 Lan Jiang
Lan Jiang Cheng Song
Cheng Song Xi Zhu
Xi Zhu Jianke Yang
Jianke Yang- 1Central Laboratory, Yijishan Hospital of Wannan Medical College, Wuhu, China
- 2Key Laboratory of Non-coding RNA Transformation Research of Anhui Higher Education Institution, Yijishan Hospital of Wannan Medical College, Wuhu, China
- 3College of Biological and Pharmaceutical Engineering, West Anhui University, Luan, China
- 4Department of Medicine III, University Hospital, LMU Munich, Munich, Germany
- 5School of Preclinical Medicine, Wannan Medical College, Wuhu, China
Tea (Camellia sinensis) is an important economic beverage crop. Its flowers and leaves could be used as healthcare tea for its medicinal value. SWEET proteins were recently identified in plants as sugar transporters, which participate in diverse physiological processes, including pathogen nutrition, seed filling, nectar secretion, and phloem loading. Although SWEET genes have been characterized and identified in model plants, such as Arabidopsis thaliana and Oryza sativa, there is very little knowledge of these genes in C. sinensis. In this study, 28 CsSWEETs were identified in C. sinensis and further phylogenetically divided into four subfamilies with A. thaliana. These identified CsSWEETs contained seven transmembrane helixes (TMHs) which were generated by an ancestral three-TMH unit with an internal duplication experience. Microsynteny analysis revealed that the large-scale duplication events were the main driving forces for members from CsSWEET family expansion in C. sinensis. The expression profiles of the 28 CsSWEETs revealed that some genes were highly expressed in reproductive tissues. Among them, CsSWEET1a might play crucial roles in the efflux of sucrose, and CsSWEET17b could control fructose content as a hexose transporter in C. sinensis. Remarkably, CsSWEET12 and CsSWEET17c were specifically expressed in flowers, indicating that these two genes might be involved in sugar transport during flower development. The expression patterns of all CsSWEETs were differentially regulated under cold and drought treatments. This work provided a systematic understanding of the members from the CsSWEET gene family, which would be helpful for further functional studies of CsSWEETs in C. sinensis.
Introduction
SWEET is a novel kind of low-affinity sugar transporter, which does not depend on the pH value of the environment and transports in two directions along the concentration gradient (Yuan and Wang, 2013). The members of the SWEET family are widely distributed, which are ubiquitous in higher eukaryotes and also distributed in protozoa, metazoa, fungi, bacteria, and archaea (Hamada et al., 2005; Saier et al., 2006; Xuan et al., 2013). The membrane proteins encoded by the SWEET have a certain number of conserved transmembrane domains, which are named MtN3/saliva (Hamada et al., 2005). This domain was first found in nodulin from the roots of Medicago sativa (Gamas et al., 1996). Recently, phylogenetic analysis has been performed for the main protein members of the SWEET family, which are divided into three branches: (i) the plant-like SWEET, most of which contain two MtN3/saliva transmembrane domains, (ii) the animal-like SWEET, which has two specific MtN3/saliva domains, and (iii) some SWEET proteins from bacteria to Archaea (cocci) and nematodes, which consist of an MtN3/saliva domain containing three transmembrane alpha helices (Yuan and Wang, 2013). A large number of SWEET family members have been found in Arabidopsis thaliana (Wipf et al., 2020), Vitis vinifera (Chong et al., 2014), Solanum lycopersicum (Shammai et al., 2018), Manihot esculenta (Cao et al., 2019), Eucalyptus grandis (Yin et al., 2020), etc., and are involved in many physiological processes. For instance, RPG1 (ATSWEET8) plays a vital role in microspore outer wall formation in Arabidopsis (Guan et al., 2008); in Oryza sativa, the SWEET family members are also involved in reproductive development (Wang et al., 2010); SAG29 (AtSWEETl5), located on the plasma membrane in Arabidopsis, can regulate cell activity in a hypersaline environment (Seo et al., 2011). The SWEETs also participate in the regulation of the aging process. The overexpression of OsSWEET5 can cause growth delay and premature aging in rice seedlings, while no phenotypic changes were observed in the knockout ones (Zhou et al., 2014). Chu et al. (2006) found that the rice harboring mutant OsSWEET11 (Xa13) can resist powdery mildew, suggesting that SWEETs not only served as a glucose transporter but also participated in the interaction between host and pathogen. However, the members of the CsSWEET gene family have not yet been identified, and the roles of CsSWEETs remain unclear in tea plant (Camellia sinensis).
Camellia sinensis, which originated from the tropical area of Southwest China, is an important economic beverage crop in China (Wei et al., 2018; Zhang et al., 2020b). Sugar transport and homeostasis contribute to plant growth and development (May et al., 1998; Lastdrager et al., 2014; Rahimi et al., 2019; Pan et al., 2020; Saddhe et al., 2020). Some published papers focused on the genes that participated in sugar metabolism, such as hexose kinase, invertase, and galactinol synthase, in C. sinensis (Yue et al., 2015; Zhou et al., 2017; Samarina et al., 2020). Due to the economic interest in C. sinensis as a beverage crop, its genome, proteome, and transcriptome were recently sequenced and released (Wei et al., 2018; Wu et al., 2018; Liu et al., 2020; Xia et al., 2020; Zhang et al., 2020b), which help us to analyze the SWEET genes in C. sinensis systematically. In the present study, 26 CsSWEETs in C. sinensis were identified, and their gene structures, phylogenetic, microsynteny, and expression were analyzed. Our study revealed the functions of these CsSWEETs and provided candidate genes for further research.
Materials and Methods
Database Search for C. sinensis Genome
The C. sinensis genome with GFF annotation, CDS, and protein files were downloaded from the Tea Plant Information Archive1 (TPIA) database (Xia et al., 2019). The AtSWEETs of A. thaliana and HsSWEET1 of Homo sapiens were obtained from TAIR2 and Genbank3, respectively. The HMM profile of MtN3/saliva domain (PF03083) was obtained from the Pfam database4 (Mistry et al., 2020), and the HMMER (version 3.1) software (Mistry et al., 2013) was used to identify CsSWEET proteins in C. sinensis genome (E-value ≤ 1e−3). The Pfam (version 33.1), SMART (version 9), and InterPro (version 5) were used to confirm the above-mentioned identified sequences with E-value ≤ 1e−3 (Jones et al., 2014; Letunic and Bork, 2018; Mistry et al., 2020). Finally, we manually discarded these sequences with a lack of complete or core MtN3/saliva domain for further analyses.
Phylogenetic Analyses
The MAFFT (version 7) software was used to execute the multiple alignments of all SWEET proteins with default parameters (Katoh et al., 2005). The best substitution model of these SWEET proteins was determined by ModelFinder software (Kalyaanamoorthy et al., 2017). The maximum likelihood (ML) tree was determined by IQ-tree (version 2.1.2) software (Nguyen et al., 2015) with an SH-aLRT test for 1,000 random addition replicates and a bootstrap test for 10,000 replicates as described by Cao et al. (2020a). The Figtree software was used to visualize this ML tree.
Internal Repeats and Topological Analyses
The ClustalX (version 2) software (Thompson et al., 2003) was used to create the multiple alignments of SWEETs from C. sinensis and A. thaliana. The HHrepID was used to identify the internal repeats in SWEET proteins (Söding et al., 2006). In addition, we also used the AveHAS to calculate the topological conservation (Zhai and Saier, 2001a,b) as well as create the hydropathy, amphipathicity, and similarity plots, respectively (Zhai and Saier, 2001a). The GFF annotation file was obtained from the TPIA (see text footnote 1) database (Xia et al., 2019). The TBtools (version 1) software was used to generate this gene structure map based on the GFF annotation file (Chen et al., 2020). The MEME online tool was used to predict the conserved motifs (Bailey et al., 2015).
Microsynteny Analysis
The MicroSyn (version 1) software was used to detect the microsynteny of CsSWEET genes in C. sinensis genome with a threshold E-value of <1e−5 (Cai et al., 2011). In this study, we determined a syntenic block, a region which include three or more conserved homolog genes that were distributed within 15 genes upstream and downstream of CsSWEET genes as described by Cao et al. (2020b).
RNA-Seq Expression Analysis
In our study, transcriptome data from various tissues in the public database NCBI contained three biological replicates. The RNA-seq reads, including seed, flower, stem, root, two and a bud, one and a bud, old leaf, mature leaf, the second leaf, the first leaf, lateral bud, apical bud, early stage lateral bud, CP24 (24 h after pollination; CP, cross-pollinated), CP48, CP72, SP24 (SP, self-pollinated), SP48, and SP72, were obtained from NCBI (PRJNA291116 and PRJNA230752). The Trimmomatic (version 0.33) was used to carried out the quality-based trimming (Bolger et al., 2014). The HISAT2 was used to map the paired reads to the C. sinensis genome with default parameters (Kim et al., 2019). The StringTie was used to calculate the fragments per kilobase of exon model per million reads mapped values of differently expressed genes (Pertea et al., 2016).
Results and Discussion
The Phylogenetic Analysis Divides CsSWEET Genes Into Four Subfamilies in C. sinensis
In the present study, we identified 26 CsSWEET genes in C. sinensis genome by using HMMER 3.0 software, similarly as in Manihot esculenta, Jatropha curcas, Ricinus communis, Vernicia fordii, Malus × domestica, Oryza sativa, Zea mays, Glycine max, and Pyrus bretschneideri (Chen et al., 2010; Yuan and Wang, 2013; Chong et al., 2014; Cao et al., 2019; Yin et al., 2020). Subsequently, we renamed the newly identified CsSWEETs based on the nomenclature of the AtSWEETs of A. thaliana. The detailed information of each CsSWEET, including chromosome position, gene identifiers, isoelectric point, molecular weight, and gene name, is shown in Table 1.
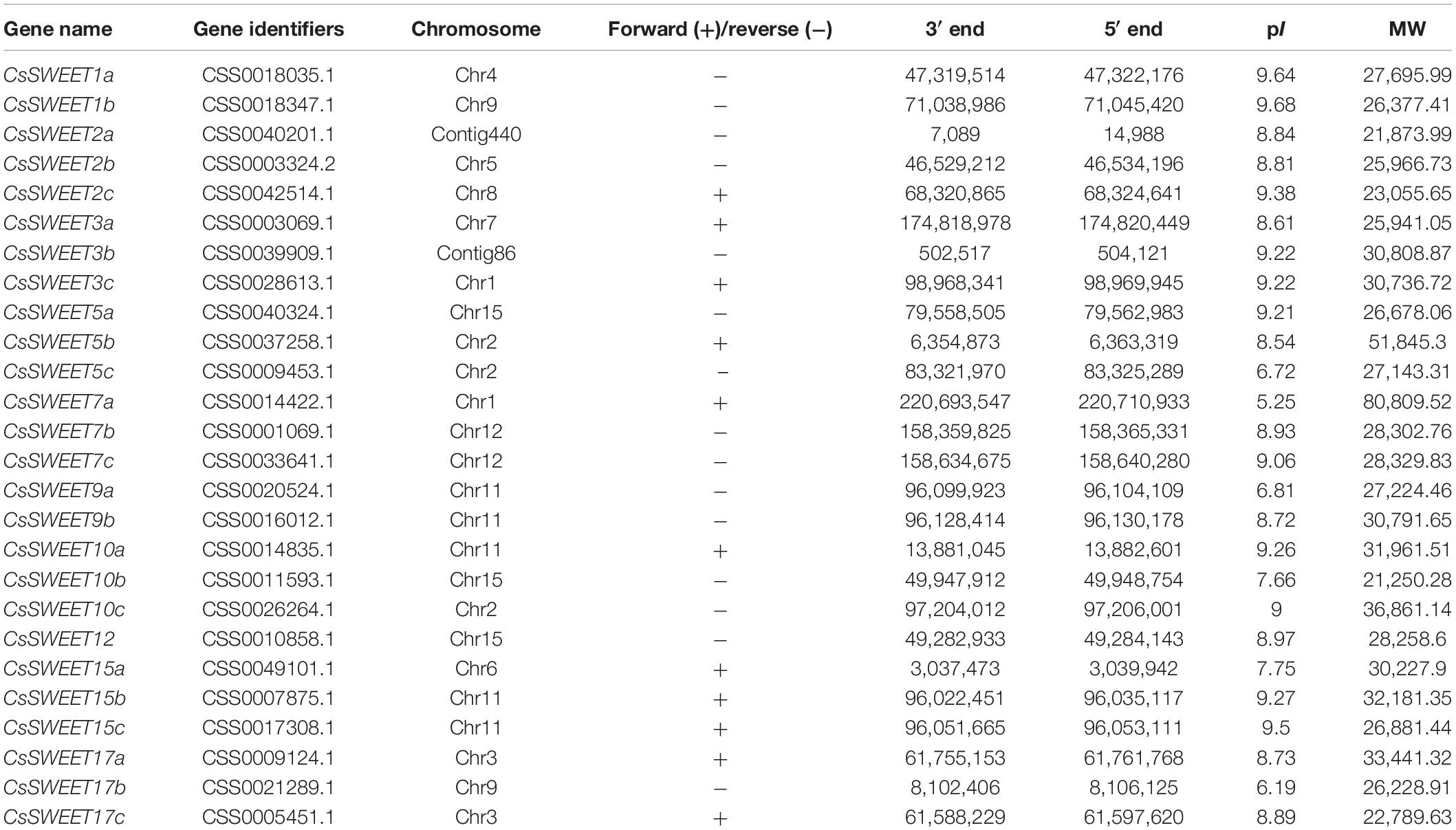
Table 1. Detailed information of SWEET family members in C. sinensis.
To gain insight into the evolutionary relationship of CsSWEETs in C. sinensis, the SWEETs from C. sinensis and A. thaliana were aligned by MAFFT software. Remarkably, HsSWEET1 from Homo sapiens was an outgroup, which has only one copy and could transport glucose in H. sapiens. In V. fordii, Cao et al. (2019) found that VfSWEET1 contributes to the balance of sucrose levels, and other VfSWEETs may play key roles in the growth and development of plants (Cao et al., 2019). In A. thaliana, AtSWEETs were identified with versatile functions, such as control fructose content and sucrose efflux (Chen et al., 2010, 2012). In our study, the SWEETs from C. sinensis and A. thaliana were clustered into four subfamilies, including subfamily I, subfamily II, subfamily III, and subfamily IV (Figure 1). Subsequently, there was at least one AtSWEET from A. thaliana in each subfamily. Previously published papers have confirmed that members from different SWEET subfamilies may have multiple biological functions, such that the members from subfamily I and subfamily II were efficient hexose transporters (Figure 1), the members from subfamily III may be responsible for sucrose transporters, and the members from subfamily IV appear to be predominantly fructose transporters (Chen et al., 2010, 2012; Hu et al., 2018; Cao et al., 2019).
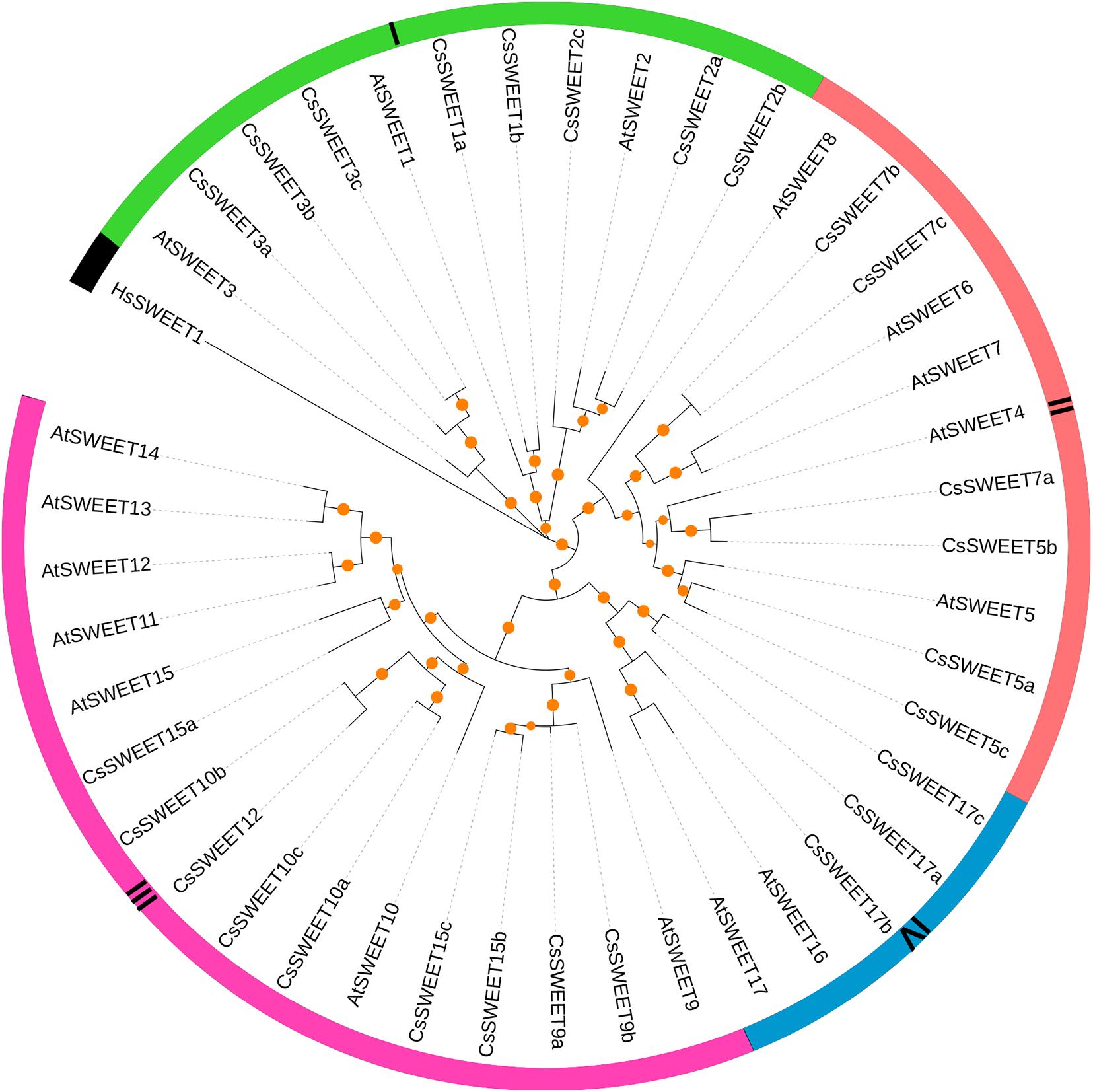
Figure 1. The phylogenetic relationships of SWEET genes in Camellia sinensis, Arabidopsis thaliana, and Homo sapiens. The maximum likelihood tree was built by IQ-tree software with bootstrap test (10,000 replicates).
The Structure Analyses Reveal the Distribution of Exon–Intron and the Conserved Composition of Motif in C. sinensis
According to the predicted sequences and GFF annotation files, we determined the distribution of exon–intron of the 26 CsSWEET genes in C. sinensis. As shown in Supplementary Figure 1, most CsSWEET genes contained five introns; however, several of the members from the CsSWEET gene family contained two, three, or four introns. Remarkably, there are extreme differences in the number of introns from the CsSWEET subfamily II, ranging from 4 to 14, indicating that these CsSWEETs may have experienced intron loss or gain some during evolution (Supplementary Figure 1). These results were also confirmed by previous studies. For example, Cao et al. (2019) found that the number of introns from VfSWEETs ranged from four to 10. Additionally, we found that, although the lengths of members from CsSWEET gene family vary, introns stepped into almost the same position of the gene open reading frame.
The MEME website was used to identify the sequence characteristics of CsSWEET genes. Subsequently, we determined 20 distinct motifs for CsSWEETs and annotated these motifs by Pfam and SMART database (Letunic and Bork, 2018; Mistry et al., 2020). The SWEET domain (MtN3/saliva) was encoded by motifs 1, 2, 3, and 4 (Supplementary Figure 2). However, the remaining motifs were not scanned for function annotations in Pfam and SMART database. Overall, the CsSWEET proteins within the same subfamily exhibited similar conserved motifs, especially in paralogs gene pairs, suggesting that these sequences might contain similar functions in C. sinensis.
Internal Repeat and Transmembrane Domain Analysis of the CsSWEETs Reveal Insight Into Key Functional Residues
In general, SWEET proteins have been confirmed to contain seven transmembrane helixes (TMHs) in eukaryotes (Feng and Frommer, 2015; Jia et al., 2017). As expected, the membrane topology of CsSWEETs and AtSWEETs revealed that these proteins contained a moderately amphipathic nature with seven well-conserved hydrophobicity peaks (TMH1–TMH7) as determined by AveHAS plot (Figures 2A,B). Compared with TMHs, the loop area between two TMHs is not conservative in this study (Figure 2C). Previously published manuscripts have shown that the internal fusion and duplication of small fragments play an important role in the SWEETs evolution, which has been the focus of discussion by researchers (Hu et al., 2018; Cao et al., 2019). Researchers have studied the internal duplication of the SWEET proteins (Li et al., 2017; Hu et al., 2018; Cao et al., 2019), but direct evidence of the CsSWEETs from C. sinensis has not been reported. To confirm the presence of internal duplication, a repeat analysis of CsSWEETs and AtSWEETs was performed by the HHrepID program (Söding et al., 2006). The results confirmed that TMH1–TMH3 and TMH5–TMH7 were located in the duplicated regions, implying that SWEETs originate from an ancestral three-TMH unit which experienced an internal repeat duplication, which is in agreement with previously published papers (Li et al., 2017; Hu et al., 2018; Cao et al., 2019).
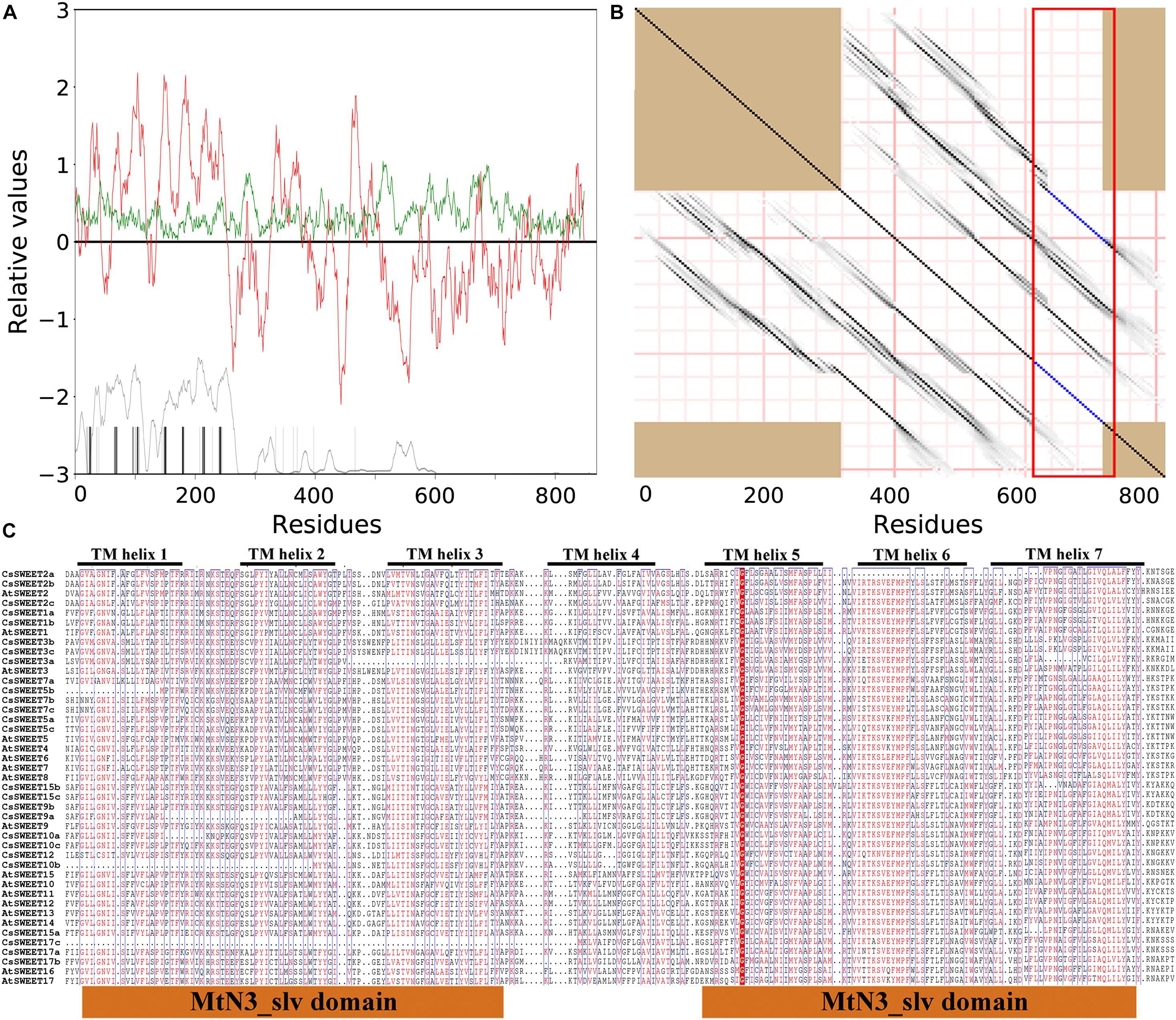
Figure 2. Sequence characteristics of SWEETs in Camellia sinensis. (A) The red upper lines and green upper lines indicate hydrophobicity and amphipathicity, respectively. (B) Internal repeat of SWEETs. Above the dark diagonal lines suggests the duplicated regions. (C) Alignment of SWEET sequences. The highly conserved residues are represented by boxes.
Li et al. (2017) revealed that Y, P, and D from TMH2, TMH5, and TMH7 were fully conserved residues in pear, respectively (Li et al., 2017). Among these three residues, Y mutation into A will lead to the loss of transport function in A. thaliana (Xuan et al., 2013). Proline (P) is a key component of TMHs and allows for dynamic processes during the transport cycle as described by Deber and Therien (2002). In the present study, we suggested that these three residues (Y, P, and D) might contain important functions within the activity of CsSWEETs in C. sinensis. In addition, the mutation of 58 amino acid residues G into D can significantly reduce AtSWEET1 activity in A. thaliana (Xuan et al., 2013). However, this amino acid is not completely conserved in C. sinensis, including the residue G in CsSWEET12 that had changed to N and in CsSWEET15a that had changed to K, but these proteins also have transport activity (Figure 2C), which has been verified by RNA-seq. We also noted that the first and the second MtN3/saliva domain included TMH1–TMH3 and TMH5–TMH7, respectively.
Microsynteny Analysis Indicates That Large-Scale Duplication Events Contribute to the Expansion for CsSWEET Genes in C. sinensis
The genome of C. sinensis experienced two rounds of whole-genome duplications (WGDs), including an ancient WGD event (∼90 to 100 Mya) and a recent WGD event (∼30 to 40 Mya) (Wei et al., 2018; Wu et al., 2018; Liu et al., 2020; Xia et al., 2020; Zhang et al., 2020b). To gain insight into the expansion mechanism of the members from CsSWEET gene family, we carried out a microsynteny analysis with E-value 10−5 as described by Cao et al. (2020b). In our study, CsSWEET3c/CsSWEET3a, CsSWEET10c/CsSWEET12, CsSWEET10c/CsSWEET10b, CsSW EET7b/CsSWEET7c, CsSWEET3c/CsSWEET3b, CsSWEET3a/C sSWEET3b, CsSWEET7a/CsSWEET5c, and CsSWEET2b/Cs SWEET2a contained more than three pairs of conserved flanking genes, suggesting that significant microsynteny might occur in the CsSWEET genes (Figure 3). These results indicated that the large-scale duplication events contribute to the expansion for CsSWEET genes in C. sinensis.
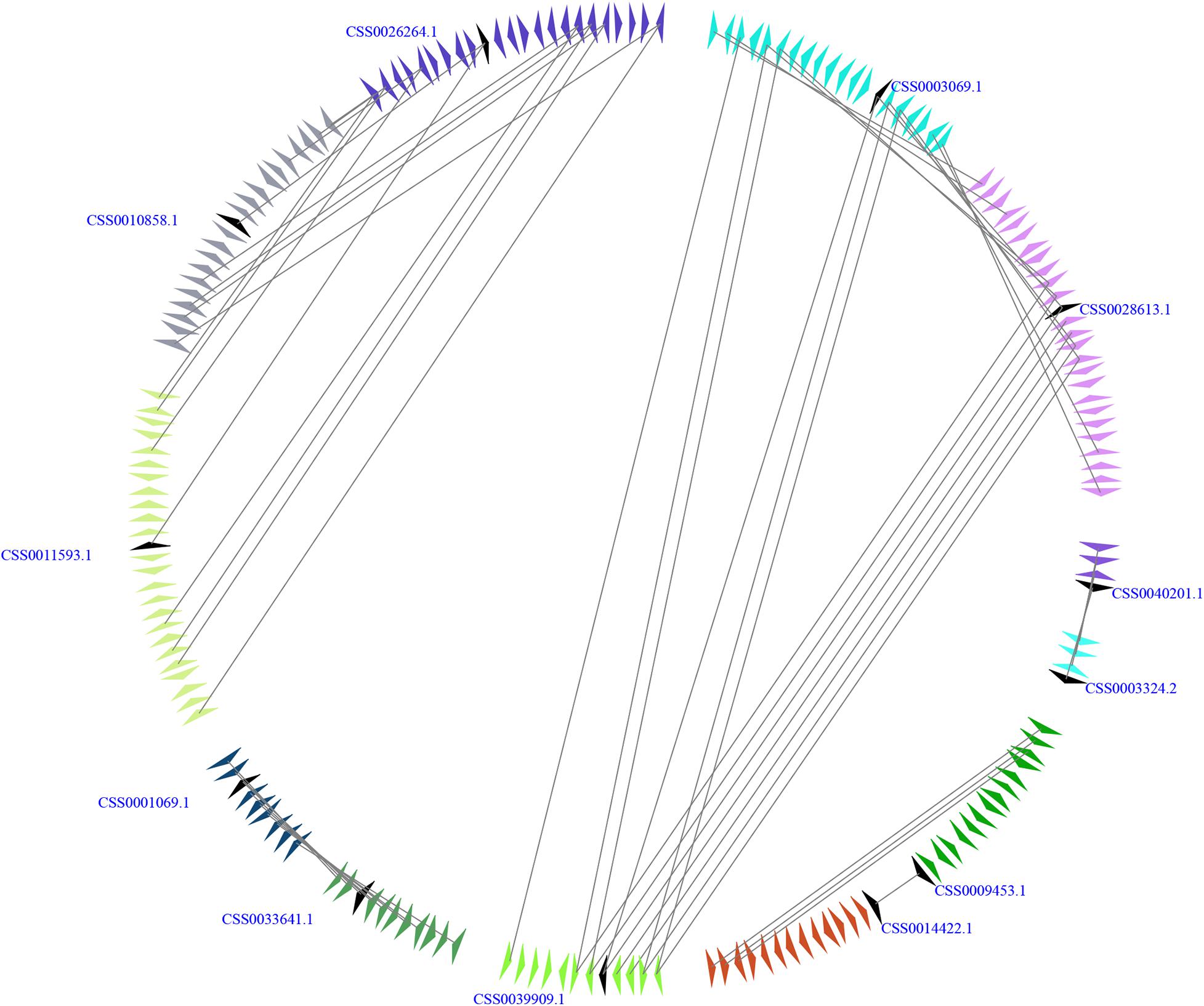
Figure 3. Microsynteny relationships of SWEET genes in Camellia sinensis. A series of triangles indicate genomic fragments and the genes’ orientations. The duplicated gene pairs on the two fragments are linked by gray lines.
Generally, the evolutionary data of WGD events were estimated by Ks values (Wang et al., 2011; Tiley et al., 2018; Zwaenepoel and Van De Peer, 2019). As shown in Table 1, we found that the Ks values of CsSWEET duplication pairs ranged from 0.0103 to 2.1158 (Supplementary Table 1 and Supplementary Figure 3). Subsequently, we found that CsSWEET3c/CsSWEET3a, CsSWEET3a/CsSWEET3b, and CsSWEET2b/CsSWEET2a might be generated through a recent WGD event, while CsSWEET3c/CsSWEET3b and CsSWEET10c/CsSWEET12 might be generated through an ancient WGD event, suggesting that these two rounds of WGD events might play key roles in the expansion for CsSWEET genes in C. sinensis.
The Expression of CsSWEETs Reveals Possible Functions in C. sinensis
Previously published work provided transcriptome data for CsSWEET genes in C. sinensis (Xia et al., 2019; Xia et al., 2020). The members from SWEET gene family are found to play diverse functional roles in various tissues and contribute to different sugar transport mechanisms in plants (Chen, 2014; Hedrich et al., 2015; Li et al., 2017; Cao et al., 2019). Differential expression analysis of CsSWEETs in C. sinensis is helpful for us to find out the specialized functions of these CsSWEETs in sugar transport from the practical application point of view. In the present study, we collected transcriptome data from 19 different tissues, including seed, flower, stem, root, two and a bud, one and a bud, old leaf, mature leaf, the second leaf, the first leaf, lateral bud, apical bud, early stage lateral bud, CP24 (24 h after pollination; CP, cross-pollinated), CP48, CP72, SP24 (SP, self-pollinated), SP48, and SP72. The published papers indicated that the expression of SWEETs in seeds pollen, flower, and embryo sacs in Sorghum bicolor, V. fordii, Litchi chinensis, O. sativa, and A. thaliana was higher (Yuan and Wang, 2013; Mizuno et al., 2016; Cao et al., 2019; Xie et al., 2019), implying that these genes may contribute to reproductive development. Here CsSWEET1a that was clustered into the same subfamily with VfSWEET1 from V. fordii, PbSWEET14 from P. bretschneideri, and AtSWEET1 from A. thaliana was extremely highly expressed in the leaf of C. sinensis. PbSWEET14 and VfSWEET1 contribute to the efflux of sucrose in leaves (Li et al., 2017; Cao et al., 2019), while AtSWEET1 plays an important role as a single glucose transporter in multiple systems. Therefore, the CsSWEET1a might play crucial roles in the efflux of sucrose and act as a glucose uniporter in the leaves of C. sinensis (Figure 4). AtSWEET16 and AtSWEET17 were shown to be vacuolar hexose transporters that controlled fructose content (Chen et al., 2010). The CsSWEET17b, which is the orthologous gene of AtSWEET16 and AtSWEET17, was extremely highly expressed in the stems of C. sinensis, suggesting that this gene could control fructose content as a hexose transporter. The expression patterns of CsSWEET2a, CsSWEET5b, and CsSWEET7a were diverse in several tissues, implying that these CsSWEETs might play a role in these tested tissues of C. sinensis (Figure 4).
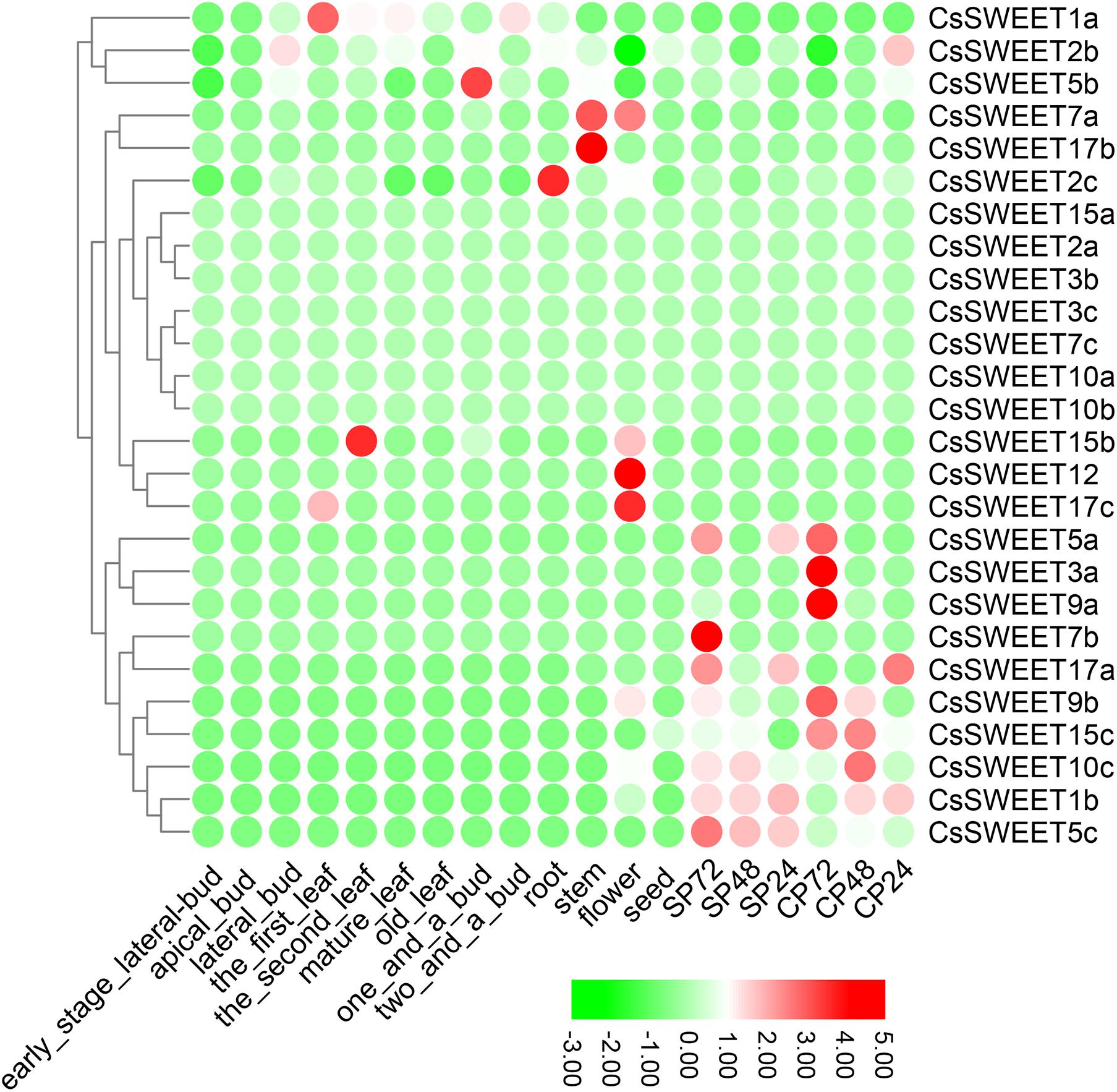
Figure 4. Expression profiles of the CsSWEETs in different tissues, including seed, flower, stem, root, two and a bud, one and a bud, old leaf, mature leaf, the second leaf, the first leaf, lateral bud, apical bud, and early stage lateral bud. CP24 = 24 h after pollination; CP, cross-pollinated – CP48, CP72; SP24 (SP, self-pollinated), SP48, and SP72.
In C. sinensis, the expression of CsSWEETs was examined in pollen development. We found that 10 CsSWEETs, including CsSWEET5a, CsSWEET3a, CsSWEET9a, CsSWEET7b, CsSWEET17a, CsSWEET9b, CsSWEET15c, CsSWEET10c, CsSWEET1b, and CsSWEET5c, were highly expressed in one or several pollen stages of C. sinensis (Figure 4). As previously reported, Chen et al. (2012) identified AtSWEET11 and AtSWEET12 that contributed to phloem loading and pollen nutrition. Gao et al. (2018) found that the OsSWEET11 from O. sativa can greatly affect the starch pollen content. In our study, CsSWEET9b, CsSWEET9a, and CsSWEET10c are the orthologous genes of AtSWEET11 and AtSWEET12, indicating that these highly expressed genes may share similar roles and functions.
Stress-Induced Expression Profiles of CsSWEETs in C. sinensis
Plants often suffer from a variety of abiotic stresses in the process of growth and development (Franco et al., 2011; Keunen et al., 2013; Etesami, 2018). Previous studies have shown that SWEETs help to control the responses to environmental stresses in plants (Li et al., 2018; Zhang et al., 2020a). Therefore, the expression patterns of CsSWEETs were investigated in response to different stresses, including cold stress and drought stress. In our study, the RNA-seq data for cold stress and drought stress were obtained from the TPIA (see text footnote 1) database (Xia et al., 2019). In response to drought, seven genes (CsSWEET3a, CsSWEET7a, CsSWEET1a, CsSWEET10a, CsSWEET2a, CsSWEET7b, and CsSWEET2c) were up-regulated under polyethylene glycol (PEG) for 72 h. Compared with control, nine genes (CsSWEET2b, CsSWEET5b, CsSWEET15c, CsSWEET1b, CsSWEET9b, CsSWEET17a, CsSWEET17c, CsSWEET7c, and CsSWEET15b) were down-regulated under all PEG treatments (Figure 5). In response to cold, four genes (CsSWEET3b, CsSWEET1a, CsSWEET1b, and CsSWEET15c) were up-regulated under all cold treatments. Compared with control, nine genes (CsSWEET5b, CsSWEET2a, CsSWEET17b, CsSWEET15b, CsSWEET17a, CsSWEET7a, CsSWEET2b, CsSWEET3a, and CsSWEET17c) were down-regulated under all cold treatments (Figure 6). Among them, the expression of CsSWEET1a was significantly up-regulated in all chosen two treatments, indicating that this gene might help tea resist environmental stresses. This phenomenon was different from previous studies. For example, the banana MaSWEET1a was not induced by osmotic, cold, and salt stresses (Miao et al., 2017). The expression level of GhSWEET1 was only upregulated at 6 h under cold stress, but it was almost not expressed when induced by drought stress (Li et al., 2018). Taken together, we believed that tea plants might have developed specialized regulatory mechanisms for different abiotic stresses.
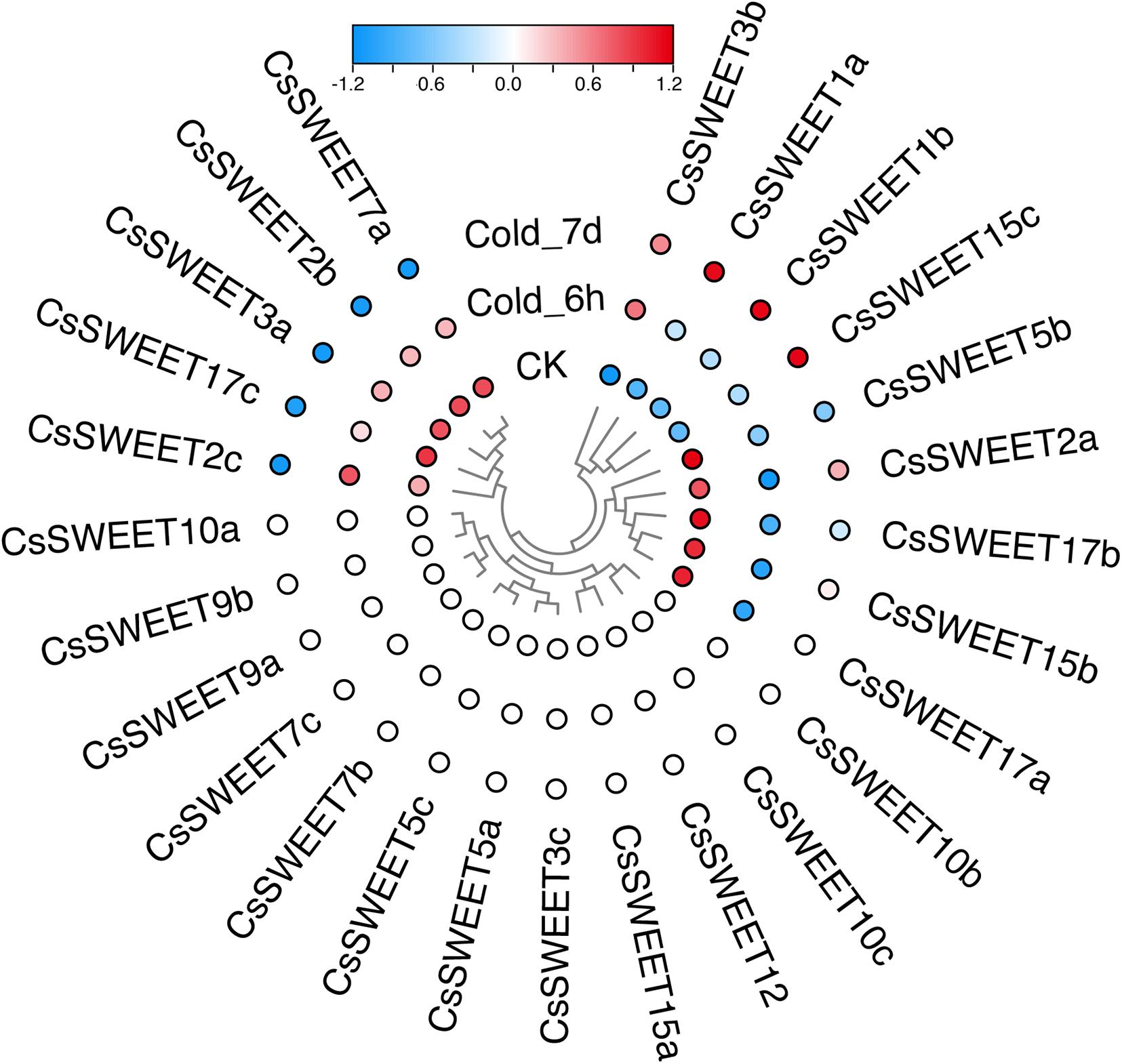
Figure 5. Expression profiles of the CsSWEETs in response to cold treatment.
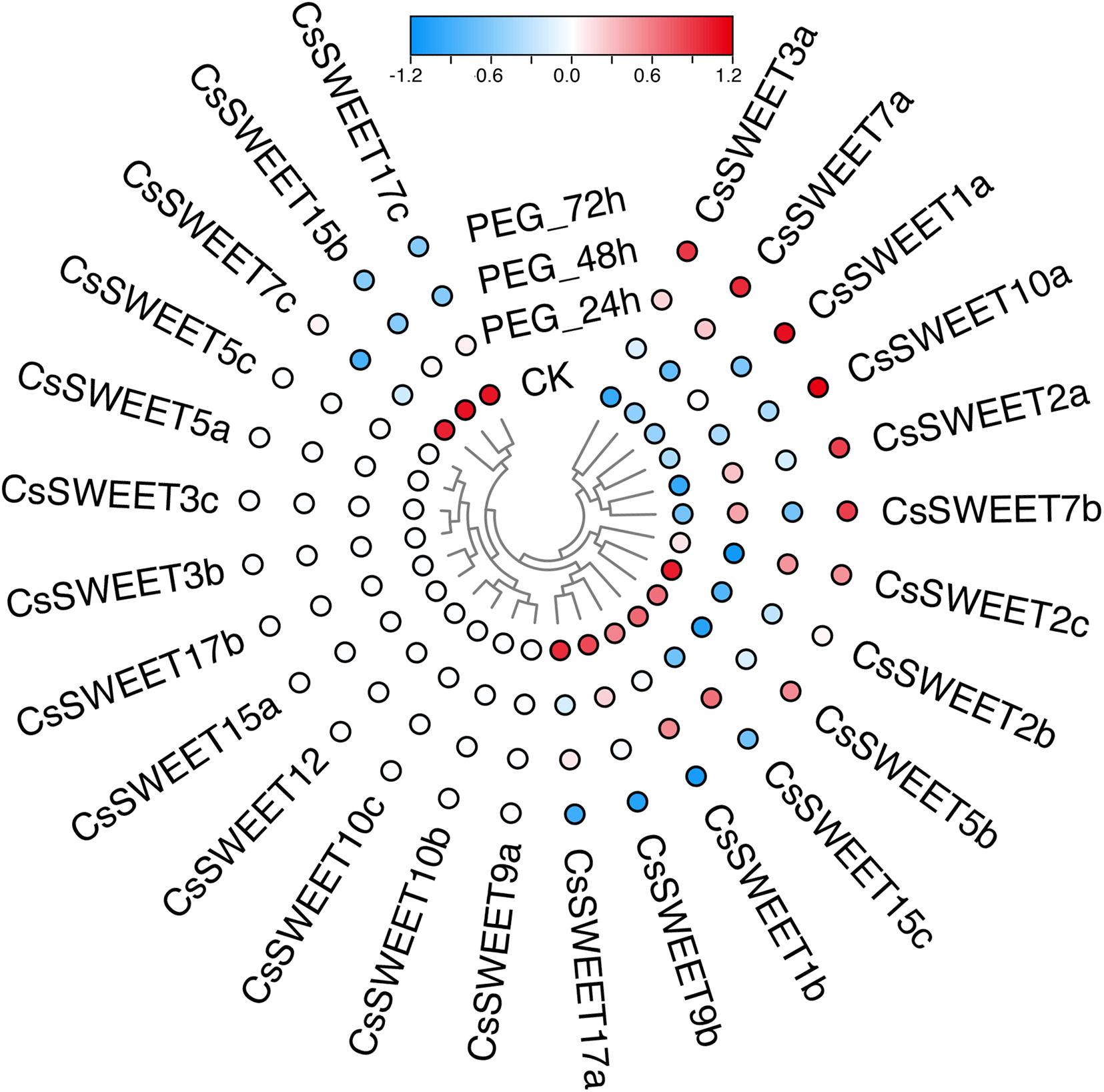
Figure 6. Expression profiles of the CsSWEETs in response to drought treatment.
Conclusion
In the present study, we identified 28 CsSWEETs in the C. sinensis genome. Further analyses for ML tree, intron–exon, and duplication suggested that the conservation of CsSWEETs was accompanied by a certain degree of divergence. The expression profiles of all CsSWEETs suggested that several genes were highly expressed in reproductive tissues, indicating that these genes played important roles in sugar transport. Additionally, 28 CsSWEETs were differently expressed for RNA-seq data under cold and drought stresses, indicating that tea plants might have developed specialized regulatory mechanisms for different abiotic stresses.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
LJ and XZ performed the experiments, analyzed the data, and wrote the manuscript. LJ, CS, XZ, and JY conceived the research and revised the manuscript. All the authors read and approved the final manuscript.
Funding
This work was supported by the Talent Scientific Research Start-up Foundation of Yijishan Hospital, Wannan Medical College (grant no. YR202001), the Opening Foundation of Key Laboratory of Non-coding RNA Transformation Research of Anhui Higher Education Institution (grant no. RNA202004), and the Key Projects of Natural Science Research of Universities in Anhui Province (grant no. KJ2020A0622).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.655843/full#supplementary-material
Supplementary Figure 1 | The gene structure of CsSWEETs in Camellia sinensis. Green boxes suggest exons, and gray lines indicate introns.
Supplementary Figure 2 | The distribution of conserved motifs in CsSWEETs of Camellia sinensis. The MEME was used to identify the motif. The different colors of the boxes indicate different motifs numbered 1–20.
Supplementary Figure 3 | Ka/Ks analysis for duplicated CsSWEETs paralogs.
Supplementary Table 1 | Ka/Ks analysis for duplicated CsSWEETs paralogs.
Footnotes
- ^ http://tpia.teaplant.org/
- ^ https://www.arabidopsis.org/
- ^ https://www.ncbi.nlm.nih.gov/
- ^ http://pfam.xfam.org
References
Bailey, T. L., Johnson, J., Grant, C. E., and Noble, W. S. (2015). The MEME suite. Nucleic Acids Res. 43, W39–W49.
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Cai, B., Yang, X., Tuskan, G. A., and Cheng, Z.-M. (2011). MicroSyn: a user friendly tool for detection of microsynteny in a gene family. BMC Bioinformatics 12:79. doi: 10.1186/1471-2105-12-79
Cao, Y., Liu, M., Long, H., Zhao, Q., Jiang, L., and Zhang, L. (2020a). Hidden in plain sight: Systematic investigation of Leucine-rich repeat containing genes unveil the their regulatory network in response to Fusarium wilt in tung tree. Int. J. Biol. Macromole. 163, 1759–1767. doi: 10.1016/j.ijbiomac.2020.09.106
Cao, Y., Liu, W., Zhao, Q., Long, H., Li, Z., Liu, M., et al. (2019). Integrative analysis reveals evolutionary patterns and potential functions of SWEET transporters in Euphorbiaceae. Int. J. Biol. Macromole. 139, 1–11. doi: 10.1016/j.ijbiomac.2019.07.102
Cao, Y., Xu, X., and Jiang, L. (2020b). Integrative analysis of the RNA interference toolbox in two Salicaceae willow species, and their roles in stress response in poplar (Populus trichocarpa Torr. & Gray). Int. J. Biol. Macromole. 162, 1127–1139. doi: 10.1016/j.ijbiomac.2020.06.235
Chen, C., Chen, H., Zhang, Y., Thomas, H. R., Frank, M. H., He, Y., et al. (2020). TBtools-an integrative toolkit developed for interactive analyses of big biological data. BioRxiv 2020:289660.
Chen, L. Q. (2014). SWEET sugar transporters for phloem transport and pathogen nutrition. N. Phytol. 201, 1150–1155. doi: 10.1111/nph.12445
Chen, L.-Q., Hou, B.-H., Lalonde, S., Takanaga, H., Hartung, M. L., Qu, X.-Q., et al. (2010). Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 468, 527–532.
Chen, L.-Q., Qu, X.-Q., Hou, B.-H., Sosso, D., Osorio, S., Fernie, A. R., et al. (2012). Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science 335, 207–211. doi: 10.1126/science.1213351
Chong, J., Piron, M.-C., Meyer, S., Merdinoglu, D., Bertsch, C., and Mestre, P. (2014). The SWEET family of sugar transporters in grapevine: VvSWEET4 is involved in the interaction with Botrytis cinerea. J. Exp. Bot. 65, 6589–6601. doi: 10.1093/jxb/eru375
Chu, Z., Yuan, M., Yao, J., Ge, X., Yuan, B., Xu, C., et al. (2006). Promoter mutations of an essential gene for pollen development result in disease resistance in rice. Genes Dev. 20, 1250–1255. doi: 10.1101/gad.1416306
Deber, C. M., and Therien, A. G. (2002). Putting the β-breaks on membrane protein misfolding. Nat. Struct. Biol. 9, 318–319. doi: 10.1038/nsb0502-318
Etesami, H. (2018). Can interaction between silicon and plant growth promoting rhizobacteria benefit in alleviating abiotic and biotic stresses in crop plants? Agricult. Ecosyst. Env. 253, 98–112. doi: 10.1016/j.agee.2017.11.007
Feng, L., and Frommer, W. B. (2015). Structure and function of SemiSWEET and SWEET sugar transporters. Trends Biochem. Sci. 40, 480–486. doi: 10.1016/j.tibs.2015.05.005
Franco, J. A., Bañón, S., Vicente, M. J., Miralles, J., and Martínez-Sánchez, J. J. (2011). Root development in horticultural plants grown under abiotic stress conditions–a review. J. Horticult. Sci. Biotechnol. 86, 543–556. doi: 10.1080/14620316.2011.11512802
Gamas, P., De Carvalho Niebel, F., Lescure, N., and Cullimore, J. V. (1996). Use of a subtractive hybridization approach to identify new Medicago truncatula genes induced during root nodule development. MPMI-Mole. Plant Microb. Interact. 9, 233–242. doi: 10.1094/mpmi-9-0233
Gao, Y., Zhang, C., Han, X., Wang, Z. Y., Ma, L., Yuan, D. P., et al. (2018). Inhibition of OsSWEET11 function in mesophyll cells improves resistance of rice to sheath blight disease. Mole. Plant Pathol. 19, 2149–2161. doi: 10.1111/mpp.12689
Guan, Y.-F., Huang, X.-Y., Zhu, J., Gao, J.-F., Zhang, H.-X., and Yang, Z.-N. (2008). RUPTURED POLLEN GRAIN1, a member of the MtN3/saliva gene family, is crucial for exine pattern formation and cell integrity of microspores in Arabidopsis. Plant Physiol. 147, 852–863. doi: 10.1104/pp.108.118026
Hamada, M., Wada, S., Kobayashi, K., and Satoh, N. (2005). Ci-Rga, a gene encoding an MtN3/saliva family transmembrane protein, is essential for tissue differentiation during embryogenesis of the ascidian Ciona intestinalis. Differentiation 73, 364–376. doi: 10.1111/j.1432-0436.2005.00037.x
Hedrich, R., Sauer, N., and Neuhaus, H. E. (2015). Sugar transport across the plant vacuolar membrane: nature and regulation of carrier proteins. Curr. Opin. Plant Biol. 25, 63–70. doi: 10.1016/j.pbi.2015.04.008
Hu, W., Hua, X., Zhang, Q., Wang, J., Shen, Q., Zhang, X., et al. (2018). New insights into the evolution and functional divergence of the SWEET family in Saccharum based on comparative genomics. BMC Plant Biol. 18:270.
Jia, B., Zhu, X. F., Pu, Z. J., Duan, Y. X., Hao, L. J., Zhang, J., et al. (2017). Integrative view of the diversity and evolution of SWEET and SemiSWEET sugar transporters. Front. Plant Sci. 8:2178.
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., Mcanulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., Von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh, K., Kuma, K.-I., Toh, H., and Miyata, T. (2005). MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518. doi: 10.1093/nar/gki198
Keunen, E. L. S., Peshev, D., Vangronsveld, J., Van Den Ende, W. I. M., and Cuypers, A. N. N. (2013). Plant sugars are crucial players in the oxidative challenge during abiotic stress: extending the traditional concept. Plant Cell Env. 36, 1242–1255. doi: 10.1111/pce.12061
Kim, D., Paggi, J. M., Park, C., Bennett, C., and Salzberg, S. L. (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. doi: 10.1038/s41587-019-0201-4
Lastdrager, J., Hanson, J., and Smeekens, S. (2014). Sugar signals and the control of plant growth and development. J. Exp. Bot. 65, 799–807. doi: 10.1093/jxb/ert474
Letunic, I., and Bork, P. (2018). 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 46, D493–D496.
Li, J., Qin, M., Qiao, X., Cheng, Y., Li, X., Zhang, H., et al. (2017). A new insight into the evolution and functional divergence of SWEET transporters in Chinese white pear (Pyrus bretschneideri). Plant Cell Physiol. 58, 839–850. doi: 10.1093/pcp/pcx025
Li, W., Ren, Z., Wang, Z., Sun, K., Pei, X., Liu, Y., et al. (2018). Evolution and stress responses of Gossypium hirsutum SWEET genes. Int. J. Mole. Sci. 19:769. doi: 10.3390/ijms19030769
Liu, Z.-W., Li, H., Liu, J.-X., Wang, Y., and Zhuang, J. (2020). Integrative transcriptome, proteome, and microRNA analysis reveals the effects of nitrogen sufficiency and deficiency conditions on theanine metabolism in the tea plant (Camellia sinensis). Horticulture Res. 7, 1–13.
May, M. J., Vernoux, T., Leaver, C., Montagu, M. V., and Inzé, D. (1998). Glutathione homeostasis in plants: implications for environmental sensing and plant development. J. Exp. Bot. 49, 649–667. doi: 10.1093/jexbot/49.321.649
Miao, H., Sun, P., Liu, Q., Miao, Y., Liu, J., Zhang, K., et al. (2017). Genome-wide analyses of SWEET family proteins reveal involvement in fruit development and abiotic/biotic stress responses in banana. Sci. Rep. 7, 1–15.
Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Salazar, G. A., Sonnhammer, E. L. L., et al. (2020). Pfam: The protein families database in 2021. Nucleic Acids Res. 8, D412–D419.
Mistry, J., Finn, R. D., Eddy, S. R., Bateman, A., and Punta, M. (2013). Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 41, e121–e121.
Mizuno, H., Kasuga, S., and Kawahigashi, H. (2016). The sorghum SWEET gene family: stem sucrose accumulation as revealed through transcriptome profiling. Biotechnol. Biofuels 9:127.
Nguyen, L.-T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mole. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Pan, J., Li, D., Zhu, J., Shu, Z., Ye, X., Xing, A., et al. (2020). Aluminum relieves fluoride stress through stimulation of organic acid production in Camellia sinensis. Physiol. Mole. Biol. Plants 26, 1127–1137. doi: 10.1007/s12298-020-00813-2
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., and Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protocols 11:1650. doi: 10.1038/nprot.2016.095
Rahimi, M., Kordrostami, M., and Mortezavi, M. (2019). Evaluation of tea (Camellia sinensis L.) biochemical traits in normal and drought stress conditions to identify drought tolerant clones. Physiol. Mole. Biol. Plants 25, 59–69. doi: 10.1007/s12298-018-0564-x
Saddhe, A. A., Manuka, R., and Penna, S. (2020). Plant sugars: homeostasis and transport under abiotic stress in plants. Physiol. Plantarum 2020:13283.
Saier, M. H. Jr., Tran, C. V., and Barabote, R. D. (2006). TCDB: the Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res. 34, D181–D186.
Samarina, L. S., Bobrovskikh, A. V., Doroshkov, A. V., Malyukova, L. S., Matskiv, A. O., Rakhmangulov, R. S., et al. (2020). Comparative Expression Analysis of Stress-Inducible Candidate Genes in Response to Cold and Drought in Tea Plant [Camellia sinensis (L.) Kuntze]. Front. Genet. 2020:11.
Seo, P. J., Park, J.-M., Kang, S. K., Kim, S.-G., and Park, C.-M. (2011). An Arabidopsis senescence-associated protein SAG29 regulates cell viability under high salinity. Planta 233, 189–200. doi: 10.1007/s00425-010-1293-8
Shammai, A., Petreikov, M., Yeselson, Y., Faigenboim, A., Moy-Komemi, M., Cohen, S., et al. (2018). Natural genetic variation for expression of a SWEET transporter among wild species of Solanum lycopersicum (tomato) determines the hexose composition of ripening tomato fruit. Plant J. 96, 343–357. doi: 10.1111/tpj.14035
Söding, J., Remmert, M., and Biegert, A. (2006). HHrep: de novo protein repeat detection and the origin of TIM barrels. Nucleic Acids Res. 34, W137–W142.
Thompson, J. D., Gibson, T. J., and Higgins, D. G. (2003). Multiple sequence alignment using ClustalW and ClustalX. Curr. Protocols Bioinform. 2003, 2–3.
Tiley, G. P., Barker, M. S., and Burleigh, J. G. (2018). Assessing the performance of Ks plots for detecting ancient whole genome duplications. Genome Biol. Evol. 10, 2882–2898.
Wang, L., Xie, W., Chen, Y., Tang, W., Yang, J., Ye, R., et al. (2010). A dynamic gene expression atlas covering the entire life cycle of rice. Plant J. 61, 752–766. doi: 10.1111/j.1365-313x.2009.04100.x
Wang, Y., Wang, X., Tang, H., Tan, X., Ficklin, S. P., Feltus, F. A., et al. (2011). Modes of gene duplication contribute differently to genetic novelty and redundancy, but show parallels across divergent angiosperms. PLoS One 6:e28150. doi: 10.1371/journal.pone.0028150
Wei, C., Yang, H., Wang, S., Zhao, J., Liu, C., Gao, L., et al. (2018). Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl. Acad. Sci. 115, E4151–E4158.
Wipf, D., Pfister, C., Mounier, A., Leborgne-Castel, N., Frommer, W. B., and Courty, P.-E. (2020). Identification of Putative Interactors of Arabidopsis Sugar Transporters. Trends Plant Sci. 26:009.
Wu, L.-Y., Fang, Z.-T., Lin, J.-K., Sun, Y., Du, Z.-Z., Guo, Y.-L., et al. (2018). Complementary iTRAQ proteomic and transcriptomic analyses of leaves in tea plant (Camellia sinensis L.) with different maturity and regulatory network of flavonoid biosynthesis. J. Proteom. Res. 18, 252–264.
Xia, E. H., Li, F. D., Tong, W., Li, P. H., Wu, Q., Zhao, H. J., et al. (2019). Tea Plant Information Archive: a comprehensive genomics and bioinformatics platform for tea plant. Plant Biotechnol. J. 17, 1938–1953. doi: 10.1111/pbi.13111
Xia, E., Tong, W., Hou, Y., An, Y., Chen, L., Wu, Q., et al. (2020). The reference genome of tea plant and resequencing of 81 diverse accessions provide insights into its genome evolution and adaptation. Mole. Plant 13, 1013–1026. doi: 10.1016/j.molp.2020.04.010
Xie, H., Wang, D., Qin, Y., Ma, A., Fu, J., Qin, Y., et al. (2019). Genome-wide identification and expression analysis of SWEET gene family in Litchi chinensis reveal the involvement of LcSWEET2a/3b in early seed development. BMC Plant Biol. 19:499.
Xuan, Y. H., Hu, Y. B., Chen, L.-Q., Sosso, D., Ducat, D. C., Hou, B.-H., et al. (2013). Functional role of oligomerization for bacterial and plant SWEET sugar transporter family. Proc. Natl. Acad. Sci. 110, E3685–E3694.
Yin, Q., Zhu, L., Du, P., Fan, C., Wang, J., Zhang, B., et al. (2020). Comprehensive analysis of SWEET family genes in Eucalyptus (Eucalyptus grandis). Biotechnol. Biotechnol. Equip. 34, 595–604. doi: 10.1080/13102818.2020.1790417
Yuan, M., and Wang, S. (2013). Rice MtN3/saliva/SWEET family genes and their homologs in cellular organisms. Mole. Plant 6, 665–674. doi: 10.1093/mp/sst035
Yue, C., Cao, H.-L., Wang, L., Zhou, Y.-H., Huang, Y.-T., Hao, X.-Y., et al. (2015). Effects of cold acclimation on sugar metabolism and sugar-related gene expression in tea plant during the winter season. Plant Mole. Biol. 88, 591–608. doi: 10.1007/s11103-015-0345-7
Zhai, Y., and Saier, M. H. (2001a). A web-based program (WHAT) for the simultaneous prediction of hydropathy, amphipathicity, secondary structure and transmembrane topology for a single protein sequence. J. Mole. Microbiol. Biotechnol. 3, 501–502.
Zhai, Y., and Saier, M. H. Jr. (2001b). A web-based program for the prediction of average hydropathy, average amphipathicity and average similarity of multiply aligned homologous proteins. J. Mole. Microbiol. Biotechnol. 3, 285–286.
Zhang, R., Niu, K., and Ma, H. (2020a). Identification and Expression Analysis of the SWEET Gene Family from Poa pratensis Under Abiotic Stresses. DNA Cell Biol. 39, 1606–1620. doi: 10.1089/dna.2020.5418
Zhang, W., Zhang, Y., Qiu, H., Guo, Y., Wan, H., Zhang, X., et al. (2020b). Genome assembly of wild tea tree DASZ reveals pedigree and selection history of tea varieties. Nat. Commun. 11, 1–12. doi: 10.1017/cbo9781316036198.002
Zhou, Y., Liu, L., Huang, W., Yuan, M., Zhou, F., Li, X., et al. (2014). Overexpression of OsSWEET5 in rice causes growth retardation and precocious senescence. PLoS One 9:e94210. doi: 10.1371/journal.pone.0094210
Zhou, Y., Liu, Y., Wang, S., Shi, C., Zhang, R., Rao, J., et al. (2017). Molecular cloning and characterization of galactinol synthases in Camellia sinensis with different responses to biotic and abiotic stressors. J. Agricult. Food Chem. 65, 2751–2759. doi: 10.1021/acs.jafc.7b00377
Keywords: SWEET, duplication, Camellia sinensis, expression, microsynteny
Citation: Jiang L, Song C, Zhu X and Yang J (2021) SWEET Transporters and the Potential Functions of These Sequences in Tea (Camellia sinensis). Front. Genet. 12:655843. doi: 10.3389/fgene.2021.655843
Received: 19 January 2021; Accepted: 15 February 2021;
Published: 31 March 2021.
Edited by:
Yunpeng Cao, Central South University Forestry and Technology, ChinaReviewed by:
Tianzhe Chen, Anhui Agricultural University, ChinaYuanyuan Jiang, South China Agricultural University, China
Chaoju Qian, Northwest Institute of Eco-Environment and Resources, Chinese Academy of Sciences (CAS), China
Copyright © 2021 Jiang, Song, Zhu and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xi Zhu, WGkuWmh1QG1lZC51bmktbXVlbmNoZW4uZGU=; Jianke Yang, YWppYW5rZWJjQHdubWMuZWR1LmNu