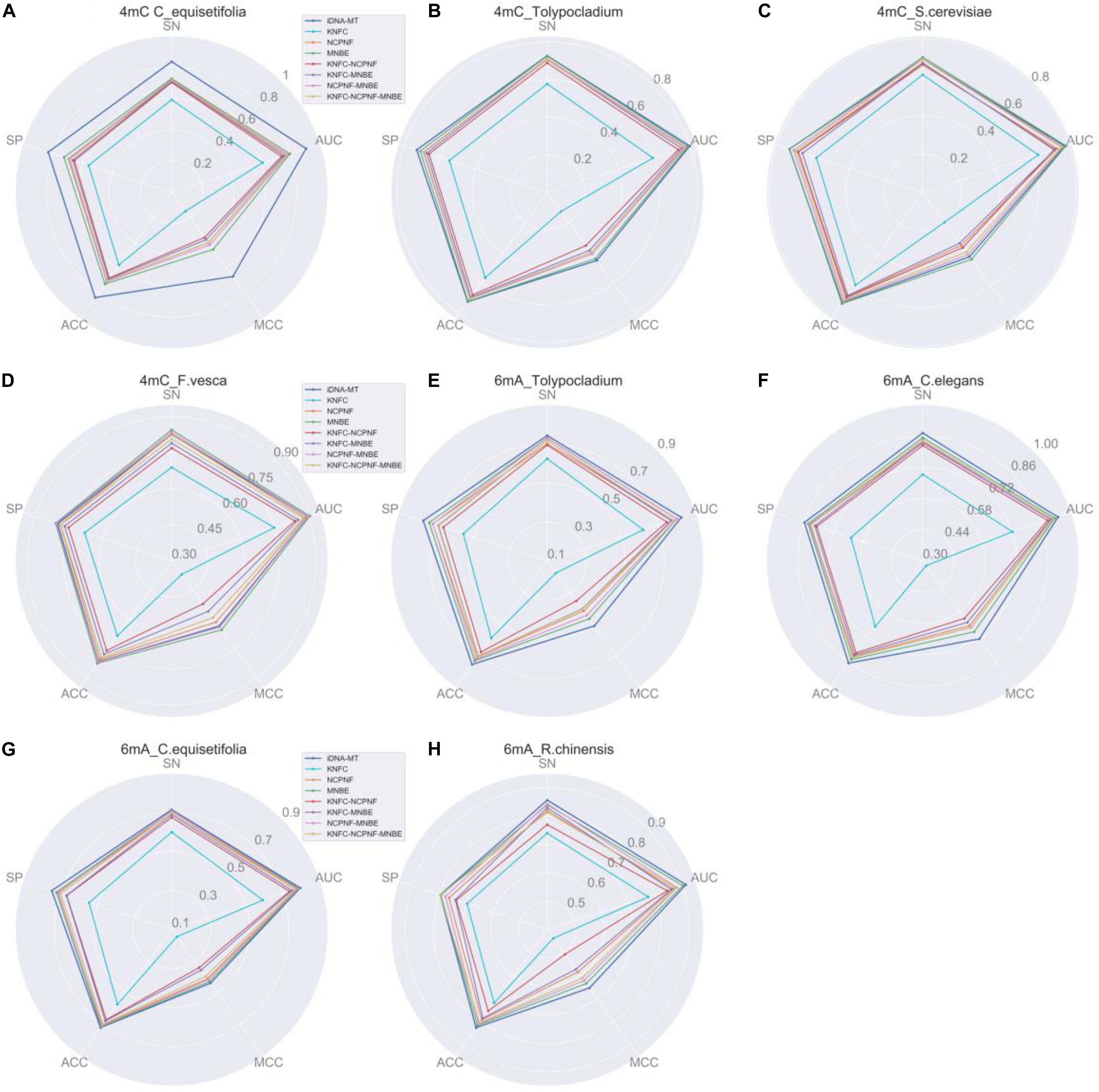
Figure 3. Performance evaluation of iDNA-MT and seven state-of-the-art methods for identifying 4mC site and 6mA site. (A–D) represents the performance of identifying 4mC site. (E,F) represents the performance of identifying 6mA site. (A–D) represent the performance of different methods for identifying 4mC site. (E–H) represent the performance of different methods for identifying 6mA site.
To evaluate whether or not introducing the multi-task learning technique can capture more discriminative features to improve the performance of DNA modification site prediction in multiple species, we compared the model considering the multi-task learning, namely iDNA-MT, with the model not considering the multi-task learning for prediction. The comparative results for 4mC site and 6mA site are illustrated in Tables 2, 3, respectively. In Tables 2, 3, we show better results in bold.
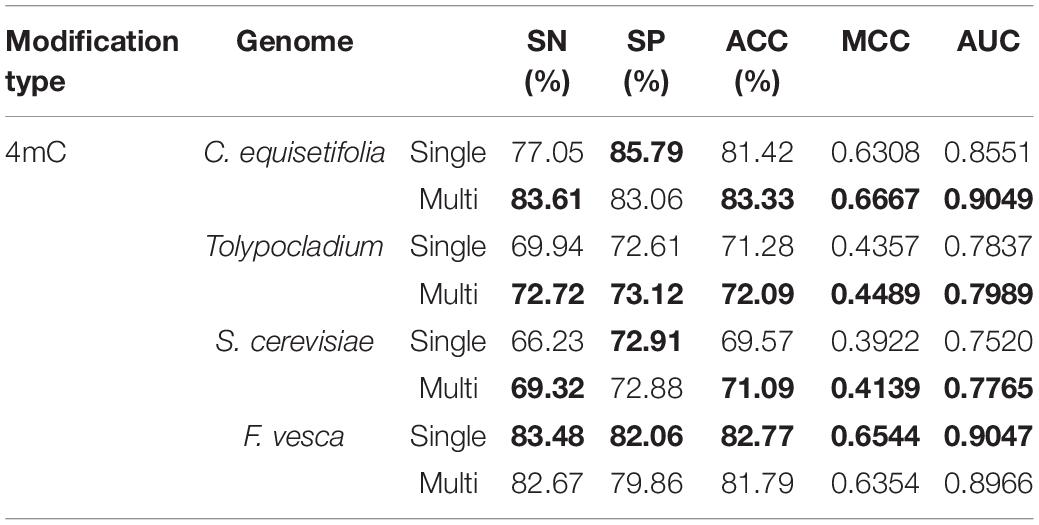
Table 2. Comparison results of the model using the multi-task learning and the model not using the multi-task learning for identifying 4mC site.
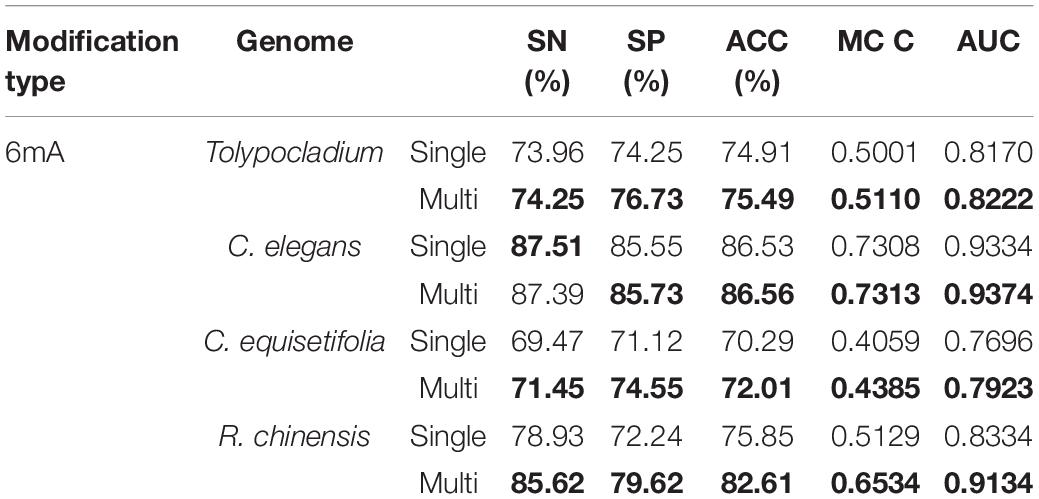
Table 3. Comparison results of the model using the multi-task learning and the model not using the multi-task learning for identifying 6mA site.
As shown in Table 2 for 4mC site prediction, we can see that training with the multi-task learning, the model achieves higher performance in three species, including C. equisetifolia, Tolypocladium, and S. cerevisiae, with only one exception in F. vesca. Specifically, the model using the multi-task learning achieves an ACC of 83.33%, an MCC of 0.6667, and an AUC of 0.9049 for species C. equisetifolia, yielding a relative improvement of 2.3%, 5.7%, and 5.8%, respectively, achieves an ACC of 72.09%, an MCC of 0.4489 and an AUC of 0.7989 for species Tolypocladium, yielding a relative improvement of 1.1%, 3.0%, and 1.9%, respectively, and achieves an ACC of 71.09%, an MCC of 0.4139 and an AUC of 0.7765 for species S. cerevisiae, yielding a relative improvement of 2.2%, 5.5%, and 3.3%, respectively. For species F. vesca, the model using multi-task learning is slightly worse than the model not using multi-task learning, which achieves 82.67%, 79.86%, 81.79%, 0.6354, and 0.8966 in terms of SN, SP, ACC, MCC, and AUC. From Table 3, we can see that for all four species (Tolypocladium, C. elegans, C. equisetifolia, and R. chinensis), the model using multi-task learning all significantly outperforms the model not using multi-task learning for identification 6mA site in terms of SN, SP, ACC, MCC, and AUC. The most significant improvement is observed in species R. chinensis, in which the model using multi-task learning improves the SN from 78.93% to 85.62%, the SP from 72.24% to 79.62%, the ACC from 75.85% to 82.61%, the MCC from 0.5129 to 0.6534 and the AUC from 0.8334 to 0.9134.
These results discussed above demonstrate that by introducing the multi-task learning, the model can achieve outstanding performance for 4mC site and 6mA site prediction in multiply species, respectively. The reason may be that multi-task learning aims to learn shared representations from multiple related tasks, which are used to share and supplement the information learned from different tasks to improve the performance of multiple related learning tasks. Therefore, there is not surprising that the model using multi-task learning significantly outperforms the model not using multi-task learning.
The sharing module of iDNA-MT mainly employed BGRU to exploit the potential information both from forward and backward and then used the max-pooling layer to extract the most significant features in subsequences, which play key roles in DNA modification identification. To evaluate the efficiency and superiority of the neural network architecture in sharing module, we replaced it with other three typical text classification methods, respectively, including:
1. TextRNN (Liu et al., 2016): It uses the long short-term memory network (LSTM) to capture long-term semantic dependencies in a sentence.
2. Att-BLSTM (Zhou et al., 2016): It utilizes both neural attention mechanism and bidirectional long short-term memory networks (BLSTM) to capture the most important semantic information in a sentence.
3. Transformer (Vaswani et al., 2017): It is a novel neural network architecture based on a self-attention mechanism.
Figure 4 shows the comparison results of the proposed iDNA-MT and the other methods using different typical text classification methods in sharing modules on two different modification sites in terms of five metrics (SN, SP, ACC, MCC, and AUC). As shown in Figure 4, we can see that for 4mC site, the performance of iDNA-MT is significantly better than the other methods using different typical text classification methods in sharing module in every species. For 6mA site, although the performance of iDNA-MT is lower than other methods in species C. equisetifolia, the performance of iDNA-MT significantly outperforms other methods in the rest species. Therefore, iDNA-MT is superior to other methods in identifying 4mC sites and 6mA sites in multiple species, respectively. The proposed iDNA-MT used BGRU to capture the dependency information of subsequences from the past and the future and added a max-pooling layer to extract the most important information hiding in every subsequence, which avoids irrelevant information from interfering with identifying results. Therefore, there is no surprise that iDNA-MT achieves the best performance when combing BGRU and a max-pooling layer.
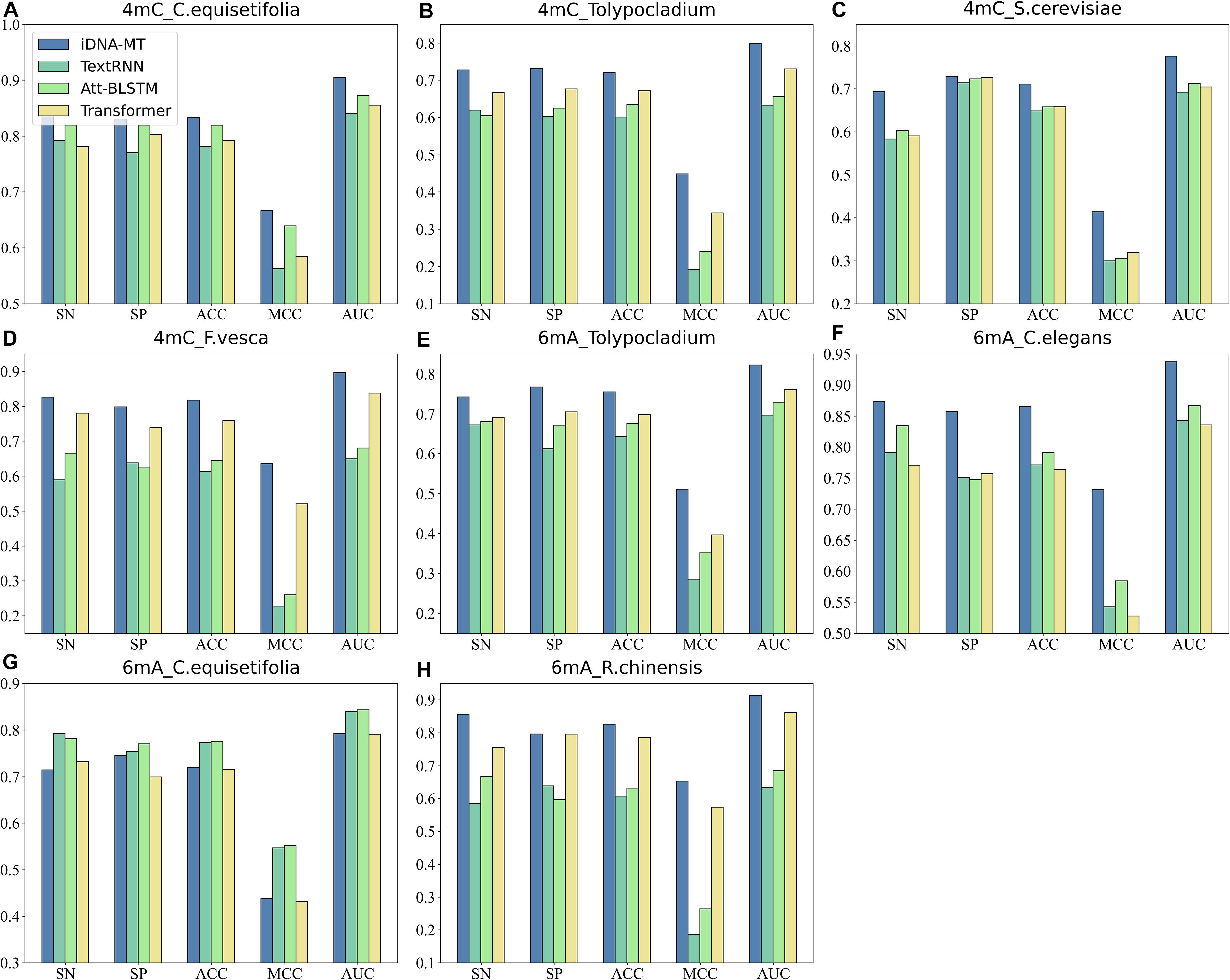
Figure 4. Performance evaluation of iDNA-MT and other methods using different typical text classification methods in sharing module (A–D) represent the performance of iDNA-MT and other methods for identifying 4mC site. (E–H) represent the performance of iDNA-MT and other methods for identifying 6mA site.
Although 4mA and 6mC are two important genetic modifications and play crucial roles in regulating a series of biological processes, their biological functions are still unclear. Therefore, the accurate identification of them is pivotal to understand specific biological functions. In this study, we proposed a multi-task learning predictor namely iDNA-MT for identifying 4mA site and 6mC site in multiple species, respectively, which can automatically extract the discriminative features for different tasks. To better represent the DNA sequences of different species, we constructed a sharing module, containing a BGRU and a max-pooling layer, to capture sharing information among different species. To evaluate the efficiency of our multi-task model, we compared it with the state-of-the-art single-task models on benchmark datasets of two different DNA modifications. Experimental results have shown that the proposed iDNA-MT achieved the top performance comparing with existing single-task models on two benchmark datasets, indicating that multi-task learning can improve the performance of multiple related tasks by leveraging useful information among them. In future work, we would like to investigate other sharing mechanisms to further improve the prediction of different DNA modifications in multiple species and apply it to other fields (Wei et al., 2017a,b,c, 2018c, 2019c,d; Zou et al., 2019).
All datasets generated for this study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
XY and LW surveyed the algorithms and implementations, preprocessed the datasets, and performed all the analyses. XCY and XL designed the benchmarking test. All the authors have written, read, and approved the manuscript.
This work was supported in part by the New Energy and Industrial Technology Development Organization 265 (NEDO) and the Japan Society for the Promotion of Science (JSPS), Grants-in-Aid for Scientific Research under Grant 18H03250, and the Natural Science Foundation of China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.663572/full#supplementary-material
Ao, C., Jin, S., Lin, Y., and Zou, Q. (2019). Review of progress in predicting protein methylation sites. Curr. Organ. Chem. 23, 1663–1670. doi: 10.2174/1385272823666190723141347
Basith, S., Manavalan, B., Shin, T. H., and Lee, G. (2019). SDM6A: a web-based integrative machine-learning framework for predicting 6mA sites in the rice genome. Mol. Therapy - Nucleic Acids 18, 131–141. doi: 10.1016/j.omtn.2019.08.011
Bergman, Y., and Cedar, H. (2013). DNA methylation dynamics in health and disease. Nat. Struct. Mol. Biol. 20, 274–281.
Cai, L., Ren, X., Fu, X., Peng, L., Gao, M., and Zeng, X. J. B. (2020). iEnhancer-XG: interpretable sequence-based enhancers and their strength predictor. Bioinformatics doi: 10.1093/bioinformatics/btaa914 Online ahead of print.
Chen, W., Lv, H., Nie, F., and Lin, H. (2019). i6mA-Pred: identifying DNA N6-methyladenine sites in the rice genome. Bioinformatics 35, 2796–2800. doi: 10.1093/bioinformatics/btz015
Chen, W., Yang, H., Feng, P., Ding, H., and Lin, H. (2017). iDNA4mC: identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics 33, 3518–3523.
Chen, J. L. J., and Zou, Q. (2019). DeepM6ASeq-EL: prediction of human N6-methyladenosine (m6A) Sites with LSTM and ensemble learning. Front. Comput. Sci. doi: 10.1007/s11704-020-0180-0
Cheng, J., and Baldi, P. (2006). A machine learning information retrieval approach to protein fold recognition. Bioinformatics 22, 1456–1463.
Cho, K., Van Merrienboer, B., Bahdanau, D., and Bengio, Y. (2014). “On the properties of neural machine translation: encoder-decoder approaches,” in Proceedings of the SSST-8, Eighth Workshop on Syntax, Semantics and Structure in Statistical Translation, (Doha: Association for Computational Linguistics).
Dao, F. Y., Lv, H., Zhang, D., Zhang, Z. M., Liu, L., and Lin, H. (2020). DeepYY1: a deep learning approach to identify YY1-mediated chromatin loops. Brief. Bioinform. doi: 10.1093/bib/bbaa356. Online ahead of print.
Ding, Y., Jiang, L., Tang, J., and Guo, F. (2020a). Identification of human microRNA-disease association via hypergraph embedded bipartite local model. Comput. Biol. Chem. 89:107369. doi: 10.1016/j.compbiolchem.2020.107369
Ding, Y., Tang, J., and Guo, F. (2020b). Human protein subcellular localization identification via fuzzy model on kernelized neighborhood representation. Appl. Soft Comput. 96:106596. doi: 10.1016/j.asoc.2020.106596
Ding, Y., Tang, J., and Guo, F. (2020c). Identification of Drug–Target interactions via dual laplacian regularized least squares with multiple kernel fusion. Knowledge-Based Systems 204:106254. doi: 10.1016/j.knosys.2020.106254
Ding, Y., Tang, J., and Guo, F. (2017). Identification of drug-target interactions via multiple information integration. Inform. Sci. 418, 546–560. doi: 10.1016/j.ins.2017.08.045
Ding, Y., Tang, J., and Guo, F. (2019a). Identification of drug-side effect association via multiple information integration with centered kernel alignment. Neurocomputing 325, 211–224. doi: 10.1016/j.neucom.2018.10.028
Ding, Y., Tang, J., and Guo, F. (2019b). Identification of drug-side effect association via semisupervised model and multiple kernel learning. IEEE J. Biomed. Health Inform. 23, 2619–2632. doi: 10.1109/jbhi.2018.2883834
Ding, Y., Tang, J., and Guo, F. (2019c). Identification of drug-target interactions via fuzzy bipartite local model. Neural Comp. Appl. 32, 10303–10319. doi: 10.1007/s00521-019-04569-z
Ding, Y., Tang, J., and Guo, F. (2019d). Protein crystallization identification via fuzzy model on linear neighborhood representation. IEEE/ACM Trans. Comput. Biol. Bioinform. Online ahead of print.
Dong, Q.-W., Wang, X.-L., and Lin, L. (2006). Application of latent semantic analysis to protein remote homology detection. Bioinformatics 22, 285–290. doi: 10.1093/bioinformatics/bti801
Dou, L. J., Li, X. L., Ding, H., Xu, L., and Xiang, H. K. (2020). Is there any sequence feature in the RNA pseudouridine modification prediction problem? Mol. Ther.-Nucl. Acids 19, 293–303. doi: 10.1016/j.omtn.2019.11.014
Fang, G., Munera, D., Friedman, D. I., Mandlik, A., Chao, M. C., Banerjee, O., et al. (2012). Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat. Biotechnol. 30, 1232–1239. doi: 10.1038/nbt.2432
Fu, X., Cai, L., Zeng, X., and Zou, Q. J. B. (2020). StackCPPred: a stacking and pairwise energy content-based prediction of cell-penetrating peptides and their uptake efficiency. Bioinformatics 36, 3028–3034. doi: 10.1093/bioinformatics/btaa131
Fu, Y., and He, C. (2012). Nucleic acid modifications with epigenetic significance. Curr. Opin. Chem. Biol 16, 516–524. doi: 10.1016/j.cbpa.2012.10.002
Greer, E. L., Blanco, M. A., Gu, L., Sendinc, E., Liu, J., Aristizábal-Corrales, D., et al. (2015). DNA Methylation on N6-Adenine in C. elegans. Cell 161, 868–878. doi: 10.1016/j.cell.2015.04.005
Guo, X. Y., Zhou, W., Shi, B., Wang, X. H., Du, A. Y., Ding, Y. J., et al. (2020a). An efficient multiple kernel support vector regression model for assessing dry weight of hemodialysis patients. Curr. Bioinform. 15, 466–469.
Guo, X. Y., Zhou, W., Yu, Y., Ding, Y. J., Tang, J. J., and Guo, F. (2020b). A novel triple matrix factorization method for detecting drug-side effect association based on kernel target alignment. BioMed Res. Int. 2020, 1–11. doi: 10.1155/2020/4675395
Guohua, W., Ximei, L., Jianan, W., Jun, W., Shuli, X., Heng, Z., et al. (2017). MeDReaders: a database for transcription factors that bind to methylated DNA. Nucleic Acids Res. 46, D146–D151.
Hasan, M. M., Manavalan, B., Khatun, M. S., and Kurata, H. (2020). i4mC-ROSE, a bioinformatics tool for the identification of DNA N4-methylcytosine sites in the Rosaceae genome. Int. J. Biol. Macromol. 157, 752–758. doi: 10.1016/j.ijbiomac.2019.12.009
He, W., Jia, C., and Zou, Q. (2019). 4mCPred: machine learning methods for DNA N4-methylcytosine sites prediction. Bioinformatics 35, 593–601. doi: 10.1093/bioinformatics/bty668
Heyn, H., and Esteller, M. (2015). An adenine code for DNA: a second life for N6-methyladenine. Cell 161, 710–713. doi: 10.1016/j.cell.2015.04.021
Hong, Z., Zeng, X., Wei, L., and Liu, X. J. B. (2020). Identifying enhancer–promoter interactions with neural network based on pre-trained DNA vectors and attention mechanism. Bioinformatics 36, 1037–1043.
Hu, L., Liu, Y., Han, S., Yang, L., Cui, X., Gao, Y., et al. (2019). Jump-seq: genome-Wide capture and amplification of 5-Hydroxymethylcytosine sites. J. Am. Chem. Soc. 141, 8694–8697. doi: 10.1021/jacs.9b02512
Iyer, L. M., Abhiman, S., and Aravind, L. (2011). Chapter 2 - natural history of eukaryotic DNA methylation systems. Prog. Mol. Biol. Transl. Sci. 101, 25–104. doi: 10.1016/b978-0-12-387685-0.00002-0
Jiang, L., Xiao, Y., Ding, Y., Tang, J., and Guo, F. (2018). FKL-Spa-LapRLS: an accurate method for identifying human microRNA-disease association. BMC Genomics 19:911. doi: 10.1186/s12864-018-5273-x
Jin, Q., Meng, Z., Tuan, D. P., Chen, Q., Wei, L., and Su, R. (2019). DUNet: a deformable network for retinal vessel segmentation. Knowledge-Based Systems 178, 149–162. doi: 10.1016/j.knosys.2019.04.025
Jin, S., Zeng, X., Xia, F., Huang, W., and Liu, X. (2020). Application of deep learning methods in biological networks. Brief. Bioinform. Online ahead of print.
Kong, L., and Zhang, L. (2019). i6mA-DNCP: computational identification of DNA N6-Methyladenine sites in the rice genome using optimized dinucleotide-based features. Genes 10:828. doi: 10.3390/genes10100828
Liu, P., Qiu, X., and Huang, X. (2016). “Recurrent neural network for text classification with multi-task learning,” in Proceedings of the Twenty-Fifth International Joint Conference on Artificial Intelligence (IJCAI-16), (Shanghai: Fudan University).
Luo, X., Wang, F., Wang, G., and Zhao, Y. (2020). Identification of methylation states of DNA regions for Illumina methylation BeadChip. BMC Genomics 21:672. doi: 10.1186/s12864-019-6019-0
Lv, H., Dao, F. Y., Zhang, D., Guan, Z. X., Yang, H., Su, W., et al. (2020a). iDNA-MS: an integrated computational tool for detecting DNA modification sites in multiple genomes. iScience 23:100991. doi: 10.1016/j.isci.2020.100991
Lv, H., Dao, F.-Y., Guan, Z.-X., Yang, H., Li, Y.-W., and Lin, H. (2020b). Deep-Kcr: accurate detection of lysine crotonylation sites using deep learning method. Brief. Bioinform. bbaa255. doi: 10.1093/bib/bbaa356
Manavalan, B., Basith, S., Shin, T. H., Lee, D. Y., Wei, L., and Lee, G. (2019a). 4mCpred-EL: an ensemble learning framework for identification of DNA N4-Methylcytosine sites in the mouse genome. Cells 8:1332. doi: 10.3390/cells8111332
Manavalan, B., Basith, S., Shin, T. H., Wei, L., and Lee, G. (2019b). Meta-4mCpred: a sequence-based meta-predictor for accurate DNA 4mC site prediction using effective feature representation. Mol. Therapy-Nucleic Acids 16, 733–744. doi: 10.1016/j.omtn.2019.04.019
Manayalan, B., Basith, S., Shin, T. H., Wei, L., and Lee, G. (2019). mAHTPred: a sequence-based meta-predictor for improving the prediction of anti-hypertensive peptides using effective feature representation. Bioinformatics 35, 2757–2765. doi: 10.1093/bioinformatics/bty1047
Mondo, S. J., Dannebaum, R. O., Kuo, R. C., Louie, K. B., Bewick, A. J., LaButti, K., et al. (2017). Widespread adenine N6-methylation of active genes in fungi. Nat. Genet. 49, 964–968. doi: 10.1038/ng.3859
Ni, P., Huang, N., Zhang, Z., Wang, D.-P., Liang, F., Miao, Y., et al. (2019). DeepSignal: detecting DNA methylation state from Nanopore sequencing reads using deep-learning. Bioinformatics 35, 4586–4595. doi: 10.1093/bioinformatics/btz276
Pohao, Y., Yizhao, L., Kaining, C., Yizhi, L., Chuanle, X., and Zhi, X. J. (2017). MethSMRT: an integrative database for DNA N6-methyladenine and N4-methylcytosine generated by single-molecular real-time sequencing. Nucleic Acids Res. 45, D85–D89.
Qiang, X., Zhou, C., Ye, X., Du, P.-F., Su, R., and Wei, L. (2020). CPPred-FL: a sequence-based predictor for large-scale identification of cell-penetrating peptides by feature representation learning. Brief. Bioinform. 21, 11–23.
Quang, D., and Xie, X. (2016). DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res. 44:e107. doi: 10.1093/nar/gkw226
Schweizer, H. P. (2008). Bacterial genetics: past achievements, present state of the field, and future challenges. Biotechniques 44, 636–641.
Shen, Z., and Zou, Q. (2020). Basic polar and hydrophobic properties are the main characteristics that affect the binding of transcription factors to methylation sites. Bioinformatics 36, 4263–4268. doi: 10.1093/bioinformatics/btaa492
Smith, Z. D., and Meissner, A. (2013). DNA methylation: roles in mammalian development. Nat. Rev. Genet. 14, 204–220. doi: 10.1038/nrg3354
Su, R., Hu, J., Zou, Q., Manavalan, B., and Wei, L. (2020a). Empirical comparison and analysis of web-based cell-penetrating peptide prediction tools. Brief. Bioinform. 21, 408–420. doi: 10.1093/bib/bby124
Su, R., Liu, X., and Wei, L. (2020b). MinE-RFE: determine the optimal subset from RFE by minimizing the subset-accuracy-defined energy. Brief. Bioinform. 21, 687–698. doi: 10.1093/bib/bbz021
Su, R., Liu, X., Xiao, G., and Wei, L. (2020c). Meta-GDBP: a high-level stacked regression model to improve anticancer drug response prediction. Brief. Bioinform. 21, 996–1005. doi: 10.1093/bib/bbz022
Su, R., Liu, X., Wei, L., and Zou, Q. (2019a). Deep-Resp-Forest: a deep forest model to predict anti-cancer drug response. Methods 166, 91–102. doi: 10.1016/j.ymeth.2019.02.009
Su, R., Wu, H., Xu, B., Liu, X., and Wei, L. (2019b). Developing a multi-dose computational model for drug-induced hepatotoxicity prediction based on toxicogenomics data. IEEE-ACM Trans. Comput. Biol. Bioinform. 16, 1231–1239. doi: 10.1109/tcbb.2018.2858756
Szulwach, K. E., Li, X., Li, Y., Song, C. X., and Jin, P. (2011). 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 14, 1607–1616. doi: 10.1038/nn.2959
Tang, W., Wan, S., Yang, Z., Teschendorff, A. E., and Zou, Q. (2018). Tumor origin detection with tissue-specific miRNA and DNA methylation markers. Bioinformatics 34, 398–406. doi: 10.1093/bioinformatics/btx622
Tao, Z., Li, Y., Teng, Z., and Zhao, Y. (2020). A method for identifying vesicle transport proteins based on LibSVM and MRMD. Comput. Mathemat. Methods Med. 2020:8926750.
Thomson, J. P., and Meehan, R. R. (2016). The application of genome-wide 5-hydroxymethylcytosine studies in cancer research. Epigenomics 9, 77–91. doi: 10.2217/epi-2016-0122
Timinskas, A., Butkus, V., and Janulaitis, A. (1995). Sequence motifs characteristic for DNA [cytosine-N4] and DNA [adenine-N6] methyltransferases. Classification of all DNA methyltransferases. Gene 157, 3–11. doi: 10.1016/0378-1119(94)00783-o
Vaswani, A., Shazeer, N., Parmar, N., Uszkoreit, J., Jones, L., Gomez, A. N., et al. (2017). Attention is all you need. arXiv [preprint].
Wang, H., Ding, Y., Tang, J., and Guo, F. (2019). Identification of membrane protein types via multivariate information fusion with Hilbert–Schmidt Independence criterion. Neurocomputing. 383, 257–269. doi: 10.1016/j.neucom.2019.11.103
Wang, H., Tang, J., Ding, Y., and Guo, F. (2021). Exploring associations of non-coding RNAs in human diseases via three-matrix factorization with hypergraph-regular terms on center kernel alignment. Brief. Bioinform. Online ahead of print.
Wang, J., Chen, S., Dong, L., and Wang, G. (2020). CHTKC: a Robust and Efficient k-mer Counting Algorithm Based on a Lock-free Chaining Hash Table. oxford: oxford university press.
Wang, Y., Ding, Y., Tang, J., Dai, Y., and Guo, F. (2019). “CrystalM: a multi-view fusion approach for protein crystallization prediction,” in Proceedings of the IEEE/ACM Transactions on Computational Biology and Bioinformatics, (Piscataway, NJ: IEEE).
Wei, L., Chen, H., and Su, R. (2018a). M6APred-EL: a sequence-based predictor for identifying N6-methyladenosine sites using ensemble learning. Mol. Therapy-Nucleic Acids 12, 635–644. doi: 10.1016/j.omtn.2018.07.004
Wei, L., Ding, Y., Su, R., Tang, J., and Zou, Q. (2018b). Prediction of human protein subcellular localization using deep learning. J. Parallel Distrib. Comput. 117, 212–217.
Wei, L., Zhou, C., Chen, H., Song, J., and Su, R. (2018c). ACPred-FL: a sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics 34, 4007–4016.
Wei, L., Hu, J., Li, F., Song, J., Su, R., and Zou, Q. (2020). Comparative analysis and prediction of quorum-sensing peptides using feature representation learning and machine learning algorithms. Brief. Bioinform. 21, 106–119.
Wei, L., Liao, M., Gao, Y., Ji, R., He, Z., and Zou, Q. (2014). Improved and promising identification of human micrornas by incorporating a high-quality negative set. IEEE/ACM Trans. Comput. Biol. Bioinform. 11, 192–201. doi: 10.1109/tcbb.2013.146
Wei, L., Luan, S., Nagai, L. A. E., Su, R., and Zou, Q. (2019a). Exploring sequence-based features for the improved prediction of DNA N4-methylcytosine sites in multiple species. Bioinformatics 35, 1326–1333. doi: 10.1093/bioinformatics/bty824
Wei, L., Su, R., Luan, S., Liao, Z., Manavalan, B., Zou, Q. et al. (2019b). Iterative feature representations improve N4-methylcytosine site prediction. Bioinformatics 35, 4930–4937. doi: 10.1093/bioinformatics/btz408
Wei, L., Su, R., Wang, B., Li, X., Zou, Q., and Gao, X. (2019c). Integration of deep feature representations and handcrafted features to improve the prediction of N-6-methyladenosine sites. Neurocomputing 324, 3–9. doi: 10.1016/j.neucom.2018.04.082
Wei, L., Xing, P., Shi, G., Ji, Z., and Zou, Q. (2019d). Fast prediction of protein methylation sites using a sequence-based feature selection technique. IEEE-ACM Trans. Comput. Biol. Bioinform. 16, 1264–1273. doi: 10.1109/tcbb.2017.2670558
Wei, L., Tang, J., and Zou, Q. (2017a). Local-DPP: an improved DNA-binding protein prediction method by exploring local evolutionary information. Inform. Sci. 384, 135–144. doi: 10.1016/j.ins.2016.06.026
Wei, L., Wan, S., Guo, J., and Wong, K. K. L. (2017b). A novel hierarchical selective ensemble classifier with bioinformatics application. Artif. Intell. Med. 83, 82–90. doi: 10.1016/j.artmed.2017.02.005
Wei, L., Xing, P., Zeng, J., Chen, J., Su, R., and Guo, F. (2017c). Improved prediction of protein-protein interactions using novel negative samples, features, and an ensemble classifier. Artif. Intell. Med. 83, 67–74. doi: 10.1016/j.artmed.2017.03.001
Wion, D., and Casadesús, J. (2006). N6-methyl-adenine: an epigenetic signal for DNA–protein interactions. Nat. Rev. Microbiol. 4, 183–192. doi: 10.1038/nrmicro1350
Wu, T. P., Wang, T., Seetin, M. G., Lai, Y., Zhu, S., Lin, K., et al. (2016). DNA methylation on N6-adenine in mammalian embryonic stem cells. Nature 532, 329–333. doi: 10.1038/nature17640
Yang, H., Yang, W., Dao, F. Y., Lv, H., Ding, H., Chen, W., et al. (2020). A comparison and assessment of computational method for identifying recombination hotspots in Saccharomyces cerevisiae. Brief. Bioinform. 21, 1568–1580. doi: 10.1093/bib/bbz123
Yang, C., Ding, Y., Meng, Q., Tang, J., and Guo, F. (2021). Granular multiple kernel learning for identifying RNA-binding protein residues via integrating sequence, and structure information. Neural Comput. Appl. 1–13. doi: 10.1007/s00521-020-05573-4
Yu, H., and Dai, Z. (2019). SNNRice6mA: a deep learning method for predicting DNA N6-Methyladenine sites in rice genome. Front. Genet. 10:1071. doi: 10.3389/fgene.2019.01071
Yuming, Z., Fang, W., and Liran, J. (2015). MicroRNA promoter identification in arabidopsis using multiple histone markers. Biomed. Res. Int. 2015:861402.
Zhang, L., He, Y., Wang, H., Liu, H., Huang, Y., Wang, X., et al. (2019). Clustering count-based RNA methylation data using a nonparametric generative model. Curr. Bioinform. 14, 11–23. doi: 10.2174/1574893613666180601080008
Zhang, Y., Kou, C., Wang, S., and Zhang, Y. (2019). Genome-wide differential-based analysis of the relationship between DNA methylation and gene expression in Cancer. Curr. Bioinform. 14, 783–792. doi: 10.2174/1574893614666190424160046
Zhang, Z. Y., Yang, Y. H., Ding, H., Wang, D., Chen, W., and Lin, H. (2020). Design powerful predictor for mRNA subcellular location prediction in Homo sapiens. Brief. Bioinform. 22, 1–10.
Zhao, X., Jiao, Q., Li, H., Wu, Y., and Wang, G. (2020). ECFS-DEA: an ensemble classifier-based feature selection for differential expression analysis on expression profiles. BMC Bioinform. 21:43. doi: 10.1186/s12859-020-3388-y
Zhou, P., Shi, W., Tian, J., Qi, Z., and Xu, B. (2016). “Attention-based bidirectional long short-term memory networks for relation classification,” in Proceedings of the 54th Annual Meeting of the Association for Computational Linguistics, (Berlin: Association for Computational Linguistics).
Zhu, T., Guan, J., Liu, H., and Zhou, S. (2019). RMDB: an integrated database of single-cytosine-resolution DNA methylation in Oryza sativa. Curr. Bioinform. 14, 524–531. doi: 10.2174/1574893614666190211161717
Zou, Q., Li, J., Song, L., Zeng, X., and Wang, G. (2016). Similarity computation strategies in the microRNA-disease network: a survey. Brief. Funct. Genom. 15, 55–64.
Zou, Q., Lin, G., Jiang, X., Liu, X., and Zeng, X. (2020a). Sequence clustering in bioinformatics: an empirical study. Brief. Bioinform. 21, 1–10.
Zou, Q., Xing, P., Wei, L., and Liu, B. (2019). Gene2vec: gene subsequence embedding for prediction of mammalian N6-Methyladenosine sites from mRNA. RNA 25, 205–218. doi: 10.1261/rna.069112.118
Zou, Y., Wu, H., Guo, X., Peng, L., Ding, Y., Tang, J., et al. (2020b). MK-FSVM-SVDD: a multiple kernel-based fuzzy SVM model for predicting DNA-binding proteins via support vector data description. Curr. Bioinform. Online ahead of print.
Keywords: multi-task learning, DNA modification, feature representation, deep learning, neural network
Citation: Yang X, Ye X, Li X and Wei L (2021) iDNA-MT: Identification DNA Modification Sites in Multiple Species by Using Multi-Task Learning Based a Neural Network Tool. Front. Genet. 12:663572. doi: 10.3389/fgene.2021.663572
Received: 03 February 2021; Accepted: 02 March 2021;
Published: 31 March 2021.
Edited by:
Xiangxiang Zeng, Hunan University, ChinaCopyright © 2021 Yang, Ye, Li and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuehong Li, bGl4dWVob25nMTk3OEAxNjMuY29t; Lesong Wei, czIwMzAxNDNAcy50c3VrdWJhLmFjLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.