 Hugo R. Martinez
Hugo R. Martinez Gary S. Beasley
Gary S. Beasley Noah Miller
Noah Miller Jason F. Goldberg1
Jason F. Goldberg1 John L. Jefferies
John L. Jefferies- 1The Heart Institute, Le Bonheur Children’s Hospital, The University of Tennessee Health Science Center, Memphis, TN, United States
- 2The Cardiovascular Institute, The University of Tennessee Health Science Center, Memphis, TN, United States
Cardiomyopathies (CMs) encompass a heterogeneous group of structural and functional abnormalities of the myocardium. The phenotypic characteristics of these myocardial diseases range from silent to symptomatic heart failure, to sudden cardiac death due to malignant tachycardias. These diseases represent a leading cause of cardiovascular morbidity, cardiac transplantation, and death. Since the discovery of the first locus associated with hypertrophic cardiomyopathy 30 years ago, multiple loci and molecular mechanisms have been associated with these cardiomyopathy phenotypes. Conversely, the disparity between the ever-growing landscape of cardiovascular genetics and the lack of awareness in this field noticeably demonstrates the necessity to update training curricula and educational pathways. This review summarizes the current understanding of heritable CMs, including the most common pathogenic gene variants associated with the morpho-functional types of cardiomyopathies: dilated, hypertrophic, arrhythmogenic, non-compaction, and restrictive. Increased understanding of the genetic/phenotypic associations of these heritable diseases would facilitate risk stratification to leveraging appropriate surveillance and management, and it would additionally provide identification of family members at risk of avoidable cardiovascular morbidity and mortality.
Introduction
Cardiomyopathies (CMs) are a group of disorders in which the muscle of the heart (myocardium) becomes structurally and functionally abnormal, leading to systolic dysfunction, diastolic dysfunction, and/or tachyarrhythmias (Arbustini et al., 2013). The global burden of genetically driven cardiomyopathies is difficult to quantify given the limited epidemiological studies representing a global framework. However, just alone, hypertrophic cardiomyopathy is considered the most common inherited cardiovascular disease, estimated to be present in 1:200 adult individuals. This information was generated after taking into account clinical profiles based on advanced imaging and familial transmission rates (Maron et al., 2018). Similarly, the prevalence of dilated cardiomyopathy has been estimated in the range of 1:250 adults (Hershberger et al., 2013). Furthermore, a recent meta-analysis incorporating more than 20 studies found strong prevalence in first-degree family members of the probands (up to 65%), which displays evidence of the high degree of heritability in these disorders (Petretta et al., 2011; Watkins et al., 2011; Ware, 2015). In the pediatric population, a cardiomyopathy registry concluded that the prevalence of heart failure amongst children with cardiomyopathy is relatively elevated, representing a leading cause of morbidity and mortality for this aged group (Wilkinson et al., 2010; McKenna et al., 2017).
Cardiomyopathies have been incorporated into multiple classifications over the past decades ranging from modest to more sophisticated schemes based on the genetic origin of the disease; in fact, the MOGES nosology incorporates a morpho-functional
phenotype (M), organ(s) involved (O), the genetic inheritance pattern (G), an etiological annotation (E) including genetic defects or underlying disease/substrates, and the functional status (S) of a particular patient based on heart failure symptoms (Maron et al., 2006; Elliott et al., 2008; Arbustini et al., 2013). However, classification systems are limited, due to the genetic and phenotypic heterogeneity expressed in myocardial diseases, as well as the limitations of a particular classification system to satisfy multiple scientific and clinical disciplines.
The common modes of inheritance for these disorders include autosomal dominant, autosomal recessive, X-linked and mitochondrial, but complex modes of Mendelian inheritance, mosaicisms, variable expressivity, and incomplete penetrance have also been described (Charron, 2006).
From the molecular point of view, CMs may originate from a pathogenic variation manifesting the disease (single-gene disorder), multiple pathogenic variations in the same gene (compound heterozygosity), or from a combination of variations in different genes (digenic heterozygosity) (Towbin, 2014; Li et al., 2019). Moreover, the incorporation of advanced DNA and RNA sequencing, has led to a comprehensive assessment of modifiers at other loci, gene promoters, loci enhancers, copy number variants (deletion/duplication) and molecular regulatory regions associated with “classic” phenotypes of cardiomyopathy, more severe phenotypic expressions, overlapping sub-types, and the co-occurrence of myocardial disease and tachyarrhythmias (Weischenfeldt et al., 2013; Towbin, 2014; Li et al., 2019; Lipshultz et al., 2019; Minoche et al., 2019). In this review, we focus on common genetic associations with the classic phenotypes of heritable cardiomyopathies, such as: dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), arrhythmogenic cardiomyopathy (ACM), non-compaction cardiomyopathy (LVNC), and restrictive cardiomyopathy (RCM); providing an opportunity to guide clinical management based upon known genetic underpinnings.
Material Content
Dilated Cardiomyopathy
Dilated cardiomyopathy is typically characterized by eccentric ventricular remodeling and decreased systolic function; and it can be detected in asymptomatic individuals, in those with heart failure symptoms, or in association with arrhythmias (Jefferies and Towbin, 2010) (Figure 1). Globally, DCM is one of the most common forms of cardiomyopathy, and it represents a leading cause of cardiac transplantation in children and adults (Lipshultz et al., 2019). The etiology of DCM can be broadly categorized into genetic, acquired, or mixed. Unfortunately, in many cases, no etiology can be found, and the disease is deemed idiopathic. Of interest, it is estimated that between 25 and 60% of patients with idiopathic DCM harbor positive family history for the disease, suggesting an underlying genetic predisposition (Petretta et al., 2011). Most genetically triggered cases of DCM are transmitted in an autosomal dominant pattern exhibiting variable penetrance. Other forms of inheritance include autosomal recessive, X-linked, and mitochondrial, have been described more frequent in the pediatric population (Arbustini et al., 2013).
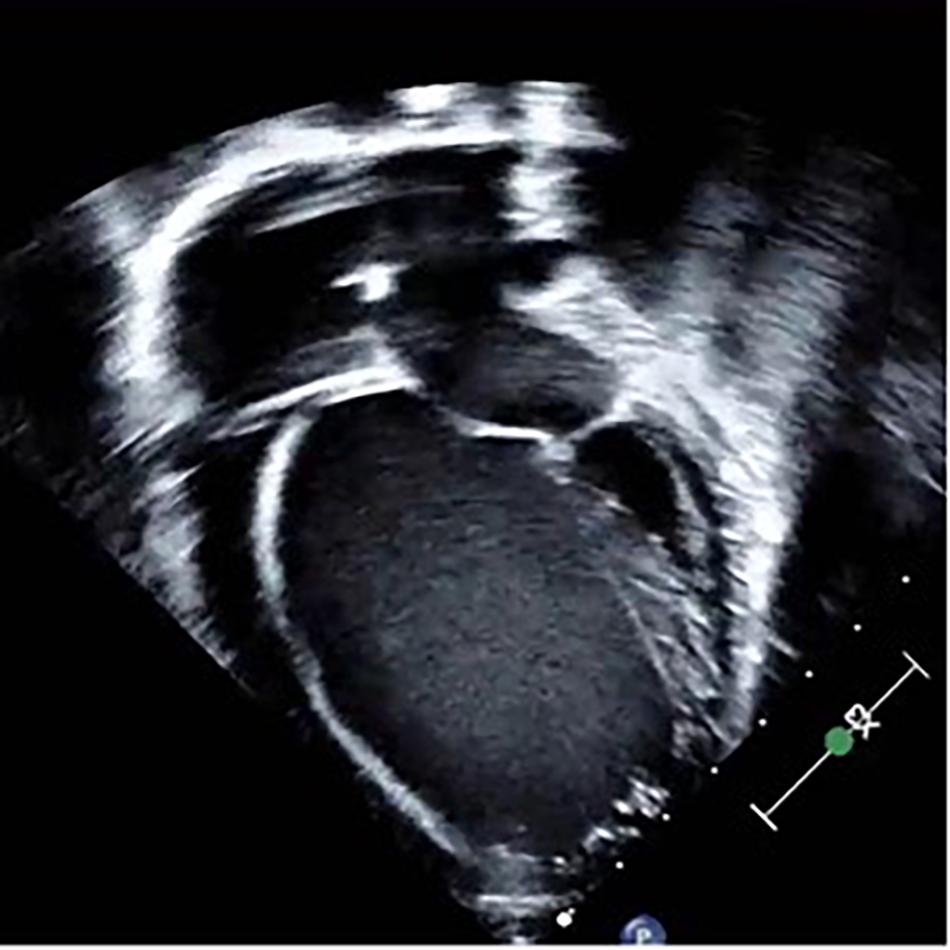
Figure 1. Two-dimensional, apical 4-chamber echocardiographic image depicting an enlarged ventricle with spherical geometry and bilateral atrial enlargement in a patient harboring dilated cardiomyopathy secondary to a pathogenic gene variant in the TTN gene.
Familial DCM occurs frequently given the high heritability of this disorder, but this approximation is subject to change as a more critical evaluation of the genes linked to DCM continues to evolve and certain gene variations shift to be certified as non-pathogenic (Petretta et al., 2011).
A conventional classification of DCM has been categorized based on the presence or absence of systemic disease. Thus, dividing DCM into syndromic and non-syndromic forms is a practical approach to evaluating this highly heterogeneous disease (Table 1). The diagnostic rate for gene testing in non-syndromic DCM is about 46–73% in tertiary centers with cardiovascular genetic expertise (Tayal et al., 2017) but it might be confounded by insufficient control for population variation. Over the past decade, over 100 genes in total have been linked to DCM in the Human Gene Mutation Database and the Online Mendelian Inheritance in Man (Fatkin et al., 2019; Online Mendelian Inheritance in Man, 2019) (Table 2). From these genes, a large-scale analysis revealed that truncating variants in the titin gene (TNN) are the most common disease-causative mutations in non-syndromic DCM (McNally et al., 2013; Nouhravesh et al., 2016). Other core genes include MYH7, TNNT2 (encoding troponin T2) and TPM1 (encoding tropomyosin 1). LMNA (encoding a nuclear envelope protein, laming A/C), has been associated with the arrhythmogenic type of DCM, as well and other skeletal myopathies such as Emery-Dreifuss, limb-girdle, and LMNA-related congenital muscular dystrophy (Maggi et al., 2016). Other rare pathogenic variants (minor allele frequency) implicated in non-syndromic DCM include genes coding for the sarcomere and Z-disk (actin, myosin-binding protein C3, myopalladin, nebulette, ZASP), cytoskeleton (dystrophin, desmin), nuclear envelope (emerin), mitochondria (tafazzin), sarcoplasmic reticulum, desmosomes, ion channels, and transcription factors (Martinez et al., 2014; McNally and Mestroni, 2017; Tayal et al., 2017; Hershberger et al., 2018a). X-linked cardiomyopathy has been reported as an isolated disease of the heart or associated with skeletal myopathies such as Duchenne and Becker muscular dystrophy (Towbin and Ortiz-Lopez, 1994; Davies and Nowak, 2006; Puckelwartz and McNally, 2011). It is worth mentioning that female carriers of X-linked dystrophinopathies with skewed inactivation of the healthy X chromosome, may present illness manifested by muscle weakness, myocardial dysfunction and heart failure. Depending on the degree of functional loss of the healthy X chromosome, female carriers may present symptoms as early as during the first decade of life (Martinez et al., 2011). Thus, starting phenotype screenings of female carriers of X-linked dystrophinopathies during childhood may be warranted.
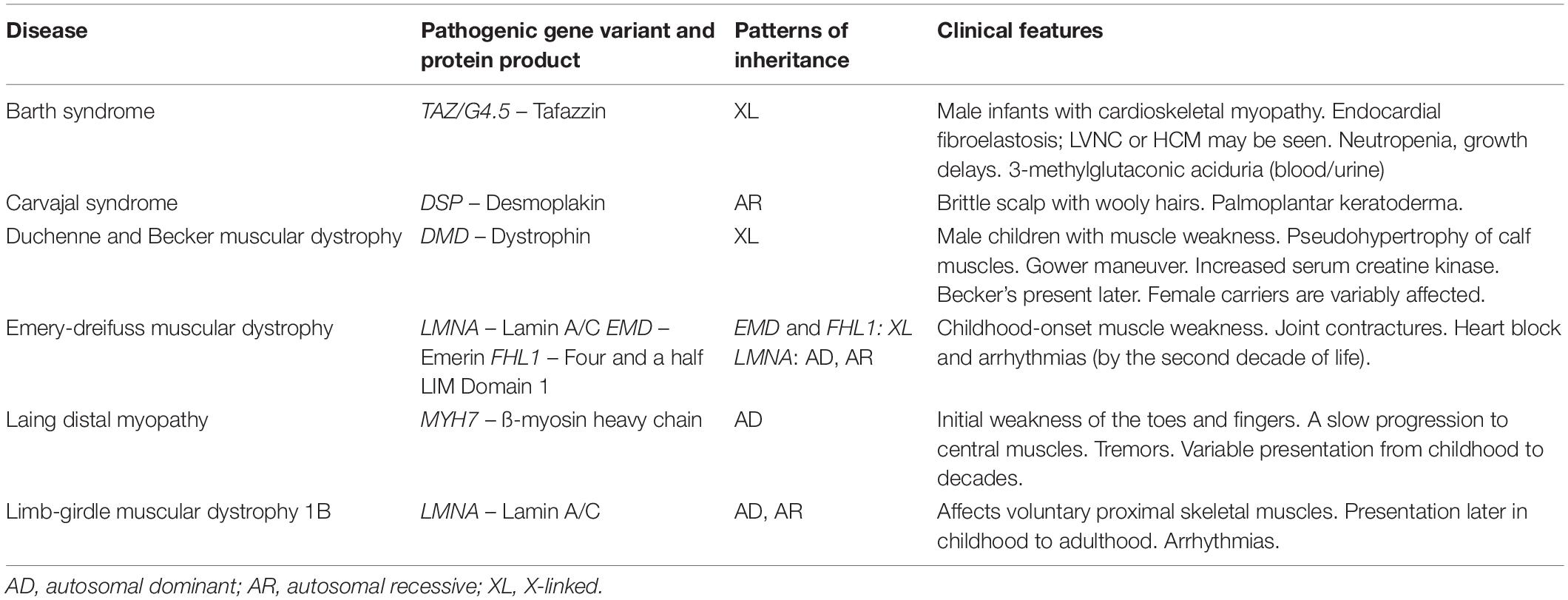
Table 1. Typical features of common types of syndromic dilated cardiomyopathy.
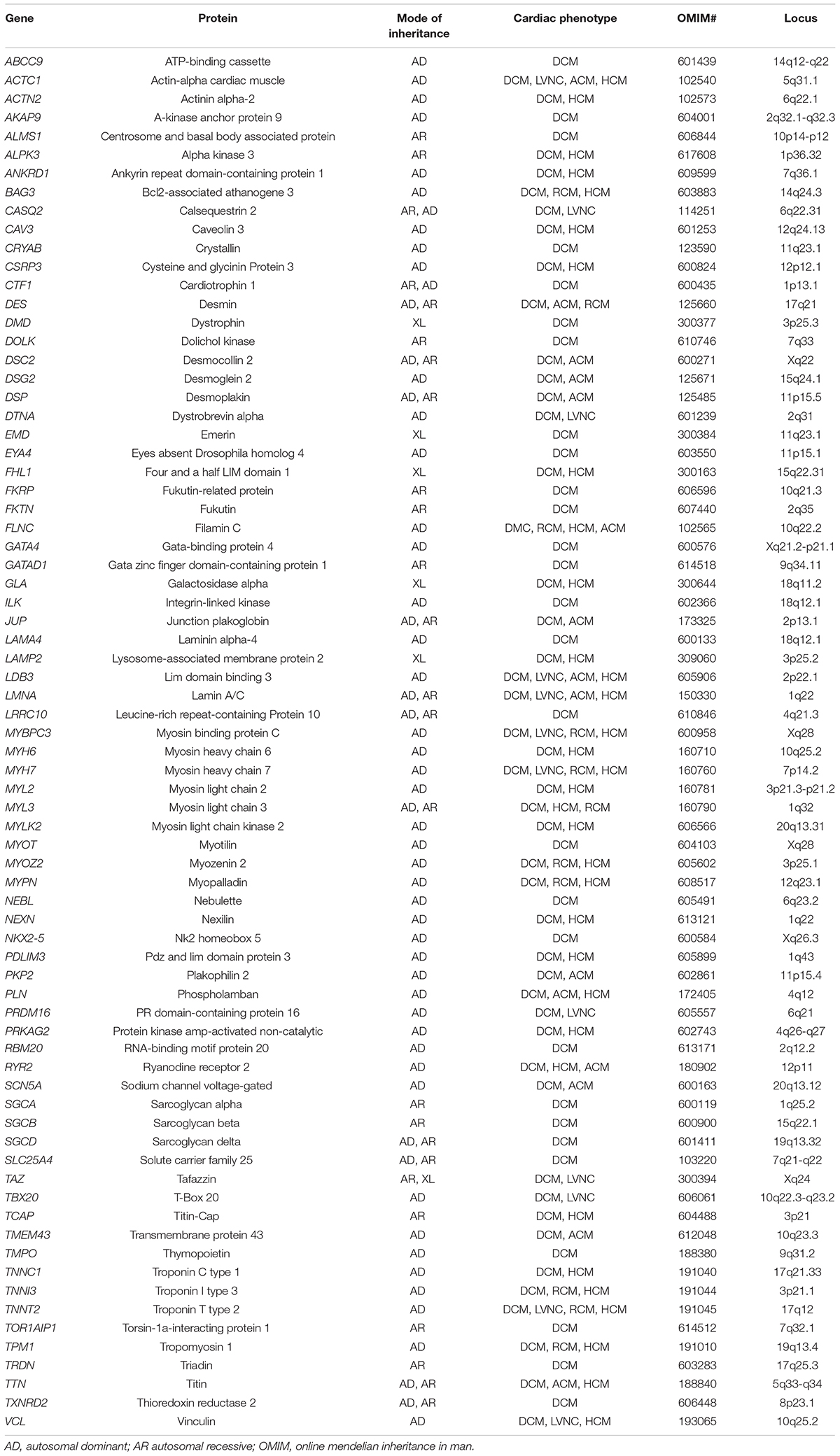
Table 2. List of common genes and patterns of inheritance associated with DCM.
Barth syndrome (BTS) is another X-linked cardiac and skeletal myopathy that encompasses abnormal mitochondrial function, short stature, cyclic neutropenia, cardiolipin deficiency, and variable degrees of 3-methylglutaconic aciduria. BTS is caused by mutations in the TAZ gene (previously called G4.5), which is located in the chromosome Xq28 region and encodes for the Tafazzin protein (Jefferies, 2013). Pathologic gene variants in this gene may result in a wide spectrum of myocardial disease including DCM, HCM, LVNC or a morpho-functional combination of these types. In many cases, affected infants succumb to heart failure, arrhythmias, or sepsis secondary to leukocyte dysfunction (Barth et al., 1983; Towbin, 2010).
Regardless of the mode of inheritance and the pathogenic steps in one or multiple molecular pathways in patients with DCM, progression to heart failure comprises subsequent complex molecular cascades leading to contractile disorganization, metabolic dysregulation, progressive cell death, inflammatory stimulation, remodeling, and heart failure (Bowles et al., 2000; Piran et al., 2012). With disruption of one or multiple proteins or factors implicated in the myocardial mechanics, molecular function and electrical pathways, the ultimate cascade of processes can precipitate the myocardium to become dysfunctional; the “common pathway” in the development of heart failure (Bowles et al., 2000).
Novel sequencing methodologies incorporating panels of genes have enhanced the knowledge of pathogenic genetic variations facilitated the creation of and access to public genomic databases, increasing the probability of finding a pathogenic gene variant. Currently the diagnostic rate of detecting a pathogenic gene variant among patients with non-syndromic DCM range around 40%; this detection rate, however, has not changed after the introduction of conservative criteria to evaluate and define pathogenic variants (Hershberger et al., 2010; van Spaendonck-Zwarts et al., 2013).
Hypertrophic Cardiomyopathy
This CM is characterized by left ventricular hypertrophy and histologic evidence of myofiber disarray and interstitial fibrosis (Rowin and Maron, 2013). A cardinal feature of HCM is the presence of asymmetrical hypertrophy, however, a diverse morphology myocardial thickening such as septal, apical and lateral, has been described in association with this disease (Chun et al., 2010) (Figure 2). Mainly, driven by the degree of left ventricular outflow track obstruction, the clinical manifestations present in a wide spectrum ranging from silent, to symptoms of heart failure with preserved ejection fraction, to arrhythmias and sudden death (Brinkley et al., 2020). Diastolic dysfunction can even be detected in individuals with a genetic HCM diagnosis who have normal LV wall thickness, suggesting that diastolic dysfunction is an early feature rather than a consequence of hypertrophy (Rakowski and Carasso, 2009). In the end-stage of the disease, HCM can also present with the “burned-out HCM” phenotype which encompass the syndrome of heart failure with reduced ejection fraction (Brinkley et al., 2020). When echocardiographic data, large-scale epidemiology studies and genetic diagnoses are taken into account, the estimated prevalence of HCM has been raised to 1 case per 200 people in the general population (Maron, 2018). Within the pediatric population, HCM is 10 times more frequent in patients under 1 year of age than in older children, with these younger children more likely to harbor HCM as a syndromic cardiomyopathy (Tables 3, 4) (Colan et al., 2007). HCM typically has an autosomal pattern of inheritance. Although genetic testing alone for the diagnosis of HCM is not recommended, it has a great value in combination of a systematic evaluation for two main reasons. First, the identification of syndromic/genetic diseases known to cause HCM (i.e., Friederichs’s ataxia, Fabry disease, Pompe disease, etc.), and second, the identification of family members of the proband harboring pathogenic gene variants.
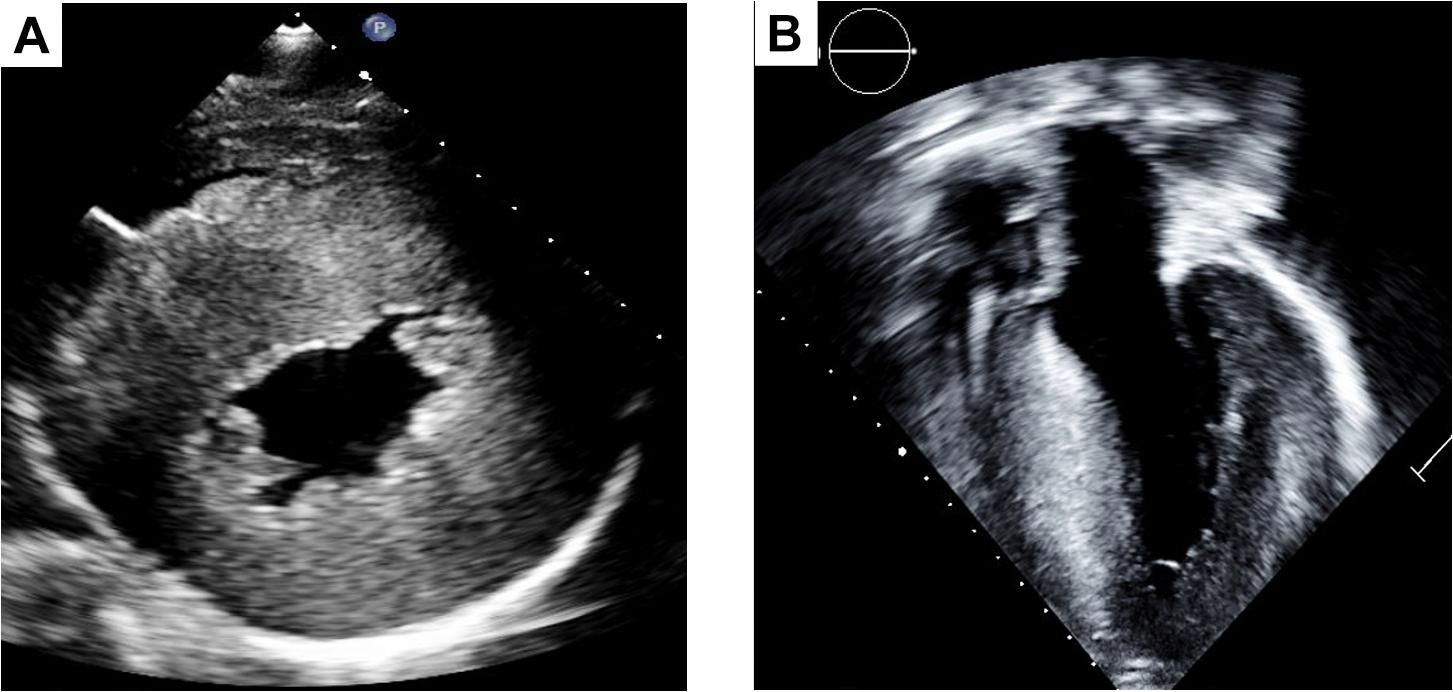
Figure 2. Two dimensional images of HCM in the parasternal short axis (A) exhibiting significant concentric hypertrophy and corroborated by the parasternal long axis view (B).
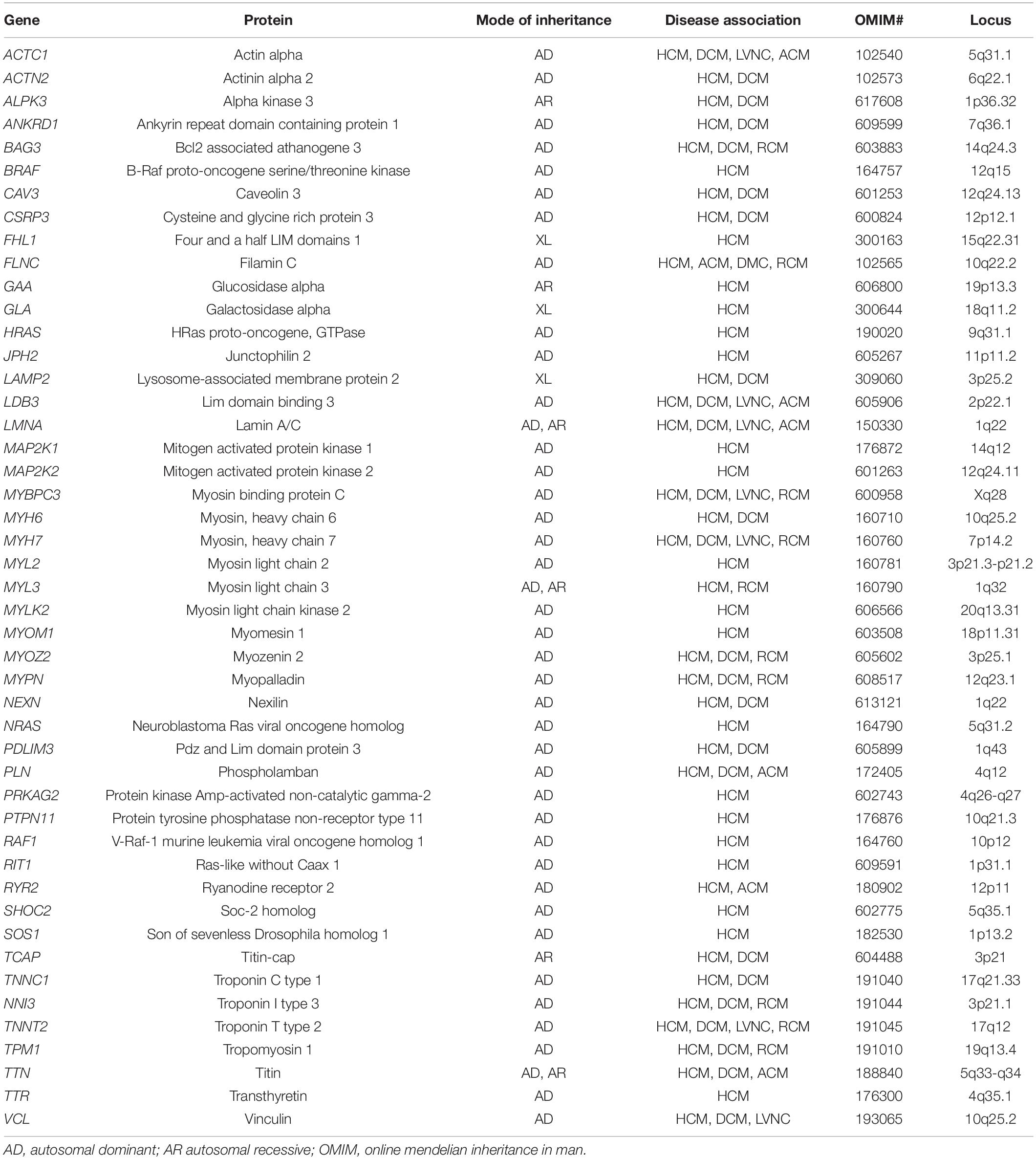
Table 3. List of common genes and patterns of inheritance associated with HCM.
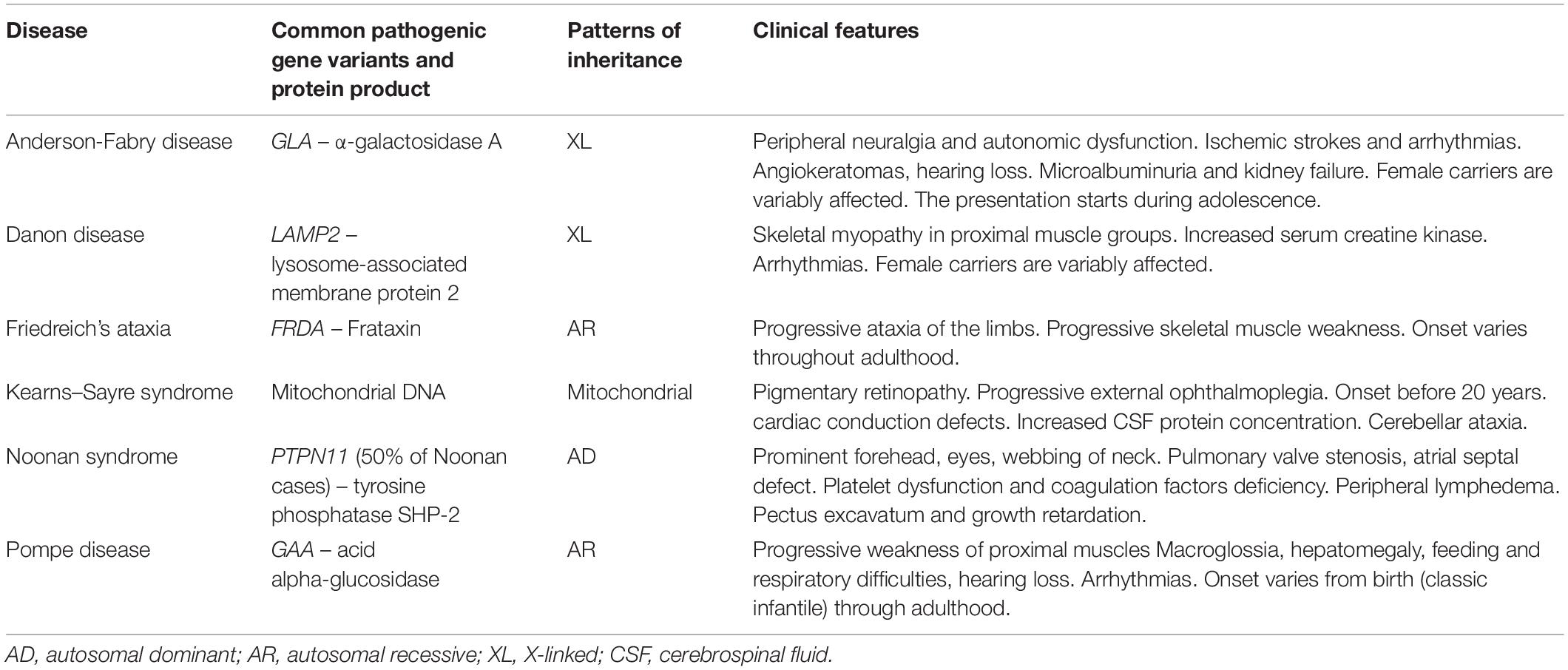
Table 4. Typical features of common types of syndromic hypertrophic cardiomyopathy (phenocopies).
Categorization of HCM includes non-syndromic HCM (without other systemic involvement) and the syndromic form of HCM (in association with inborn errors of metabolism and neuromuscular disorders), also referred as previously mentioned, phenocopies of HCM (Colan, 2010; Popa-Fotea et al., 2019). It has been estimated that 50–60% of patients who have a family member with HCM harbor a pathogenic gene variant (Alfares et al., 2015; Burns et al., 2017).
More than three decades ago, the first chromosome locus (14q11.2-q12) encoding components of the sarcomere (beta-myosin heavy chain) was elucidated as the pathogenic basis for familial HCM (Maass et al., 2002). Since the discovery, more than a thousand mutations in near 30 genes have been implicated in the development of HCM (see Table 3) (Roma-Rodrigues and Fernandes, 2014). Most forms of HCM are inherited in an autosomal dominant transmission, but mitochondrial and autosomal recessive patterns have been also described (Jarcho et al., 1989; Konno et al., 2010; Gersh et al., 2011). Most disease-causing mutations implicated in HCM include pathogenic variants in the MYH7 gene (encoding beta-myosin heavy chain) and in the MYBPC3 gene (encoding cardiac myosin-binding protein C). These mutations account individually for 40% of cases; and some other core genes include TNNT2, TPM1, ACTC1, TNNI3, TTN, and MYL2 (Tariq and Ware, 2014). Most of these variants involve missense mutations (resulting in a direct amino acid change) and frameshift-type mutations (insertions or deletions of the number of nucleotides), which alter the properties of the protein involved. Epidemiology studies and the incorporation of genetic panels in clinical practice, have estimated that approximately 70–80% of familial cases have an identified gene variant, whereas fewer mutations (approximately 20%) are identified as infantile HCM (Jarcho et al., 1989; Konno et al., 2010; Gersh et al., 2011; Lipshultz et al., 2019). Genetic defects encoding for a sarcomeric proteins can disrupt the contractile mechanics and the of calcium homeostasis in the sarcomere resulting in a remodeling process by several transcription factors and cardiomyocyte hypertrophy, which eventually results in ischemia, fibrosis, and arrhythmias (Gersh et al., 2011).
RAS/MAPK Pathway Syndromes
Since the discovery of the first gene (PTPN11) associated with Noonan syndrome in 2001, multiple genes (RAF1, SOS1, KRAS, NRAS, BRAF, MAP2K1, MAP2K2, HRAS, and SHOC2) have been identified in the RAS/mitogen-activated protein kinase pathway. An important molecular corridor managing cell proliferation and differentiation. Thus, dysregulation results in a spectrum of disorders known as “RASopathies” including Noonan and Noonan-like syndromes (Rauen et al., 2010). Children with Noonan-associated syndromes who present HCM and heart failure have a worse risk profile when compared to other children with sarcomeric HCM (Wilkinson et al., 2012).
The management of RASopathies should involve a multidisciplinary team with expertise in the assessment of cardiac structural defects, HCM, and arrhythmias (Table 4).
Congenital metabolic disorders result from absent or abnormal enzymes—or their cofactors—which can lead to accumulation or deficiency of a specific metabolite. Although these disorders exhibit different modes of inheritance, most are transmitted in an autosomal recessive or mitochondrial pattern.
Pompe disease, is a glycogen storage disorder (type II) with autosomal recessive mode of inheritance affecting neuromuscular and myocardial tissues. This disease has broadly classified into the infantile onset (within the first year of life) and the late onset (from childhood to adulthood). The underlying mechanisms include the development of autophagy and the continuous accumulation of glycogen lysosomes secondary to a deficient acid alpha-glucosidase (Hirschhorn and Huie, 1999). The clinical presentation varies in severity and it notably affects skeletal muscle, myocardium and the central nervous system. In the infantile form, musculoskeletal weakness may prevent adequate respiratory mechanics and a normal development. In the myocardium, the build-up of glycogen leads to a progressive phenocopy of hypertrophic cardiomyopathy (Ruiz-Guerrero and Barriales-Villa, 2018). Electrocardiography (ECG) reveals a short PR interval, a manifest delta wave, and giant QRS complexes in all leads, suggesting biventricular hypertrophy. Chronic troponinemia may be observed based on our clinical experience. Other findings include elevated creatine kinase, lactate dehydrogenase, and aspartate aminotransferase. The juvenile and adult forms present with variable age of onset. Enzyme replacement therapy has shown to decrease ventricular hypertrophy, LV outflow tract obstruction, and normalization of electrocardiographic findings (Levine et al., 2008).
Danon disease, is a glycogen storage disorder (type IIb) with X-linked mode of inheritance incorporating with skeletal muscle weakness and intellectual disabilities. Danon disease involves a genetic defect in the LAMP2 gene located at chromosome Xq24, which encodes the lysosome-associated membrane protein. This lysosomal storage disease mimics the phenotypic features of the sarcomeric-gene-associated HCM. However, in Danon disease the presence of ventricular preexcitation is more frequently encountered than in those individuals with sarcomeric HCM (Arad et al., 2005; Rowland et al., 2016). The cardiac degeneration is usually appreciated clinically by the presence of HCM during childhood or adolescence with subsequent transition to a DCM phenotype with progressive heart failure (Maron et al., 2009). Female carriers have also been described in this disorder and are attributed to unfavorable X chromosome inactivation (lyonization) causing the development of symptoms around the fourth decade of life (Toib et al., 2010).
Fabry disease is an X-linked lysosomal disease secondary to a deficient hydrolase alpha-galactosidase A (Brady et al., 1967). Most cases are familial and few originate from de novo mutations (Garman and Garboczi, 2004). Patients with Fabry disease may present with a multisystem involvement including neurologic (paresthesia and pain crises), dermatologic (angiokeratomas and telangiectasias), ophthalmologic (corneal dystrophy), renal (proteinuria and renal insufficiency), and cardiac manifestations by the second to fifth decades of life (Mehta et al., 2004). Cardiac disease is relatively common in Fabry disease. Patients may develop a HCM phenocopy (similar to that seen in sarcomeric HCM), arrhythmias, and valvar abnormalities. Enzyme replacement therapy has shown attenuation of cardiovascular compromise (Perrot et al., 2002; O’Mahony and Elliott, 2010).
Friedreich’s ataxia is another disease expressed as a phenocopy of HCM affecting approximately 1 in 20,000 – 750,000 individuals (Burk, 2017). From the genetic standpoint, the inheritance mode is autosomal recessive, and it is characterized by trinucleotide repeat expansion of a normal codon affecting the protein frataxin, a mitochondrial protein implicated in iron homeostasis required for the mitochondrial electron transport chain. The trinucleotide repeat expansion causes a reduction in the frataxin levels. Thus, the bigger the expansion, the earlier the age of onset and the severity of the disease (Patel and Isaya, 2001; Burk, 2017). The major clinical manifestations of Friedreich’s ataxia appear during childhood and adolescence, and include progressive neurologic dysfunction (gait ataxia, optic atrophy, loss of position and vibration sense), diabetes mellitus, and myocardial involvement. The cardiac phenotype is manifested by arrhythmias and HCM. Heart failure remains the leading cause of death in this population (Kipps et al., 2009; Peverill, 2012; Weidemann et al., 2012).
Left Ventricular Non-Compaction Cardiomyopathy
LVNC is characterized by multiple prominent ventricular trabeculations, intertrabecular deep recesses, and myocardium with two distinct layers: a non-compacted subendocardial layer and a thin compacted epicardial layer (Towbin, 2010). LVNC is presumably caused by abnormal in utero myocardial compaction, a final stage of myocardial morphogenesis (Zhang et al., 2013; Liu et al., 2018). However, this hypothesis does not explain why LVNC manifests in a wide variety of sub phenotypes (Table 5) with a heterogeneous clinical course. LVNC also shows increased incidence in certain physiologic states, such as pregnancy (Gati et al., 2014) and vigorous physical activity (Gati et al., 2013; de la Chica et al., 2020). It is also seen in association with other conditions such as in patients with congenital heart disease (CHD), neuromuscular disorders, mitochondrial disease and metabolic derangements (Finsterer et al., 2017; Martinez et al., 2017; Ross et al., 2020; Vershinina et al., 2020). Since the initial description in 1926, LVNC has been coined with various names, including spongiform myocardium, fetal or primordial myocardium, hypertrabeculation syndrome, and non-compaction cardiomyopathy (Maron et al., 2006; Engberding et al., 2007). Currently, this cardiomyopathy is categorized as a primary genetic cardiomyopathy by the American Heart Association and as a primary morphofunctional cardiomyopathy by the MOGE(s) nosology (Maron et al., 2006; Elliott et al., 2008; Arbustini et al., 2014). Because the genesis of LVNC is incompletely understood, diagnosis typically relies on non-invasive imaging, Figure 3. Since the 1990s, many diagnostic criteria have been proposed based on morphological characteristics of the myocardium seen by echocardiography and cardiac magnetic resonance. Without analytic and universally accepted criteria, the prevalence of LVNC is difficult to estimate, but the approximate range is estimated in the 0.014–1.3% range of patients undergoing echocardiography (Pignatelli et al., 2003; Stanton et al., 2009). However, among patients with heart failure and patients with severe CHD, the prevalence has been reported at around 4 and 7.5%, respectively (Kovacevic-Preradovic et al., 2009; Martinez et al., 2017). In this review, we provide an informative categorization of 9 distinct subphenotypes of LVNC (Towbin and Beasley, 2020) (Table 5). The overall clinical manifestation is heterogenous ranging from asymptomatic to a severe course accompanied by heart failure requiring heart transplant, arrhythmias, sudden cardiac death, and thromboembolic phenomena (Lee et al., 2017). Familial cases are well-documented, and autosomal dominant transmission is currently the most recognized inheritance pattern (with variable penetrance and phenotypic heterogeneity). Other heritable modes of transmission include X-linked, autosomal recessive, and mitochondrial (Roma-Rodrigues and Fernandes, 2014). In pediatric and adult cohorts, the rate of finding a pathogenic gene variant in patients manifesting LVNC ranges from 17 to 41% depending on patient selection and the cardiomyopathy gene panels (Sedaghat-Hamedani et al., 2017; van Waning et al., 2019). One of the earliest locus associations with LVNC was described in 1997 in the gene G4.5/TAZ located at chromosome Xq28 (Lee et al., 2017). Since then, multiple pathologic gene variants have been described as potential causes of LVNC. Genes encoding sarcomeric and cytoskeletal proteins (TTN, ACTN2, RBM20, LMNA, DES, DYS, DTNA, LDB3, MYH7, MYBPC3, and ACTC1) as well as genes associated with cardiac morphogenesis (FKBP12, MIB1, Tbx20, Nkx2-5, Smad7, NF-ATc, and Jarid2), ion channels (SCN5A, HCN4, and RYR2), and mitochondria (NNT, TAZ) have been implicated in the development of LVNC (Bagnall et al., 2014; Hastings et al., 2016; van Waning et al., 2018, 2019). Along with sarcomere-encoding and cytoskeleton-encoding genes, pathogenic variants in a variety of genes, including SCN5A, LMNA, RBM20, TTN, and DES, have been associated with LVNC and rhythm disturbance (Finsterer and Stollberger, 2008; Shan et al., 2008) (Table 6). Mutations in the mitochondrial genome and chromosomal abnormalities have also been linked to LVNC, including 1p36 deletion (encompassing the PRDM16 gene), 7p14.3p14.1 deletion, 22q11.2 deletion, distal 22q11.2, and other chromosomal trisomies (Towbin, 2010; Tariq and Ware, 2014; Dong et al., 2017; Miller et al., 2017; Miszalski-Jamka et al., 2017; van Waning et al., 2018).
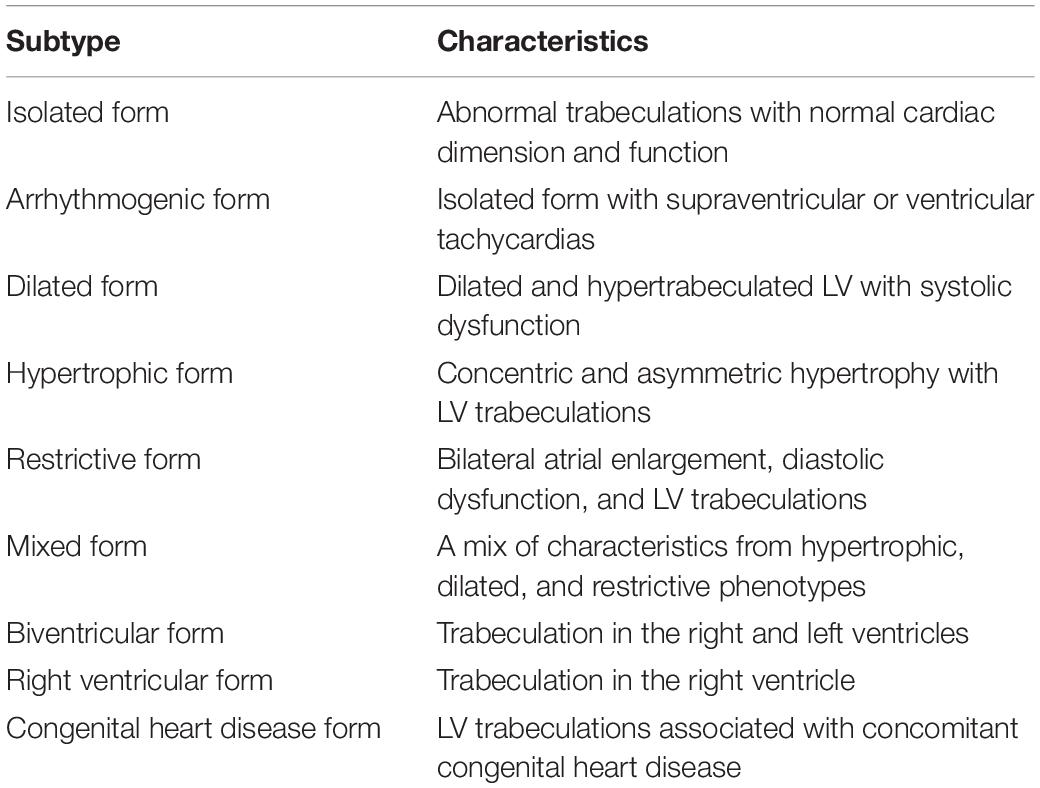
Table 5. Subphenotypes in the clinical spectrum of left ventricular non-compaction.
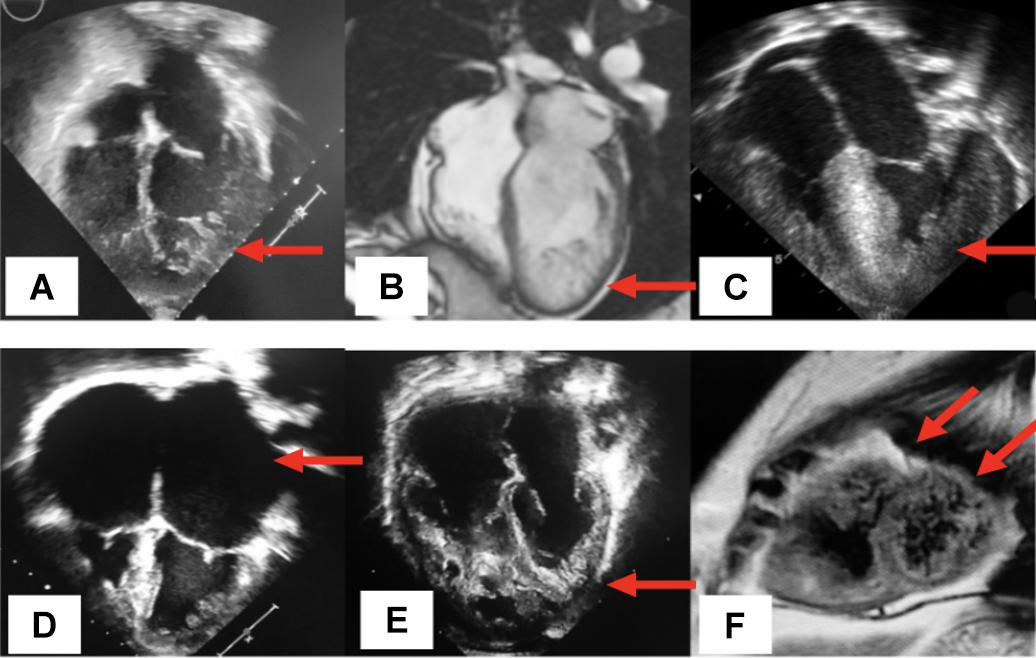
Figure 3. Distinct phenotypes of non-compaction cardiomyopathy. (A) Echocardiographic 4-chamber view displays the isolated type of LVNC illustrated by the cardinal feature of left ventricular trabeculations (arrow) with normal anatomy and function; (B) from a cardiac magnetic resonance imaging (CMRi), a 4-chamber view displays the dilated sun-type of LVNC, denoting the enlargement of the LV and the presence of inferolateral trabeculations (arrow); (C) echocardiographic 4-chamber view shows the hypertrophic type of LVNC represented by asymmetric hypertrophy of the interventricular septum and the presence of lateral LV trabeculations (arrow); (D) display of the restrictive type of LVNC, significant bilateral atrial enlargement (arrows) and the presence of left ventricular trabeculations; (E) biventricular hypertrabeculations (arrows); (F) cMRI in a short axis view displays a mixed LVNC phenotype represented by dilated and dysfunctional ventricles in a patient with ventricular arrhythmias and biventricular trabeculations secondary to a pathogenic variant in the PRDM16 gene (arrows).
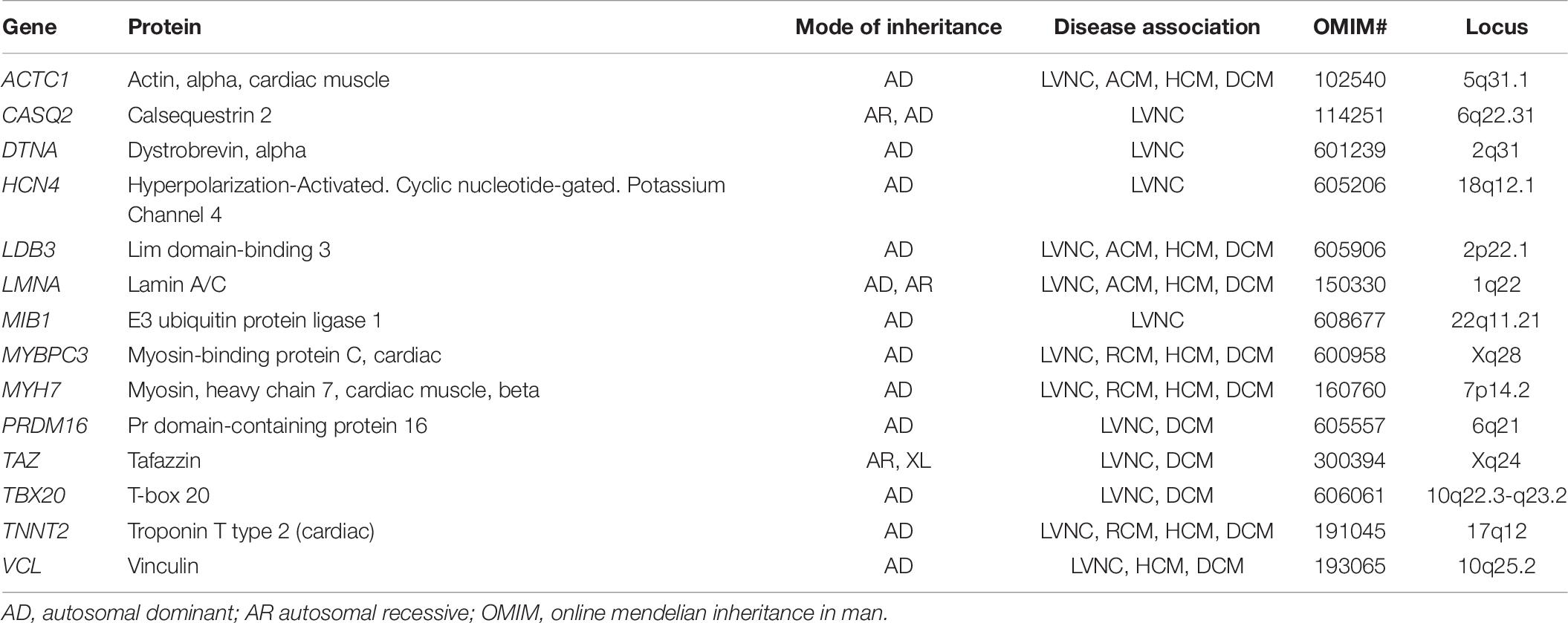
Table 6. List of common genes and patterns of inheritance associated with LVNC.
Arrhythmogenic Cardiomyopathy (ACM)
Arrhythmogenic cardiomyopathy is myocardial disorder unrelated to coronary artery disease, hypertension, or valvular heart disease. ACM was formerly known as arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/ARVC). The prevalence has been estimated at around 1 in 1,000 – 5,000 people (Peters et al., 2004). The clinical diagnosis may be supported by evidence of conduction disease, supraventricular arrhythmias, and/or ventricular arrhythmias originating from any cardiac structure. ECG abnormalities include right bundle branch block pattern, epsilon wave (a low-amplitude deflection between the end of the QRS and the onset of the T wave in the precordial leads V1 – V3), and T wave inversion in the precordial leads V1 – V4. Classically, the RV is dilated and contains fibro-fatty infiltration of the myocardium. The left ventricle is overtly affected less frequently. Thus, ACM should be categorized into right dominance, left dominance, or biventricular involvement.
Notably, ACM clinically overlaps with other CM types, particularly DCM. However, ACM is distinct because it is marked by arrhythmia at presentation with or without biventricular dilatation and/or impaired systolic function (Towbin et al., 2019). This heritable disorder is primarily transmitted in an autosomal dominant pattern, but autosomal recessive heritability has been reported in families with cardiocutaneous disease (Greece, Italy, India, Ecuador, Israel, and Turkey) (Protonotarios and Tsatsopoulou, 2004). The most notable autosomal recessive diseases include Naxos disease (a homozygous pathogenic variant in the gene encoding the protein plakoglobin, characterized by ACM, non-epidermolytic palmoplantar keratoderma, and wooly hair) and Carvajal syndrome (caused by a homozygous pathogenic gene variant that truncates the DSP protein) (Protonotarios and Tsatsopoulou, 2004; Marcus et al., 2010). Studies have suggested that up to 50% of the cases are familial (Hamid et al., 2002). Pathogenic gene variants within the desmosomal proteins are the main cause of the “classic” ACM (Bauce et al., 2010). Pathogenic gene variants in desmosomal proteins have significant implications in the development of ACM and account for up to 60% of affected patients (den Haan et al., 2009; Delmar and McKenna, 2010). Overall, the most commonly mutated gene is plakophilin, which accounted for 46–61% of patients from two different registries (Groeneweg et al., 2015). To date, about 18 causative genes involved in ACM have been identified (Hamid et al., 2002; Towbin et al., 2019) (Table 7).
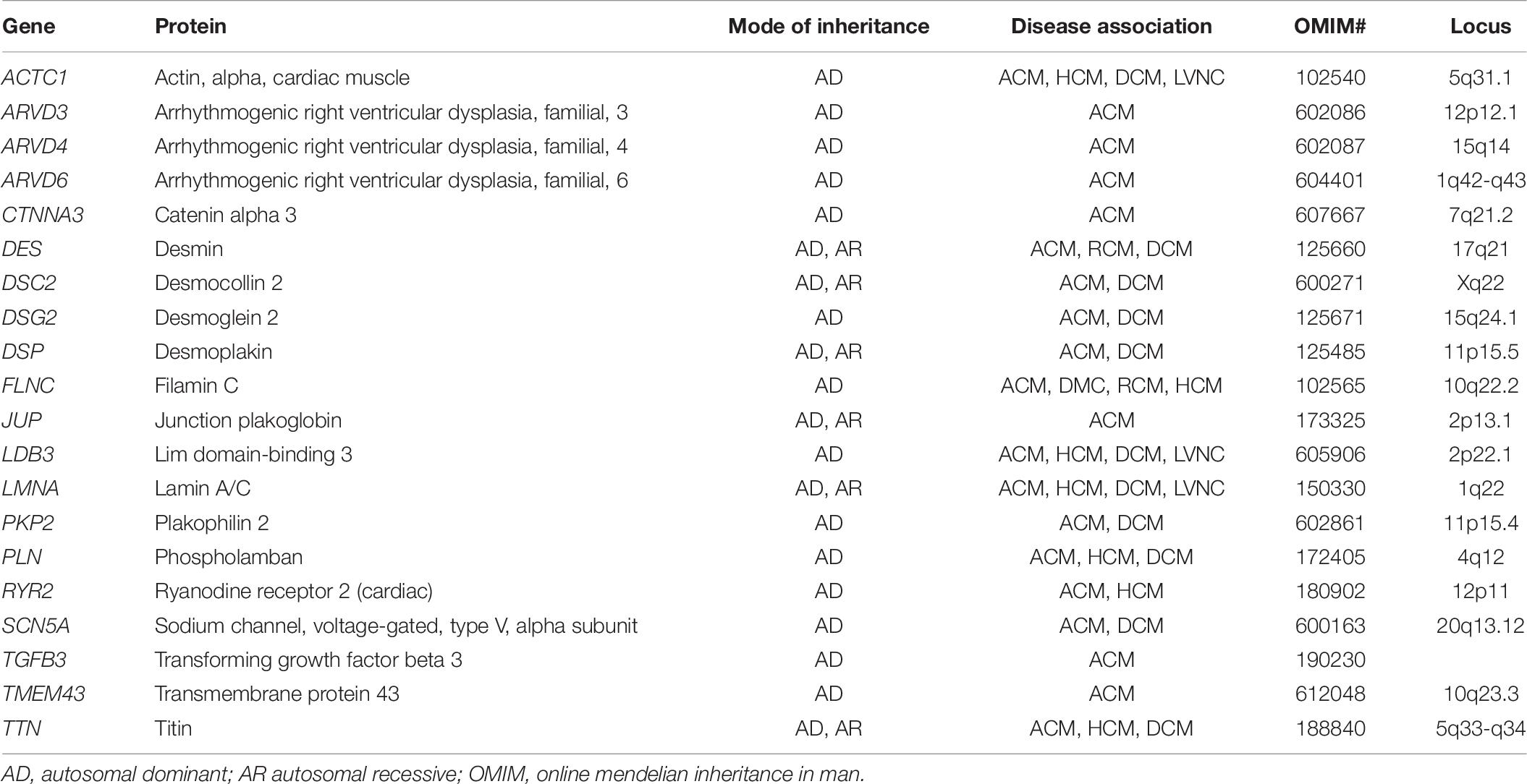
Table 7. List of common genes and patterns of inheritance associated with ACM.
Restrictive Cardiomyopathy (RCM)
Restrictive cardiomyopathy is characterized by abnormal diastology with typically preserved ejection fraction, relatively normal ventricular dimensions and the presence of atrial enlargement. The underlying hemodynamic consequence includes elevated ventricular filling pressures and a restrictive physiology (Maron et al., 2006; Denfield and Webber, 2010) (Figure 4).
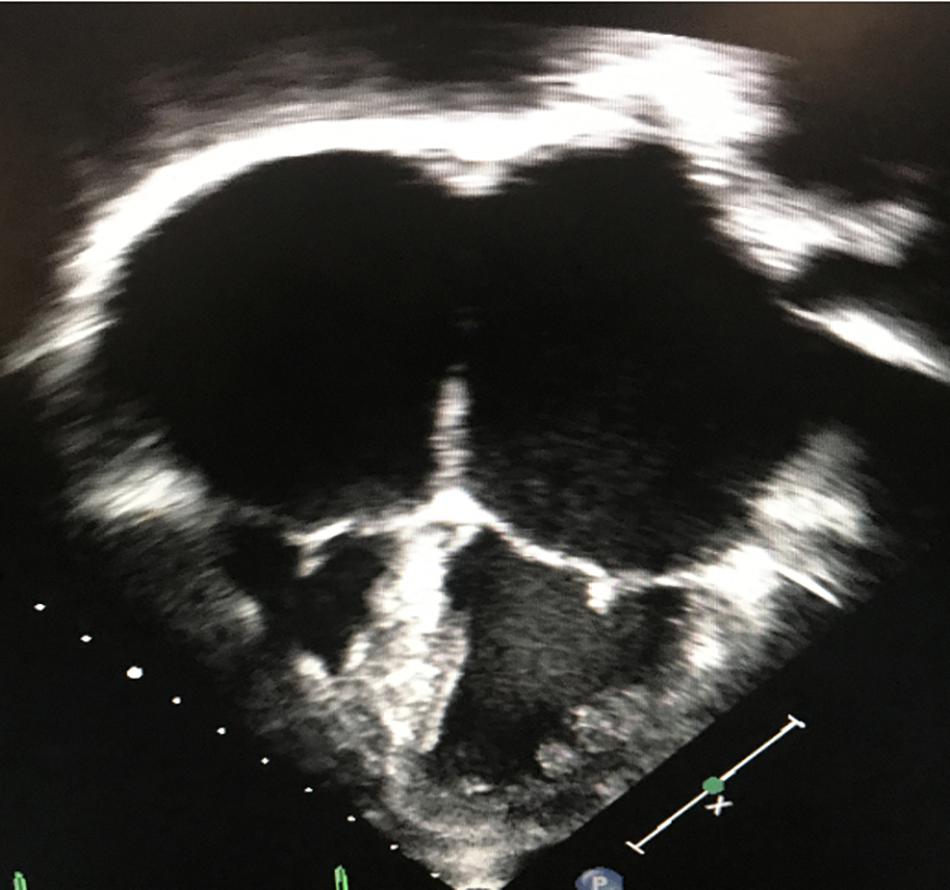
Figure 4. Two-dimensional, apical 4-chamber echocardiographic image depicting small, restrictive ventricles and significant biatrial enlargement in a patient with restrictive cardiomyopathy.
The clinical presentation is heterogenous and it relies on the degree of the inability to accommodate adequate cardiac input into the ventricles due to abnormal myocardial pliability or diastolic relaxation. Clinical symptoms may include exercise intolerance, dyspnea, volume overload, tachyarrhythmias, syncope, or sudden cardiac death. The hallmark of non-invasive imaging is atrial or bi-atrial enlargement. Normal or mild concentric hypertrophy with normal or reduced ventricular cavity can also be seen. Familial cases contribute in up to 30% of cases in RCM, and it usually exhibits autosomal dominant, autosomal recessive, X-linked, and mitochondrial transmitted modes of inheritance (Kaski et al., 2008). RCM can be classified based on the underlying process: non-infiltrative or primary; infiltrative (or systemic); associated with storage diseases (or syndromic); idiopathic; or in combination with other morpho functional types, such as DCM, HCM, and LVNC (Denfield and Webber, 2010). Primary myocardial disease or non-infiltrative types have been attributed to pathogenic gene variants in sarcomeric genes, such as TNNI3 (most common), TNNT2, MYH7, ACTC1, TPM1, MYL3, and MYL2, (Table 8) (Caleshu et al., 2011; Towbin, 2014). Scleroderma and other pathologies in the connective tissues are also considered causes of genetically triggered RCM (Worthley et al., 2001; Nguyen et al., 2006). Infiltrative causes of RCM are mainly represented by amyloidosis (the most common type), sarcoidosis and hemochromatosis. The former, encompass a group of metabolic derangements characterized by deposition of insoluble fibrillar protein-like metabolites altering organ function and structure (including the myocardium). There are approximately 20 different proteins associated with cardiac amyloidosis, with TRR, AL, ATTRm, ATTRwt, and ApoA-I representing the most common types (Rapezzi et al., 2009; Brown et al., 2021). Sarcoidosis is a multisystemic granulomatous process affecting the lungs, skin and myocardium. The incidence varies with geography and ethnic groups, for instance, it has been described to affect black > white Americans, and female > male patients. The strongest genetic associations are embedded in the major histocompatibility complex and BTNL2 gene (Spagnolo et al., 2007). Lysosomal storage disorders are characterized by abnormal lysosomal metabolism leading to the accumulation of various glycosaminoglycans, glycoproteins, or glycolipids within lysosomes of various tissues, including the myocardium. Gaucher disease and Fabry disease (two of the most common lysosomal disorders) may manifest as CM (HCM or RCM), valvular disease, coronary artery disease, and/or aortic enlargement (Lindor and Karnes, 1995). Hemochromatosis is a disorder typically characterized by increased intestinal iron absorption leading to iron overload and systemic deposition. This disease has been associated with low-penetrance autosomal dominance of pathogenic variants identified in the HFE gene (Pietrangelo, 2010, 2015). Glycogen storage disorders such Hurler disease and Hunter disease are characterized by the existence of deficient enzymes necessary for the breakdown of glycosaminoglycans. When these disorders manifest, cardiac involvement incorporate impaired ventricular filling, endocardial fibroelastosis, and cardiac valvulopathy (stenosis and/or insufficiency) (Brown et al., 2021).

Table 8. List of common genes and patterns of inheritance associated with RCM.
Clinical Genetic Discussion
A methodical approach to the genetic diagnosis of heritable cardiomyopathies should be initiated by obtaining a detailed history of present illness, a complete family history incorporating a three-generation pedigree, and a physical examination with attention to syndromic cardiovascular disease (Musunuru et al., 2020). Care should be taken when interpreting the family pedigrees, as they may not represent updated information in followed-up visits, variable expressivity in patients, bilateral genotypic predisposition and the role of epigenetics (Hershberger et al., 2013).
The review of cardiovascular imaging, electrocardiographic data, and the incorporation of functional cardiovascular assessments provides informative data to stratify disease, identify morbidity and to guide clinical and individual management of myocardial diseases (Table 9). In “classic” phenotypic characteristics, pattern-recognition of syndromic disease may guide clinicians to selecting the best targeted medical management. The selection and initiation of genetic testing should be guided by the aforementioned information. Current guidelines recommend genetic testing in children and adult patients harboring heritable cardiomyopathies (Ingles et al., 2011; Hershberger et al., 2018b).

Table 9. Initial workup of heritable cardiomyopathies.
When genetic results are interpreted, we recommend bearing in mind the following concepts. Penetrance, which describes the phenomenon by whether a phenotype can be observed on an individual harboring pathogenic genotype. For instance, most autosomal dominant cardiomyopathies (associated with sarcomeric mutations) are characterized by incomplete penetrance by adulthood. Whereas autosomal recessive cardiomyopathies are associated with complete penetrance before adulthood (Charron et al., 2010). Variable expressivity, this term entitles the description of variable phenotypic manifestations among carriers of a particular pathogenic gene variant (Charron, 2006; Balistreri et al., 2019).
Genetic testing should be perceived as a complementary study in the diagnosis of cardiomyopathy.
The major critical aspect of integrating genetic testing to study the first affected individual (proband), is to refine diagnoses in syndromic and non-syndromic cardiomyopathies, as early identification of genetically predisposed individuals allows for disease stratification, predicts morbidity, individualizes medical management, and in some cases, it provides genotype-specific therapy. Similarly, genetic testing should be routinely incorporated to screen family members of the proband harboring the risk of hosting similar pathogenic gene variants. This approach is encouraged by the existence of high prevalence of heritability in first-degree relatives.
Future Directions
The field of cardiomyopathy and genetics should continue to sum efforts to accomplish research goals and improve patient healthcare. Precision medicine from genomics and personal health history should be integrated in three areas in particular: (1) to create gene-based therapies for individual patients, (2) to enhance preventive medicine to transform health care systems from illness to prevention and wellness, and (3) by better understanding disease modifiers and epigenetics in cardiovascular disease. For those patients who harbor CM as a chronic illness, we found that the quality of life and the quality of outcomes for these survivors should be continuously assessed by healthcare providers, guardians and family members. Academic and investigational projects driving improvements in outcomes of cardiomyopathy patients are important since determinants of health wellbeing and disease exist in our communities. This topic is of importance, as there exist a tremendous relatively untapped set of opportunities to create synergy between clinicians, policy makers, advocacy groups and funding institutions.
The domains of precision medicine and artificial intelligence have brought rapid advances and alternatives in research by allowing one to manage big data from electronic medical records and collaborative databases. As a result, we have witnessed the development of molecular compounds (myosin inhibitors, calcium sensitizers, myosin activators, etc.) in the management of various types of cardiomyopathies. These methodologies have also contributed to the utilization of genotype phenotype coupling, pharmaco-genomic profiles, and individualization of therapies based on algorithms supporting the use of predictive analytics. Cardiovascular genetics is currently leading a transition from – making the diagnosis prior to applying gene testing to requesting diagnostic genome testing for an early diagnosis to avoid unnecessary harm – perhaps a natural extension for what it pertains to the cardiomyopathy genes.
Conclusion
In recent years, the number of individuals diagnosed with hereditary CMs has grown, likely due to increased awareness, advances in imaging modalities, and a better understanding of phenotypic associations and the molecular genesis of these diseases.
In our experience, genetic testing has been most beneficial in CMs associated with other cardiac features, such as LV dysfunction, LV hypertrophy, arrhythmias, and the co-occurrence of other cardiac and non-cardiac syndromic features.
The central principle supporting the incorporation of genetic testing in cardiovascular medicine relies on the allowance of practitioners to recognize at-risk family members and the advantageous strategy to delineate disease, provide adequate surveillance and tailor individual therapeutic options.
Implementation of precision medicine in the evaluation of heritable CMs likely ensures a dramatic improvement in patient outcomes and shifts the current paradigm from a disease-treatment to an early-diagnostic/preventive healthcare system.
Author Contributions
HM took the lead in writing the manuscript. All authors provided critical feedback and helped shape the manuscript. JLJ conceived the original idea of the project.
Funding
This work was supported by the St. Jude Pediatric Research Recruitment Support Fund hosted by HM (R079700270).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Jennifer Osher, MS; and Carye Hackman, BBA, for their help in preparation of this manuscript.
References
Alfares, A. A., Kelly, M. A., McDermott, G., Funke, B. H., Lebo, M. S., Baxter, S. B., et al. (2015). Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet. Med. 17, 880–888. doi: 10.1038/gim.2014.205
Arad, M., Maron, B. J., Gorham, J. M., Johnson, W. H. Jr., Saul, J. P., Perez-Atayde, A. R., et al. (2005). Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N. Engl. J. Med. 352, 362–372. doi: 10.1056/nejmoa033349
Arbustini, E., Narula, N., Dec, G. W., Reddy, K. S., Greenberg, B., Kushwaha, S., et al. (2013). The MOGE(S) Classification for a Phenotype-Genotype Nomenclature of Cardiomyopathy: Endorsed by the World Heart Federation. Glob. Heart 8, 355–382. doi: 10.1016/j.gheart.2013.11.001
Arbustini, E., Narula, N., Tavazzi, L., Serio, A., Grasso, M., Favalli, V., et al. (2014). The MOGE(S) classification of cardiomyopathy for clinicians. J. Am. Coll. Cardiol. 64, 304–318. doi: 10.1016/j.jacc.2014.05.027
Bagnall, R. D., Molloy, L. K., Kalman, J. M., and Semsarian, C. (2014). Exome sequencing identifies a mutation in the ACTN2 gene in a family with idiopathic ventricular fibrillation, left ventricular noncompaction, and sudden death. BMC Med. Genet. 15, 99. doi: 10.1186/s12881-014-0099-0
Balistreri, C. R., Forte, M., Greco, E., Paneni, F., Cavarretta, E., Frati, G., et al. (2019). An overview of the molecular mechanisms underlying development and progression of bicuspid aortic valve disease. J. Mol. Cell Cardiol. 132, 146–153. doi: 10.1016/j.yjmcc.2019.05.013
Barth, P. G., Scholte, H. R., Berden, J. A., Van der Klei-Van Moorsel, J. M., Luyt-Houwen, I. E., Van ’t Veer-Korthof, E. T., et al. (1983). An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 62, 327–355. doi: 10.1016/0022-510x(83)90209-5
Bauce, B., Nava, A., Beffagna, G., Basso, C., Lorenzon, A., Smaniotto, G., et al. (2010). Multiple mutations in desmosomal proteins encoding genes in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm 7, 22–29. doi: 10.1016/j.hrthm.2009.09.070
Bowles, N. E., Bowles, K. R., and Towbin, J. A. (2000). The “final common pathway” hypothesis and inherited cardiovascular disease. The role of cytoskeletal proteins in dilated cardiomyopathy. Herz 25, 168–175. doi: 10.1007/s000590050003
Brady, R. O., Gal, A. E., Bradley, R. M., Martensson, E., Warshaw, A. L., and Laster, L. (1967). Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N. Engl. J. Med. 276, 1163–1167. doi: 10.1056/nejm196705252762101
Brinkley, D. M., Wells, Q. S., and Stevenson, L. W. (2020). Avoiding Burnout From Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 75, 3044–3047. doi: 10.1016/j.jacc.2020.05.009
Brown, K. N., Pendela, V. S., and Diaz, R. R. (2021). Restrictive Cardiomyopathy. Treasure Island, FL: StatPearls Publishing.
Burns, C., Bagnall, R. D., Lam, L., Semsarian, C., and Ingles, J. (2017). Multiple Gene Variants in Hypertrophic Cardiomyopathy in the Era of Next-Generation Sequencing. Circ. Cardiovasc. Genet. 10:e001666.
Caleshu, C., Sakhuja, R., Nussbaum, R. L., Schiller, N. B., Ursell, P. C., Eng, C., et al. (2011). Furthering the link between the sarcomere and primary cardiomyopathies: restrictive cardiomyopathy associated with multiple mutations in genes previously associated with hypertrophic or dilated cardiomyopathy. Am. J. Med. Genet. A 155A, 2229–2235. doi: 10.1002/ajmg.a.34097
Charron, P. (2006). Clinical genetics in cardiology. Heart 92, 1172–1176. doi: 10.1136/hrt.2005.071308
Charron, P., Arad, M., Arbustini, E., Basso, C., Bilinska, Z., Elliott, P., et al. (2010). Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 31, 2715–2726. doi: 10.1093/eurheartj/ehq271
Chun, E. J., Choi, S. I., Jin, K. N., Kwag, H. J., Kim, Y. J., Choi, B. W., et al. (2010). Hypertrophic cardiomyopathy: assessment with MR imaging and multidetector CT. Radiographics 30, 1309–1328. doi: 10.1148/rg.305095074
Colan, S. D., Lipshultz, S. E., Lowe, A. M., Sleeper, L. A., Messere, J., Cox, G. F., et al. (2007). Epidemiology and cause-specific outcome of hypertrophic cardiomyopathy in children: findings from the Pediatric Cardiomyopathy Registry. Circulation 115, 773–781. doi: 10.1161/circulationaha.106.621185
Davies, K. E., and Nowak, K. J. (2006). Molecular mechanisms of muscular dystrophies: old and new players. Nat. Rev. Mol. Cell Biol. 7, 762–773. doi: 10.1038/nrm2024
de la Chica, J. A., Gomez-Talavera, S., Garcia-Ruiz, J. M., Garcia-Lunar, I., Oliva, B., Fernandez-Alvira, J. M., et al. (2020). Association Between Left Ventricular Noncompaction and Vigorous Physical Activity. J. Am. Coll. Cardiol. 76, 1723–1733. doi: 10.1016/j.jacc.2020.08.030
Delmar, M., and McKenna, W. J. (2010). The cardiac desmosome and arrhythmogenic cardiomyopathies: from gene to disease. Circ. Res. 107, 700–714. doi: 10.1161/circresaha.110.223412
den Haan, A. D., Tan, B. Y., Zikusoka, M. N., Llado, L. I., Jain, R., Daly, A., et al. (2009). Comprehensive desmosome mutation analysis in north americans with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ. Cardiovasc. Genet. 2, 428–435. doi: 10.1161/circgenetics.109.858217
Denfield, S. W., and Webber, S. A. (2010). Restrictive cardiomyopathy in childhood. Heart Fail. Clin. 6, 445–452. doi: 10.1016/j.hfc.2010.05.005
Dong, X., Fan, P., Tian, T., Yang, Y., Xiao, Y., Yang, K., et al. (2017). Recent advancements in the molecular genetics of left ventricular noncompaction cardiomyopathy. Clin. Chim. Acta 465, 40–44. doi: 10.1016/j.cca.2016.12.013
Elliott, P., Andersson, B., Arbustini, E., Bilinska, Z., Cecchi, F., Charron, P., et al. (2008). Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur. Heart J. 29, 270–276. doi: 10.1093/eurheartj/ehm342
Engberding, R., Yelbuz, T. M., and Breithardt, G. (2007). Isolated noncompaction of the left ventricular myocardium – a review of the literature two decades after the initial case description. Clin. Res. Cardiol. 96, 481–488. doi: 10.1007/s00392-007-0528-6
Fatkin, D., Huttner, I. G., Kovacic, J. C., Seidman, J. G., and Seidman, C. E. (2019). Precision Medicine in the Management of Dilated Cardiomyopathy: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 74, 2921–2938.
Finsterer, J., and Stollberger, C. (2008). Primary myopathies and the heart. Scand. Cardiovasc. J. 42, 9–24. doi: 10.1080/14017430701854953
Finsterer, J., Stollberger, C., and Towbin, J. A. (2017). Left ventricular noncompaction cardiomyopathy: cardiac, neuromuscular, and genetic factors. Nat. Rev. Cardiol. 14, 224–237. doi: 10.1038/nrcardio.2016.207
Garman, S. C., and Garboczi, D. N. (2004). The molecular defect leading to Fabry disease: structure of human alpha-galactosidase. J. Mol. Biol. 337, 319–335. doi: 10.1016/j.jmb.2004.01.035
Gati, S., Chandra, N., Bennett, R. L., Reed, M., Kervio, G., Panoulas, V. F., et al. (2013). Increased left ventricular trabeculation in highly trained athletes: do we need more stringent criteria for the diagnosis of left ventricular non-compaction in athletes? Heart 99, 401–408. doi: 10.1136/heartjnl-2012-303418
Gati, S., Papadakis, M., Papamichael, N. D., Zaidi, A., Sheikh, N., Reed, M., et al. (2014). Reversible de novo left ventricular trabeculations in pregnant women: implications for the diagnosis of left ventricular noncompaction in low-risk populations. Circulation 130, 475–483. doi: 10.1161/circulationaha.114.008554
Gersh, B. J., Maron, B. J., Bonow, R. O., Dearani, J. A., Fifer, M. A., Link, M. S., et al. (2011). 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 124, 783–831e.
Groeneweg, J. A., Bhonsale, A., James, C. A., te Riele, A. S., Dooijes, D., Tichnell, C., et al. (2015). Clinical Presentation, Long-Term Follow-Up, and Outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and Family Members. Circ. Cardiovasc. Genet. 8, 437–446.
Hamid, M. S., Norman, M., Quraishi, A., Firoozi, S., Thaman, R., Gimeno, J. R., et al. (2002). Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J. Am. Coll. Cardiol. 40, 1445–1450. doi: 10.1016/s0735-1097(02)02307-0
Hastings, R., de Villiers, C. P., Hooper, C., Ormondroyd, L., Pagnamenta, A., Lise, S., et al. (2016). Combination of Whole Genome Sequencing, Linkage, and Functional Studies Implicates a Missense Mutation in Titin as a Cause of Autosomal Dominant Cardiomyopathy With Features of Left Ventricular Noncompaction. Circ. Cardiovasc. Genet. 9, 426–435. doi: 10.1161/circgenetics.116.001431
Hershberger, R. E., Givertz, M. M., Ho, C. Y., Judge, D. P., Kantor, P. F., McBride, K. L., et al. (2018a). Genetic Evaluation of Cardiomyopathy-A Heart Failure Society of America Practice Guideline. J. Card. Fail. 24, 281–302. doi: 10.1016/j.cardfail.2018.03.004
Hershberger, R. E., Givertz, M. M., Ho, C. Y., Judge, D. P., Kantor, P. F., McBride, K. L., et al. (2018b). Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 20, 899–909. doi: 10.1038/s41436-018-0039-z
Hershberger, R. E., Hedges, D. J., and Morales, A. (2013). Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 10, 531–547. doi: 10.1038/nrcardio.2013.105
Hershberger, R. E., Norton, N., Morales, A., Li, D., Siegfried, J. D., and Gonzalez-Quintana, J. (2010). Coding sequence rare variants identified in MYBPC3, MYH6, TPM1, TNNC1, and TNNI3 from 312 patients with familial or idiopathic dilated cardiomyopathy. Circ. Cardiovasc. Genet. 3, 155–161. doi: 10.1161/circgenetics.109.912345
Hirschhorn, R., and Huie, M. L. (1999). Frequency of mutations for glycogen storage disease type II in different populations: the delta525T and deltaexon 18 mutations are not generally “common” in white populations. J. Med. Genet. 36, 85–86.
Ingles, J., Zodgekar, P. R., Yeates, L., Macciocca, I., Semsarian, C., Fatkin, D., et al. (2011). Guidelines for genetic testing of inherited cardiac disorders. Heart Lung Circ. 20, 681–687. doi: 10.1016/j.hlc.2011.07.013
Jarcho, J. A., McKenna, W., Pare, J. A., Solomon, S. D., Holcombe, R. F., Dickie, S., et al. (1989). Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N. Engl. J. Med. 321, 1372–1378. doi: 10.1056/nejm198911163212005
Kaski, J. P., Syrris, P., Burch, M., Tome-Esteban, M. T., Fenton, M., Christiansen, M., et al. (2008). Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart 94, 1478–1484. doi: 10.1136/hrt.2007.134684
Kipps, A., Alexander, M., Colan, S. D., Gauvreau, K., Smoot, L., Crawford, L., et al. (2009). The longitudinal course of cardiomyopathy in Friedreich’s ataxia during childhood. Pediatr. Cardiol. 30, 306–310.
Konno, T., Chang, S., Seidman, J. G., and Seidman, C. E. (2010). Genetics of hypertrophic cardiomyopathy. Curr. Opin. Cardiol. 25, 205–209.
Kovacevic-Preradovic, T., Jenni, R., Oechslin, E. N., Noll, G., Seifert, B., and Attenhofer Jost, C. H. (2009). Isolated left ventricular noncompaction as a cause for heart failure and heart transplantation: a single center experience. Cardiology 112, 158–164. doi: 10.1159/000147899
Lee, T. M., Hsu, D. T., Kantor, P., Towbin, J. A., Ware, S. M., Colan, S. D., et al. (2017). Pediatric Cardiomyopathies. Circ. Res. 121, 855–873.
Levine, J. C., Kishnani, P. S., Chen, Y. T., Herlong, J. R., and Li, J. S. (2008). Cardiac remodeling after enzyme replacement therapy with acid alpha-glucosidase for infants with Pompe disease. Pediatr. Cardiol. 29, 1033–1042. doi: 10.1007/s00246-008-9267-3
Li, C. J., Chen, C. S., Yiang, G. T., Tsai, A. P., Liao, W. T., and Wu, M. Y. (2019). Advanced Evolution of Pathogenesis Concepts in Cardiomyopathies. J. Clin. Med. 8, 520. doi: 10.3390/jcm8040520
Lindor, N. M., and Karnes, P. S. (1995). Initial assessment of infants and children with suspected inborn errors of metabolism. Mayo Clin. Proc. 70, 987–988. doi: 10.4065/70.10.987
Lipshultz, S. E., Law, Y. M., Asante-Korang, A., Austin, Dipchand, A. I., Everitt, M. D., et al. (2019). Cardiomyopathy in Children: Classification and Diagnosis: A Scientific Statement From the American Heart Association. Circulation 140, 9–68e.
Liu, Y., Chen, H., and Shou, W. (2018). Potential Common Pathogenic Pathways for the Left Ventricular Noncompaction Cardiomyopathy (LVNC). Pediatr. Cardiol. 39, 1099–1106. doi: 10.1007/s00246-018-1882-z
Maass, A., Konhilas, J. P., Stauffer, B. L., and Leinwand, L. A. (2002). From sarcomeric mutations to heart disease: understanding familial hypertrophic cardiomyopathy. Cold Spring Harb. Symp. Quant. Biol. 67, 409–415. doi: 10.1101/sqb.2002.67.409
Maggi, L., Carboni, N., and Bernasconi, P. (2016). Skeletal Muscle Laminopathies: A Review of Clinical and Molecular Features. Cells 5, 33. doi: 10.3390/cells5030033
Marcus, F. I., McKenna, W. J., Sherrill, D., Basso, C., Bauce, B., Bluemke, D. A., et al. (2010). Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Eur. Heart J. 31, 806–814.
Maron, B. J. (2018). Clinical Course and Management of Hypertrophic Cardiomyopathy. N. Engl. J. Med. 379, 1977.
Maron, B. J., Roberts, W. C., Arad, M., Haas, T. S., Spirito, P., Wright, G. B., et al. (2009). Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. JAMA 301, 1253–1259. doi: 10.1001/jama.2009.371
Maron, B. J., Rowin, E. J., and Maron, M. S. (2018). Global Burden of Hypertrophic Cardiomyopathy. JACC Heart Fail. 6, 376–378. doi: 10.1016/j.jchf.2018.03.004
Maron, B. J., Towbin, J. A., Thiene, G., Antzelevitch, C., Corrado, D., Arnett, D., et al. (2006). Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 113, 1807–1816. doi: 10.1161/circulationaha.106.174287
Martinez, H. R., Craigen, W. J., Ummat, M., Adesina, A. M., Lotze, T. E., and Jefferies, J. L. (2014). Novel cardiovascular findings in association with a POMT2 mutation: three siblings with alpha-dystroglycanopathy. Eur. J. Hum. Genet. 22, 486–491. doi: 10.1038/ejhg.2013.165
Martinez, H. R., Pignatelli, R., Belmont, J. W., Craigen, W. J., and Jefferies, J. L. (2011). Childhood onset of left ventricular dysfunction in a female manifesting carrier of muscular dystrophy. Am. J. Med. Genet. A 155A, 3025–3029. doi: 10.1002/ajmg.a.33784
Martinez, H. R., Ware, S. M., Schamberger, M. S., and Parent, J. J. (2017). Noncompaction cardiomyopathy and heterotaxy syndrome. Prog. Pediatr. Cardiol. 46, 23–27. doi: 10.1016/j.ppedcard.2017.06.007
McKenna, W. J., Maron, B. J., and Thiene, G. (2017). Classification, Epidemiology, and Global Burden of Cardiomyopathies. Circ. Res. 121, 722–730. doi: 10.1161/circresaha.117.309711
McNally, E. M., and Mestroni, L. (2017). Dilated Cardiomyopathy: Genetic Determinants and Mechanisms. Circ. Res. 121, 731–748. doi: 10.1161/circresaha.116.309396
McNally, E. M., Golbus, J. R., and Puckelwartz, M. J. (2013). Genetic mutations and mechanisms in dilated cardiomyopathy. J. Clin. Invest. 123, 19–26. doi: 10.1172/jci62862
Mehta, A., Ricci, R., Widmer, U., Dehout, F., Garcia de Lorenzo, A., Kampmann, C., et al. (2004). Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur. J. Clin. Invest. 34, 236–242. doi: 10.1111/j.1365-2362.2004.01309.x
Miller, E. M., Hinton, R. B., Czosek, R., Lorts, A., Parrott, A., Shikany, A. R., et al. (2017). Genetic Testing in Pediatric Left Ventricular Noncompaction. Circ. Cardiovasc. Genet. 10:e001735.
Minoche, A. E., Horvat, C., Johnson, R., Gayevskiy, V., Morton, S. U., Drew, A. P., et al. (2019). Genome sequencing as a first-line genetic test in familial dilated cardiomyopathy. Genet. Med. 21, 650–662. doi: 10.1038/s41436-018-0084-7
Miszalski-Jamka, K., Jefferies, J. L., Mazur, W., Glowacki, J., Hu, J., Lazar, M., et al. (2017). Novel Genetic Triggers and Genotype-Phenotype Correlations in Patients With Left Ventricular Noncompaction. Circ. Cardiovasc. Genet. 10:e001763.
Musunuru, K., Hershberger, R. E., Day, S. M., Klinedinst, N. J., Landstrom, A. P., Parikh, V. N., et al. (2020). Genetic Testing for Inherited Cardiovascular Diseases: A Scientific Statement From the American Heart Association. Circ. Genom. Precis. Med. 13:e000067.
Nguyen, L. D., Terbah, M., Daudon, P., and Martin, L. (2006). Left ventricular systolic and diastolic function by echocardiogram in pseudoxanthoma elasticum. Am. J. Cardiol. 97, 1535–1537. doi: 10.1016/j.amjcard.2005.11.091
Nouhravesh, N., Ahlberg, G., Ghouse, J., Andreasen, C., Svendsen, J. H., Haunso, S., et al. (2016). Analyses of more than 60,000 exomes questions the role of numerous genes previously associated with dilated cardiomyopathy. Mol. Genet. Genomic Med. 4, 617–623. doi: 10.1002/mgg3.245
O’Mahony, C., and Elliott, P. (2010). Anderson-Fabry disease and the heart. Prog. Cardiovasc. Dis. 52, 326–335.
Online Mendelian Inheritance in Man (2019). McKusick-Nathans Institute of Genetic Medicine. Baltimore, MD: Johns Hopkins University.
Patel, P. I., and Isaya, G. (2001). Friedreich ataxia: from GAA triplet-repeat expansion to frataxin deficiency. Am. J. Hum. Genet. 69, 15–24. doi: 10.1086/321283
Perrot, A., Osterziel, K. J., Beck, M., Dietz, R., and Kampmann, C. (2002). Fabry disease: focus on cardiac manifestations and molecular mechanisms. Herz 27, 699–702. doi: 10.1007/s00059-002-2429-9
Peters, S., Trummel, M., and Meyners, W. (2004). Prevalence of right ventricular dysplasia-cardiomyopathy in a non-referral hospital. Int. J. Cardiol. 97, 499–501. doi: 10.1016/j.ijcard.2003.10.037
Petretta, M., Pirozzi, F., Sasso, L., Paglia, A., and Bonaduce, D. (2011). Review and metaanalysis of the frequency of familial dilated cardiomyopathy. Am. J. Cardiol. 108, 1171–1176. doi: 10.1016/j.amjcard.2011.06.022
Peverill, R. E. (2012). Letter by Peverill regarding article, “The heart in Friedreich ataxia: definition of cardiomyopathy, disease severity, and correlation with neurological symptoms”. Circulation 126:e272.
Pietrangelo, A. (2010). Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology 139, e1–e2.
Pietrangelo, A. (2015). Genetics, Genetic Testing, and Management of Hemochromatosis: 15 Years Since Hepcidin. Gastroenterology 149, 1240–51e4.
Pignatelli, R. H., McMahon, C. J., Dreyer, W. J., Denfield, S. W., Price, J., Belmont, J. W., et al. (2003). Clinical characterization of left ventricular noncompaction in children: a relatively common form of cardiomyopathy. Circulation 108, 2672–2678. doi: 10.1161/01.cir.0000100664.10777.b8
Piran, S., Liu, P., Morales, A., and Hershberger, R. E. (2012). Where genome meets phenome: rationale for integrating genetic and protein biomarkers in the diagnosis and management of dilated cardiomyopathy and heart failure. J. Am. Coll. Cardiol. 60, 283–289. doi: 10.1016/j.jacc.2012.05.005
Popa-Fotea, N. M., Micheu, M. M., Bataila, V., Scafa-Udriste, A., Dorobantu, L., Scarlatescu, A. I., et al. (2019). Exploring the Continuum of Hypertrophic Cardiomyopathy-From DNA to Clinical Expression. Medicina 55, 299. doi: 10.3390/medicina55060299
Protonotarios, N., and Tsatsopoulou, A. (2004). Naxos disease and Carvajal syndrome: cardiocutaneous disorders that highlight the pathogenesis and broaden the spectrum of arrhythmogenic right ventricular cardiomyopathy. Cardiovasc. Pathol. 13, 185–194.
Puckelwartz, M., and McNally, E. M. (2011). Emery-Dreifuss muscular dystrophy. Handb. Clin. Neurol. 101, 155–166.
Rakowski, H., and Carasso, S. (2009). Diastolic dysfunction and histopathology in hypertrophic cardiomyopathy: is relaxation in disarray? J. Am. Soc. Echocardiogr. 22, 1335–1337. doi: 10.1016/j.echo.2009.10.006
Rapezzi, C., Merlini, G., Quarta, C. C., Riva, L., Longhi, S., Leone, O., et al. (2009). Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation 120, 1203–1212. doi: 10.1161/circulationaha.108.843334
Rauen, K. A., Schoyer, L., McCormick, F., Lin, A. E., Allanson, J. E., Stevenson, D. A., et al. (2010). Proceedings from the 2009 genetic syndromes of the Ras/MAPK pathway: From bedside to bench and back. Am. J. Med. Genet. A 152A, 4–24.
Roma-Rodrigues, C., and Fernandes, A. R. (2014). Genetics of hypertrophic cardiomyopathy: advances and pitfalls in molecular diagnosis and therapy. Appl. Clin. Genet. 7, 195–208. doi: 10.2147/tacg.s49126
Ross, S. B., Singer, E. S., Driscoll, E., Nowak, N., Yeates, L., Puranik, R., et al. (2020). Genetic architecture of left ventricular noncompaction in adults. Hum. Genome Var. 7, 33.
Rowin, E. J., and Maron, M. S. (2013). The ever expanding spectrum of phenotypic diversity in hypertrophic cardiomyopathy. Am. J. Cardiol. 112, 463–464. doi: 10.1016/j.amjcard.2013.05.045
Rowland, T. J., Sweet, M. E., Mestroni, L., and Taylor, M. R. (2016). Danon disease - dysregulation of autophagy in a multisystem disorder with cardiomyopathy. J. Cell Sci. 129, 2135–2143. doi: 10.1242/jcs.184770
Ruiz-Guerrero, L., and Barriales-Villa, R. (2018). Storage diseases with hypertrophic cardiomyopathy phenotype. Glob. Cardiol. Sci. Pract. 2018, 28.
Sedaghat-Hamedani, F., Haas, J., Zhu, F., Geier, C., Kayvanpour, E., Liss, M., et al. (2017). Clinical genetics and outcome of left ventricular non-compaction cardiomyopathy. Eur. Heart J. 38, 3449–3460.
Shan, L., Makita, N., Xing, Y., Watanabe, S., Futatani, T., Ye, F., et al. (2008). SCN5A variants in Japanese patients with left ventricular noncompaction and arrhythmia. Mol. Genet. Metab. 93, 468–474. doi: 10.1016/j.ymgme.2007.10.009
Spagnolo, P., Sato, H., Grutters, J. C., Renzoni, E. A., Marshall, S. E., Ruven, H. J., et al. (2007). Analysis of BTNL2 genetic polymorphisms in British and Dutch patients with sarcoidosis. Tissue Antigens 70, 219–227. doi: 10.1111/j.1399-0039.2007.00879.x
Stanton, C., Bruce, C., Connolly, H., Brady, P., Syed, I., Hodge, D., et al. (2009). Isolated left ventricular noncompaction syndrome. Am. J. Cardiol. 104, 1135–1138.
Tariq, M., and Ware, S. M. (2014). Importance of genetic evaluation and testing in pediatric cardiomyopathy. World J. Cardiol. 6, 1156–1165. doi: 10.4330/wjc.v6.i11.1156
Tayal, U., Prasad, S., and Cook, S. A. (2017). Genetics and genomics of dilated cardiomyopathy and systolic heart failure. Genome Med. 9, 20.
Toib, A., Grange, D. K., Kozel, B. A., Ewald, G. A., White, F. V., and Canter, C. E. (2010). Distinct clinical and histopathological presentations of Danon cardiomyopathy in young women. J. Am. Coll. Cardiol. 55, 408–410. doi: 10.1016/j.jacc.2009.11.019
Towbin, J. A. (2010). Left ventricular noncompaction: a new form of heart failure. Heart Fail. Clin. 6, 453–469. doi: 10.1016/j.hfc.2010.06.005
Towbin, J. A., and Beasley, G. (2020). Left Ventricular Noncompaction and Vigorous Physical Activity. J. Am. Coll. Cardiol. 76, 1734–1736. doi: 10.1016/j.jacc.2020.08.051
Towbin, J. A., and Ortiz-Lopez, R. (1994). X-linked dilated cardiomyopathy. N. Engl. J. Med. 330, 369–370.
Towbin, J. A., McKenna, W. J., Abrams, D. J., Ackerman, M. J., Calkins, H., Darrieux, F. C. C., et al. (2019). 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: Executive summary. Heart Rhythm 16, 373–407e.
van Spaendonck-Zwarts, K. Y., van Rijsingen, I. A., van den Berg, M. P., Lekanne Deprez, R. H., Post, J. G., van Mil, A. M., et al. (2013). Genetic analysis in 418 index patients with idiopathic dilated cardiomyopathy: overview of 10 years’ experience. Eur. J. Heart Fail. 15, 628–636. doi: 10.1093/eurjhf/hft013
van Waning, J. I., Caliskan, K., Hoedemaekers, Y. M., van Spaendonck-Zwarts, K. Y., Baas, A. F., Boekholdt, S. M., et al. (2018). Genetics, Clinical Features, and Long-Term Outcome of Noncompaction Cardiomyopathy. J. Am. Coll. Cardiol. 71, 711–722.
van Waning, J. I., Caliskan, K., Michels, M., Schinkel, A. F. L., Hirsch, A., Dalinghaus, M., et al. (2019). Cardiac Phenotypes, Genetics, and Risks in Familial Noncompaction Cardiomyopathy. J. Am. Coll. Cardiol. 73, 1601–1611. doi: 10.1016/j.jacc.2018.12.085
Vershinina, T., Fomicheva, Y., Muravyev, A., Jorholt, J., Kozyreva, A., Kiselev, A., et al. (2020). Genetic Spectrum of Left Ventricular Non-Compaction in Paediatric Patients. Cardiology 145, 746–756. doi: 10.1159/000510439
Ware, S. M. (2015). Evaluation of genetic causes of cardiomyopathy in childhood. Cardiol. Young 25(Suppl. 2), 43–50. doi: 10.1017/s1047951115000827
Watkins, H., Ashrafian, H., and Redwood, C. (2011). Inherited cardiomyopathies. N. Engl. J. Med. 364, 1643–1656.
Weidemann, F., Rummey, C., Bijnens, B., Stork, S., Jasaityte, R., Dhooge, J., et al. (2012). The heart in Friedreich ataxia: definition of cardiomyopathy, disease severity, and correlation with neurological symptoms. Circulation 125, 1626–1634. doi: 10.1161/circulationaha.111.059477
Weischenfeldt, J., Symmons, O., Spitz, F., and Korbel, J. O. (2013). Phenotypic impact of genomic structural variation: insights from and for human disease. Nat. Rev. Genet. 14, 125–138. doi: 10.1038/nrg3373
Wilkinson, J. D., Landy, D. C., Colan, S. D., Towbin, J. A., Sleeper, L. A., Orav, E. J., et al. (2010). The pediatric cardiomyopathy registry and heart failure: key results from the first 15 years. Heart Fail. Clin. 6, 401–413. doi: 10.1016/j.hfc.2010.05.002
Wilkinson, J. D., Lowe, A. M., Salbert, B. A., Sleeper, L. A., Colan, S. D., Cox, G. F., et al. (2012). Outcomes in children with Noonan syndrome and hypertrophic cardiomyopathy: a study from the Pediatric Cardiomyopathy Registry. Am. Heart J. 164, 442–448. doi: 10.1016/j.ahj.2012.04.018
Worthley, M. I., Farouque, H. M., McNeil, J. D., and Worthley, S. G. (2001). Scleroderma cardiomyopathy presenting with thromboembolism. Intern. Med. J. 31, 64–65. doi: 10.1046/j.1445-5994.2001.00009.x
Keywords: genetic expression, heritable cardiomyopathies, cardiac phenotypes, genetic testing, myocardial disease
Citation: Martinez HR, Beasley GS, Miller N, Goldberg JF and Jefferies JL (2021) Clinical Insights Into Heritable Cardiomyopathies. Front. Genet. 12:663450. doi: 10.3389/fgene.2021.663450
Received: 02 February 2021; Accepted: 06 April 2021;
Published: 28 April 2021.
Edited by:
Weinian Shou, Indiana University Bloomington, United StatesReviewed by:
Wuqiang Zhu, Mayo Clinic Arizona, United StatesJinghai Chen, Zhejiang University, China
Copyright © 2021 Martinez, Beasley, Miller, Goldberg and Jefferies. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hugo R. Martinez, aG1hcnRpbmV6QHV0aHNjLmVkdQ==