 Lin Zhang
Lin Zhang Jin Chen
Jin Chen Jiani Ma
Jiani Ma Hui Liu1,2*
Hui Liu1,2*- 1Engineering Research Center of Intelligent Control for Underground Space, Ministry of Education, China University of Mining and Technology, Xuzhou, China
- 2School of Information and Control Engineering, China University of Mining and Technology, Xuzhou, China
N7-methylguanosine (m7G) is a typical positively charged RNA modification, playing a vital role in transcriptional regulation. m7G can affect the biological processes of mRNA and tRNA and has associations with multiple diseases including cancers. Wet-lab experiments are cost and time ineffective for the identification of disease-related m7G sites. Thus, a heterogeneous network method based on Convolutional Neural Networks (HN-CNN) has been proposed to predict unknown associations between m7G sites and diseases. HN-CNN constructs a heterogeneous network with m7G site similarity, disease similarity, and disease-associated m7G sites to formulate features for m7G site-disease pairs. Next, a convolutional neural network (CNN) obtains multidimensional and irrelevant features prominently. Finally, XGBoost is adopted to predict the association between m7G sites and diseases. The performance of HN-CNN is compared with Naive Bayes (NB), Random Forest (RF), Support Vector Machine (SVM), as well as Gradient Boosting Decision Tree (GBDT) through 10-fold cross-validation. The average AUC of HN-CNN is 0.827, which is superior to others.
Introduction
N7-methylguanosine (m7G) is one of the most abundant modifications present in tRNA, rRNA, and mRNA 5′cap and plays critical roles in regulating RNA processing, metabolism, and function (Malbec et al., 2019). As an essential post-transcriptional modification, m7G plays an essential role in gene expression, processing and metabolism, protein synthesis, transcription stability and other aspects (Pandolfini et al., 2019). m7G is often enriched in the 5′UTR region and AG-enriched contexts. The internal m7G modification is dynamically regulated under both H2O2 and heat shock treatments, with remarkable accumulations in CDS and 3′UTR regions and functions in promoting mRNA translation efficiency (Malbec et al., 2019). m7G46 methylation of specific tRNA is associated with human mutation and the corresponding yeast mutation, which is m7G modification at position 46 in tRNA. Reduced m7G46 modification causes a growth deficiency phenotype in yeast, which provides a potential mechanism for primordial dwarfism associated with this lesion (Shaheen et al., 2015).
Munns et al. (1985) concluded that a specific autoimmune disorder is associated with the presence of anti-m7G autoantibodies in 50 patients’ cases. Bradrick (2017) found that mosquito-borne flaviviruses are important human pathogens, and m7G of the 5′cap structure is essential for infection. Lin et al. (2018) developed m7G methylated tRNA immunoprecipitation sequencing (MeRIP-seq) and tRNA reduction and cleavage sequencing (TRAC-seq) to conform that Mettl1-mediated tRNA m7G modification is essential for the proper expression of neural lineage genes. m7G methyltransferase complex METTL1/WDR4 causes primordial dwarfism and brain malformation. Thus, m7G sites and human diseases may show associations (Enroth et al., 2019). The study of disease-associated m7G may reveal the pathogenesis of the disease.
However, there is still a lack of systematic research on RNA modification due to technical limitations. Few studies have systematically explored the association between m7G sites and diseases. It is laborious and expensive to find disease-related m7G sites by wet-lab experiments. Recently, more and more artificial intelligence methods have been applied in the analysis of biological data. It can be regarded as a classification issue for disease-related m7G sites prediction, where the known association is denoted as 1, 0 otherwise. Some classical classifiers can be used to solve this problem, such as Naive Bayesian (NB), Support Vector Machine (SVM), Random Forest (RF), Gradient Boosting Decision Tree (GBDT), and Matrix Factorization (MF). With Bayes theorem, NB is proposed, which has a strong bias for linearity (Ting and Zheng, 2003). The prediction accuracy decreases dramatically in nonlinear scenarios. SVM is known to be suitable in small sample and nonlinear scenarios (Chang and Lin, 2011), which depends on the kernel to map data to a high-dimensional space. The data about disease-related m7G sites are high sparsity, so it is not easy to find the appropriate kernel. RF is an essential method in machine learning and has been widely used in many fields (Ham et al., 2005). However, it is not easy to obtain high precision and generalization performance simultaneously. GBDT is suitable for regression analysis, but the computation load is too high (Rao et al., 2019). Consistent with RF, it is also not suitable for sparse data. MF is the classic model of recommendation system (Lee and Seung, 1999). The low-rank matrix can be used to predict the association between m7G sites and diseases. But the higher the requirement of a low-rank matrix, the longer the training time.
In this paper, a deep learning framework based on heterogeneous networks and convolutional neural networks is proposed to find disease-associated m7G sites. The site-site similarities were calculated according to the chemical structure of m7G site, and the disease–disease similarities were achieved by miRNAs based on induced disease sets. Simultaneously, the known associations between the m7G site and the disease were incorporated into the heterogeneous network. Then, the convolutional neural network (CNN) was then adopted to extract multidimensional feature, making full use of the sparse data. Finally, XGBoost was used to predict the associations between m7G sites and various diseases.
Materials and Methods
Datasets
m7GDiseaseDB is an m7G-disease association database by taking 1218 disease-associated genetic variants as a bridge, which may lead to gain/loss of the m7G sites, with implications for disease pathogenesis involving m7G RNA methylation (Song et al., 2020). Among them, 768 associations between 741 m7G sites and 177 diseases were extracted via 741 variants with high confidence levels in m7GDiseaseDB. Specifically, the genomic locations, host genes of those sites were also included for further feature calculation.
In the mathematical view, let R ∈ ℝM×N be the association matrix consisting of M sites S = {s1,s2,⋯,sM} and N diseases D = {d1,d2,⋯,dN}. If there is an association between m7G site si and disease dj, Rij is 1, 0 otherwise.
Heterogeneous Network Based on Convolutional Neural Network
Figure 1 illustrates the framework HN-CNN. A heterogeneous network was constructed with site-site similarity, disease–disease similarity and the known m7G-disease associations to generate feature pairs. Then, each feature pair was transformed into a vector with high-dimensional hidden information by CNN. XGBoost predicts the candidate samples lastly, which chooses the regression classification tree as a base learner.
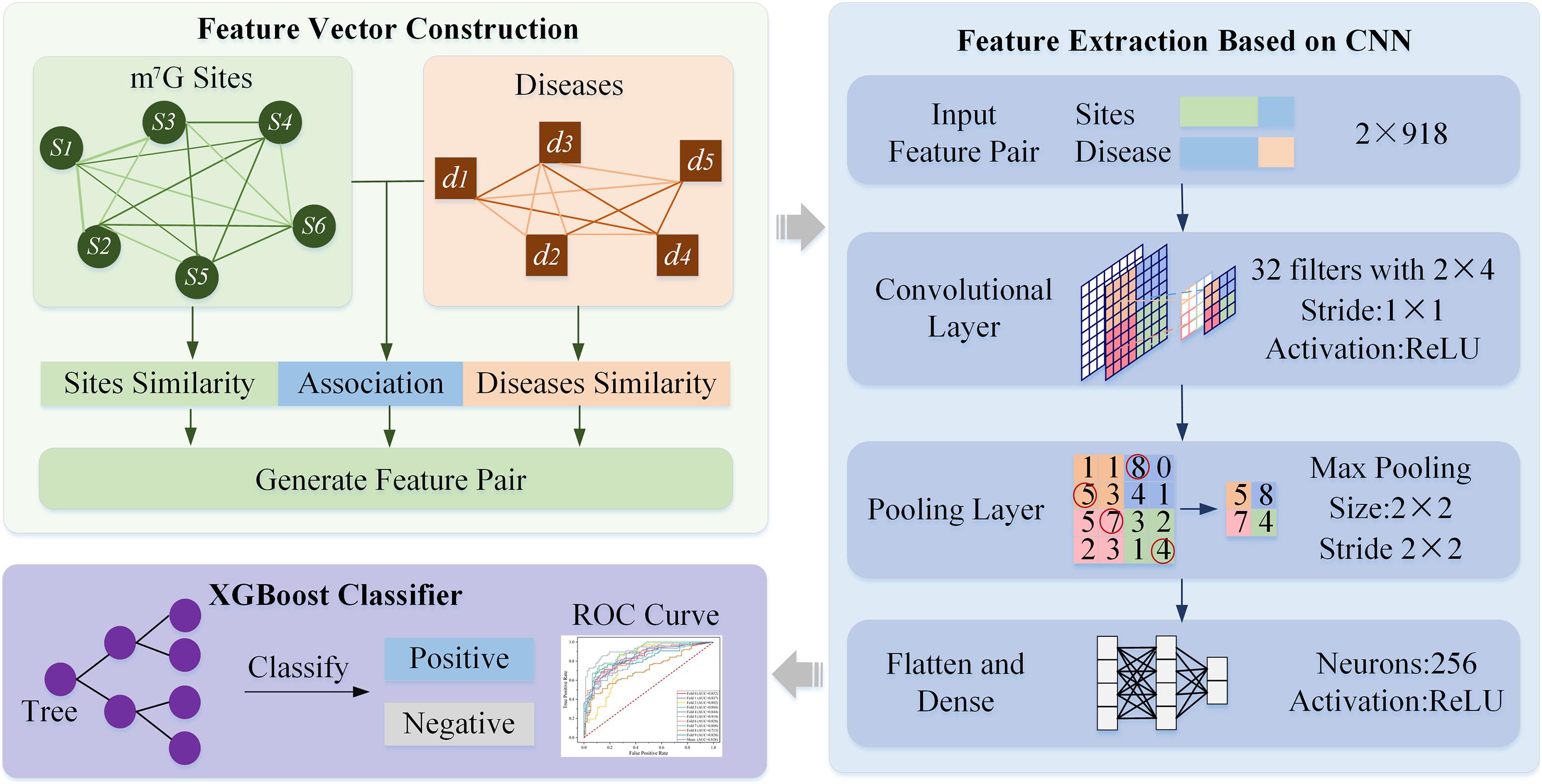
Figure 1. Framework of HN-CNN. “Feature Vector Construction” is a heterogeneous network based on feature extraction, which is constructed with similarities and known m7G-disease association. “Feature Extraction Based on CNN” is a CNN-based feature extraction followed by XGBoost. In “XGBoost Classifier,” XGBoost predicts the candidate samples, which chooses the regression classification tree as a base learner.
Feature Vector Construction
Chemical properties of m7G sites were utilized to depict the m7G feature just as previously described in similar work (Chen et al., 2019). Based on the chemical features of m7G sites, the site similarities were calculated by Jaccard coefficient which is defined as Equation as (1):
where A and B represent the chemical feature of two sites.
In addition, the disease–disease similarity is calculated by DisSetSim (Hu et al., 2017), which is an online system for calculating similarity with diseases names and open source databases. Disease-related genes, functional annotation of genes and the gene functional network of human are involved in calculating disease–disease similarity. Heterogeneous network adopts site-site similarity, disease–disease similarity, combined with the known association between m7G sites and diseases, shown directly in Figure 2A.
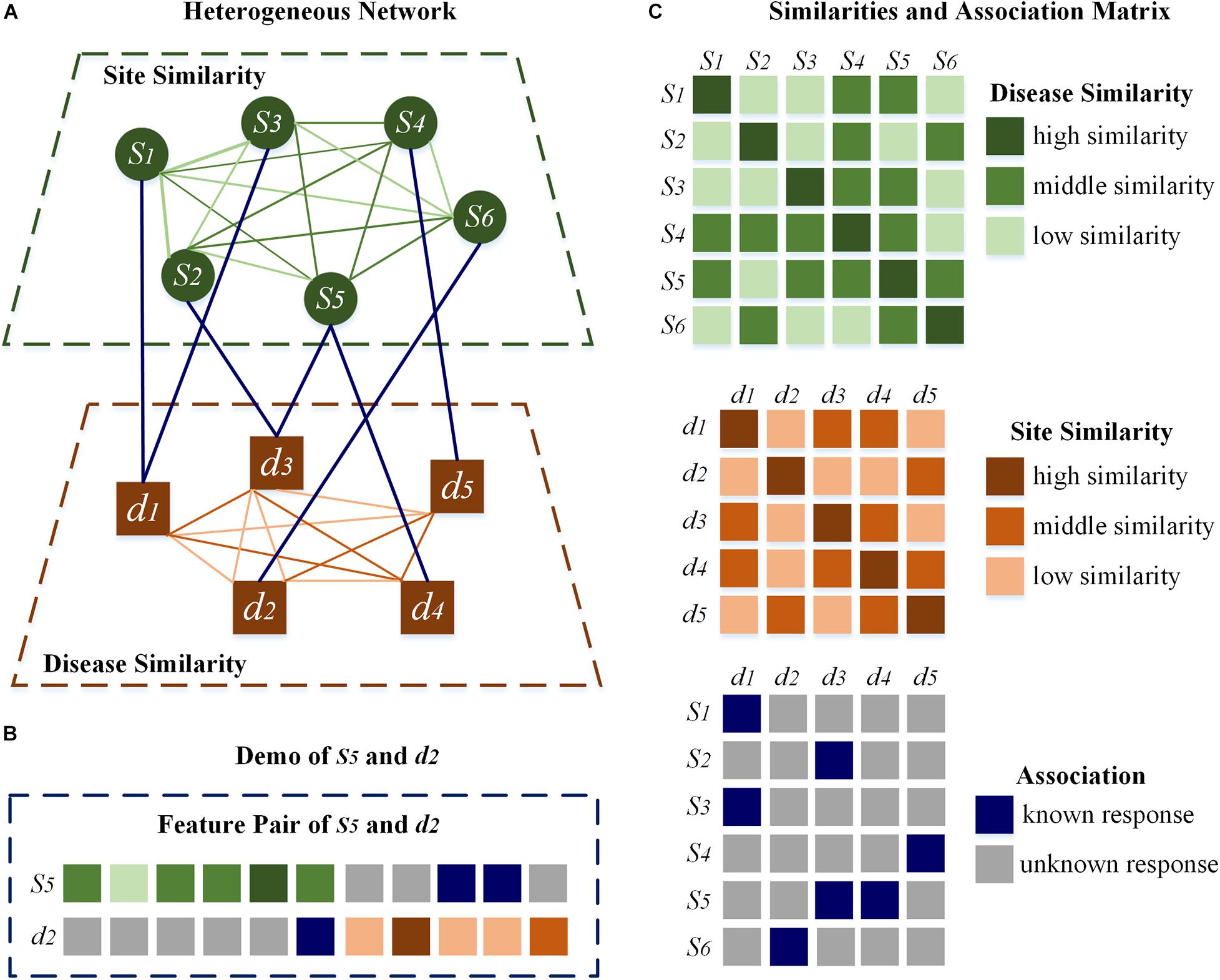
Figure 2. Heterogeneous network and feature pair. (A) The heterogeneous network. (B) The demo of s5 and d2. (C) The related matrixes directly.
HN-CNN pays more attention to the latent description of associations of m7G sites and diseases. Similarities and association are included in the heterogeneous network. Taking s5 and d2 in Figure 2B as an example, vector related to s5 is selected from the association matrix and site–site similarity, which is different from other sites. Vector related to d2 is selected from disease–disease similarity and the association matrix to form the vector of d2. Those two vectors combine to form the feature pair about s5 and d2, and each pair is unique. Therefore, the feature pair retains the commonness and the characteristics. Commonness means that the vector representing the same site or disease is invariant. Characteristics means the combination of site-disease is unique, which is different from any other feature pairs. Finally, the feature pair, which is shown in Figure 2B, is the connection between heterogeneous network and CNN.
Feature Extraction Based on CNN
Convolutional neural network (CNN) has a deep learning structure, which can mine hidden information. It is superior to the single network in terms of feature extraction and model fitting (Shin et al., 2016). The input layer becomes a multidimensional characteristic surface through the convolutional layer, and the propagation mode between the convolutional layers is shown in Equation (2). Then, features are mapped by pooling, and maximum pooling is shown in Equation (3). Finally, the selected features are flattened to form the final feature vectors:
where is the j-th feature map of the i-th layer, N is the number of the i-th layer’s kernels, is the j-th element in the i-th convolution kernel at the l layer, is the bias parameters, σ is the activation function:
Where maxp×q chooses the maximum from with the p×q-size pooling. The is the j-th pooling vector in the l-th layer.
Although the feature pairs were achieved in the previous section, the data is sparse with little information. The convolutional layer comprises multiple convolution kernels, which mine different characteristics of feature pairs. Therefore, the generated feature pairs are extracted by CNN. After that, feature vectors are formed, which contain not only various but also different information. In this paper, the associations of adjacent data in feature pairs are weak, so the convolution step size is set as 1 to make full use of each known data and mine each data’s hidden information. If the step size becomes bigger, some information will be ignored. The convolution kernel’s width was set as 2 to explore the association between m7G sites and diseases. To extract more dimensional information and mine the diverse relationships in feature pairs, the more convolution kernels are used, the better performance we have. However, the more computing resources and the longer the computation time are needed with too many kernels, along with the higher repetition rate. Considering high sparsity between the data, such as the sparsity of disease–disease similarity is 72.78%, the number of convolution kernels is set to 32. Meanwhile, the prediction accuracy is the best by experiment. If the number of convolution kernels is reduced, the accuracy will be decreased for mining the information of feature pairs deficiently. When the number of convolution kernels is increased, the accuracy is also decreased for repeated or useless features.
Then, the data are passed into the pooling layer. The pooling layer can reduce the input information dimension, keep the characteristic invariance, select the primary information, and reduce the redundancy information. In this paper, the size of maximum pooling is 2 × 2. Length 2 can screen out the data with prominent characteristics between sites and diseases; width 2 can effectively remove the duplicate data and screen out the critical information that has been expanded to the higher dimension.
Finally, feature pairs have been processed into vectors containing various kinds of information, but those vectors contain a large amount of information, with many types. The pooled vectors are compressed by full connection to integrate the feature data. The final feature vectors are formed, where n is the number of known associations, and d is the number of neurons in the full connection layer. In this paper, d is set to 256. When d is less than 256, the performance dramatically decreases due to less information in V. The performance also decreased due to too much or even useless information in V. V contains categorical information, optimizing by cross-entropy, to make V highly relevant to the original information, and V is used by subsequent classifiers.
XGBoost Classifier
XGBoost classifier is adopted to predict associations between m7G site and disease. It retains the feature information better and weakens the influence of parameters on final accuracy. As an integrated learning algorithm that optimizes distributed gradient enhancement, XGBoost has good performance in generalization by regulation and second-order Taylor expansions (Torlay et al., 2017). In this article, the regression classification tree is chosen as a base learner, whose input is V, and output is shown in Equation (4):
where is the result, vi is the i-th vector in eigenvector V, fk is the k-th decision tree, K is the number of leaf nodes, and E is the set of classification regression trees. The optimized objective function for XGBoost is shown in Equation (5):
where yi is the ground truth, and is binary cross-entropy loss and shown in Equation (6):
Ω(fk) is regularization to prevent overfitting and enhance generalization ability. Ω(fk) is shown in Equation (7):
where γ is the complexity cost by adding new leaf nodes. T is the number of leaves in a tree. ||w||2 is the sum of the square of each leaf node. λ is the regularization coefficient about the L2 norm ||w||2.
There are several hyperparameters in XGBoost such as the complexity cost of adding new leaf nodes γ and the regularization coefficient λ. To achieve better AUCs, cross validation is inlaid into XGBoost to find the best parameters with γ∈{0,0.2,0.4,0.6,0.8,1} and λ∈{0, 0.01, 0.001}. Meanwhile, early stopping is adopted to avoid overfitting.
Results
In this paper, HN-CNN is proposed to predict the association between m7G sites and diseases, and the performance is evaluated by 10-fold cross-validation. The original correlation matrix only marks the known relationship of m7G sites and diseases that can be considered positive, but the unknown does not mean negative. Thus, the same number of the negative is selected from unknown data randomly, and both the positive and the negative constitute the dataset. The set is divided into 10 parts on average, among which nine parts are used for training and the remaining 1 part for testing. The above operation should be repeated 10 times and the AUC should be recorded every time. It should be noted that the test set cannot be repeated in 10 training sets. After 10-folds, the average of 10 AUCs is the final result.
Evaluation Metrics
HN-CNN predicts the positive probability of association between m7G sites and diseases. A threshold θ is needed when validation. If the probability is more prominent than θ, the sample is considered as positive. On the other hand, it is identified as negative. True positive rates (TPR) and false positive rates (FPR) are calculated according to the prediction and the truth [Equations (8) and (9)] (Hanczar et al., 2010):
where TP is true positive, FP is false positive, TN is true negative, and FN is false negative. If θ changes, TPR and FPR will also change. The receiver operating characteristic (ROC) curve is drawn with different TPRs and FPRs (Moses et al., 1993). ROC curve can display the performance of the model intuitively, but it cannot compare models accurately. The area under the ROC curve (AUC) can be used to evaluate the performance of classifier, which ranges from 0 to 1. The more AUC is close to 1, the better performance the classifier has (Fawcett, 2006). So, we choose the ROC curve and AUC to measure the models.
The is the mean of m runs of 10-fold cross-validation, which is calculated by Equation (10):
where m is the number of experiments, AUCi is the i-th AUC in 10-fold cross-validation. In this paper, m = 10.
Comparison With Other Methods
To verify the advantages of CNN in extracting features, features that are not processed by CNN were compared with the features processed by CNN, which are classified with base classifiers such as GBDT, NB, SVM and RF. The result is shown in Figure 3A. The ordinate in the figure is the result of 10-fold cross verification, which is the average AUC. All average AUCs are calculated by 10-times of 10-fold cross-validation. The legends “Base Classifier” and “CNN and Base Classifier” are distinguished by whether the feature pair has been processed by CNN. “CNN and Base Classifier” means that feature pairs are processed with CNN, but the models of “Base Classifier” are not, which put feature pairs into classifiers directly.
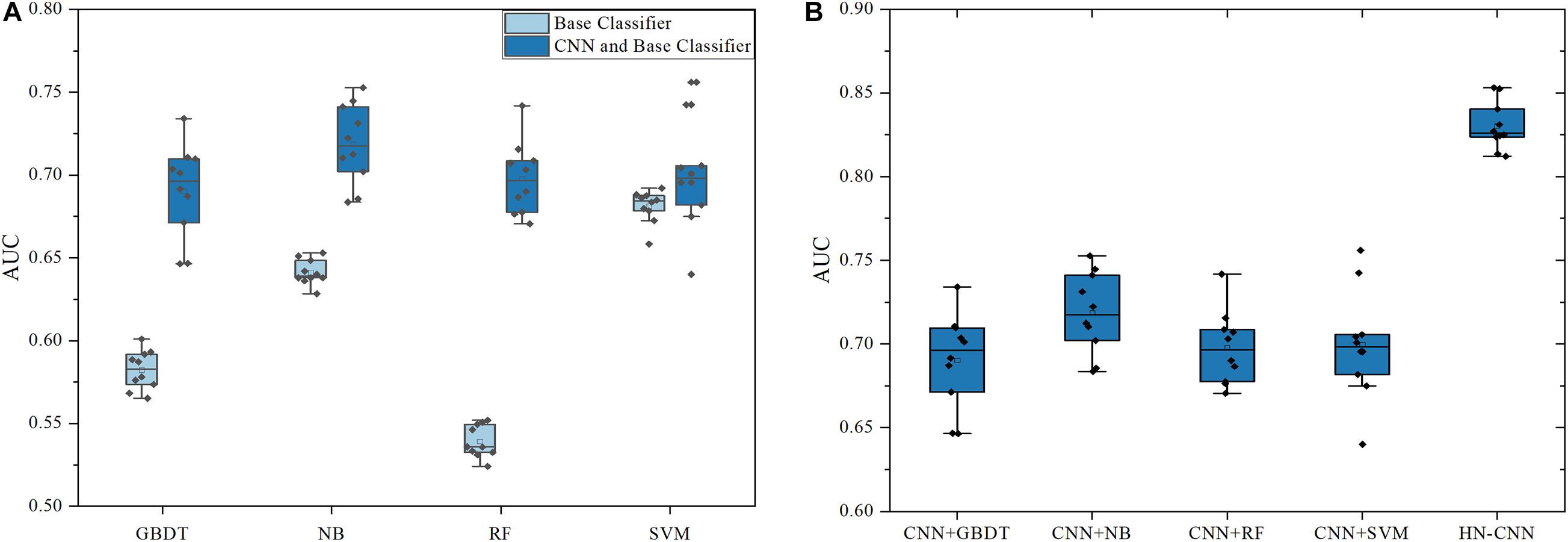
Figure 3. The AUCs of HN-CNN and other methods. (A) The AUCs of base classifiers with/without CNN. (B) The AUCs of HN-CNN and base classifiers with CNN.
According to the results in Figure 3A, it can be analyzed that the prediction accuracy is significantly improved after CNN extracts the feature with the same parameters and classifiers, which is the most obvious in the RF classifier. Without CNN, the mean AUC is 0.539 by RF. However, the average AUC is 0.698 with CNN, which increased by about 0.16. Besides, it is observed in Figure 3A that only the base classifiers without CNN have a greater impact on the prediction results. The average AUC directly predicted by SVM is 0.681, which is about 0.14 higher than that of RF. Classifiers with CNN improve the prediction effect and reduce the gap between classifiers. Therefore, CNN can effectively mine hidden data and improve classification accuracy.
The XGBoost was chosen as the final classifier for two reasons. XGBoost is an integrated machine learning algorithm based on decision trees, and its generalization performance is better than a single classifier. In other words, XGBoost finds the optimal solution within a fixed range of parameters. The results of XGBoost and other methods are shown in Figure 3B. CNN+GBDT in X-coordinate means that the features are extracted by CNN and classified by GBDT, and so on. The ordinate is the average AUC of 10-fold cross-validation. It can be analyzed that XGBoost is superior to the base classifiers. The average AUC of HN-CNN is 0.830, which is 0.111 higher than CNN+NB. Therefore, HN-CNN has the advantage in feature extraction and classification, which greatly improves the prediction accuracy.
Case Study
The number of known associations is much less than the unknown, which can also be interpreted as the positive is much less than the negative. To weaken the influence of the negative, negative samples equal to the number of positive samples were selected randomly. The highest test accuracy in the 10-fold cross-validation was selected as the final prediction model, which predicts the positive probability of all unknown samples. We selected five of the top 20 to analyze and show the results in Table 1. R. analyzes the related genes with GO based on “clusterProfiler” (Yu et al., 2012). Among the results, CC is short for cellular component, MF is the molecular function, and BP is the biological process. Each gene description is described by p-value. If the p value is close to 0, the gene description is more obvious.
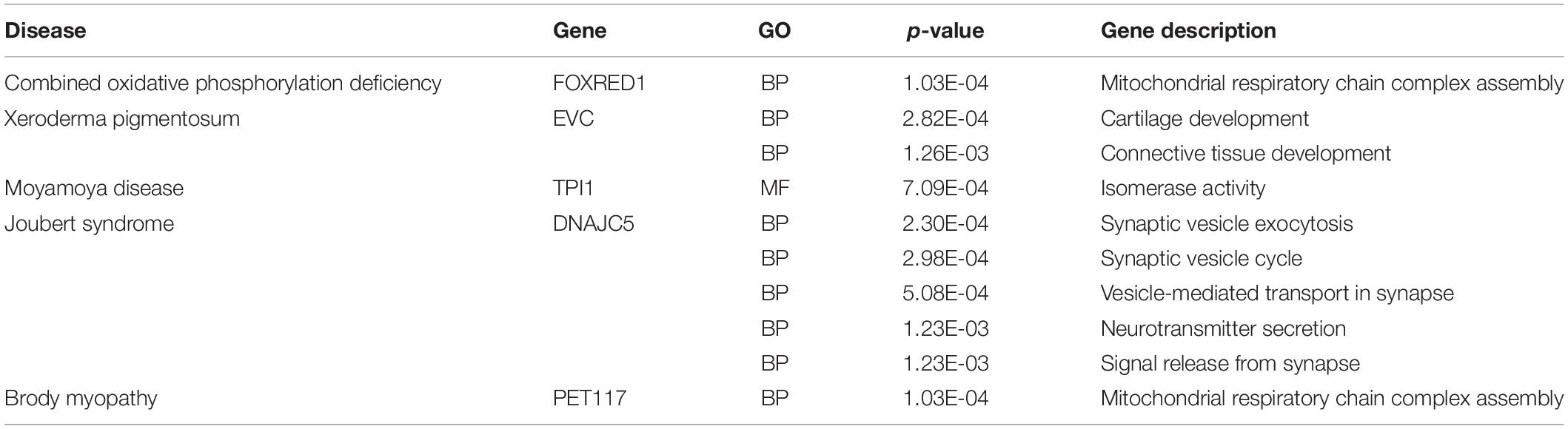
Table 1. Case study.
Combined oxidative phosphorylation deficiency is caused by homozygous or compound heterozygous mutations in the ELAC2 gene, which is a mitochondrial tRNA processing gene (Haack et al., 2013). FOXRED1 can cause complex I deficiency and effect protein function (Calvo et al., 2010). Mitochondrial respiratory chain complex assembly mainly causes mitochondrial diseases (Deutschmann et al., 2014). There is a high correlation between disease and FOXRED1, in line with the laws of biology.
Xeroderma pigmentosum is a rare genetic disease characterized by extreme photosensitivity, resulting in a higher incidence of cutaneous tumors (Cleaver et al., 1999). EVC is essential for cartilage development (Pacheco et al., 2012). The p-value of connective tissue development is 1.26E-03, whose mutations contribute to tumor formation.
Moyamoya disease is a chronic, occlusive cerebrovascular disease with unknown etiology characterized by bilateral steno-occlusive changes at the terminal portion of the internal carotid artery and an abnormal vascular network base of the brain (Sakurai et al., 2004). Moyamoya disease is associated with various diseases, like atherosclerosis, autoimmune diseases, Down syndrome. TPI1 is a crucial enzyme in carbohydrate metabolism, negatively associated with tumor size (Jiang et al., 2017). Therefore, TPI1 may inhibit the size of tumors and induce Moyamoya disease.
Inheritance of Joubert syndrome is autosomal and recessive, which is characterized by hypoplasia of the cerebellar vermis (Kendall et al., 1990; Lee et al., 2012). DNAJC5 encodes the cysteine string protein, which is a presynaptic protein implicated in neurodegeneration (Cadieux-Dion et al., 2013). It causes autosomal dominant Kufs disease (Jarrett et al., 2018). One of Kufs’ phenotypes is generalized tonic-clonic seizures, which is similar to related disorders of Joubert syndrome (Chance et al., 1999; Josephson et al., 2001).
Brody myopathy is a rare muscle disorder characterized by exercise-induced impairment of muscle relaxation and stiffness (Odermatt et al., 2000). Pet117 is shown to reside in the mitochondrial matrix, associated with the inner membrane (Taylor et al., 2017). Its gene description hence mitochondrial respiratory efficiency, which is mitochondrial respiratory chain complex assembly (Cogliati et al., 2013). So, it may be further manifested as Brody myopathy symptoms.
Discussion and Conclusion
It is efficient and time-saving to predict the association between m7G sites and diseases. HN-CNN integrates diverse information through heterogeneous networks. It adopts CNN to help extract latent relationships in feature pairs, which focuses on personalized associations between m7G sites and diseases. At last, XGBoost is used to classify whether there exists association with more generalization. In the 10-fold cross-validation, HN-CNN gets better results than the other methods. The predicted results are analyzed through R to show better demonstrated the reliability of the experimental method in case study. In the future, the data will be updated, and the sparsity will be reduced. HN-CNN will obtain better prediction results in the association prediction due to the amount of data.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
JC and LZ reviewed the resources, wrote the manuscript, and revised the manuscript. JM provided the data and revised the manuscript. HL took the lead in the work and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work is supported by the Fundamental Research Funds for the Central Universities (2019ZDPY15) for support.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.655284/full#supplementary-material
Supplementary Table 1 | The top 50 of most likely site-disease associations.
References
Bradrick, S. S. (2017). Causes and consequences of flavivirus RNA methylation. Front. Microbiol. 8:2374. doi: 10.3389/fmicb.2017.02374
Cadieux-Dion, M., Andermann, E., Lachance-Touchette, P., Ansorge, O., Meloche, C., Barnabe, A., et al. (2013). Recurrent mutations in DNAJC5 cause autosomal dominant Kufs disease. Clin. Genet. 83, 571–575. doi: 10.1111/cge.12020
Calvo, S. E., Tucker, E. J., Compton, A. G., Kirby, D. M., Crawford, G., Burtt, N. P., et al. (2010). High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat. Genet. 42, 851–858. doi: 10.1038/ng.659
Chance, P. F., Cavalier, L., Satran, D., Pellegrino, J. E., Koenig, M., and Dobyns, W. B. (1999). Clinical nosologic and genetic aspects of Joubert and related <KEYWORDS> syndromes. J. Child Neurol. 14, 660–666. doi: 10.1177/088307389901401007
Chang, C. C., and Lin, C. J. (2011). LIBSVM: a library for support vector machines. ACM Trans. Intell. Syst. Technol. 2:27. doi: 10.1145/1961189.1961199
Chen, W., Feng, P. M., Song, X. M., Lv, H., and Lin, H. (2019). iRNA-m7G: identifying N-7-methylguanosine sites by fusing multiple features. Mol. Ther. Nucleic Acids 18, 269–274. doi: 10.1016/j.omtn.2019.08.022
Cleaver, J. E., Thompson, L. H., Richardson, A. S., and States, J. C. (1999). A summary of mutations in the UV-sensitive disorders: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. Hum. Mut. 14, 9–22. doi: 10.1002/(sici)1098-1004199914:1<9::aid-humu2<3.3.co;2-y
Cogliati, S., Frezza, C., Soriano, M. E., Varanita, T., Quintana-Cabrera, R., Corrado, M., et al. (2013). Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell 155, 160–171. doi: 10.1016/j.cell.2013.08.032
Deutschmann, A. J., Amberger, A., Zavadil, C., Steinbeisser, H., Mayr, J. A., Feichtinger, R. G., et al. (2014). Mutation or knock-down of 17 beta-hydroxysteroid dehydrogenase type 10 cause loss of MRPP1 and impaired processing of mitochondrial heavy strand transcripts. Hum. Mol. Genet. 23, 3618–3628. doi: 10.1093/hmg/ddu072
Enroth, C., Poulsen, L. D., Iversen, S., Kirpekar, F., Albrechtsen, A., and Vinther, J. (2019). Detection of internal N7-methylguanosine (m7G) RNA modifications by mutational profiling sequencing. Nucleic Acids Res. 47:e126. doi: 10.1093/nar/gkz736
Fawcett, T. (2006). An introduction to ROC analysis. Pattern Recognit. Lett. 27, 861–874. doi: 10.1016/j.patrec.2005.10.010
Haack, T. B., Kopajtich, R., Freisinger, P., Wieland, T., Rorbach, J., Nicholls, T. J., et al. (2013). ELAC2 mutations cause a mitochondrial RNA processing defect associated with hypertrophic cardiomyopathy. Am. J. Hum. Genet. 93, 211–223. doi: 10.1016/j.ajhg.2013.06.006
Ham, J., Chen, Y. C., Crawford, M. M., and Ghosh, J. (2005). Investigation of the random forest framework for classification of hyperspectral data. IEEE Trans. Geosci. Remote Sens. 43, 492–501. doi: 10.1109/Tgrs.2004.842481
Hanczar, B., Hua, J. P., Sima, C., Weinstein, J., Bittner, M., and Dougherty, E. R. (2010). Small-sample precision of ROC-related estimates. Bioinformatics 26, 822–830. doi: 10.1093/bioinformatics/btq037
Hu, Y., Zhao, L. L., Liu, Z. Y., Ju, H., Shi, H. B., Xu, P. G., et al. (2017). DisSetSim: an online system for calculating similarity between disease sets. J. Biomed. Semantics 8:28. doi: 10.1186/s13326-017-0140-2
Jarrett, P., Easton, A., Rockwood, K., Dyack, S., McCollum, A., Siu, V., et al. (2018). Evidence for cholinergic dysfunction in autosomal dominant kufs disease. Can. J. Neurol. Sci. 45, 150–157. doi: 10.1017/cjn.2017.261
Jiang, H., Ma, N., Shang, Y. R., Zhou, W. T., Chen, T. W., Guan, D. X., et al. (2017). Triosephosphate isomerase 1 suppresses growth, migration and invasion of hepatocellular carcinoma cells. Biochem. Biophy. Res. Commun. 482, 1048–1053. doi: 10.1016/j.bbrc.2016.11.156
Josephson, S. A., Schmidt, R. E., Millsap, P., McManus, D. Q., and Morris, J. C. (2001). Autosomal dominant Kufs’ disease: a cause of early onset dementia. J. Neurol. Sci. 188, 51–60. doi: 10.1016/s0022-510x(01)00546-9
Kendall, B., Kingsley, D., Lambert, S. R., Taylor, D., and Finn, P. (1990). Joubert syndrome: a clinico-radiological study. Neuroradiology 31, 502–506. doi: 10.1007/bf00340131
Lee, D. D., and Seung, H. S. (1999). Learning the parts of objects by non-negative matrix factorization. Nature 401, 788–791. doi: 10.1038/44565
Lee, J. E., Silhavy, J. L., Zaki, M. S., Schroth, J., Bielas, S. L., Marsh, S. E., et al. (2012). CEP41 is mutated in Joubert syndrome and is required for tubulin glutamylation at the cilium. Nat. Genet. 44, 193–199. doi: 10.1038/ng.1078
Lin, S., Liu, Q., Lelyveld, V. S., Choe, J., Szostak, J. W., and Gregory, R. I. (2018). Mettl1/Wdr4-Mediated m(7)G tRNA Methylome Is Required for Normal mRNA Translation and Embryonic Stem Cell Self-Renewal and Differentiation. Mol Cell 71, 244–255.e5. doi: 10.1016/j.molcel.2018.06.001
Malbec, L., Zhang, T., Chen, Y. S., Zhang, Y., Sun, B. F., Shi, B. Y., et al. (2019). Dynamic methylome of internal mRNA N(7)-methylguanosine and its regulatory role in translation. Cell Res. 29, 927–941. doi: 10.1038/s41422-019-0230-z
Moses, L. E., Shapiro, D., and Littenberg, B. (1993). Combining independent studies of a diagnostic test into a summary ROC curve: data-analytic approaches and some additional considerations. Stat. Med. 12, 1293–1316. doi: 10.1002/sim.4780121203
Munns, T. W., Liszewski, M. K., Freeman, S. K., and Kaine, J. L. (1985). Detection of human autoantibodies specific for 5′-m7GMP and m7G(5′)ppp(5′)N. Biochem. Biophy. Res. Commun. 128, 1014–1019. doi: 10.1016/0006-291x(85)90148-2
Odermatt, A., Barton, K., Khanna, V. K., Mathieu, J., Escolar, D., Kuntzer, T., et al. (2000). The mutation of Pro(789) to Leu reduces the activity of the fast-twitch skeletal muscle sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA1) and is associated with Brody disease. Hum. Genet. 106, 482–491. doi: 10.1007/s004390000297
Pacheco, M., Valencia, M., Caparros-Martin, J. A., Mulero, F., Goodship, J. A., and Ruiz-Perez, V. L. (2012). Evc works in chondrocytes and osteoblasts to regulate multiple aspects of growth plate development in the appendicular skeleton and cranial base. Bone 50, 28–41. doi: 10.1016/j.bone.2011.08.025
Pandolfini, L., Barbieri, I., Bannister, A. J., Hendrick, A., Andrews, B., Webster, N., et al. (2019). METTL1 Promotes let-7 MicroRNA Processing via m7G Methylation. Mol Cell 74, 1278-1290.e9. doi: 10.1016/j.molcel.2019.03.040
Rao, H., Shi, X. Z., Rodrigue, A. K., Feng, J. J., Xia, Y. C., Elhoseny, M., et al. (2019). Feature selection based on artificial bee colony and gradient boosting decision tree. Appl. Soft Comput. 74, 634–642. doi: 10.1016/j.asoc.2018.10.036
Sakurai, K., Horiuchi, Y., Ikeda, H., Ikezaki, K., Yoshimoto, T., Fukui, M., et al. (2004). A novel susceptibility locus for moyamoya disease on chromosome 8q23. J. Hum. Genet. 49, 278–281. doi: 10.1007/s10038-004-0143-6
Shaheen, R., Abdel-Salam, G. M. H., Guy, M. P., Alomar, R., Abdel-Hamid, M. S., Afifi, H. H., et al. (2015). Mutation in WDR4 impairs tRNA m(7)G(46) methylation and causes a distinct form of microcephalic primordial dwarfism. Genome Biol. 16:210. doi: 10.1186/s13059-015-0779-x
Shin, H.-C., Roth, H. R., Gao, M., Lu, L., Xu, Z., Nogues, I., et al. (2016). Deep convolutional neural networks for computer-aided detection: CNN architectures, dataset characteristics and transfer learning. IEEE Trans. Med. Imaging 35, 1285–1298. doi: 10.1109/tmi.2016.2528162
Song, B. W., Tang, Y. J., Chen, K. Q., Wei, Z., Rong, R., Lu, Z. L., et al. (2020). m7GHub: deciphering the location, regulation and pathogenesis of internal mRNA N7-methylguanosine (m(7)G) sites in human. Bioinformatics 36, 3528–3536. doi: 10.1093/bioinformatics/btaa178
Taylor, N. G., Swenson, S., Harris, N. J., Germany, E. M., Fox, J. L., and Khalimonchuk, O. (2017). The assembly factor Pet117 couples heme a synthase activity to cytochrome oxidase assembly. J. Biol. Chem. 292, 1815–1825. doi: 10.1074/jbc.M116.766980
Ting, K. M., and Zheng, Z. J. (2003). A study of AdaBoost with naive Bayesian classifiers: weakness and improvement. Comput. Intell. 19, 186–200. doi: 10.1111/1467-8640.00219
Torlay, L., Perrone-Bertolotti, M., Thomas, E., and Baciu, M. (2017). Machine learning-XGBoost analysis of language networks to classify patients with epilepsy. Brain Inform. 4, 159–169. doi: 10.1007/s40708-017-0065-7
Keywords: m7G sites, diseases, heterogeneous network, convolutional neural network, XGBoost
Citation: Zhang L, Chen J, Ma J and Liu H (2021) HN-CNN: A Heterogeneous Network Based on Convolutional Neural Network for m7 G Site Disease Association Prediction. Front. Genet. 12:655284. doi: 10.3389/fgene.2021.655284
Received: 18 January 2021; Accepted: 15 February 2021;
Published: 04 March 2021.
Edited by:
Giovanni Nigita, The Ohio State University, United StatesReviewed by:
Zhang Shaowu, Northwestern Polytechnical University, ChinaQuan Zou, University of Electronic Science and Technology of China, China
Wei Chen, North China University of Science and Technology, China
Copyright © 2021 Zhang, Chen, Ma and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hui Liu, aHVpLmxpdUBjdW10LmVkdS5jbg==; Lin Zhang, bGluLnpoYW5nQGN1bXQuZWR1LmNu