
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
DATA REPORT article
Front. Genet., 17 June 2021
Sec. Computational Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.662928
This article is part of the Research TopicBioinformatics of Genome Regulation, Volume IIView all 18 articles
 Ekaterina M. Dvorianinova1,2†
Ekaterina M. Dvorianinova1,2† Elena N. Pushkova1†
Elena N. Pushkova1† Roman O. Novakovskiy1†
Roman O. Novakovskiy1† Liubov V. Povkhova1,2
Liubov V. Povkhova1,2 Nadezhda L. Bolsheva1
Nadezhda L. Bolsheva1 Ludmila P. Kudryavtseva3Tatiana A. Rozhmina3
Ludmila P. Kudryavtseva3Tatiana A. Rozhmina3 Nataliya V. Melnikova1
Nataliya V. Melnikova1 Alexey A. Dmitriev1*
Alexey A. Dmitriev1*Fusarium oxysporum f. sp. lini is the one of notoriously known pathogens of flax (Linum usitatissimum L.), causing wilt. In the case of young seedlings, the disease may lead to complete yield losses (Kommedahl et al., 1970). At the same time, flax is an important crop used for manufacturing oil of high nutritional value, food supplements, industrial products, and fiber (Jhala and Hall, 2010; Czemplik et al., 2011; Kezimana et al., 2018; Parikh and Pierce, 2019). However, flax production is dependent on the resistance of flax varieties to phytopathogens, whereas F. oxysporum demonstrates considerable genetic diversity (Edel et al., 2001; Michielse and Rep, 2009).
At present, phytopathogenic strains of F. oxysporum are classified into numerous formae speciales according to their ability to colonize different hosts (Armstrong and Armstrong, 1981). Unfortunately, this system cannot provide researchers with enough information on a type of the pathogen and the severity of the infection it causes (Edel-Hermann and Lecomte, 2019). Like other representatives of the species, forma specialis lini is morphologically and genetically heterogeneous. Strains of the pathogen differ in such characteristics as the type of sporulation, conidia formation, pigment production, and the rate of growth on different media. Moreover, the degree of pathogenicity varies within the forma and depends on the infected crop variety (Kommedahl et al., 1970).
For molecular classification of F. oxysporum, numerous markers were used (Baayen et al., 2000; Lievens et al., 2008; Baysal et al., 2010; Sharma et al., 2014; van Dam et al., 2018; Srinivas et al., 2019; Sasseron et al., 2020), and genes associated with virulence were considered as targets for molecular discrimination of strains of the fungus (Lievens et al., 2008; van Dam et al., 2018). It was shown that secreted in xylem (SIX) genes are associated with pathogenicity of F. oxysporum, and the majority of them are distributed within the sequence of one chromosome (Rep et al., 2004; Houterman et al., 2007; Kashiwa et al., 2017; Carvalhais et al., 2019). Importantly, the combination of these genes differs between and within formae (Lievens et al., 2009). Secreted in xylem are also responsible for the resistance of cultivars to certain pathogen races due to gene-for-gene interaction between SIX genes of a pathogen and R genes (resistance genes) of a plant. Breaking the resistance of plant lineages to wild-type F. oxysporum could be accomplished by deletion of a certain SIX gene, which is recognized by the immune system of a plant (Houterman et al., 2008). However, in the case of F. oxysporum f. sp. lini, the connection between the degree of pathogenicity of a strain and the set of its SIX genes is not investigated deeply, and gaining insights into the mechanisms of pathogenicity offers an opportunity for effective disease control.
Besides, F. oxysporum representatives differ not only in the content of genes and their sequences but also in the number of chromosomes due to chromosomal rearrangements and the mobility of lineage-specific chromosomes (Davière et al., 2001; Ma et al., 2010; Schmidt et al., 2013; Vlaardingerbroek et al., 2016; Wang et al., 2020). Having learned the statistics of deposited F. oxysporum assemblies, one may conclude that genome sizes vary from about 50 to 70 Mb and differ between many formae (data of the NCBI Genome database, https://www.ncbi.nlm.nih.gov/genome/browse/#!/eukaryotes/707/).
Summing up, F. oxysporum f. sp. lini appears to be heterogeneous. However, there is a lack of next-generation sequencing data concerning the genome structure of the flax pathogen and the molecular basis of pathogenicity in relation to diverse virulence of the strains. In this work, we have chosen six strains of F. oxysporum f. sp. lini of low, medium, and high virulence (two strains per each degree of pathogenicity), performed genome sequencing on Oxford Nanopore Technologies (ONT) and Illumina platforms, and obtained de novo assemblies of the sequenced strains.
F. oxysporum f. sp. lini samples were provided by the Institute for Flax (Torzhok, Russia). The strains were of the following pathogenicity degrees: the low (strains #456, #482), the medium (#476, #525), and the high one (#483, #39). Mycelium was grown in test tubes on potato dextrose agar medium for 3 weeks (Alpha Biosciences, USA).
Following the previously developed protocol, pure high-molecular-weight DNA of the fungal samples was obtained (Krasnov et al., 2020). In brief, the DNA was extracted using the CTAB method followed by its isolation with Blood and Cell Culture DNA Mini Kit (Qiagen, USA). The quality and concentration of the DNA were evaluated on a NanoDrop 2000C spectrophotometer (Thermo Fisher Scientific, USA) and a Qubit 2.0 fluorometer (Life Technologies, USA). The assessment of DNA length and the control of RNA absence were performed by electrophoresis in a 0.8% agarose gel (Lonza, Switzerland).
Before library preparation, DNA fragments up to 10 kb were removed from the samples with a Short Read Eliminator Kit (Circulomics, USA), and the remaining DNA was purified with AMPure XP beads (Beckman Coulter, USA) in a ratio of 1:0.7 (sample:beads). SQK-LSK109 Ligation Sequencing Kit (ONT, UK) for 1D genomic DNA sequencing was used to prepare the library. During this procedure, minor modifications were introduced to the recommended protocol that included sample barcoding with the EXP-NBD103 (Native Barcoding Expansion) kit (ONT). Namely, at the steps of DNA recovery at 20°C and ligation the time of incubation was increased to 20 and 60 min respectively. Sequencing was performed on a MinION (ONT) instrument with a FLO-MIN-106D (R9.4.1) flow cell (ONT).
Upon DNA shearing on a S220 ultrasonic homogenizer (Covaris, USA), the library was prepared from 1 μg of fragmented DNA with the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs, UK) according to the manufacturer's protocol with size selection of adaptor-ligated DNA of about 600–800 bp. A 2100 Bioanalyzer instrument (Agilent Technologies, USA) and a Qubit 2.0 fluorometer (Life Technologies) were used to evaluate the quality and concentration of the DNA library respectively. The DNA library was sequenced on a MiSeq instrument (Illumina, USA) with a read length of 300+300 bp.
On genome sequencing of five strains (#456, #476, #482, #483, #525), we obtained 2.7 Gb of ONT data (374–756 Mb per strain, N50 = 32–44 kb) and 26.7 million Illumina paired-end reads (2.9–7.9 million 300+300 bp reads per strain). Since F. oxysporum f. sp. lini genome size is about 60–70 Mb (Kanapin et al., 2020; Krasnov et al., 2020), the obtained data corresponded to 10x genome coverage with ONT reads and 50x coverage with Illumina reads on average. The Nanopore and Illumina sequencing data for these five strains and isolate #39, which was sequenced by us earlier, were deposited in NCBI under the BioProject accession number PRJNA721899. For further use in performing de novo genome assemblies of the five sequenced strains, ONT fast5 files were basecalled with Guppy 3.6.1 (https://community.nanoporetech.com/protocols/Guppy-protocol/v/GPB_2003_v1_revU_14Dec2018) using the dna_r9.4.1_450bps_hac.cfg config file. Adapter removal and demultiplexing were carried out with Porechop 0.2.4 (https://github.com/rrwick/Porechop). Reads with an average quality below 6 (Q < 6) were discarded with Trimmomatic 0.39 (Bolger et al., 2014). To perform the initial genome assemblies, we chose the Canu tool (version 2.1) developed for long-read datasets (Koren et al., 2017), as it provided us with the best results when assembling the genome of isolate #39 in our previous study (Krasnov et al., 2020). The approximate size of the genomes was set as 60 Mb, and the other Canu parameters were kept default. The resulting quality of the assemblies was judged by QUAST features (version 5.0.2) (Mikheenko et al., 2018) and BUSCO completeness (version 4.1.2, hypocreales_odb10 dataset) (Seppey et al., 2019). BUSCO and QUAST statistics for the Canu genome assemblies of the five strains were the following: completeness laid in a range of 61.9–94.5%, N50 was 0.2–2.0 Mb, L50 varied between 9 and 82 (Table 1, Canu). These assemblies were not good enough for further analysis that can be explained by the low coverage of genomes with ONT reads (about 6–13x). So, we decided to perform hybrid assemblies of the genomes of the five strains with MaSuRCA (version 3.4.2, CA algorithm) (Zimin et al., 2013) using both ONT and Illumina data, as the obtained coverage with Illumina reads was high (30–80x) and MaSuRCA showed decent results for isolate #39 earlier (Krasnov et al., 2020). The N50 parameter of the hybrid assemblies of strains #456, #476, #482, #483, and #525 laid in a range of 1.0–2.5 Mb, L50 was 7–21, and the BUSCO completeness varied from 96.5 to 99.7% (Table 1, MaSuRCA). These statistics indicate fairly high contiguity and completeness of the obtained MaSuRCA assemblies. They were deposited in NCBI under the BioProject accession number PRJNA721899. Besides, we tested if ONT reads were crucial for assembly quality and assembled the genome of strain #525 from only Illumina reads using MaSuRCA, as the smallest amount of ONT data was obtained for this strain (0.37 Mb). It appeared that even 5–10x coverage of a genome with long ONT reads significantly improved assembly contiguity—N50 = 961 kb and L50 = 21 for the hybrid assembly against N50 = 92 kb and L50 = 145 for the assembly from Illumina reads only. Each of the received assemblies of the five strains contained a circular contig representing a complete mitochondrial genome which can be used in phylogenetic studies of Fusarium species. The obtained assemblies are a useful resource for comparative genomic studies of the flax pathogen strains.
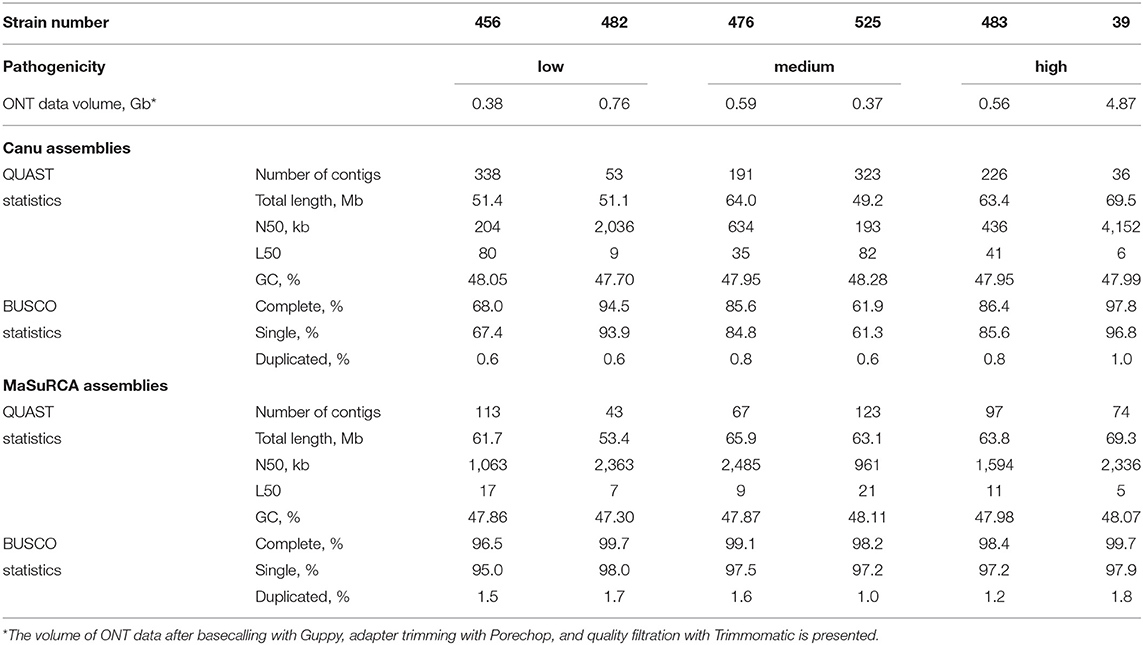
Table 1. QUAST and BUSCO statistics for Canu and MaSuRCA genome assemblies of six F. oxysporum f. sp. lini strains.
The comparison of the F. oxysporum f. sp. lini strains studied in the present work was also performed based on Illumina reads mapped to reference genomes. We trimmed Illumina reads of the six strains with Trimmomatic 0.39 (trailing:28) and discarded the short ones (minlen:50); the remaining data were mapped to reference assemblies of F. oxysporum using Bowtie 2 (version 2.3.5.1) (Langmead et al., 2019), which also reported the overall alignment rate, and the alignments were sorted with samtools (version 1.10) (Li et al., 2009). Samtools was also used to determine the coverage of contigs by Illumina reads in each assembly, and the overall coverage was calculated as the sum of covered bases in each contig divided by the number of all bases in an assembly. The only two complete F. oxysporum genomes deposited in GenBank were downloaded to be used for mapping—the assembly of strain Fo5176 (GCA_013112355.1, https://www.ncbi.nlm.nih.gov/genome/707?genome_assembly_id=1472496) by Fokkens et al. (2020) and the assembly of strain Fo47 (GCA_013085055.1, https://www.ncbi.nlm.nih.gov/genome/707?genome_assembly_id=910449) by Wang et al. (2020). Besides, the genome of F. oxysporum f. sp. lini isolate #39 was reassembled with Canu, using the earlier obtained data (Krasnov et al., 2020) and according to the same strategy as described above (ONT read processing with Guppy, Porechop, Trimmomatic, assembling with Canu), and with MaSuRCA (basecalling with Guppy, assembling from ONT and Illumina reads with MaSuRCA) (Table 1). Polishing of the Canu assembly was conducted using ONT reads according to the previously optimized scheme—two iterations with the Racon polisher and one with Medaka (Dmitriev et al., 2020). The accuracy of the Canu assembly was also improved by polishing with POLCA (from MaSuRCA) using Illumina reads (Zimin and Salzberg, 2020). The MaSuRCA assembly does not need additional polishing (Zimin et al., 2017). The resulting Canu assembly consisted of 41 contigs with N50 of 4.15 Mb, had a length of 69.5 Mb, and completeness of 99.8%. As expected, the polished Canu assembly outperformed the MaSuRCA one in terms of QUAST and BUSCO statistics, so the Canu-assembled and polished genome of F. oxysporum f. sp. lini isolate #39 was used for further mapping.
Illumina reads of the six differently virulent strains of F. oxysporum f. sp. lini were separately mapped to the two complete genomes of strains Fo5176 and Fo47 and also the assembly of isolate #39 obtained anew (Table 2). The overall alignment of the mapped reads corresponding to the six strains against the genomes of isolate #39 and strains Fo5176 and Fo47 as well as the percentages of the genome covered were higher for isolate #39 than for Fo5176 and Fo47. Fo5176 infects Arabidopsis thaliana (Fokkens et al., 2020) and has a comparable to isolate #39 genome size (68.0 and 69.5 Mb respectively), while Fo47 is classified as an endophyte and has a significantly smaller genome size−50.4 Mb (Wang et al., 2020). The percentages of aligned reads and covered genome fractions varied between F. oxysporum f. sp. lini strains; however, we did not reveal the similarity between genomes of the strains with the same pathogenicity degree. For example, high-virulent isolate #39 had less resemblance to another high-virulent strain #483 than to strains #476 and #525 with medium virulence. At the same time, low-virulent strain #482 had the most significant difference from isolate #39. Thus, mapping our Illumina sequencing data of differently virulent strains enabled us to evaluate broad-scale differences in genomes. Besides, using the present Illumina data, one can also reveal single nucleotide polymorphisms (SNPs) or small insertions/deletions in the loci of interest and perform a comparison of the F. oxysporum f. sp. lini strains with F. oxysporum strains of other formae speciales. At the same time, the genomes of the six studied strains, even of those ones with similar virulence, differed in the amount of mapped Illumina data, and, therefore, mapping Illumina reads to a reference genome can result in the loss of significant part of information. Besides, chromosomal rearrangements may be implicated in gene regulation, and, for evaluation of the role of such variations in fungal pathogenicity, de novo genome assemblies obtained from long reads are indispensable (Demené et al., 2021). Thus, both Illumina and ONT data obtained by us for F. oxysporum f. sp. lini strains of different virulence are essential for a comprehensive comparative analysis of the genomes and identification of genetic differences associated with pathogenicity.
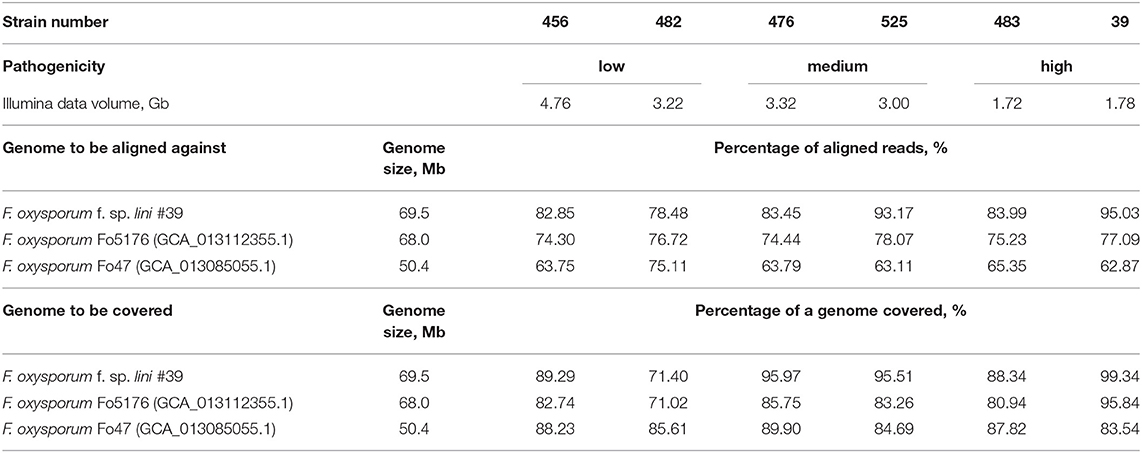
Table 2. Percentages of aligned Illumina reads against the assembly of F. oxysporum f. sp. lini isolate #39 and the complete genomes of F. oxysporum strains Fo47 and Fo5176 and percentages of the genomes covered by Illumina reads for six F. oxysporum f. sp. lini strains.
Another example of the use of the obtained dataset is the identification of genes of interest and their further analysis. For instance, we performed the search for genes involved in a process of plant colonization. The blastn analysis showed that the obtained assemblies of all the strains but low-virulent #482 contained regions with high homology (e-value < 10−10, identity of 99% on average) to partial coding sequences of virulence genes found in forma specialis lini: SIX1 (NCBI: KM893920.1), SIX7 (KM893928.1), SIX10 (KP964982.1), SIX12 (KP964992.1), SIX13 (KP964998.1) (Laurence et al., 2015; Taylor et al., 2016). The identified SIX sequences were found arranged in clusters. For example, in the genome of isolate #39, partial coding sequences of SIX1, SIX7, SIX10, SIX13 mapped completely against the locus of 400 kb of tig00000022, and SIX7 mapped twice against this locus. Besides, homologs of SIX7, SIX12 sequences were present in tig00000001, and those of SIX7, SIX10, SIX12, SIX13 were found in a 100 kb locus of tig00000029. Similar clustering of SIX sequences was observed in the assemblies of strains #456, #476, #483, and #525. Thus, the obtained data are valuable for identifying genes of a particular family, including those responsible for pathogenicity and playing an important role in the interaction between F. oxysporum and plants.
One more illustration of a way to use our dataset is the study of the role of DNA modifications in the regulation of F. oxysporum genome. DNA methylation is implicated in gene expression regulation, repression of transposable elements, and chromatin remodeling. To date, it is known that cytosines in fungi can be methylated within the context of CpG sites, CN (N for any nucleotide) pairs, and long clusters of cytosines; the type of the most methylated motif varies between fungal species, while the overall level of methylation in fungal genomes is relatively low (He et al., 2020). Several approaches of the ONT data use for the evaluation of methylation level throughout the genome of an object were developed (Xu and Seki, 2020; Tourancheau et al., 2021). We assessed DNA methylation levels in the genome of isolate #39, for which high coverage with ONT reads was obtained (about 70x), using the nanopolish tool (https://nanopolish.readthedocs.io/en/latest/quickstart_call_methylation.html), which is trained for methylation evaluation within the CpG context. The basecalled ONT reads were mapped against the assembly of isolate #39 with minimap2, alignments were derived using samtools, and methylation levels of CpG sites were estimated with nanopolish. Methylation across the whole genome was low (Supplementary Data 1) that is in concordance with other studies on DNA methylation in fungi (Bewick et al., 2019). Nevertheless, in the assembly of F. oxysporum f. sp. lini isolate #39, more than 500 CpG sites (with coverage >20 ONT reads) had methylation levels ≥0.5. Then, due to poor annotation of F. oxysporum genomes, we performed a blast-search for the regions containing these CpG sites in fungi taxa. Most of the methylated CpG sites were not assigned to specific functional elements of the genome; however, there were also CpG sites that were located in promoter regions, transposons, and gene bodies. Therefore, our dataset is useful for the evaluation of the role of DNA methylation in genome regulation of F. oxysporum f. sp. lini.
Finally, the obtained genome assemblies of F. oxysporum f. sp. lini strains can be used in gene expression studies, for example, as a reference for transcriptome assembly with further expression analysis or in selection of conservative regions suitable for primer design for the assessment of gene expression by quantitative PCR in different F. oxysporum strains or even different Fusarium species.
This work mainly focused on genome sequencing of strains of the flax pathogen F. oxysporum f. sp. lini, possessing diverse pathogenicity degrees, on two platforms—ONT and Illumina. The collected data allowed us to assemble the genomes of five strains and reassemble the genome of isolate #39 (the used data were obtained in our previous work), which lay the basis for further investigation of F. oxysporum virulence mechanisms and contribute to understanding the general structure of the pathogen population. Due to F. oxysporum f. sp. lini includes a vast number of genotypes, it is of high significance to study the origins of pathogenicity at molecular level. Our dataset can be of great use for researchers working on breeding resistant flax varieties and developing methods to prevent the disease and economic losses.
The obtained data can be found in NCBI under the BioProject accession number PRJNA721899.
TR, NM, and AD conceived and designed the work. ED, EP, RN, LP, NB, and LK performed the experiments. ED, RN, TR, NM, and AD analyzed the data. ED, NM, and AD wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work (sequencing and analysis of genomes of F. oxysporum f. sp. lini strains) was funded by RFBR according to the research project 19-34-90055. The maintenance of flax pathogen collection is carried out under the financial support of the Ministry of Science and Higher Education of the Russian Federation, state assignment number 075-00853-19-00.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the Center for Precision Genome Editing and Genetic Technologies for Biomedicine, EIMB RAS for providing genome sequencing techniques and computing power. This work was performed using the equipment of EIMB RAS Genome center (http://www.eimb.ru/ru1/ckp/ccu_genome_ce.php).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.662928/full#supplementary-material
Supplementary Data 1. Methylation levels of CpG sites in the genome assembly of F. oxysporum f. sp. lini isolate #39. Data derived from Nanopore sequencing data using nanopolish. Sites with at least 20x coverage are presented.
Armstrong, G. M., and Armstrong, J. K. (1981). “Formae speciales and races of Fusarium oxysporum causing wilt diseases,” in Fusarium: Disease, Biology, and Taxonomy, eds P. E. Nelson, T. A. Toussoun, and R. J. Cook (Pennsylvania, PA: Pennsylvania State University Press), 391–399.
Baayen, R. P., O'Donnell, K., Bonants, P. J., Cigelnik, E., Kroon, L. P., Roebroeck, E. J., et al. (2000). Gene genealogies and AFLP analyses in the Fusarium oxysporum complex identify monophyletic and nonmonophyletic formae speciales causing wilt and rot disease. Phytopathology 90, 891–900. doi: 10.1094/PHYTO.2000.90.8.891
Baysal, O., Siragusa, M., Gumrukcu, E., Zengin, S., Carimi, F., Sajeva, M., et al. (2010). Molecular characterization of Fusarium oxysporum f. melongenae by ISSR and RAPD markers on eggplant. Biochem. Genet. 48, 524–537. doi: 10.1007/s10528-010-9336-1
Bewick, A. J., Hofmeister, B. T., Powers, R. A., Mondo, S. J., Grigoriev, I. V., James, T. Y., et al. (2019). Diversity of cytosine methylation across the fungal tree of life. Nat. Ecol. Evol. 3, 479–490. doi: 10.1038/s41559-019-0810-9
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Carvalhais, L. C., Henderson, J., Rincon-Florez, V. A., O'Dwyer, C., Czislowski, E., Aitken, E. A. B., et al. (2019). Molecular diagnostics of banana Fusarium wilt targeting secreted-in-xylem genes. Front. Plant Sci. 10:547. doi: 10.3389/fpls.2019.00547
Czemplik, M., Boba, A., Kostyn, K., Kulma, A., Mituła, A., Sztajnert, M., et al. (2011). “Flax engineering for biomedical application,” in Biomedical Engineering, Trends, Research and Technologies, ed S. Olsztynska (London: IntechOpen), 407–434. doi: 10.5772/13570
Davière, J.-M., Langin, T., and Daboussi, M.-J. (2001). Potential role of transposable elements in the rapid reorganization of the Fusarium oxysporum genome. Fungal Genet. Biol. 34, 177–192. doi: 10.1006/fgbi.2001.1296
Demené, A., Laurent, B., Cros-Arteil, S., Boury, C., and Dutech, C. (2021). Chromosomal rearrangements but no change of genes and transposable elements repertoires in an invasive forest-pathogenic fungus. bioRxiv 2021.03.09.434572. doi: 10.1101/2021.03.09.434572
Dmitriev, A. A., Pushkova, E. N., Novakovskiy, R. O., Beniaminov, A. D., Rozhmina, T. A., Zhuchenko, A. A., et al. (2020). Genome sequencing of fiber flax cultivar atlant using oxford nanopore and illumina platforms. Front. Genet. 11:590282. doi: 10.3389/fgene.2020.590282
Edel, V., Christian, S., Gautheron, N., Recorbet, G., and Alabouvette, C. (2001). Genetic diversity of Fusarium oxysporum populations isolated from different soils in France. FEMS Microbiol. Ecol. 36, 61–71. doi: 10.1111/j.1574-6941.2001.tb00826.x
Edel-Hermann, V., and Lecomte, C. (2019). Current status of Fusarium oxysporum formae speciales and races. Phytopathology 109, 512–530. doi: 10.1094/PHYTO-08-18-0320-RVW
Fokkens, L., Guo, L., Dora, S., Wang, B., Ye, K., Sanchez-Rodriguez, C., et al. (2020). A chromosome-scale genome assembly for the Fusarium oxysporum strain Fo5176 to establish a model Arabidopsis-fungal pathosystem. G3 (Bethesda) 10, 3549–3555. doi: 10.1534/g3.120.401375
He, C., Zhang, Z., Li, B., and Tian, S. (2020). The pattern and function of DNA methylation in fungal plant pathogens. Microorganisms 8:227. doi: 10.3390/microorganisms8020227
Houterman, P. M., Cornelissen, B. J., and Rep, M. (2008). Suppression of plant resistance gene-based immunity by a fungal effector. PLoS Pathog. 4:e1000061. doi: 10.1371/journal.ppat.1000061
Houterman, P. M., Speijer, D., Dekker, H. L., CG, D.E. K., Cornelissen, B. J., and Rep, M. (2007). The mixed xylem sap proteome of Fusarium oxysporum-infected tomato plants. Mol. Plant Pathol. 8, 215–221. doi: 10.1111/j.1364-3703.2007.00384.x
Jhala, A. J., and Hall, L. M. (2010). Flax (Linum usitatissimum L.): current uses and future applications. Aust. J. Basic Appl. Sci. 4, 4304–4312.
Kanapin, A., Samsonova, A., Rozhmina, T., Bankin, M., Logachev, A., and Samsonova, M. (2020). The genome sequence of five highly pathogenic isolates of Fusarium oxysporum f. sp. lini. Mol. Plant Microbe Interact. 33, 1112–1115. doi: 10.1094/MPMI-05-20-0130-SC
Kashiwa, T., Kozaki, T., Ishii, K., Turgeon, B. G., Teraoka, T., Komatsu, K., et al. (2017). Sequencing of individual chromosomes of plant pathogenic Fusarium oxysporum. Fungal Genet. Biol. 98, 46–51. doi: 10.1016/j.fgb.2016.12.001
Kezimana, P., Dmitriev, A. A., Kudryavtseva, A. V., Romanova, E. V., and Melnikova, N. V. (2018). Secoisolariciresinol diglucoside of flaxseed and its metabolites: biosynthesis and potential for nutraceuticals. Front. Genet. 9:641. doi: 10.3389/fgene.2018.00641
Kommedahl, T., Christensen, J. J., and Frederiksen, R. A. (1970). A Half Century of Research in Minnesota on Flax Wilt Caused by Fusarium oxysporum. Minneapolis, MN: University of Minnesota.
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Krasnov, G. S., Pushkova, E. N., Novakovskiy, R. O., Kudryavtseva, L. P., Rozhmina, T. A., Dvorianinova, E. M., et al. (2020). High-quality genome assembly of Fusarium oxysporum f. sp. lini. Front. Genet. 11:959. doi: 10.3389/fgene.2020.00959
Langmead, B., Wilks, C., Antonescu, V., and Charles, R. (2019). Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 35, 421–432. doi: 10.1093/bioinformatics/bty648
Laurence, M. H., Summerell, B. A., and Liew, E. C. Y. (2015). Fusarium oxysporum f. sp. canariensis: evidence for horizontal gene transfer of putative pathogenicity genes. Plant Pathol. 64, 1068–1075. doi: 10.1111/ppa.12350
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Lievens, B., Houterman, P. M., and Rep, M. (2009). Effector gene screening allows unambiguous identification of Fusarium oxysporum f. sp. lycopersici races and discrimination from other formae speciales. FEMS Microbiol. Lett. 300, 201–215. doi: 10.1111/j.1574-6968.2009.01783.x
Lievens, B., Rep, M., and Thomma, B. P. (2008). Recent developments in the molecular discrimination of formae speciales of Fusarium oxysporum. Pest Manag. Sci. 64, 781–788. doi: 10.1002/ps.1564
Ma, L.-J., Van Der Does, H. C., Borkovich, K. A., Coleman, J. J., Daboussi, M.-J., Di Pietro, A., et al. (2010). Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464, 367–373. doi: 10.1038/nature08850
Michielse, C. B., and Rep, M. (2009). Pathogen profile update: Fusarium oxysporum. Mol. Plant Pathol. 10, 311–324. doi: 10.1111/j.1364-3703.2009.00538.x
Mikheenko, A., Prjibelski, A., Saveliev, V., Antipov, D., and Gurevich, A. (2018). Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34, i142–i150. doi: 10.1093/bioinformatics/bty266
Parikh, M., and Pierce, G. N. (2019). Dietary flaxseed: what we know and don't know about its effects on cardiovascular disease. Can. J. Physiol. Pharmacol. 97, 75–81. doi: 10.1139/cjpp-2018-0547
Rep, M., van der Does, H. C., Meijer, M., van Wijk, R., Houterman, P. M., Dekker, H. L., et al. (2004). A small, cysteine-rich protein secreted by Fusarium oxysporum during colonization of xylem vessels is required for I-3-mediated resistance in tomato. Mol. Microbiol. 53, 1373–1383. doi: 10.1111/j.1365-2958.2004.04177.x
Sasseron, G. R., Benchimol-Reis, L. L., Perseguini, J., Paulino, J. F. C., Bajay, M. M., Carbonell, S. A. M., et al. (2020). Fusarium oxysporum f. sp. phaseoli genetic variability assessed by new developed microsatellites. Genet. Mol. Biol. 43:e20190267. doi: 10.1590/1678-4685-gmb-2019-0267
Schmidt, S. M., Houterman, P. M., Schreiver, I., Ma, L., Amyotte, S., Chellappan, B., et al. (2013). MITEs in the promoters of effector genes allow prediction of novel virulence genes in Fusarium oxysporum. BMC Genomics 14:119. doi: 10.1186/1471-2164-14-119
Seppey, M., Manni, M., and Zdobnov, E. M. (2019). “BUSCO: assessing genome assembly and annotation completeness,” in Gene Prediction: Methods and Protocols, ed M. Kollmar (New York, NY: Springer New York), 227–245. doi: 10.1007/978-1-4939-9173-0_14
Sharma, M., Nagavardhini, A., Thudi, M., Ghosh, R., Pande, S., and Varshney, R. K. (2014). Development of DArT markers and assessment of diversity in Fusarium oxysporum f. sp. ciceris, wilt pathogen of chickpea (Cicer arietinum L.). BMC Genomics 15:454. doi: 10.1186/1471-2164-15-454
Srinivas, C., Nirmala Devi, D., Narasimha Murthy, K., Mohan, C. D., Lakshmeesha, T. R., Singh, B., et al. (2019). Fusarium oxysporum f. sp. lycopersici causal agent of vascular wilt disease of tomato: Biology to diversity - a review. Saudi J. Biol. Sci. 26, 1315–1324. doi: 10.1016/j.sjbs.2019.06.002
Taylor, A., Vágány, V., Jackson, A. C., Harrison, R. J., Rainoni, A., and Clarkson, J. P. (2016). Identification of pathogenicity-related genes in Fusarium oxysporum f. sp. cepae. Mol. Plant Pathol. 17, 1032–1047. doi: 10.1111/mpp.12346
Tourancheau, A., Mead, E. A., Zhang, X. S., and Fang, G. (2021). Discovering multiple types of DNA methylation from bacteria and microbiome using nanopore sequencing. Nat Methods 18, 491–498. doi: 10.1038/s41592-021-01109-3
van Dam, P., de Sain, M., Ter Horst, A., van der Gragt, M., and Rep, M. (2018). Use of comparative genomics-based markers for discrimination of host specificity in Fusarium oxysporum. Appl. Environ. Microbiol. 84:e01868-17. doi: 10.1128/AEM.01868-17
Vlaardingerbroek, I., Beerens, B., Rose, L., Fokkens, L., Cornelissen, B. J., and Rep, M. (2016). Exchange of core chromosomes and horizontal transfer of lineage-specific chromosomes in Fusarium oxysporum. Environ. Microbiol. 18, 3702–3713. doi: 10.1111/1462-2920.13281
Wang, B., Yu, H., Jia, Y., Dong, Q., Steinberg, C., Alabouvette, C., et al. (2020). Chromosome-scale genome assembly of Fusarium oxysporum strain Fo47, a fungal endophyte and biocontrol agent. Mol. Plant Microbe Interact. 33, 1108–1111. doi: 10.1094/MPMI-05-20-0116-A
Xu, L., and Seki, M. (2020). Recent advances in the detection of base modifications using the nanopore sequencer. J. Hum. Genet. 65, 25–33. doi: 10.1038/s10038-019-0679-0
Zimin, A. V., Marçais, G., Puiu, D., Roberts, M., Salzberg, S. L., and Yorke, J. A. (2013). The MaSuRCA genome assembler. Bioinformatics 29, 2669–2677. doi: 10.1093/bioinformatics/btt476
Zimin, A. V., Puiu, D., Luo, M. C., Zhu, T., Koren, S., Marcais, G., et al. (2017). Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res. 27, 787–792. doi: 10.1101/gr.213405.116
Keywords: Fusarium oxysporum, flax pathogen, different virulence, genome sequencing, nanopore sequencing, de novo genome assembly
Citation: Dvorianinova EM, Pushkova EN, Novakovskiy RO, Povkhova LV, Bolsheva NL, Kudryavtseva LP, Rozhmina TA, Melnikova NV and Dmitriev AA (2021) Nanopore and Illumina Genome Sequencing of Fusarium oxysporum f. sp. lini Strains of Different Virulence. Front. Genet. 12:662928. doi: 10.3389/fgene.2021.662928
Received: 01 February 2021; Accepted: 06 May 2021;
Published: 17 June 2021.
Edited by:
Yuriy L. Orlov, I. M. Sechenov First Moscow State Medical University, RussiaReviewed by:
Izabela Makałowska, Adam Mickiewicz University, PolandCopyright © 2021 Dvorianinova, Pushkova, Novakovskiy, Povkhova, Bolsheva, Kudryavtseva, Rozhmina, Melnikova and Dmitriev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexey A. Dmitriev, YWxleF8yNDVAbWFpbC5ydQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.