 Yaqi Wang
Yaqi Wang Wei Cao
Wei Cao Justin Merritt
Justin Merritt Zhoujie Xie
Zhoujie Xie Hao Liu
Hao Liu- 1MOE Key Laboratory of Industrial Fermentation Microbiology, Tianjin Key Laboratory of Industrial Microbiology, College of Biotechnology, Tianjin University of Science & Technology, Tianjin, China
- 2Department of Restorative Dentistry, Oregon Health & Science University, Portland, OR, United States
- 3Department of Molecular Microbiology and Immunology, Oregon Health & Science University, Portland, OR, United States
FtsH belongs to the AAA+ ATP-dependent family of proteases, which participate in diverse cellular processes and are ubiquitous among bacteria, chloroplasts, and mitochondria. FtsH is poorly characterized in most organisms, especially compared to other major housekeeping proteases. In the current study, we examined the source of FtsH essentiality in the human oral microbiome species Streptococcus mutans, one of the primary etiological agents of dental caries. By creating a conditionally lethal ftsH mutant, we were able to identify a secondary suppressor missense mutation in the vicR gene, encoding the response regulator of the essential VicRK two-component system (TCS). Transcriptomic analysis of the vicR (G195R) mutant revealed significantly reduced expression of 46 genes, many of which were located within the genomic island Tnsmu2, which harbors the mutanobactin biosynthetic gene cluster. In agreement with the transcriptomic data, deletion of the mutanobactin biosynthetic gene cluster suppressed ftsH essentiality in S. mutans. We also explored the role of FtsH in S. mutans physiology and demonstrated its critical role in stress tolerance, especially acid stress. The presented results reveal the first insights within S. mutans for the pleiotropic regulatory function of this poorly understood global regulator.
Introduction
ATP-dependent proteases play essential roles in cellular physiology and are universally conserved in both prokaryotes and eukaryotes. These proteins are commonly associated with the degradation of damaged or misfolded proteins, but they also play central regulatory roles in genetic networks by directly controlling the concentration of numerous proteins (Goldberg, 2003; Sauer and Baker, 2011). The 26S proteasome is primarily responsible for these activities in eukaryotic cells, whereas HslUV, ClpAP, ClpXP, Lon, FtsH, and their homologs comprise the principal AAA+ ATP-dependent proteases in bacteria. Clp and FtsH are particularly common among eubacteria. The two-component Clp proteases are composed of multiple subunits of an invariable proteolytic subunit (ClpP) and one AAA+ ATPase subunit (such as ClpA, ClpX, ClpC, or ClpE). The Clp proteases play central roles in stress tolerance and global genetic regulation (Bhandari et al., 2018). As an integral membrane protease, FtsH is unique among the AAA+ proteases. It is anchored to the membrane by two N-terminal transmembrane segments, and it is also widely encoded by prokaryotes as well as eukaryotes, where it functions in both mitochondria and chloroplasts (Ito and Akiyama, 2005). The broad phylogenetic conservation of ftsH attests to its fundamental role in cellular physiology. Extensive structural studies have been reported for the FtsH cytoplasmic domains in Escherichia coli (Krzywda et al., 2002), Thermus thermophilus (Niwa et al., 2002), Thermotoga maritima (Bieniossek et al., 2006), and Aquifex aeolicus (Suno et al., 2006). The structure of the full-length FtsH from A. aeolicus was also recently determined by cryo-electron microscopy (Carvalho et al., 2020). FtsH is comprised of an N-terminal transmembrane segment and a C-terminal cytoplasmic region, consisting of ATPase and protease domains. FtsH monomers assemble into a sixfold symmetric ring structure, with 12 transmembrane helices inserting into the lipid bilayer. The ATPase component of the protease unfolds target proteins and then translocates them through a central pore into the chamber of the protease complex where catalytic sites protrude from its inner surface. Following ATP hydrolysis, a large conformational change occurs within the ATPase complex, which couples substrate unfolding and translocation (Bieniossek et al., 2009).
FtsH is best characterized in E. coli, where it regulates a diverse assortment of cellular processes, such as phage infection, stress responses, and lipid biosynthesis by targeting both soluble and integral membrane proteins (Bittner et al., 2017). The ftsH gene is essential in many species, such as E. coli, Bradyrhizobium japonicum, Helicobacter pylori, and Borrelia burgdorferi, whereas it is dispensable in Bacillus subtilis, Lactococcus lactis, Caulobacter crescentus, and Staphylococcus aureus (Ito and Akiyama, 2005; Langklotz et al., 2012; Chu et al., 2016). This variability in ftsH essentiality likely reflects the diverse assortment of prokaryotic genetic pathways regulated by FtsH. It was recently reported that the magnesium transporter MgtE was directly degraded by FtsH under the help of the substrate adaptor YqgP in B. subtilis (Began et al., 2020). A membrane-bound transcription factor was also recently identified as an FtsH substrate in S. aureus (Yeo et al., 2020). However, the total number of confirmed FtsH substrates remains far below that of the Clp protease and little is still known regarding FtsH substrate recognition.
Streptococcus mutans is a Gram-positive member of the human oral microbiome. The growth environment of the oral biofilm is quite challenging for many organisms due to its rapid and substantial fluctuations in pH, temperature, redox potential, osmolarity, and nutrient availability (Lemos et al., 2005). To cope with this challenging environment, S. mutans is particularly dependent upon the function of two-component signal transduction systems and AAA+ proteases to properly control its stress tolerance mechanisms (Lemos et al., 2005). Streptococcus mutans encodes at least four distinct AAA+ proteases: the ClpP-dependent proteases (ClpXP, ClpCP, and ClpEP) and FtsH (Kajfasz et al., 2009; Liu et al., 2017; Bhandari et al., 2018). Previous studies in S. mutans demonstrated that ClpP plays a critical role in global protein quality control, and not surprisingly, ClpP mutant strains exhibit pleiotropic effects, such as stress sensitivity, slow growth, long chain formation, aggregation in liquid culture, reduced autolysis, altered biofilm formation, and virulence defects (Kajfasz et al., 2009, 2011; Chattoraj et al., 2010). Several Clp-dependent degrons have been identified in S. mutans as well (Niu et al., 2010; Tao and Biswas, 2015; Liu et al., 2017). In stark contrast, nothing is yet known about the phenotypic and/or regulatory roles of FtsH in S. mutans. Only a single streptococcal FtsH degron has been reported and this was recently identified in S. mutans (Liu et al., 2017), but little else is known about this conserved housekeeping protease.
We report here that, unlike the Clp-dependent proteases, transcriptional depletion of ftsH results in a lethal phenotype, indicating its essentiality in S. mutans. Using a forward genetic screen, we identified a suppressor of lethality point mutation in the vicR (G195R) gene, encoding the response regulator of the VicRK two-component system (TCS). Further transcriptomic and mutagenesis studies revealed that ftsH essentiality is connected to its regulation of the mutanobactin biosynthetic gene cluster. Similar to the Clp proteases, FtsH is also critical for the control of S. mutans stress tolerance. The presented results provide some of the first insights into the major FtsH phenotypes in S. mutans.
Materials and Methods
Bacterial Strains and Culture Conditions
The bacterial strains and plasmids used in this study are listed in Supplementary Table S1. Streptococcus mutans UA159 and its derivatives were grown in brain heart infusion (BHI) broth (Difco) or on BHI agar plates. All S. mutans strains were grown anaerobically (in a candle extinction jar) at 37°C. When performing transformation assays, cells were grown in Todd–Hewitt medium (Difco) with 0.3% (w/v) yeast extract (THYE). For the selection of antibiotic-resistant colonies, cultures were supplemented with 12.5 μg ml−1 erythromycin (Sigma), 1 mg ml−1 spectinomycin (Sigma), or 800 μg ml−1 kanamycin (Sigma). For counterselection, BHI plates were supplemented with 0.02 M p-chlorophenylalanine [4-CP] (Sigma). Escherichia coli JM109 cells were grown in Luria-Bertani (LB; Difco) medium with aeration at 37°C. Escherichia coli strains carrying plasmids were grown in LB medium containing spectinomycin (Sigma; 100 μg ml−1).
Streptococcus mutans Transformation
Streptococcus mutans was transformed using the natural transformation assay as previously described (Xie et al., 2013a). Briefly, S. mutans strains were grown overnight in THYE medium. Overnight cultures were diluted 1:20 in THYE and incubated at 37°C until the OD600 reached 0.2–0.3 (about 2–3 h). For each 500 μl transformation reaction, 1 μg of transforming DNA and 1 μg ml−1 final concentration of CSP-Sm (SGSLSTFFRLFNRSFTQA; GenScript, Nanjing, China) were added and incubated for an additional 2 h before plating on selective media. The reactions were incubated at 37°C until colonies were visible.
Construction of ftsH Deletion Strains
The primers used in this study are listed in Supplementary Table S2.
The ftsH deletion mutant was constructed using marked allelic replacement mutagenesis of the wild-type S. mutans strain UA159. Briefly, two fragments corresponding to approximately 1 kb of the upstream and downstream flanking sequences of ftsH (SMU_15) were generated by PCR with the primer pairs ftsHupF/ftsHupR-erm and ftsHdnF-erm/ftsHdnR. The primers ftsHupR-erm and ftsHdnF-erm incorporated 18 bases complementary to the erythromycin resistance cassette ermAM. The 1 kb ermAM cassette was amplified by PCR using the primers ermF and ermR. All three PCR amplicons were mixed in a 1:1:1 ratio. The mixture served as the template for a second PCR with the primer pair ftsHupF/ftsHdnR. The resulting amplicon was designated as ∆ftsH::ermAM, transformed into recipient stains, and selected on BHI agar supplemented with erythromycin. This resulted in no transformants when UA159 was used as the recipient strain. The conditional lethal ftsH deletion strain pXylS1ftsH/∆ftsH was obtained by transforming the ftsH deletion construct into strain pXylS1ftsH/UA159 and selecting on medium supplemented with erythromycin and 0.5% (w/v) xylose. Similarly, strain S877 was used as the recipient to obtain the ftsH deletion strain S878.
Construction of Plasmid pXylS1ftsH
pXylS1ftsH (streptococcal replicon and ftsH driven by the xylose-inducible expression cassette Xyl-S1) was constructed via POE-PCR (Xie et al., 2013a). Using pZX9 as a template (Xie et al., 2013b), a fragment containing the streptococcal replicon and Xyl-S1 cassette was amplified with the primer pair ISF-ftsH and ISR-ftsH. The ftsH open reading frame was amplified with the primer pair ftsHF-IS and ftsHR-IS using the genomic DNA of S. mutans UA159 as the template. Concatemers were generated by POE-PCR using the two amplicons. A 5 μl volume of the resulting PCR mixture was transformed directly into S. mutans UA159 to obtain plasmid pXylS1ftsH. Randomly selected transformants were chosen for plasmid extraction and confirmed by PCR.
Plasmid Extraction From S. mutans
Plasmid was extracted from S. mutans using a previously described methodology (Xie et al., 2013a). Briefly, 3 ml of stationary phase cultures were resuspended in 500 μl buffer (10 mM pH 7.5 Tris-HCl, 1 mM EDTA, and 10 mg/ml lysozyme). The cells were incubated for 4 h at 37°C. Afterward, the plasmids were extracted using the Qiagen Plasmid Miniprep kit beginning with the lysis step and following the manufacturer’s protocol.
Construction of S. mutans Strain S875
The inducible ftsH expression strain (S875) was created by placing the chromosomal copy of ftsH under the control of the xylose-inducible expression cassette (Xyl-S1) using a markerless mutagenesis approach (Xie et al., 2011). Briefly, we first inserted the counter-selectable IFDC2 cassette in the intergenic region upstream of ftsH. Using UA159 genomic DNA as a template, two fragments corresponding to the upstream and downstream regions of the insert site were amplified with the primer pairs ftsHupF/ftsHupR-ldh and ftsHF-erm/ftsHR, respectively. The IFDC2 cassette was amplified using the primer pair ldhF/ermR. The three fragments were mixed and used as the template for overlap extension PCR (OE-PCR) with the primer pair ftsHupF/ftsHR. The resulting OE-PCR amplicon was transformed into UA159 and selected on medium supplemented with erythromycin to insert the IFDC2 cassette. Next, using the genomic DNA of the resulting IFDC2 mutant strain as the template, a 1 kb DNA fragment corresponding to the upstream region of ftsH was amplified with the primer pair ftsHupF/ftsHupR-xylR. Using the plasmid pXylS1ftsH as the template, a 3.3 kb DNA fragment containing Xyl-S1 cassette and partial open reading frame region of ftsH was amplified using the primer pair xylRR-up/ftsHR. The two fragments were mixed and assembled with OE-PCR using the primer pair ftsHupF/ftsHR. The resulting PCR product was transformed into the IFDC2 mutant strain mentioned above and selected on medium containing 4-CP and 0.5% (w/v) xylose to obtain strain S875.
Isolation of Spontaneous ftsH Suppressor Mutant S876
The conditionally lethal inducible ftsH strain S875 was first cultured in BHI containing 0.5% (w/v) xylose. Stationary phase cultures were collected and washed with PBS buffer and subsequently incubated on BHI agar plates lacking xylose for approximately 48 h. Large colonies on BHI agar were restreaked onto BHI agar without xylose before preparing frozen stocks of candidate spontaneous ftsH suppressor mutants. A confirmed isolate in which the ftsH gene could be successfully deleted was designated as S. mutans strain S876.
Genome DNA Sequencing and SNP Analysis
Genomic DNA was extracted from late log cultures of the strain S876 grown in BHI. DNA Samples were then submitted to Majorbio Bio-pharm Technology Co., Ltd. (Shanghai, China) for genome sequencing on the Illumina Hiseq platform. The sequences obtained were trimmed by setting the quality score limit at 20 and discarding reads <25 bp. A total of 9,836,300 reads were generated with a 724.07-fold coverage. To analyze and identify single nucleotide polymorphisms (SNPs) and other sequence changes, the Illumina reads were aligned to the published reference strain UA159 (NC_004350.2) using the BWA program (Li and Durbin, 2010), and SNP calling was conducted using GATK software package with the standard filter method (McKenna et al., 2010). Two main parameters were used to filter SNPs: (1) sequencing depth > 10 and (2) SNP quality score > 50.
Co-Transformation Assay
Eight missense SNPs from seven genes were identified in strain S876 (Table 1). To identify the ftsH suppressor mutation, seven primer pairs (448F/448R, 565F/565R, 593F/593R, 1517F/1517R, 1565F/1565R, 1740F/1740R, and 1799F/1799R) were designed. Each primer pair targets 1 kb upstream and downstream of each mutation site. Using strain S876 as a template, seven 2 kb DNA fragments were PCR amplified and designated as SMU448(F59S), SMU565(S186G), SMU593(C106F), SMU1517(G195R), SMU1565F(G376F), SMU1740(V108A), and SMU1799(R101C), respectively. The seven DNA amplicons were co-transformed with the ∆ftsH::ermAM construct into strain UA159 and selected on medium supplemented with erythromycin. Only the SMU1517(G195R) + ∆ftsH::ermAM co-transformation generated a viable ftsH deletion mutant.
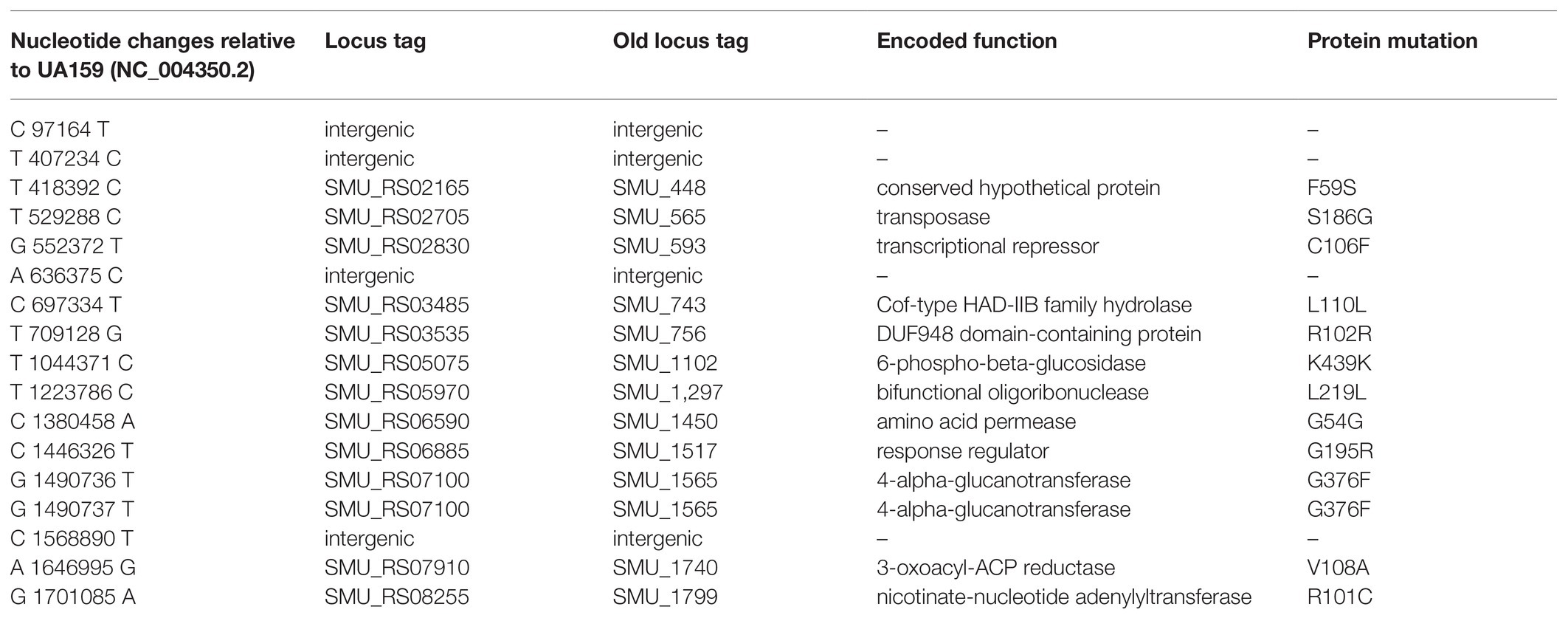
Table 1. Summary of single nucleotide polymorphisms (SNPs) detected in the ftsH suppressor strain (S876).
Construction of S. mutans S877
The strain S877 was constructed by a previously described markerless mutagenesis approach (McKenna et al., 2010). Briefly, a 1.2 kb fragment upstream of vicK was PCR amplified with the primer pair 1517F/1516upR-ldh, while a 0.5 kb region downstream of vicK was PCR amplified with the primer pair 1516F-erm/1517R. The 2.2 kb IFDC2 cassette was PCR amplified with the primer pair ldhF and ermR. The three amplicons were assembled via OE-PCR using the primer pair 1517F/1517R. The resulting 3.9 kb amplicon was transformed into UA159, and selected on BHI plates containing erythromycin to isolate transformants containing the IFDC2 cassette. Subsequently, a 2 kb region containing the vicR(G195R) point mutation was generated by PCR using the primer pair 1517F/1517R and the genomic DNA of strain S876. The resulting amplicon was transformed into the IFDC2-containing intermediate strain and selected on medium supplemented with 4-CP to excise the IFDC2 cassette. A resulting strain containing the expected point mutation was confirmed by sequencing the vicR locus and designated as S. mutans S877.
RNA Deep-Sequencing and Transcriptome Analysis
Total RNA was extracted using the RNeasy Mini Kit (Qiagen) from mid-log cultures of strain S877 and wild type UA159 grown in BHI. rRNA removal, library preparation, and quality control were performed by GENEWIZ (Suzhou, China). RNA-seq was performed using an Illumina Hiseq with a 2 × 150 paired-end (PE) configuration. Three biological replicates for each group were sequenced. The sequences were processed and analyzed by GENEWIZ. Differential expression analysis was performed using the DESeq2 Bioconductor package, a model based on negative binomial distribution. After adjusting with the Benjamini and Hochberg’s approach to control for false discovery rate, Padj of genes were set <0.05 to measure differential expression.
Quantitative Real-Time RT-PCR
To confirm the RNA-seq results, real-time quantitative reverse transcription PCR (qRT-PCR) was performed on seven genes selected from the RNA-seq dataset. RNA was extracted from cell pellets using the E.Z.N.A. Total RNA Kit (Omega Bio-Tek). Total RNA (300 ng) was used for complementary DNA (cDNA) synthesis using the PrimeScript RT Reagent Kit (Takara, RR047A) according to the manufacturer’s protocol. For real-time RT-PCR, the reactions were prepared using SYBR PremixEx TaqII kit (Takara, RR820A) and run in a StepOnePlus Real-Time PCR System (Applied Biosystems). Relative changes in gene expression were calculated using the “delta-delta threshold cycle” method described previously (Niu et al., 2008). Total cDNA abundance between samples was normalized using primers specific to the gyrA gene. All primers used for qRT-PCR are listed in Supplementary Table S2.
Construction of ∆1329–1367, ∆1402–1405, and Other Gene Deletion Strains
The mutanobactin biosynthetic gene cluster deletion strain (∆1329–1367) was constructed using marked allelic replacement mutagenesis. Briefly, a 1-kb fragment upstream of SMU_1367c and a 1-kb fragment downstream of SMU_1329c were generated by PCR using the primer pairs 1367upF/1367upR-kan and 1329dnF-kan/1329dnR. The kanamycin resistance cassette aphAIII was PCR amplified with the primer pair kanF/kanR. The three amplicons were assembled via OE-PCR using the primer pair 1367upF and 1329dnR. The resulting amplicon was transformed into S. mutans UA159, and transformants were selected on BHI plates containing kanamycin. A transformant confirmed to contain the expected mutant genotype was named as S. mutans strain ∆1329–1367. The cas gene deletion strain (∆1402–1405) was constructed using the same strategy as ∆1329–1367. A 1-kb fragment upstream of the cas9 gene (SMU_1405c) and a 1-kb fragment downstream of csn2 (SMU_1402c) were generated by PCR using the primer pairs 1405upF/1405upR-kan and 1402dnF-kan/1402dnR. Both amplicons have complementary regions with the aphAIII cassette, which was PCR amplified with primer pair kanF/kanR. The three amplicons were mixed and assembled via OE-PCR with the primer pair 1405upF/1402dnR. The resulting OE-PCR amplicon was transformed into S. mutans UA159, and selected on BHI plates containing kanamycin. A transformant having the expected genotype was named as S. mutans strain ∆1402–1405. This same strategy was also used for the deletion of SMU_277 (∆277), SMU_284 (∆284), SMU_503c (∆503), SMU_510c (∆510), SMU_609 (∆609), SMU_1322 (∆1322), SMU_1803c (∆1803), SMU_1882c (∆1882), and SMU_2035 (∆2035). The primers used for the construction of these mutant strains are included in Supplementary Table S2.
Growth Curve Analysis
The test strains were grown overnight to stationary phase in BHI broth at 37°C. The overnight cultures were diluted to an optical density OD600 ~0.01 in fresh BHI broth and incubated at 37°C. Growth kinetics was measured using an Infinite 200PRO multiplate reader.
Stress Tolerance Assays
Stress tolerance was assessed by measuring cell viability after stress exposure. To measure acid tolerance, cells were first grown 16–18 h in BHI and then diluted to an optical density of OD600 0.02 in 5 ml of fresh BHI. Cultures were then grown to mid-log phase before harvesting via centrifugation. Cell pellets were resuspended in 5 ml of 0.1 M glycine buffer at a pH of 3.5, and incubated at 37°C for 20 min. Replicate cultures incubated in 0.1 M pH 7.0 glycine buffer served as controls. Samples were serially diluted before being plated on BHI agar. Total colony forming units (CFUs) were then enumerated after 48 h growth at 37°C. Analyses of strain sensitivities to osmotic and oxidative stresses were performed similarly, except that osmotic stress was assessed using 2 M NaCl for 3 h and oxidative stress was assessed using 5 mM H2O2 for 1 h. Replicate cultures not exposed to stress were used as controls. Data are presented as the means ± SDs from three biological replicates performed in triplicate.
Results
FtsH is Essential in S. mutans UA159
To study the role of FtsH in S. mutans, we initially attempted to construct an ftsH deletion strain by transforming the ftsH deletion construct ∆ftsH::ermAM into S. mutans UA159 (Figure 1A). However, multiple attempts were unsuccessful, suggesting that this gene may be essential in S. mutans. To confirm this hypothesis, we created a conditional lethal ftsH mutation. We first constructed the plasmid pXylS1ftsH, in which the expression of ftsH was placed under the control of a xylose inducible expression cassette Xyl-S1 (Xie et al., 2013b; Figure 1B). This plasmid was transformed into strain UA159 before performing a second transformation with the ftsH deletion construct ∆ftsH::ermAM. Unlike the original transformation, this reaction generated >2,000 CFU/ml on antibiotic medium containing the inducer xylose. The deletion of the wild-type copy of ftsH was confirmed by PCR with primer pairs flanking the ftsH locus (Figure 1C). The confirmed chromosomal ftsH deletion strain was designated as pXylS1ftsH/∆ftsH. Furthermore, as shown in Figure 1D, the growth of strain pXylS1ftsH/∆ftsH is critically dependent on the presence of xylose in the medium, which confirmed the essentiality of FtsH in S. mutans. This is also consistent with recent Tn-seq and CRISPRi screening studies in S. mutans, which suggested ftsH to be essential (Shields et al., 2018, 2020).
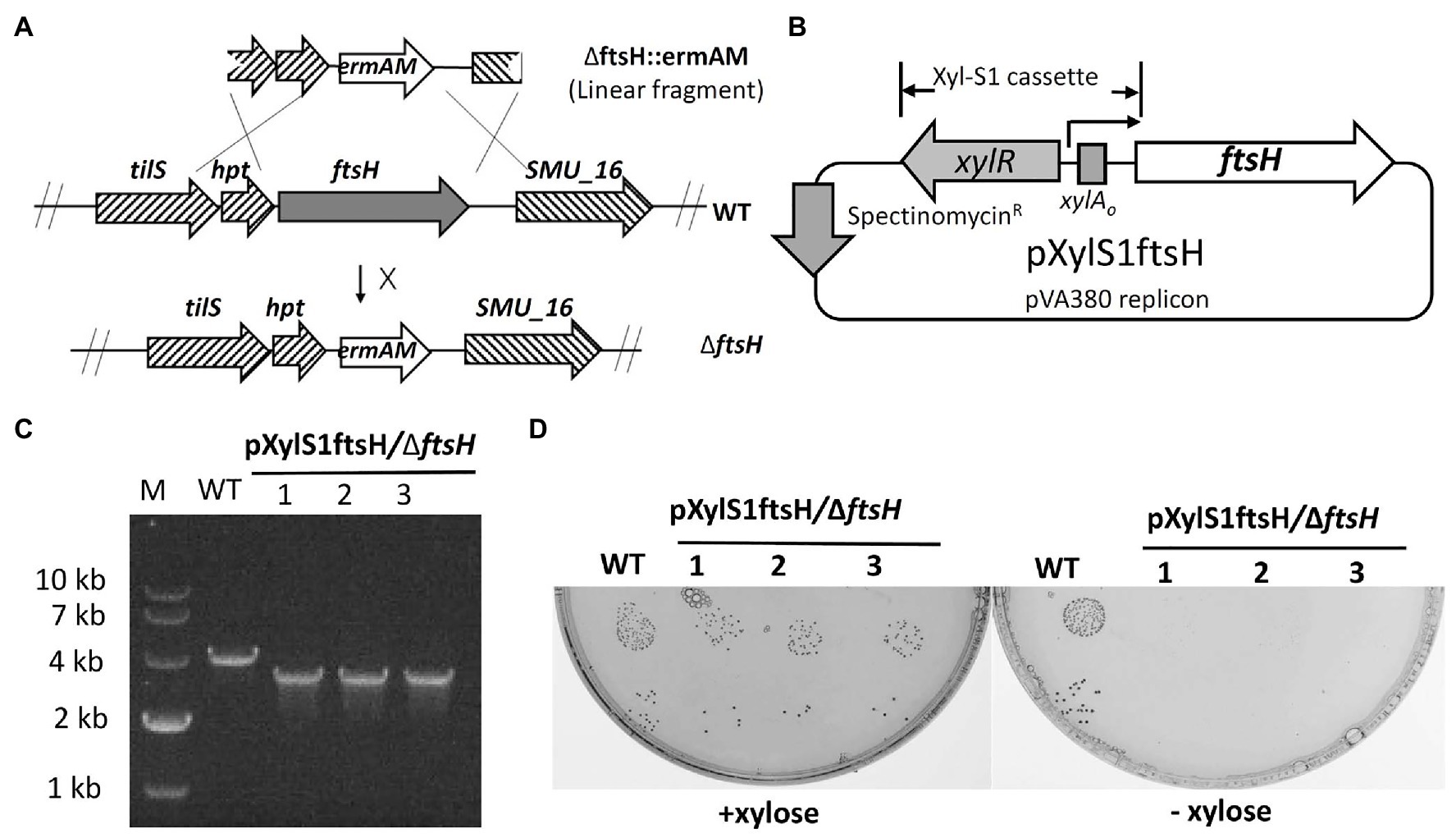
Figure 1. Mutagenesis of ftsH. (A) Illustration of the allelic replacement scheme for ftsH mutagenesis. ftsH is the last gene of a seven-gene operon that also includes hpt (encoding a hypoxanthine phosphoribosyltransferase) and tils (encoding a tRNA lysidine synthetase). The X located adjacent to the arrow indicates an unsuccessful reaction. (B) Map of the inducible ftsH expression plasmid pXylS1ftsH. Expression of the ftsH gene in pXylS1ftsH is controlled by the xylose induction cassette Xyl-S1 (Xie et al., 2013b). (C) Three randomly selected transformants of strain pXylS1ftsH/UA159 were PCR amplified using primers flanking the ftsH locus to confirm the ftsH deletion genotype. The expected PCR amplicon from the wild type is approximately 4 kb, while the expected ftsH mutant amplicon is approximately 3 kb. (D) Three randomly selected colonies from the strain pXylS1ftsH/∆ftsH were grown in BHI broth containing 0.5% (w/v) xylose overnight at 37°C. The overnight cultures were washed and serially diluted with fresh BHI. The 10−5 and 10−6 dilutions (10 μl each) were spotted onto BHI agar plates ± xylose and incubated for 24 h at 37°C. Wild type strain UA159 (WT) was cultivated in parallel in the same conditions as a control.
Isolation of Spontaneous Point Mutation Suppressors of Lethality
We reasoned that positive selection could be used to identify a spontaneous mutation to suppress FtsH lethality and reveal the source of its mutant lethal phenotype. To facilitate the screening process, we constructed strain S875, in which the expression of ftsH was placed under the control of the xylose induction cassette Xyl-S1 (Figure 2A). As expected, growth of strain S875 in BHI medium required xylose supplementation (Figure 2B). Using the inducible ftsH strain S875, cultures were passaged in BHI broth supplemented with xylose and then plated on BHI agar medium lacking xylose. Large colonies with enhanced growth without xylose were selected as candidate suppressor mutant strains (Figures 2A,B). One such putative suppressor mutant (strain S876) was subsequently transformed with the ftsH deletion construct ∆ftsH::ermAM. As shown in Figure 2C, this transformation reaction yielded >2,000 CFU/ml, and resulted in the expected ftsH deletion (Supplementary Figure S1). As expected, no transformants were detected when strain S875 or UA159 were used as recipient strains (Figure 2C). These results confirmed that strain S876 indeed contains a suppressor of lethality mutation that relieves its critical dependence upon FtsH for survival.
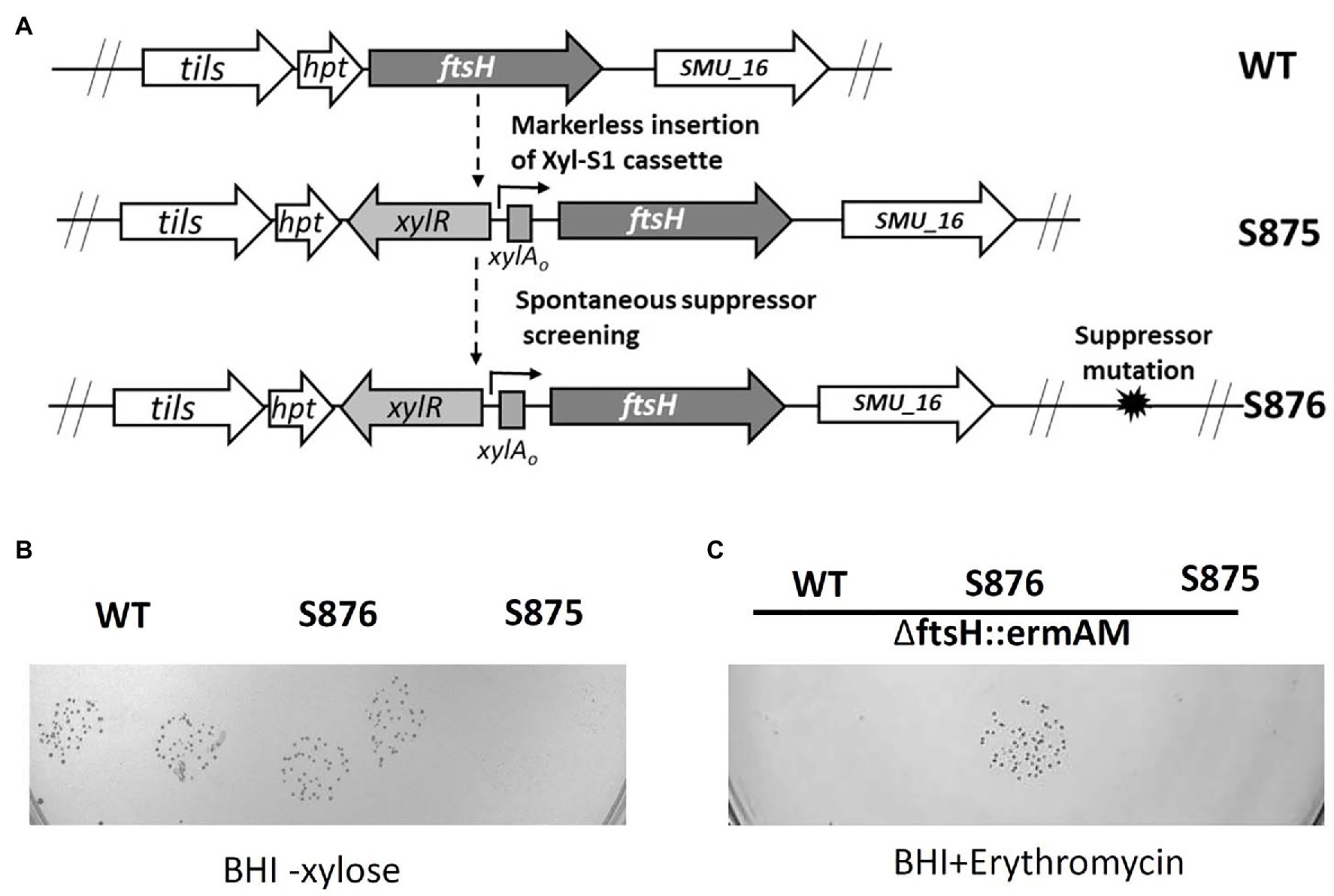
Figure 2. ftsH suppressor mutant screening. (A) Illustration of the scheme to obtain the chromosomal inducible ftsH strain S875 and the suppressor mutant strain S876. (B) S. mutans UA159 (WT), the suppressor mutant strain (S876) and the inducible ftsH expression strain (S875) were all cultured in BHI containing 0.5% (w/v) xylose. Each overnight culture was washed, serially diluted to 10−5, spotted in duplicate onto BHI agar lacking xylose, and then incubated overnight at 37°C. (C) The ftsH deletion construct ∆ftsH::ermAM was transformed into following strains: wild type strain UA159 (WT), ftsH suppressor mutant strain (S876), and inducible ftsH expression strain (S875). About 10 μl of each transformation reaction without dilution was spotted onto BHI medium containing 12.5 μg ml−1 erythromycin and incubated for 24 h at 37°C. This experiment was performed three times with similar results.
Suppressor Identification and Confirmation
To identify this suppressor mutation, we performed whole genome sequencing of strain S876 to identify all of its SNPs. Seventeen SNPs were found in strain S876 (Table 1), four of which mapped to intergenic regions, five were silent mutations, and the remaining eight created missense mutations in seven candidate genes. To identify the critical suppressor mutation(s) among the seven candidates, we individually co-transformed each together with the ∆ftsH::ermAM construct and selected on BHI agar containing erythromycin. Seven 2 kb DNA fragments named as SMU448(F59S), SMU565(S186G), SMU593(C106F), SMU1517(G195R), SMU1565F(G376F), SMU1740(V108A), and SMU1799(R101C) were each PCR amplified from the genome of strain S876. All primer pairs targeting these mutations were designed to evenly span the mutation site, which was located near the center of each PCR amplicon. As shown in Figure 3, only the co-transformation of ∆ftsH::ermAM with SMU_1517(G195R) yielded viable transformants, which were later confirmed to exhibit the expected genotype. The ORF targeted by SMU_1517(G195R) encodes a response regulator referred to as VicR. The VicRK TCS has been thoroughly investigated in several Firmicutes species, including the streptococci Streptococcus pneumoniae (Wayne et al., 2012), Streptococcus pyogenes (Liu et al., 2006), and S. mutans (Lei et al., 2018). Consistent with our results, the vicR gene is also essential in streptococci (Winkler and Hoch, 2008). As an independent confirmation of these results, we constructed the vicR point mutant strain S877 (conferring VicRG195R) using markerless mutagenesis system (Xie et al., 2011) and then successfully transformed the ∆ftsH::ermAM construct into strain S877 (Figure 4A). S877 transformants were confirmed by PCR to exhibit the expected ftsH deletion genotype (Figure 4B), and were designated as S878. From these results, we conclude that vicR (G195R) is the principal mutation responsible for suppressing the lethal phenotype of an ftsH deletion.

Figure 3. Suppressor mutation identification. Seven DNA fragments, corresponding to the seven missense mutations identified in Table 1, were generate by PCR amplification of strain S876 and designated as SMU448(F59S), SMU565(S186G), SMU593(C106F), SMU1517(G195R), SMU1565F(G376F), SMU1740(V108A), and SMU1799(R101C). Each of these amplicons was co-transformed into wild-type strain UA159 together with the ftsH deletion construct ∆ftsH::ermAM. About 100 μl of each transformation reaction were spread onto selective medium.
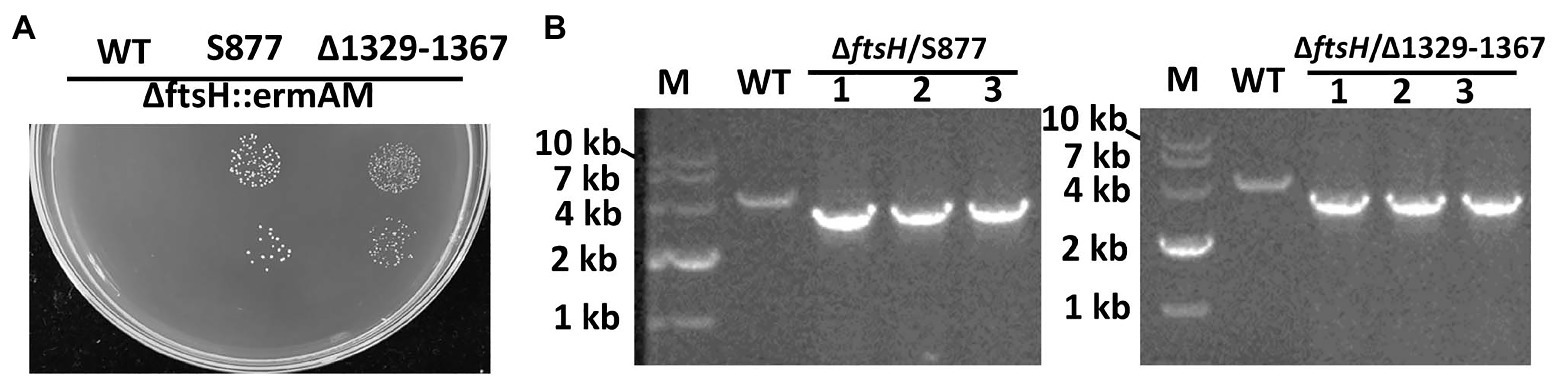
Figure 4. Both vicR(G195R) point mutation and Tnsmu2 deletion can suppress ftsH mutant lethality. (A) The ftsH deletion construct ∆ftsH::ermAM was transformed into following strains: wild type strain UA159, the markerless vicR(G195R) point mutation strain (S877), and the markerless TnSmu2 deletion strain (∆1329–1367). About 10 μl of each transformation reaction (100 and 10−1 dilutions) was spotted onto selective medium. This experiment was performed three times with similar results. (B) Three randomly selected transformants of strain (S877) and strain (∆1329–1367) were PCR amplified using primers flanking the ftsH locus to confirm the ftsH deletion genotype. The expected PCR amplicons from the wild type (WT) is approximately 4 kb, while the expected ftsH mutant amplicon is approximately 3 kb.
Transcriptome Analysis of Strain S877 Reveals an Altered Regulatory Function of VicRG195R
VicR belongs to the OmpR family of response regulators (Narayanan et al., 2014). Therefore, we were curious whether altered gene expression mediated by VicRG195R may explain its viability in an ftsH deletion background. The transcriptomes of S. mutans S877 and S. mutans UA159 were compared using RNA-seq and 46 downregulated genes and one upregulated gene of strain S877 were found to exhibit ≥2-fold changes in expression (Supplementary Table S3). Interestingly, 23 of these genes can be grouped into just two genomic loci (the TnSmu2 genomic island and the CRISPR locus, Table 2). TnSmu2 is the largest genomic island (>57 kb) found in the S. mutans UA159 genome (Ajdic et al., 2002), and it encodes a mixture of nonribosomal peptide synthases, polyketide synthases, and accessory proteins responsible for the biosynthesis of mutanobactins, a group of secondary metabolites involved in oxidative stress tolerance, and biofilm formation (Wu et al., 2010). RNA-seq data indicated the majority of the mutanobactin biosynthetic gene cluster is downregulated in strain S877 (Table 2). It has been reported that the four CRISPR associated (cas) genes (from SMU_1405c to SMU_1402c) are required for both immunity against foreign DNA invasion and the regulation of stress responses (Serbanescu et al., 2015). Each of these four cas genes were downregulated in the strain S877 (Table 2). To further validate the RNA-seq results, qRT-PCR was performed on seven genes selected from both the mutanobactin and CRISPR loci (Table 2). The qRT-PCR results mirrored those of the RNA-seq experiment, confirming their differential regulation by VicRG195R (Table 2).
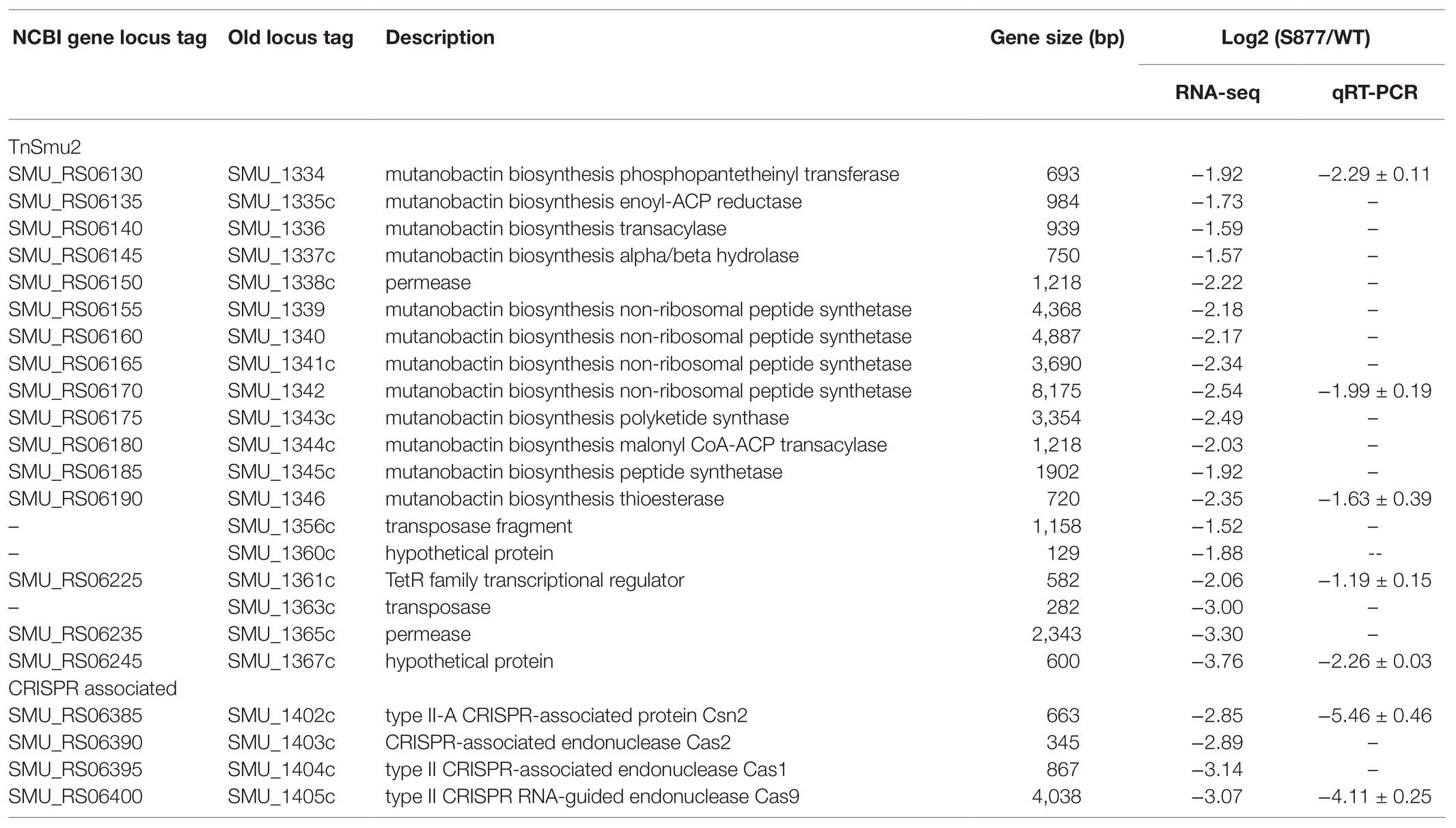
Table 2. Differentially expressed genes in Streptococcus mutans strain S877.
The results of the S877 transcriptome analysis suggested that reduced expression of target genes may be responsible for the lethality suppressor phenotype of VicRG195R. To test this hypothesis and identify the target gene(s) responsible for bypassing ftsH essentiality, 27 genes with expression changes >4-fold (log2 Fold Change < −2; Supplementary Table S3) were selected for targeted mutagenesis. About 18 of the 27 genes grouped into the two aforementioned genomic loci (mutanobactin and CRISPR loci). Therefore, we created multi-ORF deletion mutant strains of both loci (∆1329–1367 and ∆1402–1405) in addition to individual deletions of the nine other genes located in separate loci (∆277, ∆284, ∆503, ∆510, ∆609, ∆1322, ∆1803, ∆1882, and ∆2035). Next, the ftsH deletion construct ∆ftsH::ermAM was transformed into these 11 deletion mutant strains. Surprisingly, only the ∆1329–1367 mutant remained viable after deleting ftsH (Figure 4), indicating these genes likely play a central role in the lethal phenotype triggered by an ftsH mutation.
FtsH is Required to Survive Environmental Stress Growth Conditions
To gain further insight into the global regulatory role of FtsH in S. mutans, we compared a variety of growth phenotypes of the ftsH deletion strain (S878). We first measured its growth kinetics under normal laboratory growth conditions. As shown in Figure 5A, strain S878 exhibits a substantially longer lag phase relative to strain S877 and yields a lower terminal optical density, suggesting that FtsH has additional important functions involved in the regulation of cellular growth. Consequently, we compared the survival abilities of the two strains after exposure to different environmental stressors (acid, osmotic, and oxidative stresses). As shown in Figure 5B, the ftsH deletion strain (S878) is more sensitive to each of the three stress conditions, with a particular hypersensitivity to acid stress. Thus, in addition to its essential function as a regulator of the mutanobactin biosynthetic gene cluster, FtsH plays a major pleiotropic role required for normal growth and protection against environmental stress.
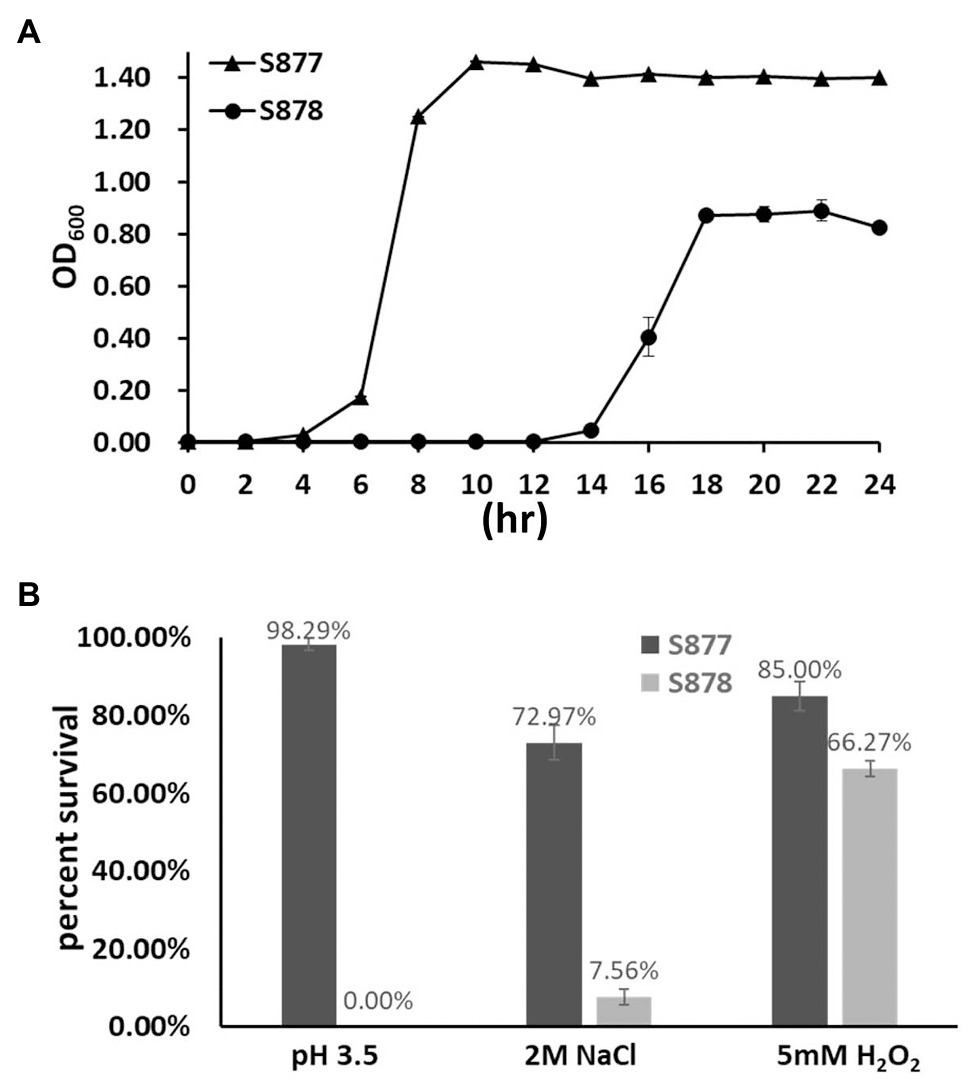
Figure 5. Characterization of the ftsH deletion strain. (A) Growth kinetics of the markerless vicR(G195R) point mutation strain (S877) and its derivative ftsH deletion strain S878 under normal culture conditions at pH 7.0. (B) Tolerance of strains S877 and S878 to acid, osmotic, and oxidative stresses. Exponential cultures of S. mutans S877 and S878 were exposed to acid stress at pH 3.5 for 20 min., osmotic stress with 2 M NaCl for 3 h., and oxidative stress with 5 mM H2O2 for 1 h. After stress exposure, serial dilutions of the cultures were plated onto BHI agar to determine the number of viable cells. Similar cultures not exposed to stress were used as controls. Colony forming unit (CFU) were counted and percent survival of strains S877 (black bars) and S878 (gray bars) was calculated relative to the control samples. Data are presented as the means ± standard deviations from three biological replicates.
Discussion
In the current study, we provide the first insights into the role of FtsH in S. mutans, including the source of its essentiality. It has been reported that FtsH is essential in E. coli due to its degradation of the lipopolysaccharide (LPS) biosynthetic enzymes LpxC and KdtA, which results in lipid imbalances and the lethal overproduction of LPS (Ogura et al., 1999; Katz and Ron, 2008). As a Gram-positive organism, S. mutans does not synthesize LPS and lacks orthologs of both lpxC and kdtA. Therefore, ftsH essentiality in S. mutans must occur through a mechanism distinct from that of E. coli. Considering that ftsH is dispensable in a variety of organisms, we reasoned that it should be possible to isolate suppressor mutations supporting the growth of the S. mutans ftsH mutant. A spontaneous suppressor mutant strain (S876) was obtained through classical genetic screening in this study. Seventeen SNPs including vicR (G195R) were found in strain S876 (Table 1). A single point mutation within the vicR (G195R) gene was found to confer the suppressor phenotype of S876 using a co-transformation approach (Figure 3). This result was further validated by constructing a VicRG195R mutant strain (S877; Figure 4). The four SNPs located in intergenic regions as well as the five SNPs causing silent mutations were not examined further, because no potential ORFs or non-coding RNAs were found in these intergenic regions. While one or more of these SNPs may partially contribute to the suppressor phenotype of strain S876, we can conclude that vicR (G195R) is the principal mutation responsible for suppressing the lethal phenotype of an ftsH deletion in S. mutans.
The VicR response regulator of the VicRK TCS (also designated as YycFG in B. subtilis) is widely conserved among the low-GC Gram-positive bacteria (Winkler and Hoch, 2008). This TCS regulates a diverse set of genes in various bacterial species and controls multiple functions, such as murein biosynthesis, cell division, lipid integrity, biofilm formation, etc. (Winkler and Hoch, 2008). The vicR gene has also been shown to be essential for most species studied thus far, including S. mutans (Senadheera et al., 2005). It has been reported for S. pneumoniae that its lethal vicR mutant phenotype is due to VicR stimulation of pcsB gene expression (Ng et al., 2003, 2005). PcsB is an essential putative hydrolase required for S. pneumoniae murein biosynthesis and cell division. Similarly, it has been reported that transcription of gbpB (pcsB ortholog) is also directly activated by VicR in S. mutans and required for its viability as well (Senadheera et al., 2005; Duque et al., 2011). In this study, we reconfirmed that vicR mutagenesis in S. mutans is lethal, unless gbpB expression is controlled by a constitutive promoter (Supplementary Figure S2A). Based upon these results as well as those of the vicR (G195R) strain, we speculated that FtsH regulation of GbpB might be responsible for the ftsH mutant lethal phenotype. However, our results were not supportive of this, as constitutive gbpB expression was unable to suppress ftsH mutant lethality, unlike the VicRG195R mutation (Supplementary Figure S2B). Therefore, we employed RNA-seq to examine the vicR (G195R) strain transcriptome and identify the potential regulatory impact of this mutation. Of the 36 genes exhibiting >2-fold reduction in transcript levels, a large percentage were located in the genomic island Tnsmu2. Consistent with expectations, a deletion of this locus did suppress the lethal ftsH mutant phenotype. However, single gene deletions in this locus did not suppress lethality (data not shown), which suggests that the suppressor function of VicRG195R is likely due to the combined effect of simultaneously downregulating multiple Tnsmu2 genes. Since the Tnsmu2 locus encodes a biosynthetic gene cluster responsible for the biosynthesis of mutanobactin [43], our results suggest that multiple PKS and NRPS enzymes encoded by this gene cluster could be targeted by FtsH. It is currently unclear why this activity would be essential for viability. The 15 genes in the mutanobactin locus (SMU_1348c–SMU_1334c) are organized into the largest operon (>36 kb) in the S. mutans genome (Wu et al., 2010). The genes encoding these PKS/NRPS are also among the largest individual genes of S. mutans. For example, the SMU_1342 ORF is >8 kb. Since the mutanobactin locus is not essential (Wu et al., 2010), we suspect that the mutanobactin proteins probably accumulate to lethal levels in an ftsH background, perhaps due to defects in their normal turnover (Figure 6). In agreement with this notion, the VicRG195R mutation reduces the expression of the entire mutanobactin gene locus (Table 2), which could conceivably reduce the toxicity triggered by the overabundance of these proteins, resulting in the observed suppression of ftsH mutant lethality. Further studies will be required to specifically test this hypothesis.
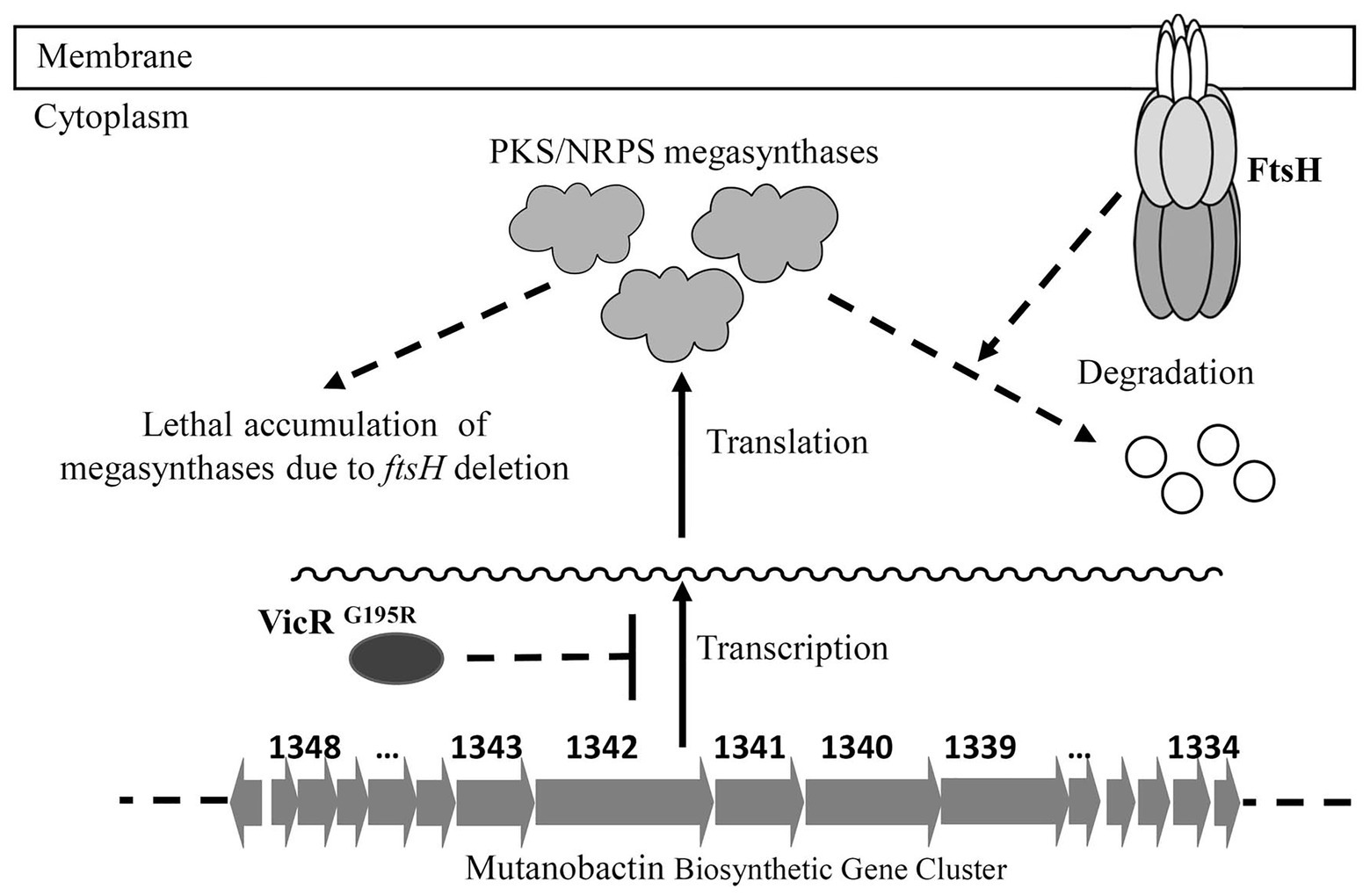
Figure 6. Model of FtsH essentiality in S. mutans. The mutanobactin biosynthetic gene cluster contains the largest operon in the genome of S. mutans. The protein products of this locus are PKS/NRPS megasynthases, which are also among the largest S. mutans proteins. It is hypothesized that FtsH is required for the turnover of these megasynthases, and the accumulation of these proteins in ftsH deletion strain is lethal to the bacterial cell. The VicRG195R mutation results in reduced transcription of the entire mutanobactin biosynthetic gene cluster, leading to reduced ftsH mutant toxicity and a suppression of the FtsH mutant lethal phenotype.
In addition, by isolating the VicRG195R suppressor mutation, it was possible to explore the global physiological role of FtsH. Similar to other bacteria; our results clearly demonstrate a major role in S. mutans physiology, which is the likely source of the ftsH mutant stress tolerance phenotypes. There was a particularly strong ftsH mutant sensitivity to acid stress, implying that key proteins involved in the acid tolerance response could be regulated via FtsH proteolysis. As the first phenotypic study of FtsH in S. mutans, this study sets the stage for future mechanistic studies to reveal how FtsH proteolysis is controlled and its regulatory impact upon different genetic pathways of S. mutans.
Data Availability Statement
The datasets generated for this study were deposited with the Sequence Read Archive (SRA) at the National Center for Biotechnology Institute under the accession PRJNA715382.
Author Contributions
JM, WC, ZX, and HL designed the experiments. YW and ZX conducted most of the studies. YW, JM, WC, ZX, and HL analyzed data. JM, ZX, and HL wrote the paper with the contributions from all other authors. All authors contributed to the article and approved the submitted version.
Funding
The study was funded in part by National Natural Science Foundation of China (31570050 and 21706192) and by a grant from Tianjin Science and Technology Committee of China (18YFZCSY01360).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.659220/full#supplementary-material
References
Ajdic, D., McShan, W. M., McLaughlin, R. E., Savic, G., Chang, J., Carson, M. B., et al. (2002). Genome sequence of Streptococcus mutans UA159, a cariogenic dental pathogen. Proc. Natl. Acad. Sci. U. S. A. 99, 14434–14439. doi: 10.1073/pnas.172501299
Began, J., Cordier, B., Brezinova, J., Delisle, J., Hexnerova, R., Srb, P., et al. (2020). Rhomboid intramembrane protease YqgP licenses bacterial membrane protein quality control as adaptor of FtsH AAA protease. EMBO J. 39:e102935. doi: 10.15252/embj.2019102935
Bhandari, V., Wong, K. S., Zhou, J. L., Mabanglo, M. F., Batey, R. A., and Houry, W. A. (2018). The role of ClpP protease in bacterial pathogenesis and human diseases. ACS Chem. Biol. 13, 1413–1425. doi: 10.1021/acschembio.8b00124
Bieniossek, C., Niederhauser, B., and Baumann, U. M. (2009). The crystal structure of apo-FtsH reveals domain movements necessary for substrate unfolding and translocation. Proc. Natl. Acad. Sci. U. S. A. 106, 21579–21584. doi: 10.1073/pnas.0910708106
Bieniossek, C., Schalch, T., Bumann, M., Meister, M., Meier, R., and Baumann, U. (2006). The molecular architecture of the metalloprotease FtsH. Proc. Natl. Acad. Sci. U. S. A. 103, 3066–3071. doi: 10.1073/pnas.0600031103
Bittner, L. M., Arends, J., and Narberhaus, F. (2017). When, how and why? Regulated proteolysis by the essential FtsH protease in Escherichia coli. Biol. Chem. 398, 625–635. doi: 10.1515/hsz-2016-0302
Carvalho, V., Prabudiansyah, I., Kovacik, L., Chami, M., Kieffer, R., van der Valk, R., et al. (2020). The cytoplasmic domain of the AAA+ protease FtsH is tilted with respect to the membrane to facilitate substrate entry. J. Biol. Chem. 296:100029. doi: 10.1074/jbc.RA120.014739
Chattoraj, P., Banerjee, A., Biswas, S., and Biswas, I. (2010). ClpP of Streptococcus mutans differentially regulates expression of genomic islands, mutacin production, and antibiotic tolerance. J. Bacteriol. 192, 1312–1323. doi: 10.1128/JB.01350-09
Chu, C. Y., Stewart, P. E., Bestor, A., Hansen, B., Lin, T., Gao, L., et al. (2016). Function of the Borrelia burgdorferi FtsH homolog is essential for viability both in vitro and in vivo and independent of HflK/C. MBio 7, e00404–e00416. doi: 10.1128/mBio.00404-16
Duque, C., Stipp, R. N., Wang, B., Smith, D. J., Hofling, J. F., Kuramitsu, H. K., et al. (2011). Downregulation of GbpB, a component of the VicRK regulon, affects biofilm formation and cell surface characteristics of Streptococcus mutans. Infect. Immun. 79, 786–796. doi: 10.1128/IAI.00725-10
Goldberg, A. L. (2003). Protein degradation and protection against misfolded or damaged proteins. Nature 426, 895–899. doi: 10.1038/nature02263
Ito, K., and Akiyama, Y. (2005). Cellular functions, mechanism of action, and regulation of FtsH protease. Annu. Rev. Microbiol. 59, 211–231. doi: 10.1146/annurev.micro.59.030804.121316
Kajfasz, J. K., Abranches, J., and Lemos, J. A. (2011). Transcriptome analysis reveals that ClpXP proteolysis controls key virulence properties of Streptococcus mutans. Microbiology 157, 2880–2890. doi: 10.1099/mic.0.052407-0
Kajfasz, J. K., Martinez, A. R., Rivera-Ramos, I., Abranches, J., Koo, H., Quivey, R. G. Jr., et al. (2009). Role of Clp proteins in expression of virulence properties of Streptococcus mutans. J. Bacteriol. 191, 2060–2068. doi: 10.1128/JB.01609-08
Katz, C., and Ron, E. Z. (2008). Dual role of FtsH in regulating lipopolysaccharide biosynthesis in Escherichia coli. J. Bacteriol. 190, 7117–7122. doi: 10.1128/JB.00871-08
Krzywda, S., Brzozowski, A. M., Verma, C., Karata, K., Ogura, T., and Wilkinson, A. J. (2002). The crystal structure of the AAA domain of the ATP-dependent protease FtsH of Escherichia coli at 1.5 A resolution. Structure 10, 1073–1083. doi: 10.1016/S0969-2126(02)00806-7
Langklotz, S., Baumann, U., and Narberhaus, F. (2012). Structure and function of the bacterial AAA protease FtsH. Biochim. Biophys. Acta 1823, 40–48. doi: 10.1016/j.bbamcr.2011.08.015
Lei, L., Stipp, R. N., Chen, T., Wu, S. Z., Hu, T., and Duncan, M. J. (2018). Activity of Streptococcus mutans VicR is modulated by antisense RNA. J. Dent. Res. 97, 1477–1484. doi: 10.1177/0022034518781765
Lemos, J. A., Abranches, J., and Burne, R. A. (2005). Responses of cariogenic streptococci to environmental stresses. Curr. Issues Mol. Biol. 7, 95–107.
Li, H., and Durbin, R. (2010). Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 26, 589–595. doi: 10.1093/bioinformatics/btp698
Liu, N., Chaudhry, M. T., Xie, Z., Kreth, J., and Merritt, J. (2017). Identification of new degrons in Streptococcus mutans reveals a novel strategy for engineering targeted, controllable proteolysis. Front. Microbiol. 8:2572. doi: 10.3389/fmicb.2017.02572
Liu, M., Hanks, T. S., Zhang, J., McClure, M. J., Siemsen, D. W., Elser, J. L., et al. (2006). Defects in ex vivo and in vivo growth and sensitivity to osmotic stress of group A Streptococcus caused by interruption of response regulator gene vicR. Microbiology 152, 967–978. doi: 10.1099/mic.0.28706-0
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Narayanan, A., Kumar, S., Evrard, A. N., Paul, L. N., and Yernool, D. A. (2014). An asymmetric heterodomain interface stabilizes a response regulator-DNA complex. Nat. Commun. 5:3282. doi: 10.1038/ncomms4282
Ng, W. L., Robertson, G. T., Kazmierczak, K. M., Zhao, J., Gilmour, R., and Winkler, M. E. (2003). Constitutive expression of PcsB suppresses the requirement for the essential VicR (YycF) response regulator in Streptococcus pneumoniae R6. Mol. Microbiol. 50, 1647–1663. doi: 10.1046/j.1365-2958.2003.03806.x
Ng, W. L., Tsui, H. C., and Winkler, M. E. (2005). Regulation of the pspA virulence factor and essential pcsB murein biosynthetic genes by the phosphorylated VicR (YycF) response regulator in Streptococcus pneumoniae. J. Bacteriol. 187, 7444–7459. doi: 10.1128/JB.187.21.7444-7459.2005
Niu, G., Okinaga, T., Qi, F., and Merritt, J. (2010). The Streptococcus mutans IrvR repressor is a CI-like regulator that functions through autocleavage and Clp-dependent proteolysis. J. Bacteriol. 192, 1586–1595. doi: 10.1128/JB.01261-09
Niu, G., Okinaga, T., Zhu, L., Banas, J., Qi, F., and Merritt, J. (2008). Characterization of irvR, a novel regulator of the irvA-dependent pathway required for genetic competence and dextran-dependent aggregation in Streptococcus mutans. J. Bacteriol. 190, 7268–7274. doi: 10.1128/JB.00967-08
Niwa, H., Tsuchiya, D., Makyio, H., Yoshida, M., and Morikawa, K. (2002). Hexameric ring structure of the ATPase domain of the membrane-integrated metalloprotease FtsH from Thermus thermophilus HB8. Structure 10, 1415–1423. doi: 10.1016/S0969-2126(02)00855-9
Ogura, T., Inoue, K., Tatsuta, T., Suzaki, T., Karata, K., Young, K., et al. (1999). Balanced biosynthesis of major membrane components through regulated degradation of the committed enzyme of lipid A biosynthesis by the AAA protease FtsH (HflB) in Escherichia coli. Mol. Microbiol. 31, 833–844. doi: 10.1046/j.1365-2958.1999.01221.x
Sauer, R. T., and Baker, T. A. (2011). AAA+ proteases: ATP-fueled machines of protein destruction. Annu. Rev. Biochem. 80, 587–612. doi: 10.1146/annurev-biochem-060408-172623
Senadheera, M. D., Guggenheim, B., Spatafora, G. A., Huang, Y. C., Choi, J., Hung, D. C., et al. (2005). A VicRK signal transduction system in Streptococcus mutans affects gtfBCD, gbpB, and ftf expression, biofilm formation, and genetic competence development. J. Bacteriol. 187, 4064–4076. doi: 10.1128/JB.187.12.4064-4076.2005
Serbanescu, M. A., Cordova, M., Krastel, K., Flick, R., Beloglazova, N., Latos, A., et al. (2015). Role of the Streptococcus mutans CRISPR-Cas systems in immunity and cell physiology. J. Bacteriol. 197, 749–761. doi: 10.1128/JB.02333-14
Shields, R. C., Walker, A. R., Maricic, N., Chakraborty, B., Underhill, S. A. M., and Burne, R. A. (2020). Repurposing the Streptococcus mutans CRISPR-Cas9 system to understand essential gene function. PLoS Pathog. 16:e1008344. doi: 10.1371/journal.ppat.1008344
Shields, R. C., Zeng, L., Culp, D. J., and Burne, R. A. (2018). Genomewide identification of essential genes and fitness determinants of Streptococcus mutans UA159. mSphere 3, e00031–e00018. doi: 10.1128/mSphere.00031-18
Suno, R., Niwa, H., Tsuchiya, D., Zhang, X., Yoshida, M., and Morikawa, K. (2006). Structure of the whole cytosolic region of ATP-dependent protease FtsH. Mol. Cell 22, 575–585. doi: 10.1016/j.molcel.2006.04.020
Tao, L., and Biswas, I. (2015). Degradation of SsrA-tagged proteins in streptococci. Microbiology 161, 884–894. doi: 10.1099/mic.0.000048
Wayne, K. J., Li, S., Kazmierczak, K. M., Tsui, H. C., and Winkler, M. E. (2012). Involvement of WalK (VicK) phosphatase activity in setting WalR (VicR) response regulator phosphorylation level and limiting cross-talk in Streptococcus pneumoniae D39 cells. Mol. Microbiol. 86, 645–660. doi: 10.1111/mmi.12006
Winkler, M. E., and Hoch, J. A. (2008). Essentiality, bypass, and targeting of the YycFG (VicRK) two-component regulatory system in gram-positive bacteria. J. Bacteriol. 190, 2645–2648. doi: 10.1128/JB.01682-07
Wu, C., Cichewicz, R., Li, Y., Liu, J., Roe, B., Ferretti, J., et al. (2010). Genomic island TnSmu2 of Streptococcus mutans harbors a nonribosomal peptide synthetase-polyketide synthase gene cluster responsible for the biosynthesis of pigments involved in oxygen and H2O2 tolerance. Appl. Environ. Microbiol. 76, 5815–5826. doi: 10.1128/AEM.03079-09
Xie, Z., Okinaga, T., Qi, F., Zhang, Z., and Merritt, J. (2011). Cloning-independent and counterselectable markerless mutagenesis system in Streptococcus mutans. Appl. Environ. Microbiol. 77, 8025–8033. doi: 10.1128/AEM.06362-11
Xie, Z., Qi, F., and Merritt, J. (2013a). Cloning-independent plasmid construction for genetic studies in streptococci. J. Microbiol. Methods 94, 77–82. doi: 10.1016/j.mimet.2013.05.005
Xie, Z., Qi, F., and Merritt, J. (2013b). Development of a tunable wide-range gene induction system useful for the study of streptococcal toxin-antitoxin systems. Appl. Environ. Microbiol. 79, 6375–6384. doi: 10.1128/AEM.02320-13
Keywords: Streptococcus, AAA+ protease, FtsH, suppressor mutation, VicRK two-component system
Citation: Wang Y, Cao W, Merritt J, Xie Z and Liu H (2021) Characterization of FtsH Essentiality in Streptococcus mutans via Genetic Suppression. Front. Genet. 12:659220. doi: 10.3389/fgene.2021.659220
Edited by:
David W. Ussery, University of Arkansas for Medical Sciences, United StatesReviewed by:
Longbiao Guo, China National Rice Research Institute, Chinese Academy of Agricultural Sciences, ChinaRobert Colquhoun Shields, University of Florida, United States
Delphine Dufour, University of Toronto, Canada
Copyright © 2021 Wang, Cao, Merritt, Xie and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhoujie Xie, zhoujiexie@tust.edu.cn; Hao Liu, liuhao@tust.edu.cn