
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 11 June 2021
Sec. Computational Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.656114
This article is part of the Research Topic Computational Approaches for Biomarker Detection and Precision Therapeutics in Cancers View all 21 articles
 Bingzhou Guo1†
Bingzhou Guo1† Hongliang Zhang2†
Hongliang Zhang2† Jinliang Wang3
Jinliang Wang3 Rilige Wu4,5
Rilige Wu4,5 Junyan Zhang4,5Qiqin Zhang6
Junyan Zhang4,5Qiqin Zhang6 Lu Xu7
Lu Xu7 Ming Shen7
Ming Shen7 Zhibo Zhang8Fangyan Gu9
Zhibo Zhang8Fangyan Gu9 Weiliang Zeng1*
Weiliang Zeng1* Xiaodong Jia10*
Xiaodong Jia10* Chengliang Yin11*
Chengliang Yin11*Background: N6-methyladenosine (m6A) RNA modification is vital for cancers because methylation can alter gene expression and even affect some functional modification. Our study aimed to analyze m6A RNA methylation regulators and m6A-related genes to understand the prognosis of early lung adenocarcinoma.
Methods: The relevant datasets were utilized to analyze 21 m6A RNA methylation regulators and 5,486 m6A-related genes in m6Avar. Univariate Cox regression analysis, random survival forest analysis, Kaplan–Meier analysis, Chi-square analysis, and multivariate cox analysis were carried out on the datasets, and a risk prognostic model based on three feature genes was constructed.
Results: Respectively, we treated GSE31210 (n = 226) as the training set, GSE50081 (n = 128) and TCGA data (n = 400) as the test set. By performing univariable cox regression analysis and random survival forest algorithm in the training group, 218 genes were significant and three prognosis-related genes (ZCRB1, ADH1C, and YTHDC2) were screened out, which could divide LUAD patients into low and high-risk group (P < 0.0001). The predictive efficacy of the model was confirmed in the test group GSE50081 (P = 0.0018) and the TCGA datasets (P = 0.014). Multivariable cox manifested that the three-gene signature was an independent risk factor in LUAD. Furthermore, genes in the signature were also externally validated using the online database. Moreover, YTHDC2 was the important gene in the risk score model and played a vital role in readers of m6A methylation.
Conclusion: The findings of this study suggested that associated with m6A RNA methylation regulators and m6A-related genes, the three-gene signature was a reliable prognostic indicator for LUAD patients, indicating a clinical application prospect to serve as a potential therapeutic target.
Lung adenocarcinoma (LUAD) is a type of non-small cell cancer. In the 2018 Global Cancer Report, lung cancer ranked top 1 with the highest incidence and mortality among all cancers (Bray et al., 2018).
N6-methyladenosine (m6A) RNA methylation is the most abundant epigenetic modification in eukaryotic mRNA. M6A methylation regulators of each modified RNA require a writer to place, an eraser to erase, and a reader to read. Based on these proteins, m6A affected RNA splicing (He et al., 2019), translation, and RNA stability (Wang et al., 2014; He et al., 2019). Evidence is now mounting that m6A methylation underlies the progression of tumors and affects specific biological processes through non-coding RNA modification (Xiao et al., 2019). Moreover, the over-expression of YTHDF1 in the reader might affect the prognosis of ovarian cancer patients (Liu et al., 2020). In the writer family, high expression of METTL3 promoted the proliferation of bladder cancer (Cheng et al., 2019) and led to a poor prognosis (Han et al., 2019). Over-expression knockdown of ALKBH5 could effectively reduce cell proliferation in pancreatic cancer in erasers family (Tang et al., 2020). Meanwhile, m6A has many functions in cancer (He et al., 2019; Ma et al., 2019; Ma and Ji, 2020), such as reduced m6A has a relationship with phenotypes of gastric cancer (Zhang et al., 2019), KIAA1429 is associated with prognosis of liver cancer (Lan et al., 2019), and FTO could facilitate the development of breast cancer (Niu et al., 2019). However, to our knowledge, there are few studies related to m6A methylation in early LUAD, and this may be a novel treatment strategy for patients with early LUAD.
In this study, GEO and TCGA data were used to explore the influence of m6A methylation genes and their regulated genes on the prognosis of early lung adenocarcinoma. The signature was conducted for identifying new therapeutic biomarkers and treatment strategy development.
Data was downloaded from Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA) public databases. GSE31210 (n = 226) was used as the training set, GSE50081 (n = 128) as the validation set 1, and TCGA (n = 400) data as the validation set 2. Three independent datasets were used for model construction and model verification. Each independent dataset included the clinical characteristics: survival status, survival time, age, sex, and clinical TNM stage. In GEO data, TNM clinical stage was divided into stages I and II, which were shown in Table 1. Besides, the GPL570 chip platform was re-annotated by the probe to get the final expression profile of the GEO data (Fan et al., 2018). Only mRNA probes were selected, and 8,597 mRNA expression profiles were obtained.
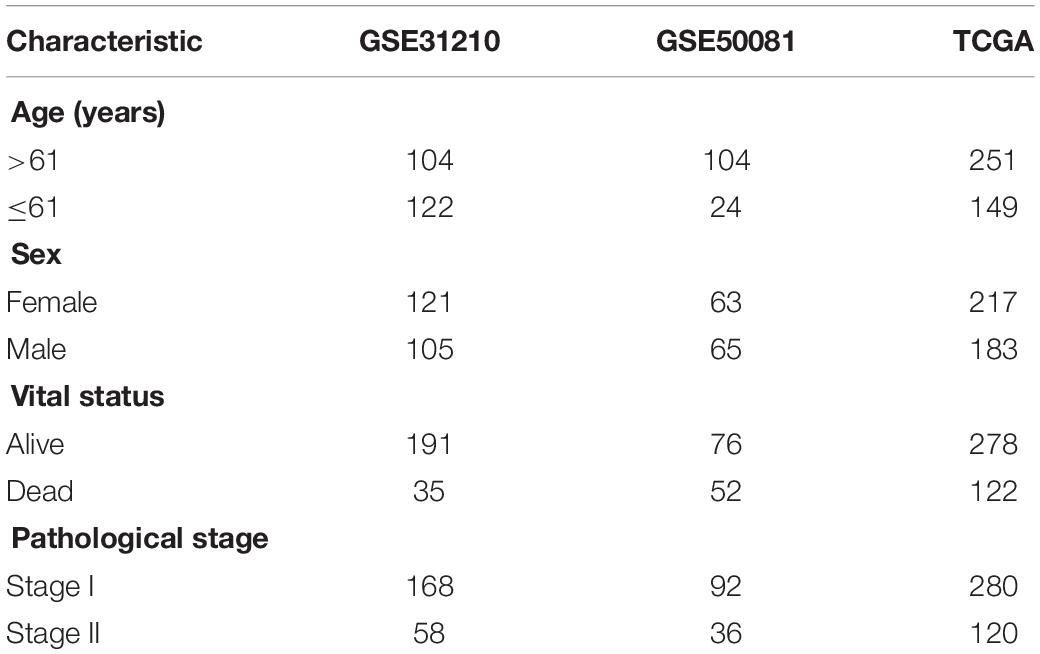
Table 1. Clinical information of the Gene Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA) datasets.
We collected 21 m6A methylated genes through literature investigation (Supplementary Table 1) (Zhang et al., 2020). We found that among these 21 genes, 14 genes were expressed in the training set (GSE31210). In LUAD, a total of 5,486 m6A-regulated genes were downloaded from the m6Avar database1 (Zheng et al., 2018). Among the 5,507 genes, 2,615 genes were expressed in GSE31210.
Discovery of the m6A RNA Methylation Regulators and m6A-Related Genes and Establishment of the m6A Methylation Risk Score Model.
We obtained prognostic-related gene sets through survival analysis [univariate cox and Kaplan–Meier (KM)] and receiver operating characteristic (ROC) curve. In the training set GSE31210, we used the random survival forest (RSF) (Ishwaran et al., 2008) to establish a prognostic model related to patient overall survival (OS). Methods of analyzing survival data were often parametric, nonlinear effects of variables, and modeled by expanding matrix for specialized functions. Identifying multiple variable interactions was also problematic. These difficulties could be effectively solved using RSF (Ishwaran et al., 2008). Its basic formula is:
The meanings of the parameters in this formula are: RS is the risk score, N is the number of genes, Exp is the expression amount of genes in the data, and Coef is the coefficient of cox analysis for the genes resulting from the random survival forest. We used gene combinations to select the largest AUC to construct a prognostic model. Based on the median of the risk scores, the data were divided into two groups: the high-risk group and the low-risk group. The KM curve was used to compare the difference between the high and low-risk groups. In the three datasets, using the median score divided two groups.
To assess the characteristics of the patients’ prognosis and its relationship with clinical features, we used chi-square analysis to judge the correlation between the model and clinical data. KM survival curve and log-rank test were used to describe the relationship between the model and OS. Furthermore, multivariate Cox regression analysis was used to study whether the clinical data (age, gender, and pathological stage) were related to OS in the training set and validation set. We used univariate Cox analysis to judge whether the clinical information had prognostic value.
Furthermore, four online tools were used to verify the gene expression levels (Oncomine database2; TIMER database3, and GEPIA database, Gene Expression Profiling Interactive Analysis4) and protein levels [The Human Protein Atlas (HPA) database5 ] in the model. Meanwhile, online public databases (The cBioPortal for Cancer Genomics6) were used to analyze and understand the gene influence on early treatment of LUAD.
The R package ‘‘clusterProfiler’’ was used to annotate the function and select the statistically significant pathways. The relationship between proteins was analyzed by using the online website STRING7 (Szklarczyk et al., 2019). Cytoscape was used to visualize the network. Then the main networks were chosen by a degree of the gene in the net analysis.
In three independent datasets, all KM and cox analyses were performed using the R package “survival”. Cox analysis was used to select prognostic genes and test models. “ROC” and “TimeROC” were available to verify the feasibility of the model. Functional annotations were made using the R package “Clusterprofiler.” All of our analyses (besides online website analysis) were performed in the R language. R packages were used as follows: “pROC,” “TimeROC,” “survival,” “clusterProfiler’,” and “randomForestSRC.” The P values of the above analyses were all <0.05 as statistically significant.
All 226, 128, and 400 patients diagnosed with LUAD were collected from the GEO (GSE31210 and GSE50081) and TCGA database, respectively. A total of 2,615 m6A-related genes out of the genes expressed were identified in the GSE31210 dataset. In Table 1, the median age of the enrolled samples was 61 years. The ratio of male vs. female was 1.15:1, with 191 live cases and 35 death cases. The longest survival was 10 years. Each sample data was only distributed in stages I–II of LUAD. The study workflow is demonstrated in Supplementary Figure 1.
After we used univariate cox analysis and ROC curve, 218 prognostic-related genes were selected, and the screening criteria were P < 0.01 and AUC > 0.6 in Supplementary Table 2. Furthermore, gene screening was performed by the importance scores of the random survival forest analysis. We permuted and combined the eight genes selected from the random survival forest, obtaining 28 – 1 = 255 prediction models (Figures 1A,B). The 255 models were evaluated by AUC, and the optimal predictive ability was found in the combination of three genes, ZCRB1, ADH1C, and YTHDC2. As a prediction model, the AUC of the three-gene model was 0.762 (Figure 1C). The risk score of the model was RSF = (1.725151 × ZCRB1) + (−1.964326 × ADH1C) + (−2.015378 × YTHDC2). Each gene name represented its expression level in a certain sample.
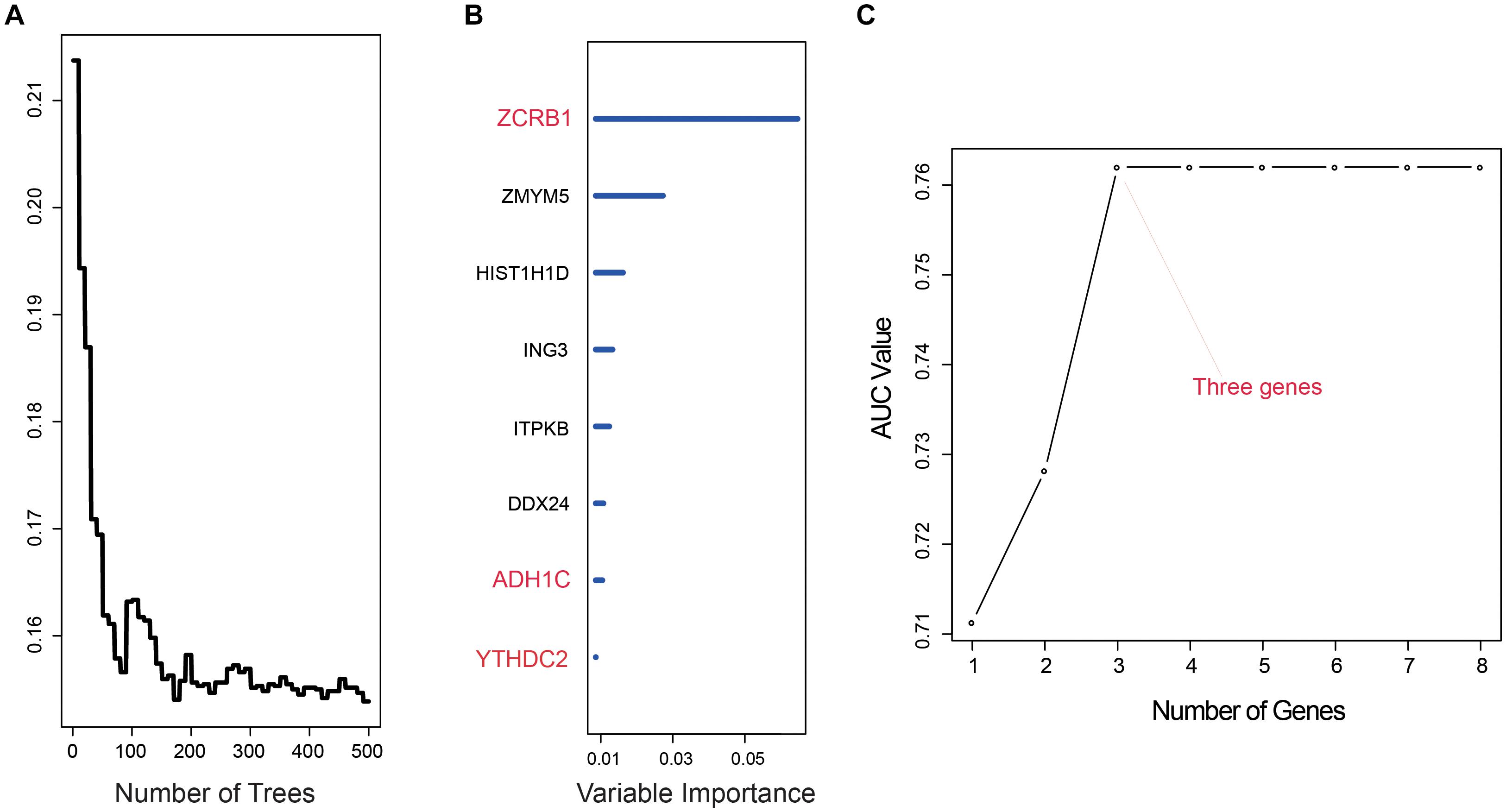
Figure 1. Random survival forest analysis. (A,B) random survival forests variable hunting analysis reveals the error rate for the data as a function of trees and uses the associated score to filter N6-methyladenosine (m6A) RNA methylation regulators and m6A-related genes. (C) Receiver operating characteristic (ROC) for selected prognostic signature from all 255 signatures.
We used the RSF formula to calculate the risk score of each sample and plotted the heat map of the three genes (Figures 2A–C), finding that in the high-risk group, ADH1C and YTHDC2 basically had low expression, while ZCRB1 obviously had high expression. This was particularly evident in the training group (GSE31210) (Figure 2A).
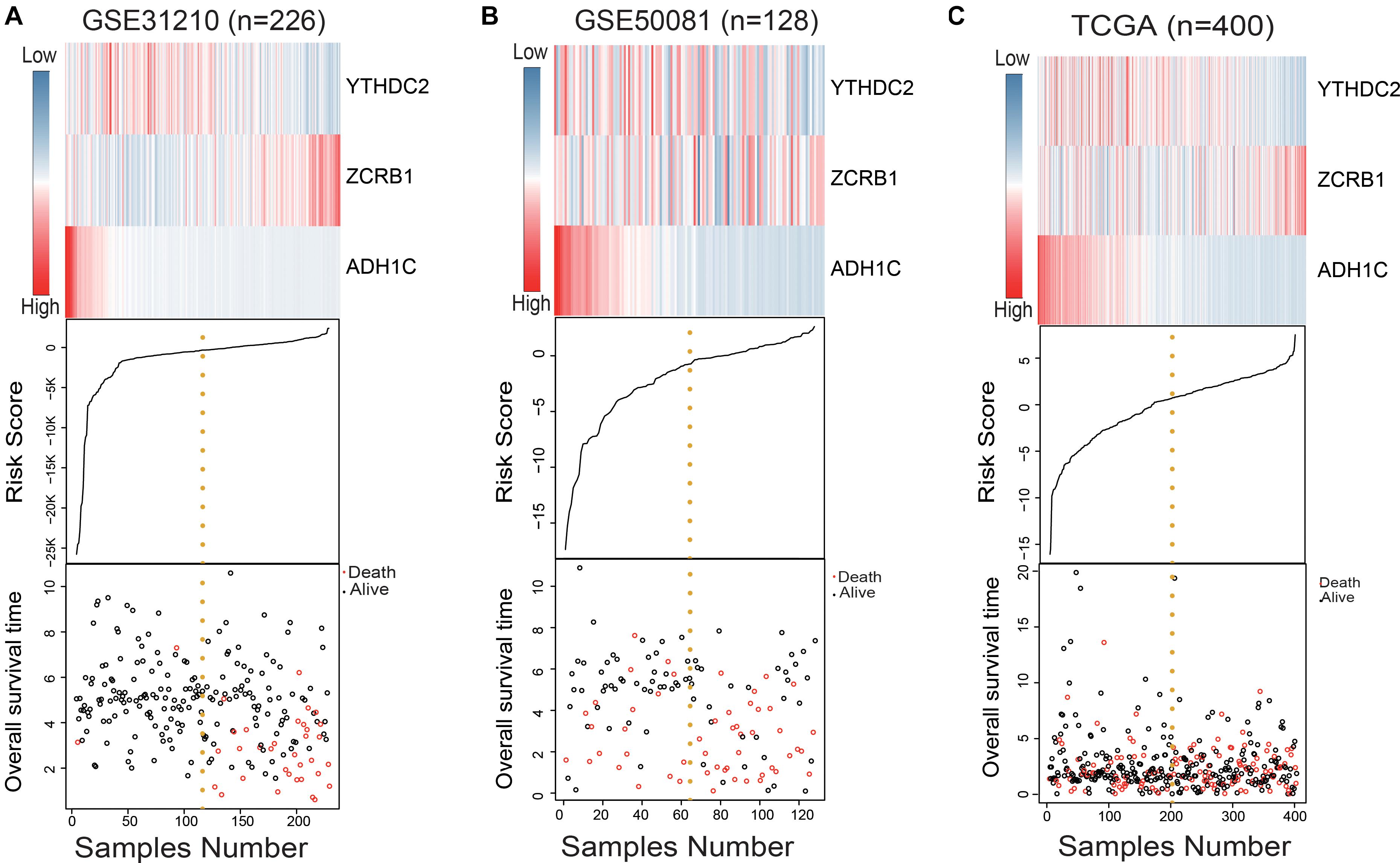
Figure 2. Evaluation of the risk predictive model in the training set and test set. (A–C) The distribution of m6A RNA methylation regulators and m6A-related gene expression level, patients’ survival status, and risk score between high- and low-risk group.
The results made it clear that ADH1C had high expression in the low-risk group as a protection factor by cox analysis (Table 2).

Table 2. Prognosis of the three genes in the signature.
In the training set, the median risk score divided all patients into two groups: high-risk group (n = 113) and low-risk group (n = 113) (Figure 3A). The KM survival curve and log rank test showed that our model had an excellent predictive power. In the validation set, the median risk score was also used to divide the patients into two groups in GSE50081 (n = 128), and there were 64 patients in the high-risk group and 64 patients in the low-risk group (Figure 3B). The KM survival curve showed that there was a significant difference between the high-risk group and low-risk group (Log rank P = 0.0018). Grouped by median risk score in TCGA (n = 400), there were 200 patients in the high-risk group and 200 samples in the low-risk group, with log rank P = 0.014 (Figure 3C).
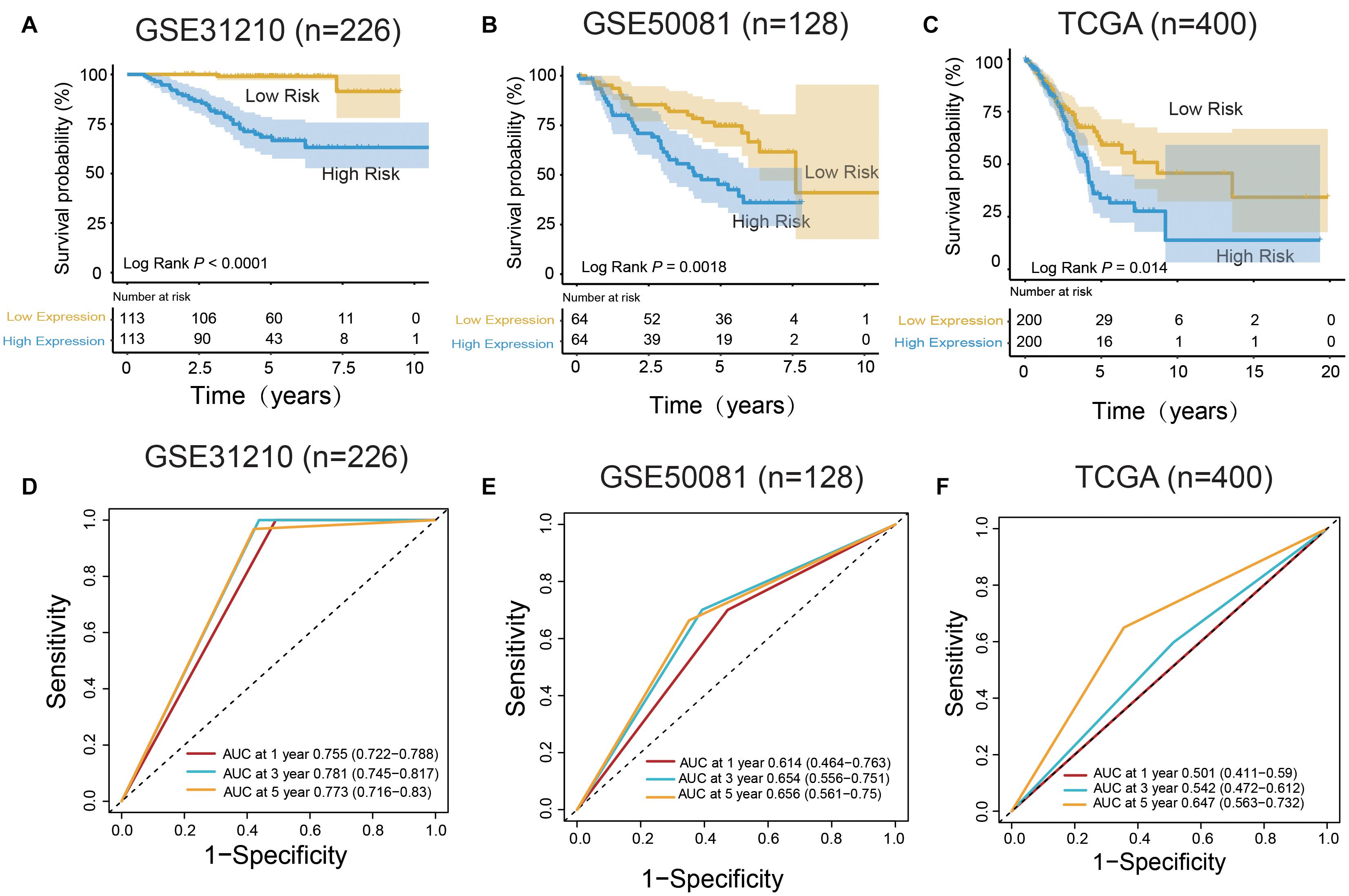
Figure 3. m6A RNA methylation regulators and m6A-related genes signature predict overall survival of patients of LUAD. (A–C) Kaplan–Meier survival curves classify patients into high- and low-risk groups by the m6A RNA methylation regulators and m6A-related genes signature in the training dataset (GSE31210), and test dataset (GSE50081 and TCGA). P values were calculated by log-rank test. (D–F) m6A RNA methylation regulators and m6A-related genes signatures were used for predicting survival in 1, 3, and 5 years by TimeROC analysis.
In the training group (GSE31210), the 5-years survival rate was 53.10% in the low-risk group and 38.05% in the high-risk group (Figure 3A). Additionally, the overall survival rate was 45.58%, indicating that the risk score could differentiate the data correctly. Survival was significantly improved in the two independent validation data (GSE50081 and TCGA). Moreover, in GSE50081, the low-risk group was 56.25% and the high-risk group was 29.69% (Figure 3B). The overall survival rate was 42.97% and the grouping label was also evident. Meanwhile, in TCGA, we selected a sample data of TNM stage (I+II) (a total of 400 cases) (Figure 3C). Five-years survival rate was calculated in the high- and low-risk groups, and the rates were 14.5 and 8%, respectively. The overall 5-years survival rate was 11.25% in both low- and high-risk groups, and the survival rate in the low-risk group had markedly improved. Using time ROC in 5-years survival circumstances, we found that the label had an excellent prediction effect (Figures 3D–F). In GSE31210 and GSE50081, the AUC was 0.773 and 0.656, respectively, and the AUC was 0.647 in the TCGA.
The association was demonstrated between the model and clinical information through the chi-square test in Table 3. There was a significant relationship between the pathological stage and the model (P < 0.05) in the GEO independent dataset rather than the TCGA dataset. Besides, there were 401 females in 754 cases, accounting for 53.18% of the total. A multivariate Cox test was utilized to determine if the signature had an independent prognostic value as a factor. The results in Table 4 showed that the signature was a risk factor, and it was statistically significant (high- vs. low-risk, GSE31210, HR = 16.24, 95% CI 3.85–68.58, P < 0.001, n = 226; GSE50081, HR = 2.23, 95% CI 1.24–4.02, P = 0.008, n = 128; TCGA, HR = 1.50, 95% CI 1.03–2.18, P = 0.036, n = 400). Univariate Cox also indicated that the signature was a risk factor.
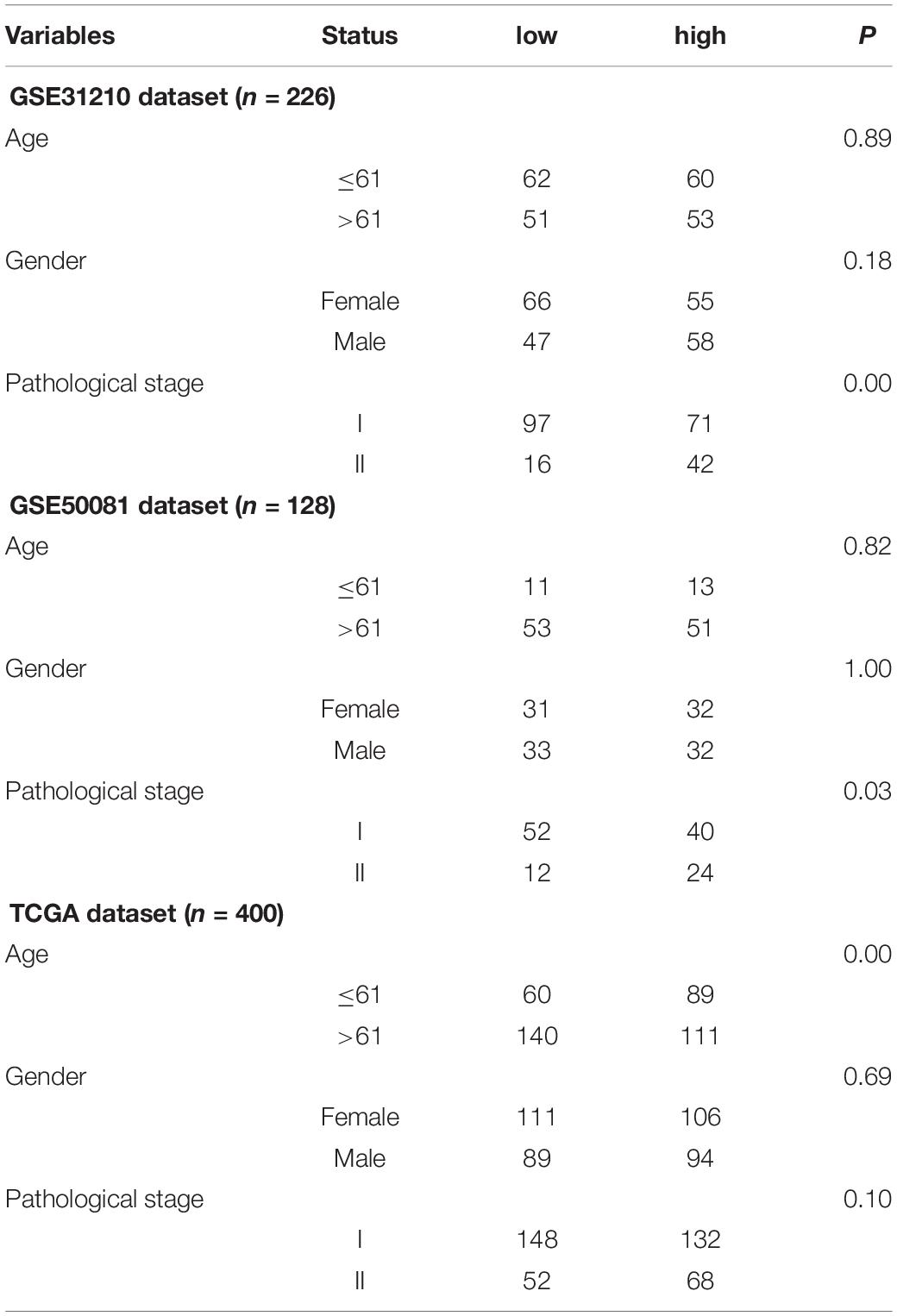
Table 3. Clinical information and signature Chi-square table.
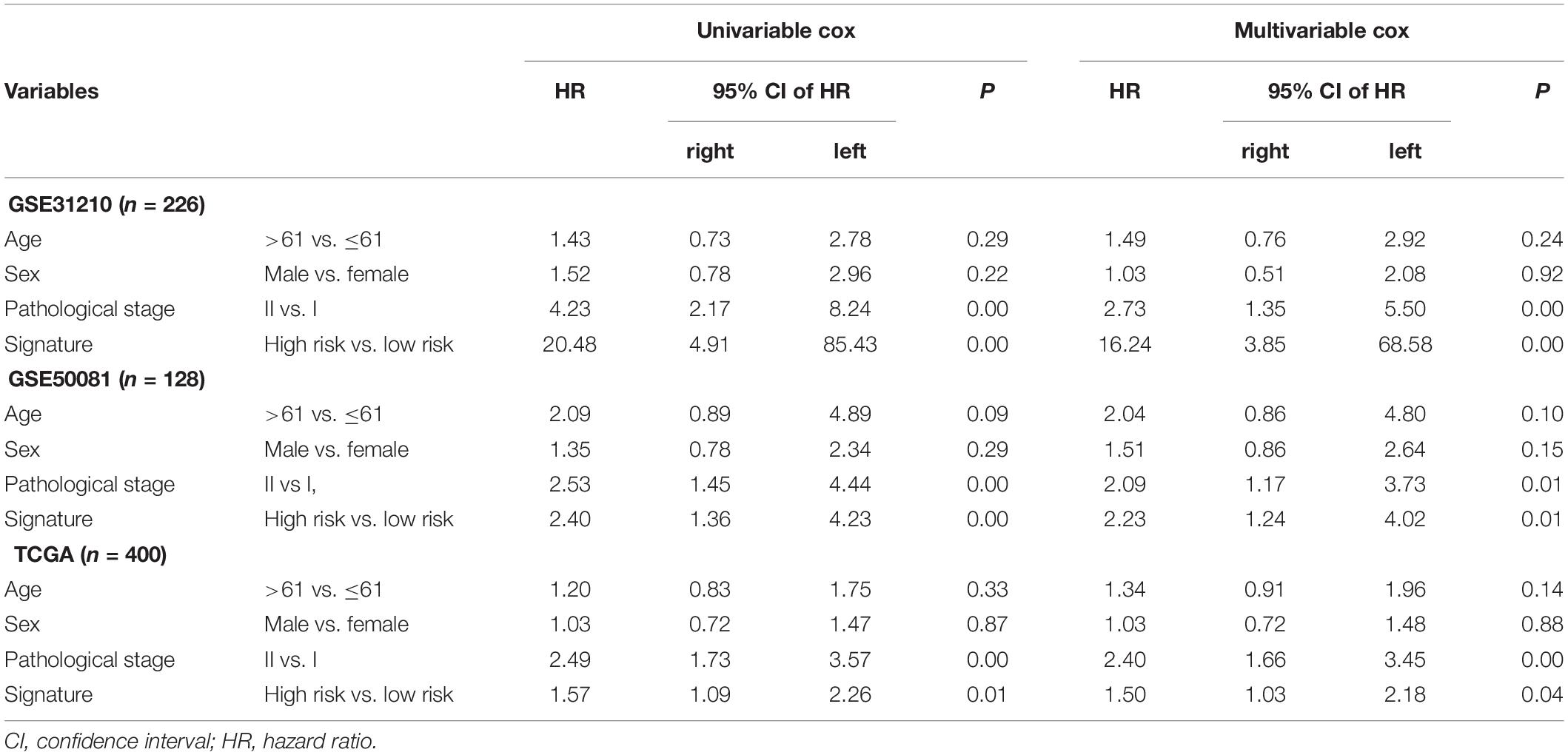
Table 4. Univariable and multivariable Cox regression analysis of the signature and clinical information with lung adenocarcinoma (LUAD) survival.
A 218 gene set, obtained from survival analysis and AUC analysis, was used for functional annotation and PPI network analysis. Among the 218 genes, including m6A RNA methylation regulatory factors, ElAVL1, METTL14, and YTHDC2 were significantly associated with OS in LUAD. Top 10 biological processes (BPs) and cellular components (CCs) were selected by functional annotation of 218 genes, among which several results of BPs were related to division (nuclear division, organelle fission) and regulation (regulation of mRNA metabolic process and regulation of chromosome organization). The primary outcome of CCs was linked to the chromosome (Figures 4A,B).
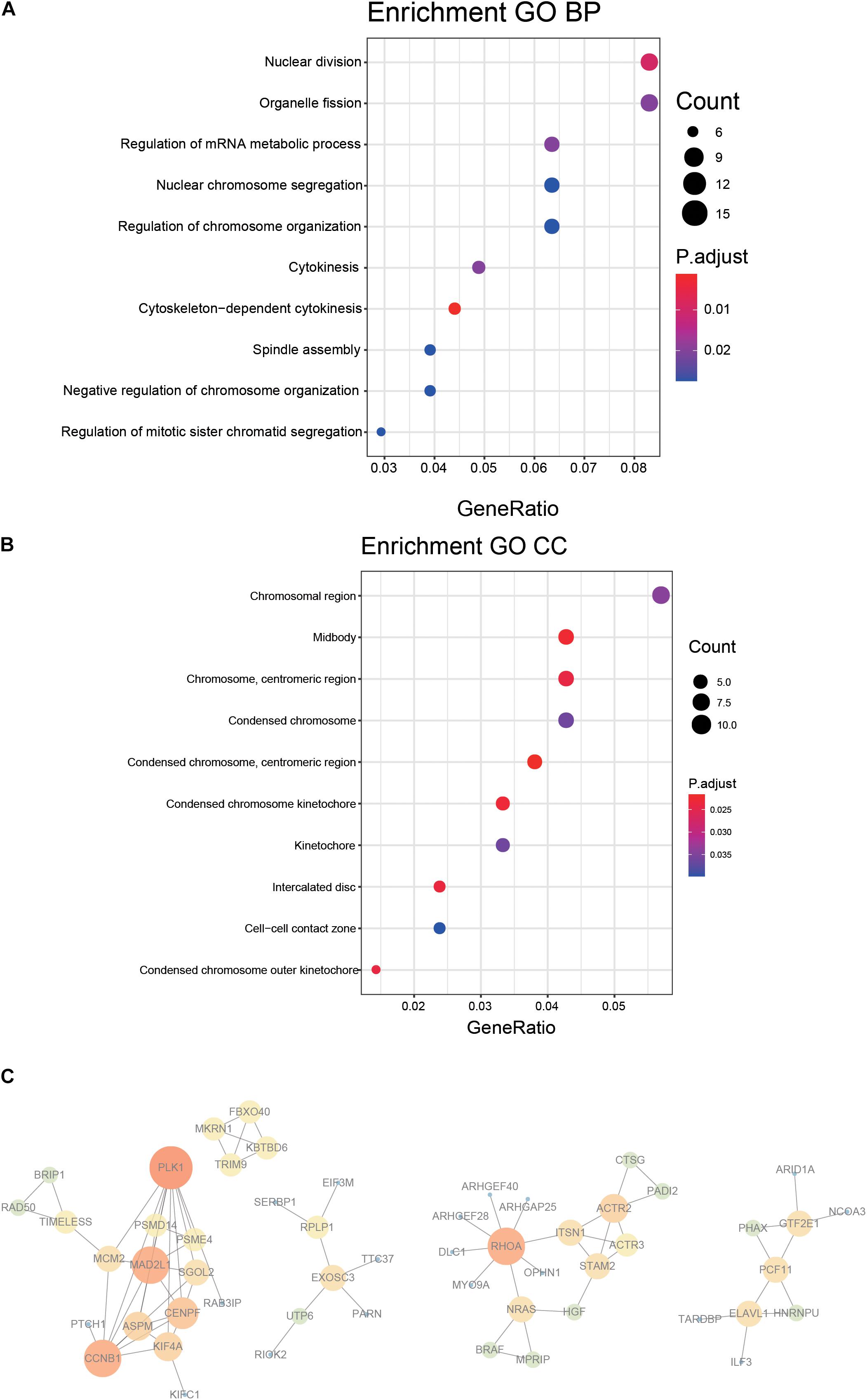
Figure 4. Functional annotation and protein–protein interaction for the genes with significant prognosis. (A,B) Function prediction (BP, biological process; CC, cellular component). (C) Protein–protein interaction.
PPI network was constructed by STRING, generated and visualized in Cytoscape. The combined score of the PPI criteria was >0.9. The PPI network had 88 relationships, and some genes were removed that were not part of the network (Figure 4C). Many key genes were observed in the network, such as PLK1, CCNB1, MAD2L1, RHOA, and ACTR2.
The results of external validation data were consistent with our results. In LUAD, two genes YTHDC2 and ADH1C were lowly expressed in the three sets of independent data (Figure 5A), which was almost the same in both the TIMER database (Figure 6) and the GEPIA database (Supplementary Figure 2). Interestingly, the aberrant expression of the three genes were frequently observed in various cancers and showed some tissue-dependent patterns. For example, ZCRB1 was overexpressed in lymphoma, and ADH1C in cervical cancer and esophageal cancer, and YTHDC2 in breast cancer, gastric cancer, head and neck cancer, myeloma, and sarcoma (Figure 5A).
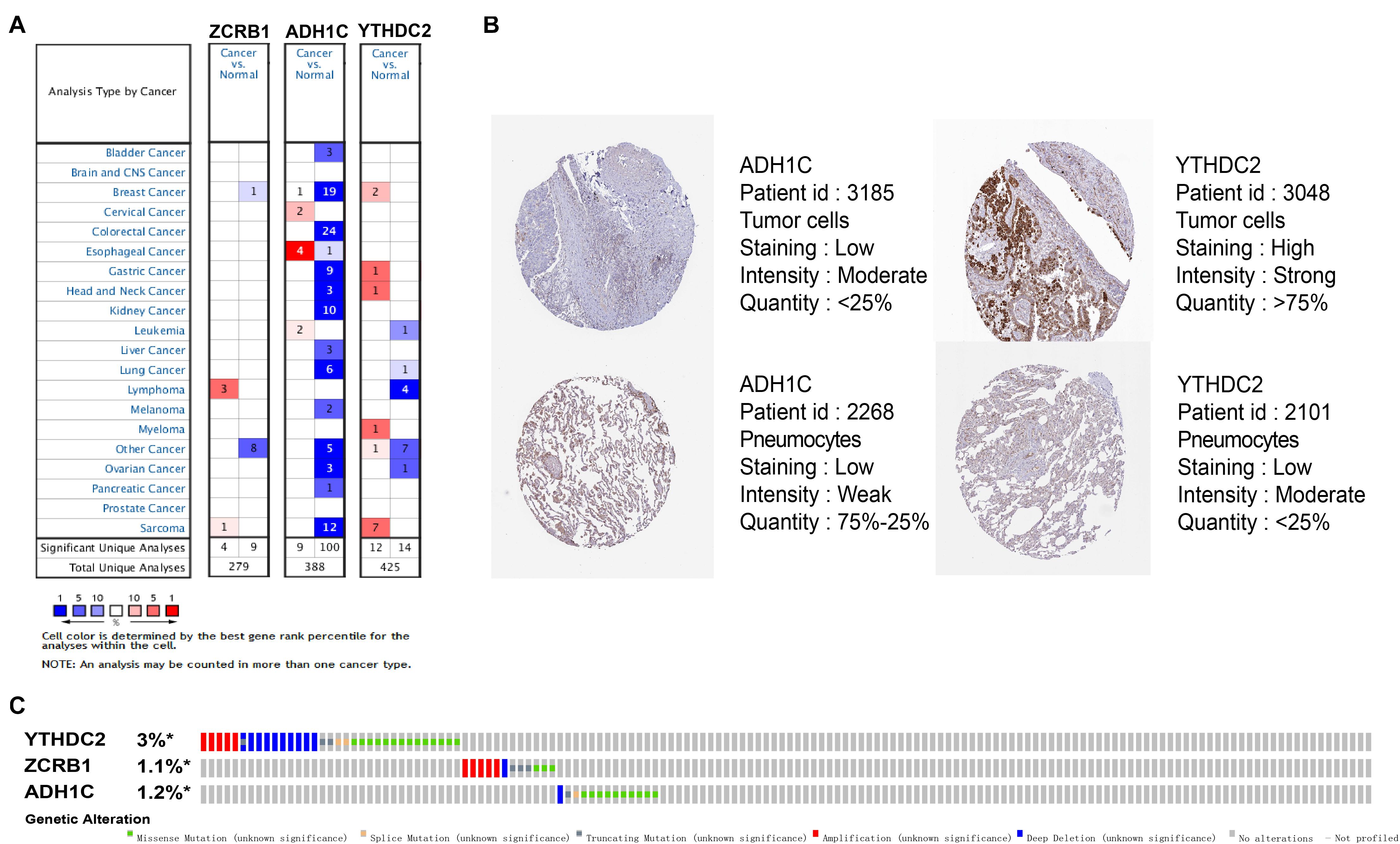
Figure 5. Expression and genetic alterations of the three predictive genes. (A) The expression profiles of the three genes in the Oncomine database. (B) The representative protein expression of the three genes in LUAD and normal lung tissue in the Human Protein Atlas database. Data of ZCRB1 were not found in the database. (C) Genetic alterations of the three genes in LUAD in the cBioportal for Cancer Genomics.
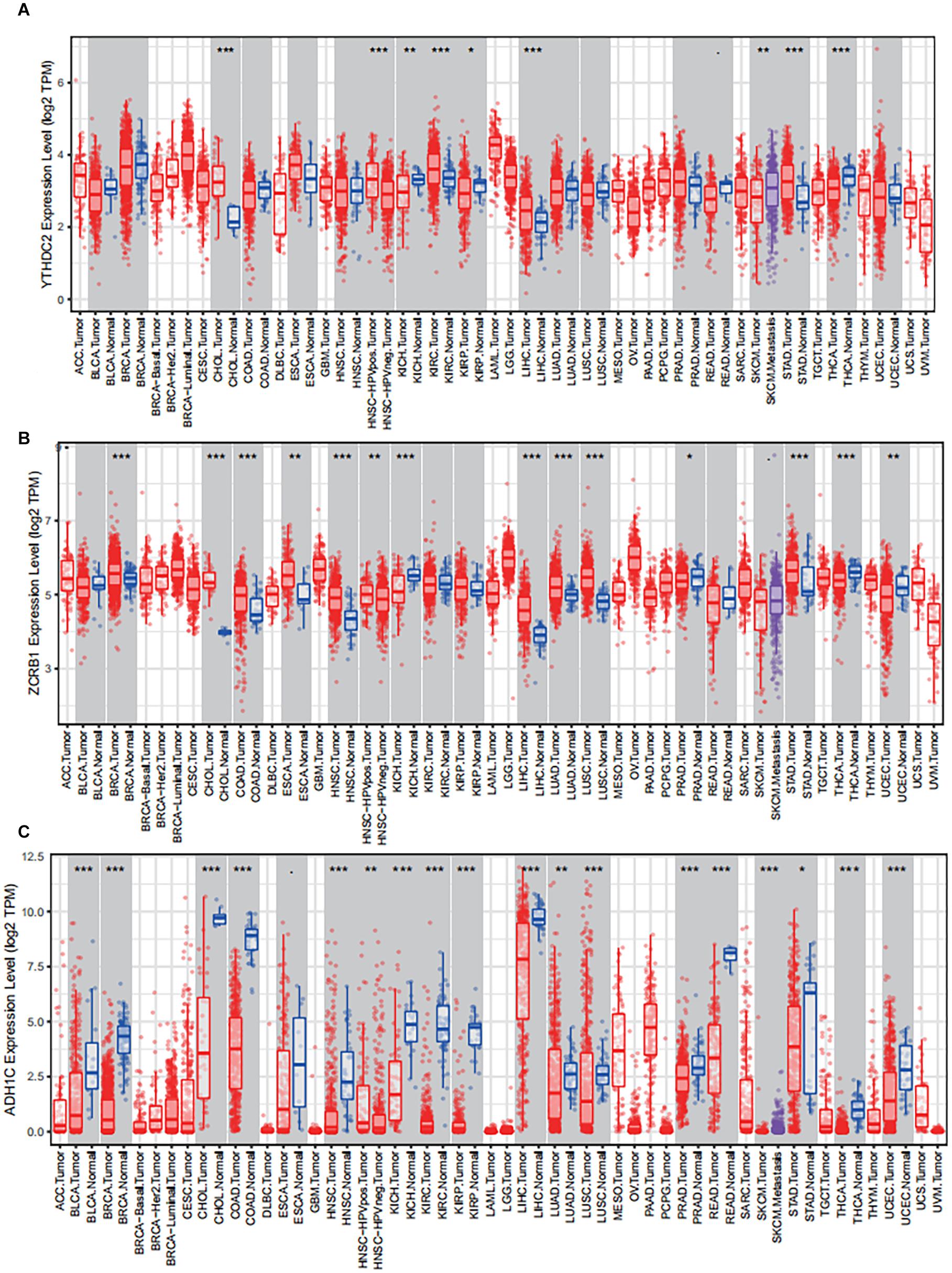
Figure 6. The expression of the three predictive genes in cancers via Tumor IMmune Estimation Resource (TIMER, https://cistrome.shinyapps.io/timer/). (A) YTHDC2 expression level in tumor tissues vs normal tissues. (B) ZCRB1 expression level in tumor tissues vs normal tissues. (C) ADH1C expression level in tumor tissues vs normal tissues. ACC, adrenocortical carcinoma; BLCA, bladder carcinoma; BRCA, breast carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, cholangio carcinoma; COAD, colon adenocarcinoma; DLBC, lymphoid neoplasm diffuse large B-cell lymphoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LAML, acute myeloid leukemia; LGG, brain lower grade glioma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUAC, lung squamous cell carcinoma; MESO, mesothelioma; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; TGCT, testicular germ cell tumors; THCA, thyroid carcinoma; THYM, thymoma; UCEC, uterine corpus endometrial carcinoma; UCS, uterine carcinosarcoma; UVM, uveal melanoma.
Survival analyses for each gene in the signature (ZCRB1, ADH1C, and YTHDC2) were performed in the cohorts of GSE31210, GSE50081, and TCGA datasets (Figure 7). ZCRB1 low-expression patient group displayed more OS than ZCRB1 high-expression patient group in GSE31210. While, ADH1C and YTHDC2 high-expression patient group displayed more OS than low-expression patient group not only in GSE31210 but also in GSE50081 and TCGA dataset.
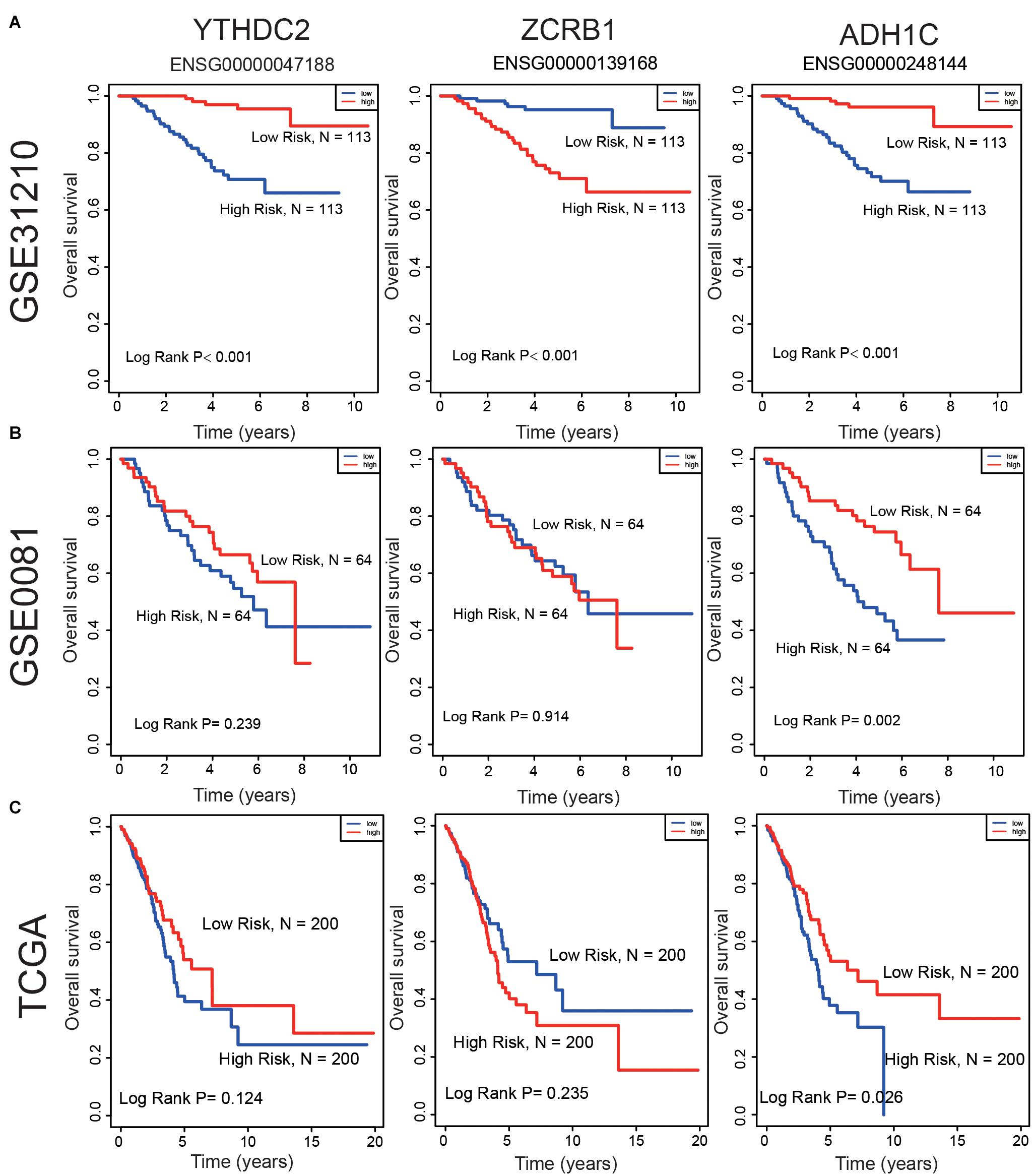
Figure 7. Compare the low and high expression of the three predictive genes in overall survival in (A) GSE31210 dataset, (B) GSE50081 dataset, and (C) TCGA dataset.
We then reviewed the proteomic data and found YTHDC2 protein was reported significantly underexpressed in non-small cell lung cancer (Sun et al., 2020). The representative protein expression of ADH1C and YTHDC2 was explored in the human protein profiles and is shown in Figure 5B. Nevertheless, ZCRB1 was not found in the HPA website. YTHDC2 possessed the most frequent genetic alterations (3%) among the three genes. Meanwhile, amplification mutation, missense mutation, and deep deletion were the most common alterations among the three genes (Figure 5C). In summary, we further verified the abnormal expression of these three genes in LUAD, and genetic changes may help explain the aberrant expression of these genes to a certain extent.
At the post-transcriptional level, more than 160 kinds of chemical modifications were discovered in various RNAs (Roundtree et al., 2017; Boccaletto et al., 2018). Among these modifications, more and more evidence showed that m6A modification had an essential effect on some underlying diseases and prognosis of tumors. Therefore, the identification of m6A RNA methylation regulators and m6A-related genes in fatal LUAD may offer valuable therapeutic targets to us and clinicians. Doctors usually diagnosed LUAD as advanced, and there was a high death rate with it. Many studies illuminated that the m6A process was linked to lung cancer, which made m6A RNA methylation regulators and m6A-related genes potential biomarkers for clinical practice. According to our research, the classification of m6A-related genes in LUAD patients was in association with prognosis. We identified a signature that consisted of one m6A RNA methylation regulator (YTHDC2) and two m6A-related genes (ZCRB1 and ADH1C) using different statistical and machine learning methods.
Up to now, little is known about the role of YTHDC2 in tumorigenesis. Even less so in LUAD, two studies were found on the role of YTHDC2 in LUAD in recent studies. In a mouse model, low YTHDC2 expression was associated with poor prognosis in LUAD patients, and YTHDC2 improves the prognosis of LUAD patients by inhibiting the independent antioxidant function of SLC7A11 (Ma et al., 2021). In non-small cell lung cancer, a research analyzed a series of publicly available online databases and found that low YTHDC2 expression was associated with lymph node metastasis and poor prognosis (Sun et al., 2020).
ZCRB1 is a zinc finger CCHC-type and RNA binding motif containing 1. A previous study found that it was U12-type splicing playing a pivotal role by RefSeq analysis. However, the function of ZCRB1 was rarely reported in cancer, only in two studies. For example, ZCRB1’s high expression can improve viral replication and transcription (Tan et al., 2012). Through genome-wide analysis of lung adenocarcinoma and healthy subjects, it was found that ZCRB1 may encode viral receptors. COVID-19 has infected plenty of people around the world, and ZCRB1 high expression may impact patients’ prognosis (Cotroneo et al., 2021).
ADH1C is alcohol dehydrogenase 1C (class I). Many reports showed that drinking had an effect on some diseases. High expression of ADH1C was found to protect patients of non-small cell lung cancer (Wang et al., 2018). Using machine learning algorithms, the researchers found that ADH1C could be a prognostic marker (Shen et al., 2019).
For the reversible effect of m6A on mRNA expression, we believe that m6A-related genes may have different functional patterns and networks when participating in malignant tumors. Thus, m6A-related genes may have different expression patterns in LUAD. In previous research, little was known about the interaction of m6A-related genes. Moreover, m6A RNA methylation regulators (WTAP, RBM15, KIAA1429, YTHDF1, and YTHDF2) were linked with TP53 and highly expressed in TP53 mutant LUAD (Zhuang et al., 2020). However, it is worth nothing that whether the TP53 mutant affects the expression of ZCRB1, ADH1C, and YTHDC2 is still unclear, and more evidence is needed to clarify their mechanism.
In conclusion, our study systematically analyzed the expression, prognostic value, protein–protein interaction, and potential function of m6A RNA methylation regulators and m6A-related genes. We found that the expression of m6A RNA methylation regulators and m6A-related genes was closely related to the clinicopathologial characteristics of LUAD. A three-gene signature was identified that might effectively identify new therapeutic targets or strategies for LUAD. In summary, our study provided important clues for further studies on the role of RNA m6A methylation regulators and m6A-related genes in LUAD.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
CY, XJ, and WZ: study conception and design. BG, HZ, and CY: manuscript writing. BG, RW, and CY: literature review. All authors: data interpretation, discussion, final editing, and approval of the manuscript in its present form.
This work was supported by Medical Big Data and AI R&D Project of the Chinese PLA General Hospital (2019MBD-025 and 2019MBD-001), National Natural Science Foundation of China (81902495), and Beijing Natural Science Foundation (7212099).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank all individuals who took part in this research.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.656114/full#supplementary-material
Supplementary Figure 1 | Flowchart of the study.
Supplementary Figure 2 | The three predictive genes expression levels in LUAD. Data was from the GEPIA database. T, tumor; N, normal tissue.
Supplementary Table 1 | The list of the 21 m6A RNA methylation regulators from publications.
Supplementary Table 2 | m6A methylation regulators and m6A-related genes set of univariate cox regression analysis in the GSE31210 dataset (P < 0.01, AUC > 0.6, n = 226).
Supplementary Table 3 | 255 signatures in the GSE31210 dataset (n = 226).
m6A, N6-methyladenosine; LUAD, lung adenocarcinoma; ROC, receiver operating characteristic; GEO, Gene Expression Omnibus; TCGA, The Cancer Genome Atlas; m1A, N1-methyladenosine; RSFVH, random survival forest algorithm; lncRNAs, long chain non-coding RNA; OS, overall survival; GO, gene ontology; HR, hazard ratio; CI, confidence interval; KM, Kaplan–Meier.
Boccaletto, P., Machnicka, M. A., Purta, E., Piatkowski, P., Baginski, B., Wirecki, T. K., et al. (2018). MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 46, D303–D307. doi: 10.1093/nar/gkx1030
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424. doi: 10.3322/caac.21492
Cheng, M., Sheng, L., Gao, Q., Xiong, Q., Zhang, H., Wu, M., et al. (2019). The m(6)A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-kappaB/MYC signaling network. Oncogene 38, 3667–3680.
Cotroneo, C. E., Mangano, N., Dragani, T. A., and Colombo, F. (2021). Lung expression of genes putatively involved in SARS-CoV-2 infection is modulated in cis by germline variants. Eur. J. Hum. Genet. 1–18. doi: 10.1038/s41431-021-00831-y
Fan, Z., Gao, S., Chen, Y., Xu, B., Yu, C., Yue, M., et al. (2018). Integrative analysis of competing endogenous RNA networks reveals the functional lncRNAs in heart failure. J. Cell. Mol. Med. 22, 4818–4829. doi: 10.1111/jcmm.13739
Han, J., Wang, J. Z., Yang, X., Yu, H., Zhou, R., Lu, H. C., et al. (2019). METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol. Cancer 18:110. doi: 10.1186/s12943-019-1036-9
He, L., Li, H., Wu, A., Peng, Y., Shu, G., and Yin, G. (2019). Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 18:176. doi: 10.1186/s12943-019-1109-9
Ishwaran, H., Kogalur, U. B., Blackstone, E. H., and Lauer, M. S. (2008). Random survival forests. Ann. Appl. Stat. 2, 841–860. doi: 10.1214/08-aoas169
Lan, T., Li, H., Zhang, D., Xu, L., Liu, H., Hao, X., et al. (2019). KIAA1429 contributes to liver cancer progression through N6-methyladenosine-dependent post-transcriptional modification of GATA3. Mol. Cancer 18:186. doi: 10.1186/s12943-019-1106-z
Liu, T., Wei, Q., Jin, J., Luo, Q., Liu, Y., Yang, Y., et al. (2020). The m6A reader YTHDF1 promotes ovarian cancer progression via augmenting EIF3C translation. Nucleic Acids Res. 48, 3816–3831. doi: 10.1093/nar/gkaa048
Ma, L., Chen, T., Zhang, X., Miao, Y., Tian, X., Yu, K., et al. (2021). The m(6)A reader YTHDC2 inhibits lung adenocarcinoma tumorigenesis by suppressing SLC7A11-dependent antioxidant function. Redox Biol. 38:101801. doi: 10.1016/j.redox.2020.101801
Ma, S., Chen, C., Ji, X., Liu, J., Zhou, Q., Wang, G., et al. (2019). The interplay between m6A RNA methylation and noncoding RNA in cancer. J. Hematol. Oncol. 12:121. doi: 10.1186/s13045-019-0805-7
Ma, Z., and Ji, J. (2020). N6-methyladenosine (m6A) RNA modification in cancer stem cells. Stem Cells. 1–9. doi: 10.1002/stem.3279
Niu, Y., Lin, Z., Wan, A., Chen, H., Liang, H., Sun, L., et al. (2019). RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol. Cancer 18:46. doi: 10.1186/s12943-019-1004-4
Roundtree, I. A., Evans, M. E., Pan, T., and He, C. (2017). Dynamic RNA Modifications in Gene Expression Regulation. Cell 169, 1187–1200. doi: 10.1016/j.cell.2017.05.045
Shen, X. Y., Liu, X. P., Song, C. K., Wang, Y. J., Li, S., and Hu, W. D. (2019). Genome-wide analysis reveals alcohol dehydrogenase 1C and secreted phosphoprotein 1 for prognostic biomarkers in lung adenocarcinoma. J. Cell Physiol. 234, 22311–22320. doi: 10.1002/jcp.28797
Sun, S., Han, Q., Liang, M., Zhang, Q., Zhang, J., and Cao, J. (2020). Downregulation of m(6) A reader YTHDC2 promotes tumor progression and predicts poor prognosis in non-small cell lung cancer. Thorac. Cancer 11, 3269–3279. doi: 10.1111/1759-7714.13667
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. doi: 10.1093/nar/gky1131
Tan, Y. W., Hong, W., and Liu, D. X. (2012). Binding of the 5′-untranslated region of coronavirus RNA to zinc finger CCHC-type and RNA-binding motif 1 enhances viral replication and transcription. Nucleic Acids Res. 40, 5065–5077. doi: 10.1093/nar/gks165
Tang, B., Yang, Y., Kang, M., Wang, Y., Wang, Y., Bi, Y., et al. (2020). m(6)A demethylase ALKBH5 inhibits pancreatic cancer tumorigenesis by decreasing WIF-1 RNA methylation and mediating Wnt signaling. Mol. Cancer 19:3. doi: 10.1186/s12943-019-1128-6
Wang, P., Zhang, L., Huang, C., Huang, P., and Zhang, J. (2018). Distinct prognostic values of alcohol dehydrogenase family members for non-small cell lung cancer. Med. Sci. Monit. 24, 3578–3590. doi: 10.12659/MSM.910026
Wang, X., Lu, Z., Gomez, A., Hon, G. C., Yue, Y., Han, D., et al. (2014). N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120. doi: 10.1038/nature12730
Xiao, S., Cao, S., Huang, Q., Xia, L., Deng, M., Yang, M., et al. (2019). The RNA N(6)-methyladenosine modification landscape of human fetal tissues. Nat. Cell Biol. 21, 651–661. doi: 10.1038/s41556-019-0315-4
Zhang, B., Wu, Q., Li, B., Wang, D., Wang, L., and Zhou, Y. L. (2020). m(6)A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol. Cancer 19:53. doi: 10.1186/s12943-020-01170-0
Zhang, C., Zhang, M., Ge, S., Huang, W., Lin, X., Gao, J., et al. (2019). Reduced m6A modification predicts malignant phenotypes and augmented Wnt/PI3K-Akt signaling in gastric cancer. Cancer Med. 8, 4766–4781. doi: 10.1002/cam4.2360
Zheng, Y., Nie, P., Peng, D., He, Z., Liu, M., Xie, Y., et al. (2018). m6AVar: a database of functional variants involved in m6A modification. Nucleic Acids Res. 46, D139–D145. doi: 10.1093/nar/gkx895
Keywords: lung adenocarcinoma, m6A, prognostic signature, m6A-related genes, RNA methylation regulators
Citation: Guo B, Zhang H, Wang J, Wu R, Zhang J, Zhang Q, Xu L, Shen M, Zhang Z, Gu F, Zeng W, Jia X and Yin C (2021) Identification of the Signature Associated With m6A RNA Methylation Regulators and m6A-Related Genes and Construction of the Risk Score for Prognostication in Early-Stage Lung Adenocarcinoma. Front. Genet. 12:656114. doi: 10.3389/fgene.2021.656114
Received: 20 January 2021; Accepted: 21 April 2021;
Published: 11 June 2021.
Edited by:
Suman Ghosal, National Institutes of Health (NIH), United StatesReviewed by:
Rituparno Sen, Leipzig University, GermanyCopyright © 2021 Guo, Zhang, Wang, Wu, Zhang, Zhang, Xu, Shen, Zhang, Gu, Zeng, Jia and Yin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chengliang Yin, Y2hlbmdsaWFuZ3lpbkAxNjMuY29t; Xiaodong Jia, ZmVpeGlhbmcuNTQyMEAxNjMuY29t; Weiliang Zeng, emVuZ3dsQGhyYm51LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.