
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 09 July 2021
Sec. Evolutionary and Population Genetics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.654256
This article is part of the Research Topic Gene Regulation as a Driver of Adaptation and Speciation View all 11 articles
 Katia Aviña-Padilla1,2
Katia Aviña-Padilla1,2 José Antonio Ramírez-Rafael3
José Antonio Ramírez-Rafael3 Gabriel Emilio Herrera-Oropeza1,4
Gabriel Emilio Herrera-Oropeza1,4 Vijaykumar Yogesh Muley1
Vijaykumar Yogesh Muley1 Dulce I. Valdivia2
Dulce I. Valdivia2 Erik Díaz-Valenzuela2
Erik Díaz-Valenzuela2 Andrés García-García3
Andrés García-García3 Alfredo Varela-Echavarría1*
Alfredo Varela-Echavarría1* Maribel Hernández-Rosales2*
Maribel Hernández-Rosales2*The structure of eukaryotic genes is generally a combination of exons interrupted by intragenic non-coding DNA regions (introns) removed by RNA splicing to generate the mature mRNA. A fraction of genes, however, comprise a single coding exon with introns in their untranslated regions or are intronless genes (IGs), lacking introns entirely. The latter code for essential proteins involved in development, growth, and cell proliferation and their expression has been proposed to be highly specialized for neuro-specific functions and linked to cancer, neuropathies, and developmental disorders. The abundant presence of introns in eukaryotic genomes is pivotal for the precise control of gene expression. Notwithstanding, IGs exempting splicing events entail a higher transcriptional fidelity, making them even more valuable for regulatory roles. This work aimed to infer the functional role and evolutionary history of IGs centered on the mouse genome. IGs consist of a subgroup of genes with one exon including coding genes, non-coding genes, and pseudogenes, which conform approximately 6% of a total of 21,527 genes. To understand their prevalence, biological relevance, and evolution, we identified and studied 1,116 IG functional proteins validating their differential expression in transcriptomic data of embryonic mouse telencephalon. Our results showed that overall expression levels of IGs are lower than those of MEGs. However, strongly up-regulated IGs include transcription factors (TFs) such as the class 3 of POU (HMG Box), Neurog1, Olig1, and BHLHe22, BHLHe23, among other essential genes including the β-cluster of protocadherins. Most striking was the finding that IG-encoded BHLH TFs fit the criteria to be classified as microproteins. Finally, predicted protein orthologs in other six genomes confirmed high conservation of IGs associated with regulating neural processes and with chromatin organization and epigenetic regulation in Vertebrata. Moreover, this study highlights that IGs are essential modulators of regulatory processes, such as the Wnt signaling pathway and biological processes as pivotal as sensory organ developing at a transcriptional and post-translational level. Overall, our results suggest that IG proteins have specialized, prevalent, and unique biological roles and that functional divergence between IGs and MEGs is likely to be the result of specific evolutionary constraints.
Most eukaryotic genes contain introns, nucleotide DNA sequences that after transcription as part of the messenger RNA are removed by splicing during its maturation. Since the introns interrupt the multiple exonic sequences, these genes are thus termed multiple exon genes (MEGs). Eukaryotic genomes, however, also contain an important proportion of genes in which the coding sequence is contained within a single exon. Diverse studies of genes of this type have been performed over the past decades and have been variously referred to as “single-exon genes” (SEGs) and “intronless genes” (IGs), both terms carrying some ambiguity as genes containing an intron in their 5′ UTR are often included among them (Sunahara et al., 1990; Gentles and Karlin, 1999; Sakharkar et al., 2002, 2005a, 2006; Tine et al., 2011; Zou et al., 2011; Yan et al., 2014). For example, a recent ontology defines SEGs as nuclear genes with functional protein-coding capacity whose coding sequence comprises only one exon, thus including genes with introns in their untranslated regions termed uiSEGs, as well as those lacking introns entirely, termed “Intronless Genes” (Jorquera et al., 2018). Pseudogenes, functional RNAs, tRNA, rRNA, ribozyme long non-coding RNAs, and miRNAs are excluded from this definition. To avoid any possible ambiguity, in this article we use the term “Intronless genes” in the narrow definition of Jorquera et al. (2018) as protein-coding nuclear genes completely devoid of introns.
Owing to their prokaryotic-like architecture, IGs in eukaryotic genomes, provide interesting datasets for computational analysis in comparative genomics and for the study of evolutionary trajectories. Comparative analysis of their sequences in different genomes could allow the identification of their unique and conserved features, thus providing insights into the role of introns in gene evolution leading to a better understanding of genome architecture and arrangement.
The abundant presence of introns in most genes of multicellular organisms entails regulatory processes associated with the generation of multiple splice variants missing in intronless genes. The absence of splicing events in IGs represents a higher transcriptional fidelity, making them even more valuable for regulatory roles. To date, more than 2000 genes with a single coding exon in the human genome have been classified (Jorquera et al., 2016). Among them, a considerable fraction of IGs encode G-protein-coupled receptors (GPCRs), core canonical histones which are integral part of nucleosomes and often confer specific structural and functional features, transcription factors, proteins involved in signal transduction, regulation of development, growth, and cell proliferation (Sakharkar et al., 2005a; Grzybowska, 2012).
The expression of IGs has been proposed to be highly specialized for neural functions and linked to diseases such as cancer, neuropathies, and developmental disorders. Examples of IGs with clinical relevance are the RPRM gene related to gastric cancer which causes increased cell proliferation and possesses tumor suppression activity (Amigo et al., 2018) and the protein kinase CK2α gene which is up-regulated in all human cancers (Hung et al., 2010). Other IGs linked to cancer include CLDN8 in colorectal carcinoma and renal cell tumors, ARLTS1 in melanoma, and PURA and TAL2 in leukemia (Grzybowska, 2012). IGs have also been associated with neuropathies, such as ECDR1, a cerebellar degeneration-related protein, and NPBWR2, a neuropeptide B/W receptor type (Louhichi et al., 2011).
Regarding their role in the diseases described above, IGs in humans are potential clinical biomarkers and drug targets that deserve careful consideration (Ohki et al., 2000; Grzybowska, 2012; Liu et al., 2017). Their functional role and their evolutionary conservation in other genomes, however, remains poorly understood. Furthermore, there is a current debate between related theories that place the origin of introns, early or late during the evolutionary history of eukaryotes (de Souza, 2003; Fedorova and Fedorov, 2003).
With this backdrop, our work aimed to characterize the functional role of mouse IGs and to infer their evolutionary pattern across six additional vertebrate genomes. We have analyzed their expression, particularly during brain development at early embryonic stages, and their potential as transcriptional as well as post-translational modulators.
Overall, this study sheds light on the concerted role played by this peculiar group of genes and helps contrast the functional features of intron-containing and intronless genes across vertebrate species and their collective evolutionary roadmaps.
Data were extracted using Python scripts1 and Ensembl APIs. Seven vertebrate genomes including Mus musculus, Homo sapiens, Pan troglodytes, Monodelphis domestica, Rattus norvegicus, Gallus gallus, and Danio rerio assembled at a chromosome level were accessed at the Ensembl REST API platform2 (using Python with the ensembl_rest package). For an explanation of the species choice see section “Search for Orthologs of Mouse IGs.” The pipeline process was as follows: protein-coding genes with CDS identifiers for transcripts for all chromosomes were retrieved and classified into a temporary dataset that contained genes with a single coding exon (temp-DS1) and a dataset containing “multiple exon genes” (MEGs) depending on exon and transcript count (Figure 1). The former was then submitted to the Intron DB3 to filter out genes with UTR introns. The output of the pipeline was a second temporary dataset containing protein-coding genes that did not contain introns in their entire length (temp-DS2). After data extraction, a manual curation step was performed to assess their nuclear nature and protein-coding transcript biotype, which allowed us to discard proteins encoded in the mitochondrial genome, hence yielding the final dataset containing only protein-coding nuclear genes completely devoid of introns, or “Intronless genes” (IG) (Figure 1). The final MEG dataset contained 20,694 protein-coding genes with two or more exons and the IG dataset contained 1,116 protein-coding genes with only one exon and one transcript.
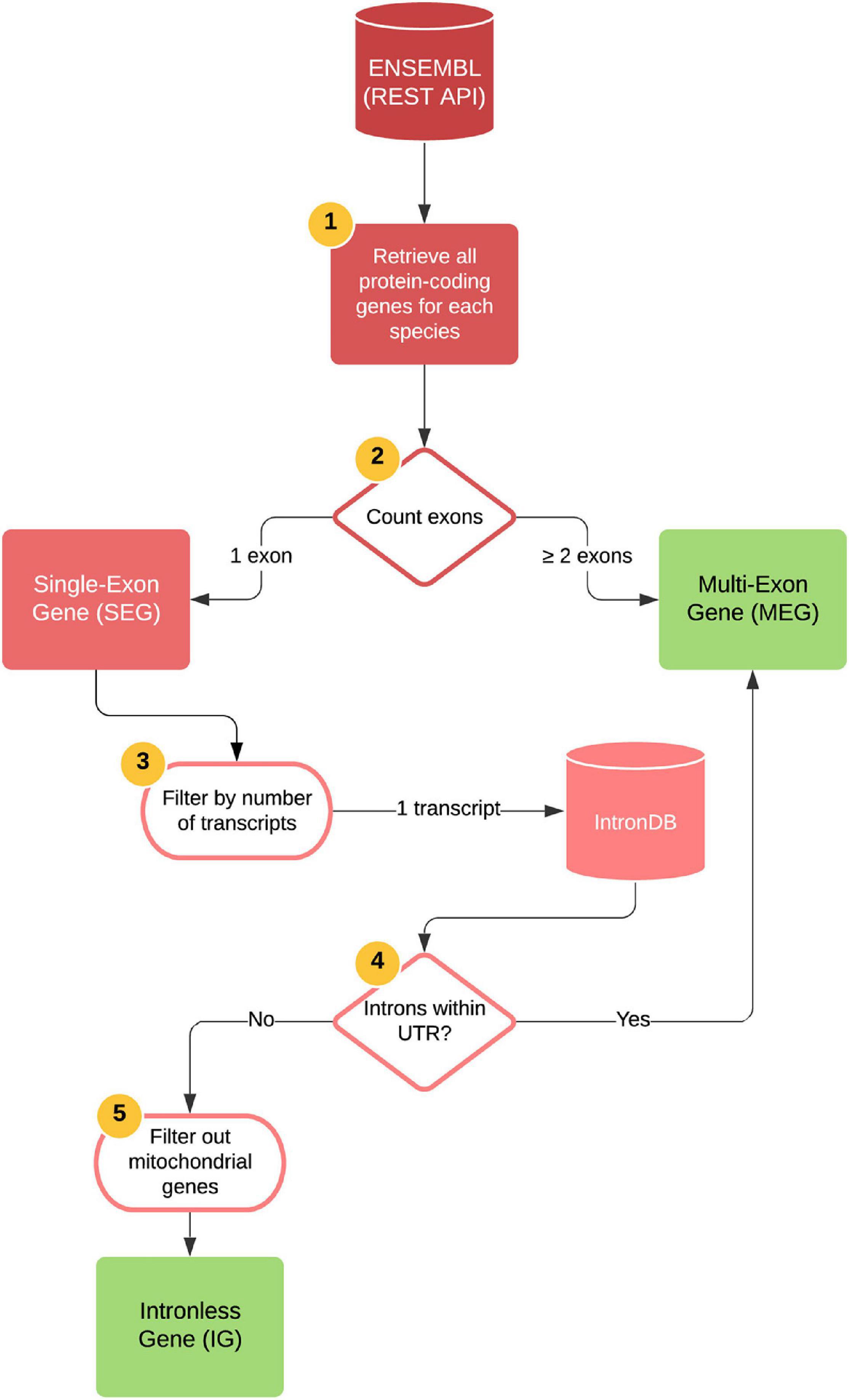
Figure 1. Bioinformatics pipeline for generating IG and MEG datasets. Automatized steps (represented as 1–4): all protein-coding genes were retrieved for each species by accessing the Ensembl REST API platform. The number of exons for each gene were counted, and genes with two or more exons were identified as MEGs. Genes with one single exon were identified as SEGs and were subsequently filtered by their number of transcripts, keeping only those with one transcript. Using the IntronDB, we then classified these genes according to the presence of introns within their UTRs. Genes with introns within these regions were identified as MEGs and those without, as IGs. Manual curation was performed to filter out mitochondrial genes and genes with incomplete protein annotations (step 5). Output files are depicted as green squares.
The mouse IG and MEG datasets were used to perform an over-representation analysis of functional assignment using the following databases: SUPERFAMILY4 (proteins of known three-dimensional structure); Pfam5 (protein domains); and PROSITE6 (biologically meaningful signatures or motifs). All tests used MEGs as controls to determine unique, shared or overrepresented features among both types of genes. For data visualization of SUPERFAMILY and Pfam results we employed the ClusterProfiler R package (Yu et al., 2012), and Python scripts using a hypergeometric test for PROSITE enrichment.
The functional enrichment analyses were performed using Metascape7 for the biological process category, including KEGG and Reactome pathways. First, the functional enrichment of the 1,116 mouse IG proteins was performed using all mouse proteins as a background “universe” (selecting input as species: M. musculus, analysis as species: M. musculus). Then, in a second approach the meta-analysis workflow was used to compare enriched terms for the list of orthologs of mouse IGs regarding their grouping into five “ages,” each of them corresponding to one of the taxonomic categories: Vertebrata, Tetrapoda, Theria, Eutheria, and Muridae.
Out of the 1,116 IGs, 543 have orthologs across the analyzed species. Mouse IG orthologs that conserved the IG structure were compared against MEGs conserved as MEGs in other organisms. The meta-analysis workflow was used to compare enriched terms for the list of mouse orthologs in the aforementioned genomes to the pathways of three random samples of the same size of multi-exon genes to confirm that we obtained similar results. For this analysis, orthologs were clustered regarding their previously inferred “age” in five groups (selecting input as species: any species, analysis as species: M. musculus).
Finally, we performed a third approach to determine the conservation of the functional role of IGs. First, we determined the overrepresented GO terms for biological processes and molecular function of the orthologs from the seven genomes using AmiGO28 were obtained. Then, GO terms with their corresponding p-values were clusterized using REVIGO which finds a representative subset of the terms using an algorithm that relies on semantic similarity measures (Supek et al., 2011).
Read counts from a previous transcriptomic analysis of mouse embryonic telencephalon were used to identify differentially expressed genes (Muley et al., 2020). The transcriptomes were obtained using the Illumina HiSeq RNA sequencing (RNA-seq) platform. The procedure for read-counts normalization, and to calculate differential expression analysis is described in https://data.mendeley.com/datasets/rdt5757cbw/1. A gene was considered expressed if its count-per-million (CPM) value was above 5.66-7. Mouse IG and MEG datasets were submitted to analysis to determine the directionality of the change in expression at developmental stage A (E.9.5) compared to stage B (E.10.5). Genes having significant p-values with positive log2–fold change represent an increased expression (UP), those with negative log2-fold change are considered with decreased expression (DN), while gene expression with p-values above 0.05 represents no change between stages (NC), and read-count lower than five in less than four samples out of eight are considered not expressed (NE).
The functional enrichment was assessed using the over-representation analysis of the functional assignment. Genes with differential expression up to two log2-fold change values were considered as up-regulated with a p- and q-value set at 0.05 and 0.10, respectively. The ClusterProfiler R package (Yu et al., 2012) was employed for data analysis and visualization.
For post-translational modification assignments of IG and MEG proteins, the dbPTM9 was used. A two proportion Z-test was used to assess whether the proportions of each post-translational modification among IG and MEG proteins were similar. The p-value was set at 0.05. When the resulting p-value was not significant, meaning that the proportions of IG and MEG proteins were similar for a specific post-translational modification, this was classified as “similar.” On the other hand, when the resulting p-value was <0.05 the post-translational modification was classified as more abundant in ‘‘IG’’ or ‘‘MEG’’ depending on which one had a higher relative percentage of such modification. Post-translational modifications exclusive of either IG or MEG proteins were classified as ‘‘unique.’’ Then, using the miPFinder program10, we determined the mouse gene candidates for IG-encoding microproteins.
Mouse peptide sequences were submitted to Proteinortho (Lechner et al., 2011, 2014) to infer orthologous genes in the genomes of rat, human, chimp, opossum, chicken, and zebrafish. As a first step, Proteinortho performs sequence comparison between each pair of genomes and reports best bidirectional hits (BBHs) for alignments with equal or above fifty percent of sequence identity. In a second step, it represents each gene or protein as a node of a graph and places an edge between two genes if they were identified as a BBH, then, it applies a clustering algorithm and finally reports orthogroups and the orthology relations as pairs of genes in two different genomes.
Each of the species used in this study is a model organism of a different taxonomic level, and therefore, the conservation of mouse orthologs in close or distant related species reveals the “age” of the gene. The conservation was measured by gradually including more species, and the resulting groups were labeled with the name of the largest taxonomical category that includes all species within a group. Therefore, for orthologs of mouse IG genes that were identified in rat, they are said to be conserved in Muridae; those present in Muridae, in human, and in chimp are said to be conserved in the group Eutheria; those found in all the previous species and in opossum as well are conserved in the group Theria; those also present in chick are conserved in Tetrapoda; and finally, those also conserved in zebrafish are conserved in Vertebrata. This classification, however, is only used to refer to the conservation among the species analyzed in the present study.
From the ProteinOrtho predictions, orthology graphs were constructed, and an in-house developed method for the evolutionary reconstruction of gene families was used. This method implements the theory reported in Hernandez-Rosales et al. (2012) and Hellmuth et al. (2013), and it can be found at https://gitlab.com/jarr.tecn/revolutionh-tl. This tool starts by performing a modular decomposition (Tedder et al., 2008) on orthology graphs and then inferring the corresponding gene trees. Each internal node of these trees represents an evolutionary event: duplication or speciation. Subsequently, the gene trees are reconciled with the species tree to determine in which branch of the species tree duplication events occur and, at the same time, infer gene losses. This method allows us to infer how ancestral a gene is, determined by its orthologs in the other species, as well as to identify species-specific genes. Finally, we identified the orthologs of the 1,116 mouse IG proteins that were conserved as IGs, or that were identified as MEGs in the abovementioned genomes.
To determine the syntenic conservation of the mouse protocadherin IG members of the beta cluster across the selected genomes, the genomic coordinates of orthologs genes were retrieved from GTF files employing custom R scripts and plotted using the genoPlotR R package.
The origin of IGs has been explained mostly by retrotransposition, which occurs by homologous recombination between the genomic copy of a gene and an intronless cDNA (Kaessmann et al., 2009). Mouse IGs represent 6% of the total number of one-exon genes, while retrotransposed single-exon pseudogenes with lost molecular function constitute almost half of them (coding, non-coding, pseudogenes) (Supplementary Figure 1).
Computational analysis was performed to identify mouse IGs. Then, based on the comparative annotation of IG and MEG datasets, a study was performed to identify their unique and shared molecular and biological features.
The grouping of IGs by protein domains that have an evolutionary relationship (SUPERFAMILY database) revealed a higher enrichment of the histone fold, 4-helical cytokine family of signal transducers, and transcription factor families including the Poxvirus and Zinc finger (POZ) domain, “Winged helix” DNA-binding domain, High Mobility Box group (HMG-box), and A DNA-binding domain in eukaryotes, as well as the transmembrane protein families Cadherin-like, and Frizzled cysteine-rich domain. MEG-encoded proteins, in contrast, are enriched in protein kinase-like, immunoglobulin, Krüppel associated box (KRAB) domain, and Armadillo repeat motifs (ARM) repeat families. The top enriched structural families of IG and MEG groups are shown in Supplementary Figure 2.
The analysis of the conserved functional domains (Pfam database) among the enriched protein families encoded by mouse IGs, revealed 598 hits of three main classes: 253 were transmembrane protein receptors, 101 core histones, 84 transcription factors, and 160 that belong to other classes (Figure 2A). Among the transmembrane protein receptors, the most enriched domains were Taste 2 receptors (TAS2R), Vomer-nasal 1 receptors (V1R), and seven transmembrane group 1 (7tm_1), common in GPCR and vomeronasal receptors (Figure 2B). Other domains identified were cadherin, Pheripheral myelin protein 22 (PMP22-claudin), (Desintegrin and metalloproteinase domain (ADAM), Disintegrin, and Frizzled (FZ). In the transcription factor group, Broad-Complex, tramtrack, and bric- -brac (BTB), Myb DNA-binding, HMG-Box, and forkhead were enriched protein domains in the mouse IGs compared to MEGs (Figure 2C). Meanwhile, in the histone group, four domains were observed: Histone, Histone H2A 1363 C-terminal (H2AC), Centromere kinetochore component CENP-T histone (CENPTC), and Linker histone (Figure 2D). Finally, in the other groups we found among others Keratin, Interferon, Ubiquitin, Actin, and FYTT enriched domains (Figure 2E). The classification of IGs in functional groups mentioned above was then used for the transcriptional analysis (Figure 2).
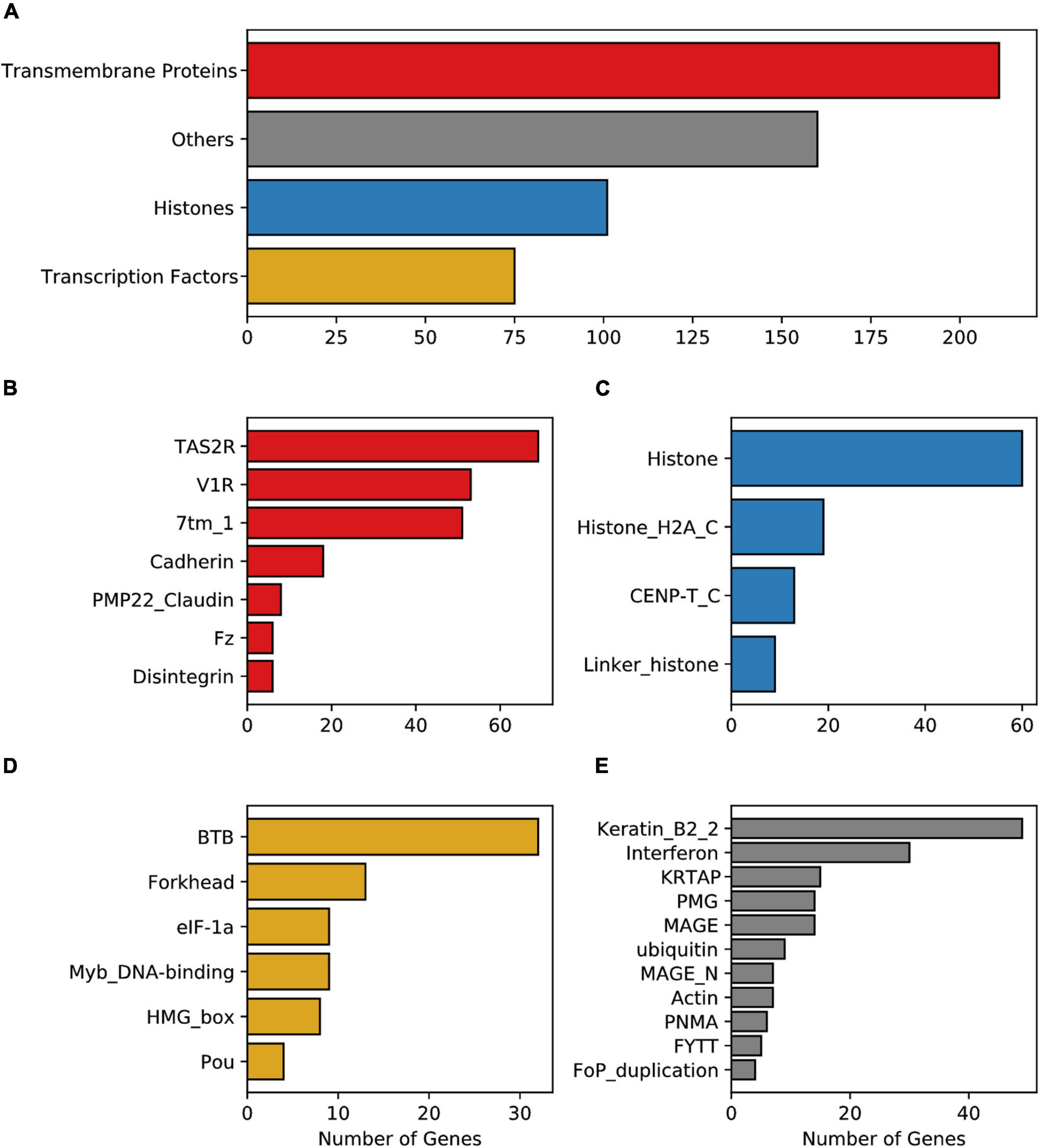
Figure 2. Enrichment of the Pfam protein domains in mouse IG proteins. (A) Enriched domains of proteins grouped into four main classes, transmembrane proteins (red), histones (blue), transcription factors (yellow), and others (gray). Enrichment of domain terms was calculated with a significant background list associated with the Pfam domain and p-adjusted values above 0.05. (B) Pfam domains grouped in the transmembrane proteins class, (C) Pfam domains grouped in the histone class, (D) Pfam domains grouped in the transcription factors class, (E) Pfam domains considered as others.
Analysis of biologically significant motifs (PROSITE database) among MEG and IG proteins identified a total of 1,239 (12,546 hits) and 144 (634 hits) distinct protein signatures, respectively. Interestingly, among the most abundant motifs in the mouse IGs are GPCR, leucine-rich repeat, histone, transcription factor Forkhead domain, Myc-type basic helix-loop-helix (bhlh) motifs, ankyrins, and cadherin domains which were found to be infrequent in MEG proteins (Figure 3A). It is noteworthy, however, that among the top motifs that were unique to IG proteins H2B signature was the largest group (Figure 3B). Hence, these results show that most of the top predictions of IGs signatures are characteristic of transmembrane receptors, histones, and specific transcription factors, having a unique signature for histones.
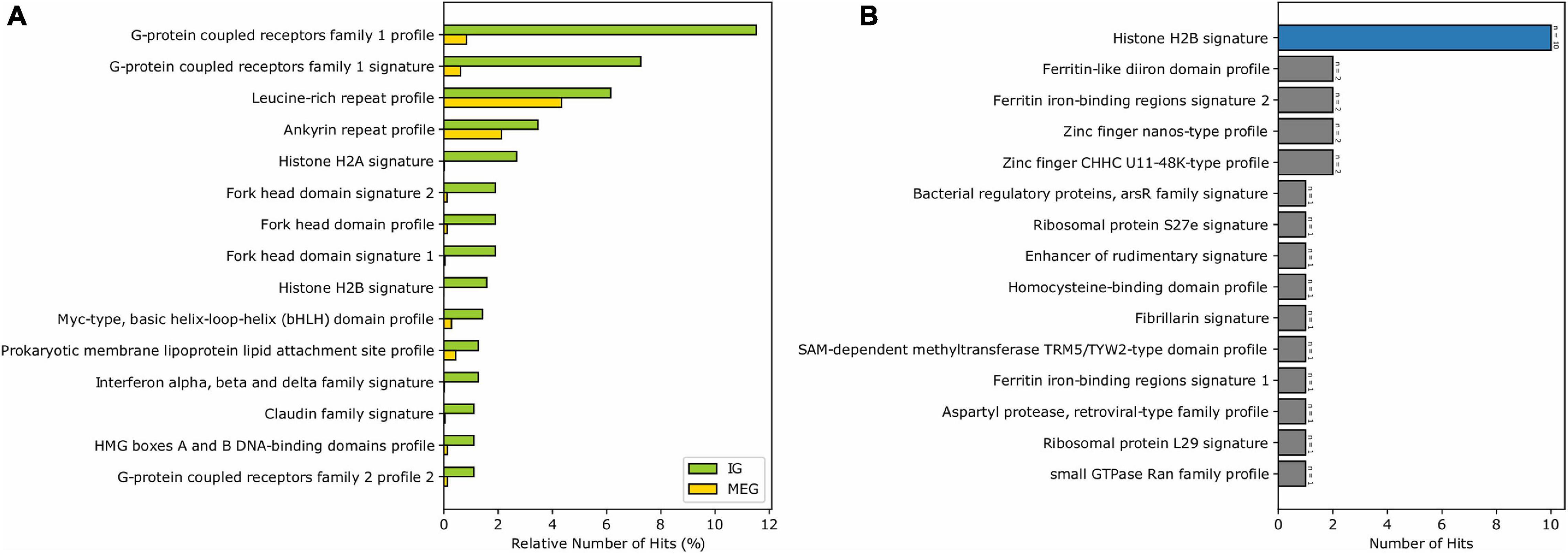
Figure 3. Enrichment of the PROSITE signatures in mouse IG proteins compared to MEG proteins. (A) The 15 most abundant PROSITE signatures of IG proteins. (B) PROSITE signatures exclusive of IG proteins (signatures belonging to the Histones class are shown in blue).
Finally, the functional enrichment of mouse IGs revealed biological pathways associated with genetic and protein regulatory processes including detection of chemical stimulus involved in sensory perception of the bitter taste, chromatin silencing, positive regulation of peptidyl-serine phosphorylation of STAT proteins, and nucleosome positioning (–log10–34.23 > −3.27). Other functions detected were immune, neuro-specific, and development processes such as mmu05322-Systemic lupus erythematosus, R-MMU-6805567 Keratinization, R-MMU-500792 GPCR ligand binding, R-MMU-1266695 Interleukin 7 signaling, hard palate development, and noradrenergic neuron differentiation (–log10–29.94 > −3.75) (Figure 4).
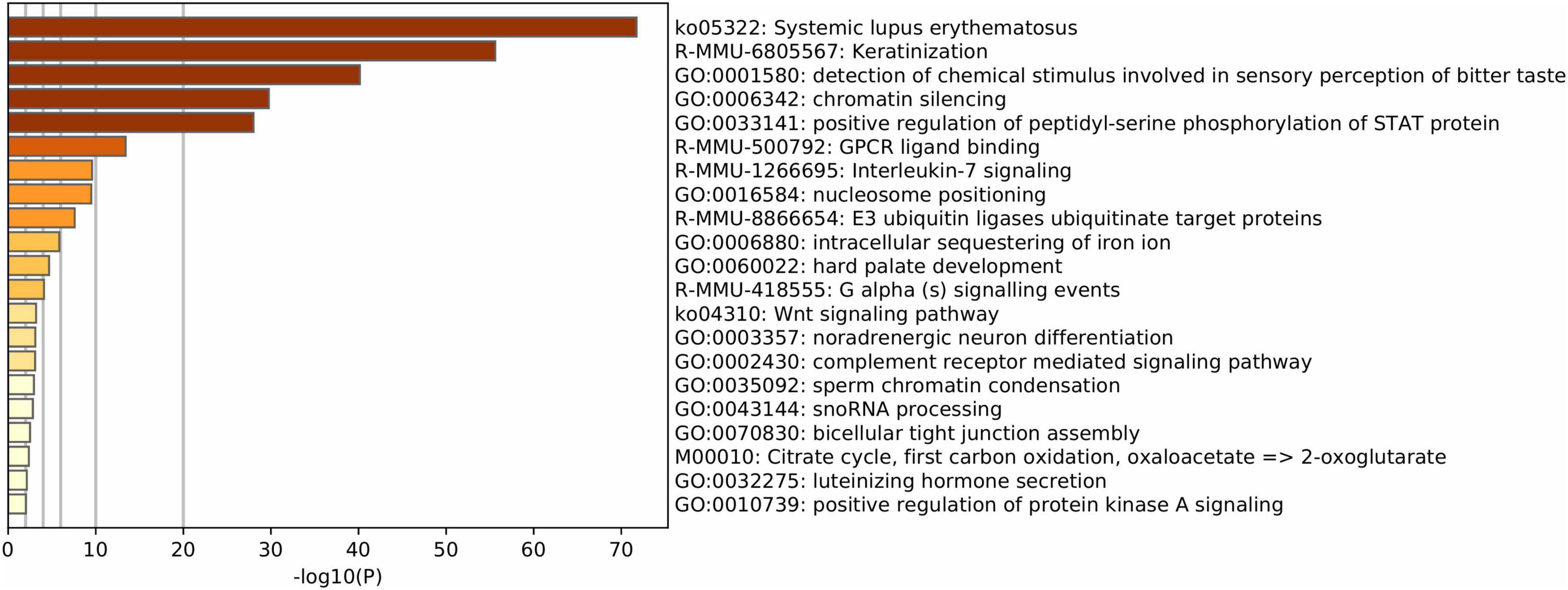
Figure 4. Functional enrichment of mouse IG proteins. Gene ontology enrichment and pathways for IG proteins. The color key from yellow to brown indicates high to low p-values, respectively.
Altogether, functional assignment analysis suggests that IGs have distinct biological roles in comparison to MEGs.
Previous studies detected enrichment of neural-related functions among IGs (Grzybowska, 2012). Moreover, since the expression of MEGs is modulated by the balance between the rate of transcription elongation and the alternative splicing of exons (Fong et al., 2014), we hypothesized that the natural absence of splicing on IG mRNAs could confer them differential regulatory roles in complex biological processes. Therefore, it was our interest to identify and analyze IGs that are expressed in mice during brain development. For that purpose, we analyzed expression data from the developing mouse telencephalon at stages in which its patterning is taking place (E9.5 and E10.5) (Muley et al., 2020).
Overall, the expression of IGs was lower than that of MEGs (Figure 5A), which is consistent with previous in silico observations (Sakharkar et al., 2005a). Out of 1,116 transcripts, differential expression analysis was performed for 1,087, with 37 of them (3.4%) showing up-regulation and nine down-regulation (0.82%) from gestational day E9.5 to E10.5 Moreover, 387 (35.63%) did not change expression, and 653 (60.12%) were not expressed during the analyzed stages (Figure 5B). Meanwhile, among MEGs, 1247 were up-regulated (6.13%), 789 were down-regulated (3.88%), 13,198 had no expression changes (64.93%), and 5090 did not show expression (25.04%) (Figure 5B). It is noteworthy that an inverse expression pattern of genes with no expression changes (a higher percentage of MEGs than of IGs) and those not expressed (a higher percentage of IGs than of MEGs) was found in this comparison.
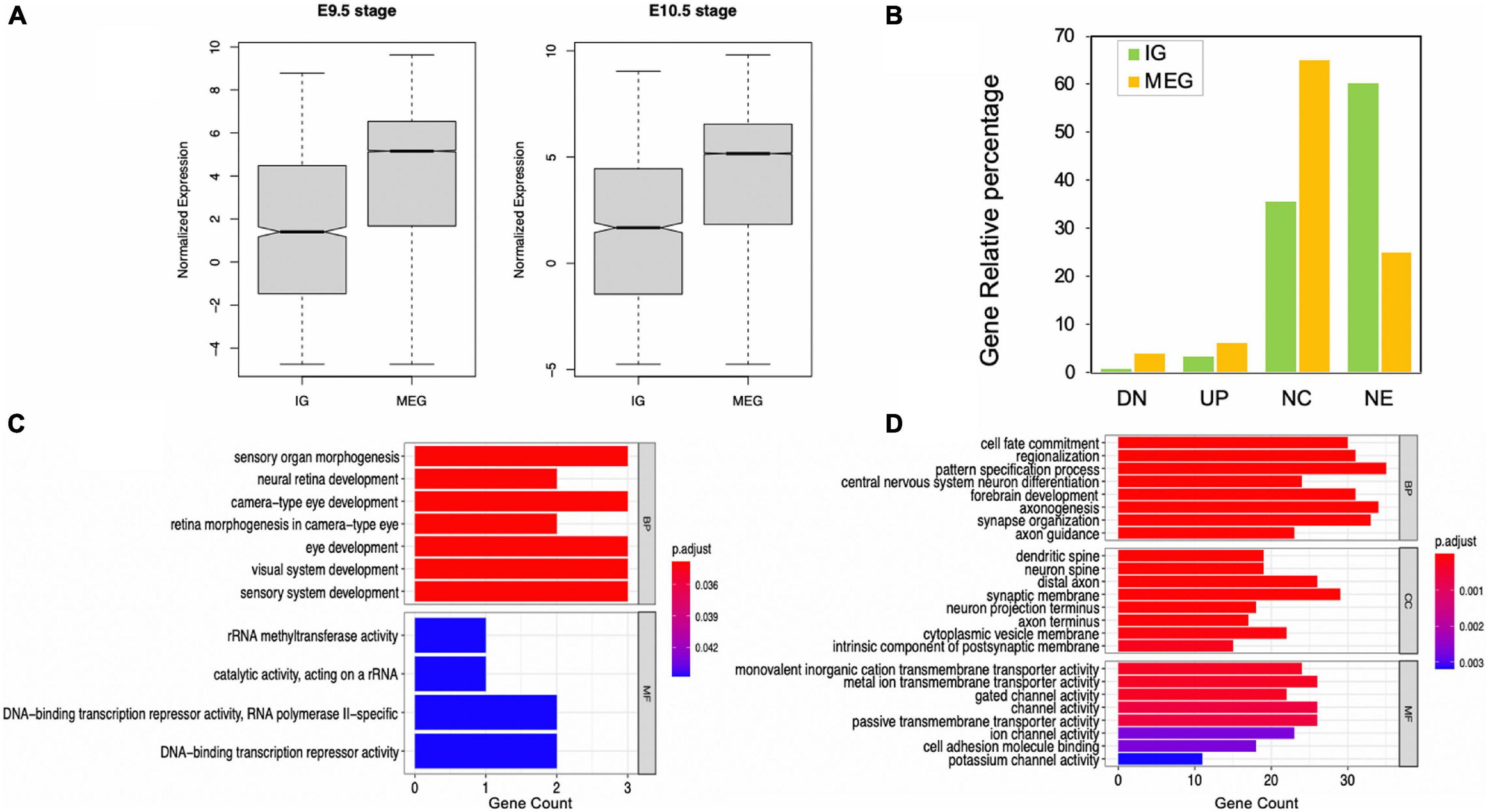
Figure 5. Expression levels of IGs in mouse embryonic telencephalon compared to MEGs. (A) IG and MEG normalized expression at 9.5 and 10.5 embryonic stages; (B) IG and MEG, gene differential expression groups in 9.5 and 10.5 stages, (UP) upregulated, (DN) downregulated, (NC) no expression changes, (NE) not expressed. Enrichment of functions in upregulated IGs (C) and MEGs (D). Each barplot shows significantly enriched gene ontology (GO) terms on the y-axis, colored by their p-adjusted values, while gene count is represented on the x-axis. Test gene sets for enrichment analysis were the up-regulated genes in each dataset, and the background set was all up-regulated genes. GO terms are grouped by BP (biological process), CC (cellular component), and MF (molecular function).
Our analysis revealed that all up-regulated IGs are exclusively enriched in biological pathways in eye and sensory organ development processes compared to MEGs also involved in other developmental and neural function pathways (Figures 5C,D). Moreover, significantly enriched terms in molecular function found for up-regulated IGs are consistent with their regulatory role, including rRNA methyltransferase and DNA-binding transcription repressor activities. In contrast, in the up-regulated MEGs the molecular function terms are highly enriched for transmembrane transporters and channel voltage activities (Figure 5D).
From the IG transmembrane protein group, transcripts for Tram1l1, Cdk5r2, Nrarp, Kcnf1, Fzd7, Fzd8, Fzd10, and Cldn5 were up-regulated. Strikingly, from the cluster of 22 β-protocadherins (pcdhbs), which contains 18 IGs, 9 of these were among those up-regulated in the E10.5 telencephalon (Pcdhb3, Pcdhb4, Pcdhb7, Pcdhb10, Pcdhb11, Pcdhb17, Pcdhb19, Pcdhb20, and Pcdhb21) (Figures 6A, 7A). It is important to note that all but one of all protocadherins of this cluster were expressed in the developing telencephalon. Our expression analysis additionally revealed up-regulation of Olig1, Bhlhe22, Bhlhe23, Pou3f1, Pou3f2, Pou3f4, Foxq1, and Neurog1, most of which are BHLH transcription factors crucial for the regulation of brain development and neuro-specific functions (Figure 6B). Moreover, regarding IGs within the histone group, H2bc21, H2bu2, H2aw were up-regulated during the mouse embryonic stages (Figure 6C). Finally, IGs from other groups with up-regulation were also observed (Figure 6D).
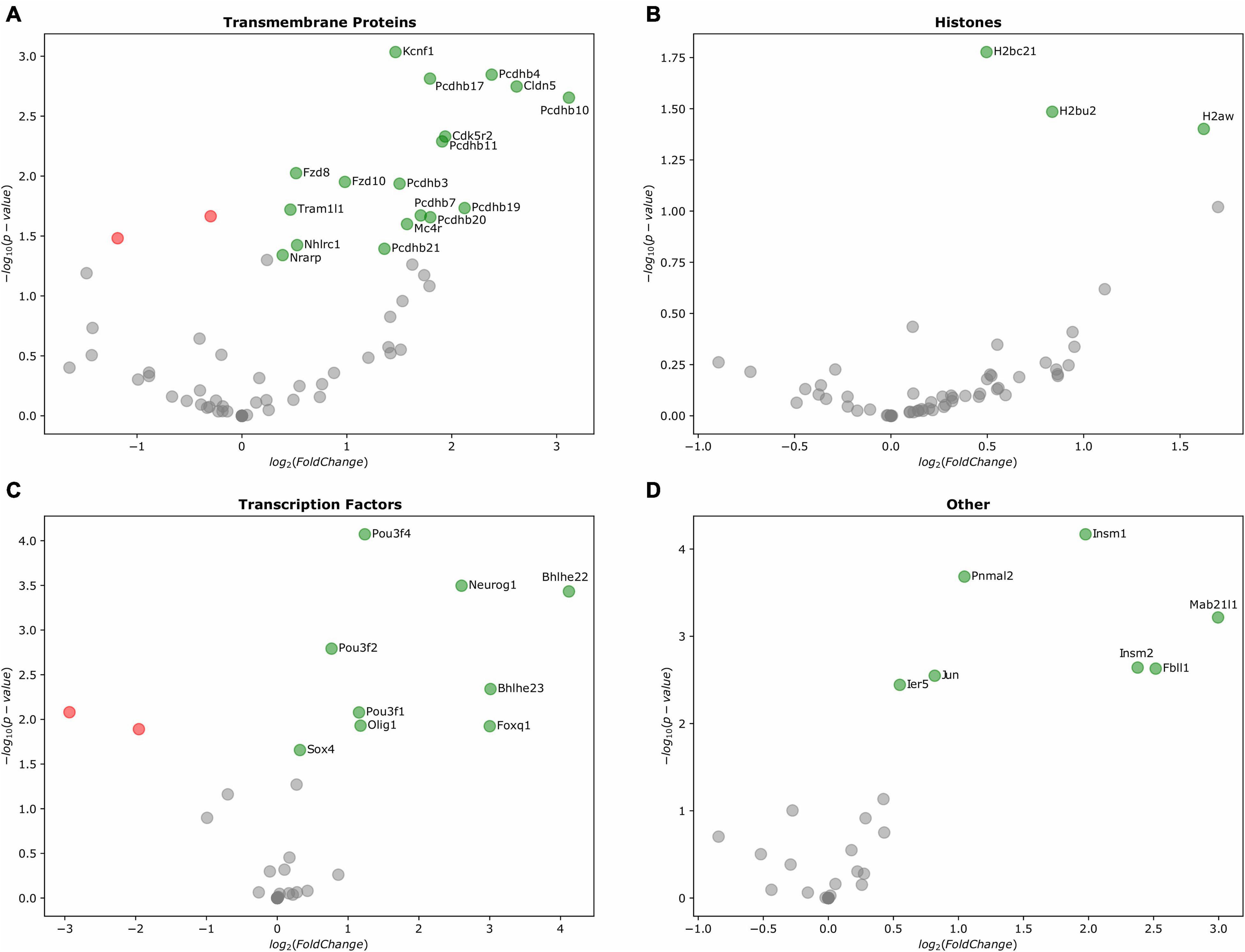
Figure 6. Differentially expressed IGs in the mouse embryonic telencephalon. Expression of genes grouped by their Pfam assignment, determined by their log2-fold change values. Up-regulated genes are highlighted in green, while downregulated genes are colored in red. (A) Gene expression of transmembrane proteins, (B) histones, (C) transcription factors, and (D) other protein families.
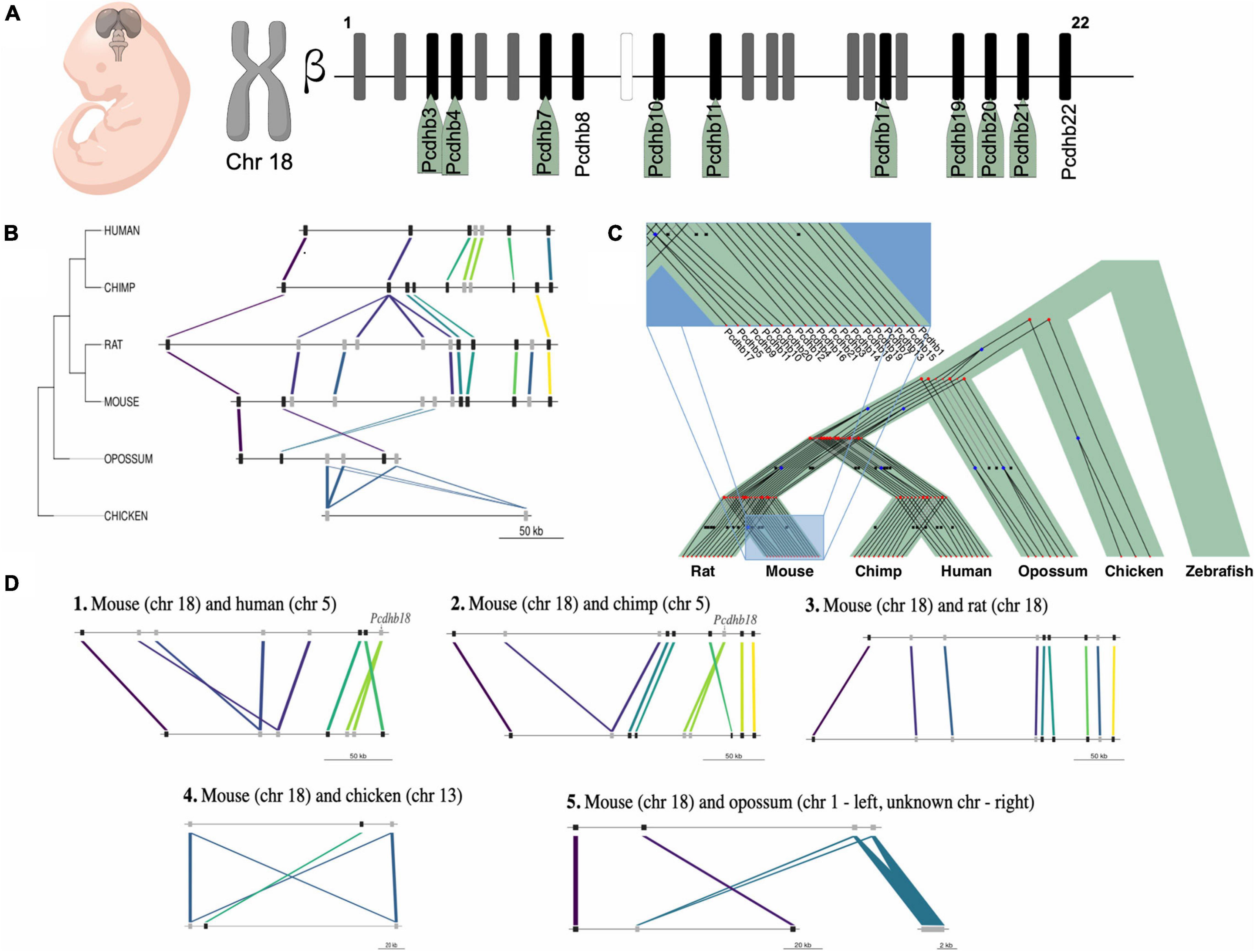
Figure 7. Expression and evolution analysis of the mouse cluster of β-protocadherins. (A) Cluster of the 22 β-protocadherins in the chromosome 18 depicting in black up-regulated genes (names highlighted in green) in telencephalon between embryonic stages 9.5 and 10.5, in gray expressed genes with no relative changes between stages, and in white not expressed genes; (B) Syntenic map of the approximately 300 kb β-protocadherin locus across five mammalian lineages and chicken as an outgroup. Colored lines depict orthology relationships across the phylogenetic tree. Genes connected by the same color belong to the same orthogroup identified by ProteinOrtho. Genes are shown as black squares for single-copy orthologs and gray squares for expanded genes; (C) Reconciliation tree of the protocadherin gene trees and the species tree. Each gene tree represents an orthogroup, internal nodes represent evolutionary events (blue squares represent duplications, red bullets represent speciations) and black crosses in leaves represent inferred gene loss events; (D) Pairwise syntenic comparisons of the mouse β-protocadherin locus to four mammalian genomes and chick highlighting the lineage-specific loss and expansions of the Pcdhb18 gene.
To gain further insight on the evolutionary conservation of IGs with a functional role in telencephalon patterning, we studied the syntenic conservation of the ortholog genes of the 18 mouse IG β-protocadherins (Figure 7A) in our set of seven species. We determined that human, chimp, rat, opossum, and chick contain orthologs of the mouse single exon pcdhb genes, which are absent in zebrafish. Overall, all the orthologous genes of the cluster are located in a single locus in their respective genomes, with varying lengths ranging from ∼128 to 310 kb, displaying a few local inversions (Figure 7B). These results are consistent with previous studies that have explored the syntenic conservation of pcdhb genes across other vertebrate species (Noonan et al., 2004; Yu et al., 2008). Even though we found syntenic conservation of some members of the β-protocadherin cluster, we observed slight disruptions of the order of genes due to gene expansions, which can be either gene duplications or de novo formation. These gains are most notorious in the mammalian genomes (Figure 7B), suggesting gene expansion of the intronless β-protocadherins could be relevant for their role in neurogenesis, as well as other neuro-specific functions associated with the Wnt canonical pathway.
We looked in more detail at the evolutionary histories of β-protocadherins, by reconciling the gene trees of this gene cluster with the taxonomic species tree (Figure 7C). First, we observed that none of these genes is conserved in zebrafish. Moreover, some genes are gained in specific lineages, for example, Pcdhb17 is only observed in mice and rats, while eight genes are shared across the mammals in the study. Three β-protocadherins appear to be shared among primates and the marsupial opossum, while only Pcdhb7, Pcdhb15, and Pcdhb19 are shared between mouse and chick, and across other intermediate species, suggesting that these are the oldest β-protocadherins that give origin to the rest of them.
Then, by assessing syntenic conservation in a pair-wise fashion, we found relevant lineage-specific gene losses (Figure 7D). For instance, Pcdhb18 is absent in rats while it is present in mice and duplicated in primates. This evidence suggests that the complexity of nervous system characteristic of mammals could also be associated with the duplication of single exon genes besides splicing-derived protein isoform diversity.
In addition to alternative splicing, and mRNA editing, post-translational modifications (PTMs) constitute a defining factor of the complexity of proteomes by increasing structural and functional diversity of each proteoform, the set of multiple protein molecules encoded by one gene. Hence, protein PTMs have an essential role in protein structure-function, including activity, stability, folding, and turnover (Uversky, 2015). Since IGs fit the “one gene—one protein” concept we aimed to determine whether PTMs represent exclusive mechanisms of regulation for these genes. In our analysis, we observed that Succinylation and S-nitrosylation had similar prevalence in IG and MEG groups. These were followed with much lower frequency by Glutathionylation, Glutarylation, Palmitoylation, and Oxidation (Figure 8A). In contrast, PTMs with a unique presence in MEGs were Nitration, Myristoylation, Sulfation, Carboxylation, GPI-anchor, and Pyrrolidone carboxylic acid (Figure 8B).
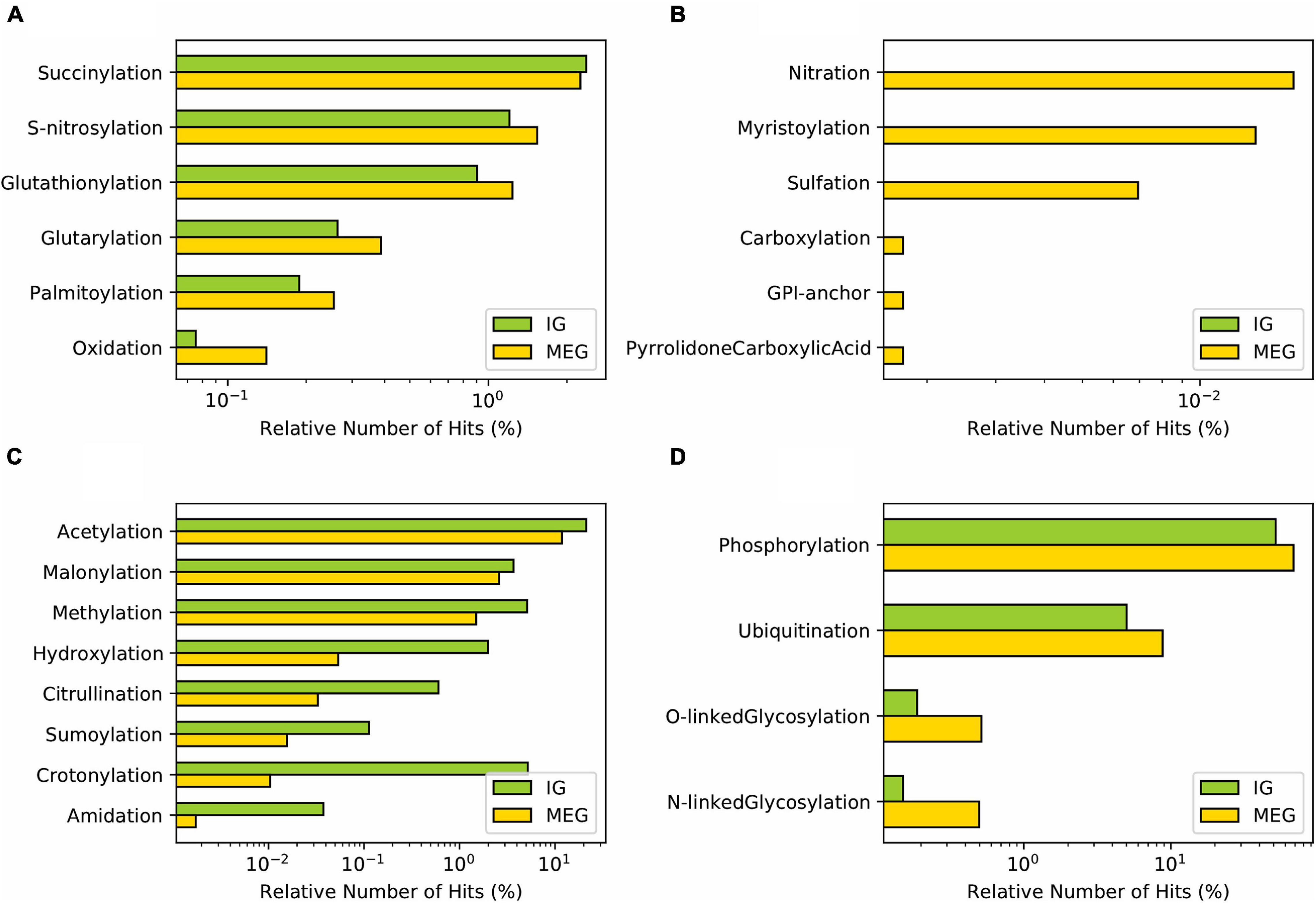
Figure 8. Distribution of post-translational modifications in mouse IG proteins compared to MEG proteins. (A) PTMs with similar distribution between IG and MEG proteins. (B) PTMs exclusive to MEGs. (C) PTMs predominant in IG, and (D) in MEG proteins.
Notably, in accordance with their protein assignment, a differential enrichment for IG proteins was observed of Acetylation, Crotonylation, Methylation, Malonylation, and Hydroxylation (Figure 8C) which are characteristic features of the core histones. Other PTMs of IG proteins, albeit at lower frequency, were Citrullination, Sumoylation, and Amidation (Figure 8C). The complementary group of PTMs more enriched in MEG proteins included Phosphorylation, followed by O-linked Glycosylation, Ubiquitination, and N-linked Glycosylation (Figure 8D). PTMs classified based on the modification-enabled functionality for membrane localization such as Myristoylation and GPI-anchor were found to be more frequent for MEGs (Figure 8D).
The regulation of multidomain proteins at the post-translational level can be mediated by microproteins (miPs) (Staudt and Wenkel, 2011) which are small proteins containing a single domain that form heterodimers with their targets and exert dominant-negative regulatory effects (de Klein et al., 2015; Eguen et al., 2015). In Eukarya, microproteins have been found to have a remarkable influence on diverse biological processes.
Aware of the differential occurrence of PTMs on IG proteins, the DNA binding repressor activity molecular function of up-regulated IGs during mouse brain development, and due to the remarkable role of miPs, we assessed whether this group of genes encoded proteins fitting the miP definition. Characteristic features of miPs are the short length of their primary structure, a homodimer domain, and negative modulating activity of protein multi-complexes. Our first approach was to analyze the peptide length of IG and MEG proteins. The highest length-frequency for IG peptides was in the range of 200–400 amino acids, compared to that of MEG peptides which was 300–500. Then, using the miPfinder tool (Straub and Wenkel, 2017), we identified the following IGs as microprotein candidates: the BHLH transcription factors Bhlha9, Msg1, Ferd3l, Bhlhe23, and Ascl5 (e-value 4.6E–30), as well as the histones H1f0, H1f1, Hils1, H1f2 H1f3, H1f4, H1f5, H1f6, and H1f10 (e-value 7.4E–09), corresponding to the H1 linker histone group.
To infer the evolutionary age of genes we implemented a bioinformatics method to assess the extent and patterns of distribution of each gene’s orthologs and paralogs in different species. The rationale of this approach is that widespread conservation of the orthologs of a gene in the different vertebrate taxa is an indication of old age for that particular gene. This approach allowed us to determine the conservation of IGs across 7 genomes, as well as to identify species-specific mouse IGs (Figure 9). In this analysis, we found that 543 out of the 1,116 mouse IGs have orthologs in at least one of the other species. For the mammalian genomes, we found 442 genes conserved as IGs out of 501 orthologs in the rat genome, 335 orthologs in chimp with 250 conserved as IGs, 397 with 262 IGs in human, and 258 with 167 IGs in opossum (Table 1). Meanwhile, we found 133 orthologs in chick with 78 conserved as IGs, and 220 in zebrafish with 91 conserved as IGs (Table 1). We also identified out-paralogs of mouse IGs (genes that arose via duplication before a speciation) that are conserved in the other species: 36 in rat, 16 in human, 11 in chimp, 9 in opossum, 2 in chick and none in zebrafish. Finally, we identified 573 IGs with no orthologs in the other species, suggesting that these are species-specific mouse IGs.
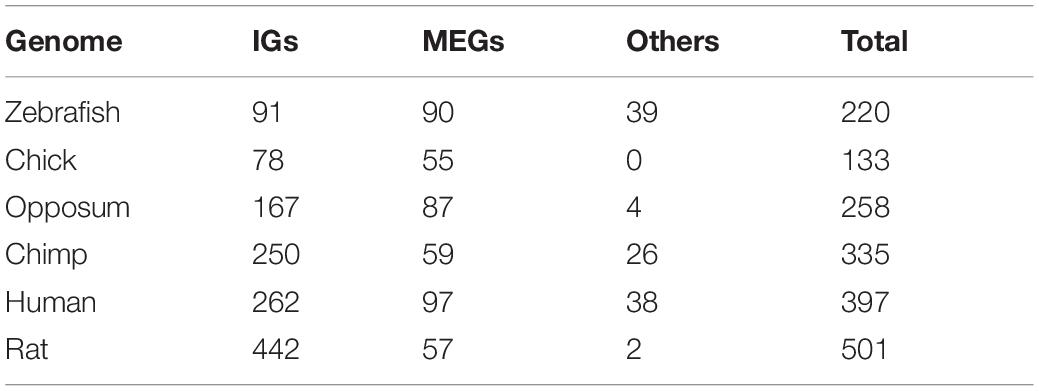
Table 1. Summary of mouse IG orthologs in selected genomes.
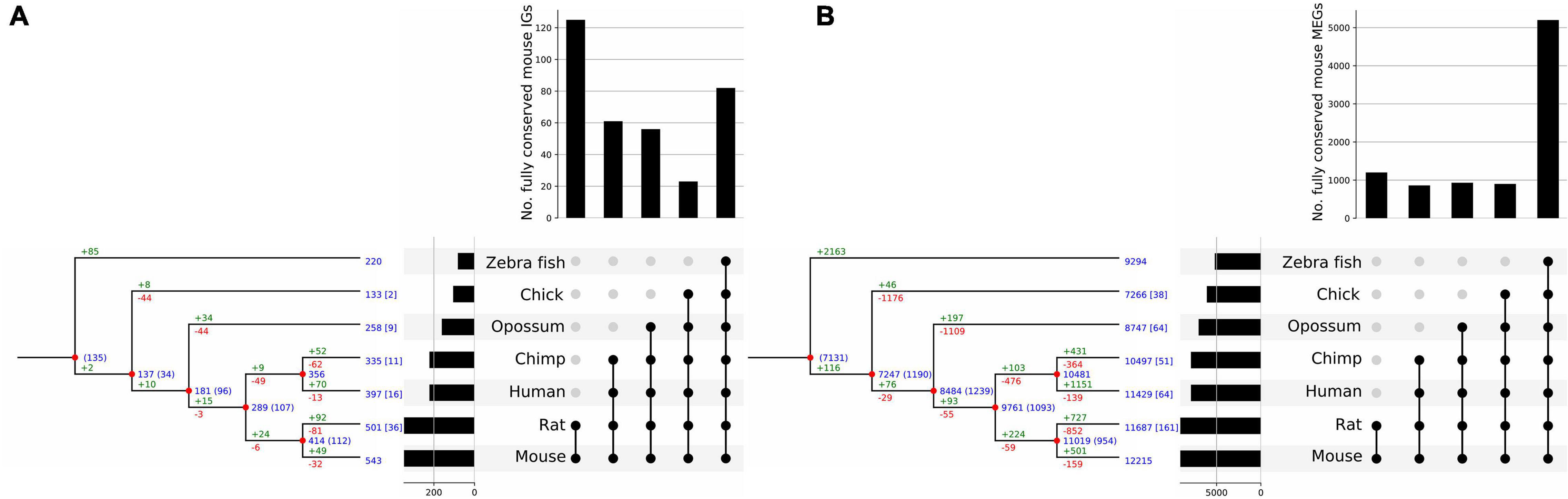
Figure 9. Clusterization of conserved mouse IG and MEG orthologs. Reconciliation trees representing the evolutionary history of mouse IG (A), and MEG (B) gene families. Blue numbers at each internal node represent the number of ancestral genes found in that clade that were inherited by an older ancestor. Blue numbers in brackets represent ancestral genes that might be generated at that evolutionary point, since they are not found in an outgroup of the clade. Green numbers represent gene gains due to duplication events and red numbers represent gene losses. Numbers at the tip of the branches represent the number of orthologous genes in other species, and numbers in square brackets represent the number of out-paralogs (genes that arose via duplication before speciation) in other species in the study. Orthologous genes are grouped by age determined by the clade they are conserved in; the histogram on top of the upset plot shows the number of genes that are specific to each clade; the histogram to the left of the upset plot represents the number of mouse orthologs for each species. Moreover, the bar for mouse shows the number of IGs for which an ortholog in another species was found.
Overall, we found that 70% of the IG orthologs are IGs as well, and 30% are MEGs (Table 1). As for MEGs (Figure 9), less than 5% of their orthologs are IGs and the rest are MEGs (Tables 1, 2). As expected, due to its evolutionary closeness with the mouse, the genome with the highest conservation in gene architecture is the rat, with approximately 88% of conserved IGs, while the largest difference was found for the zebrafish genome with only 41% of conserved IGs. Furthermore, at the superfamily level as identified by SUPERFAMILY, we found that 24% of the IG superfamilies are conserved as IG-only, while 76% are predominantly IGs but contain at least one MEG ortholog in another species. Similarly, for MEG superfamilies, 35% were conserved as MEG-only, while 65% were predominantly MEGs with at least one IG ortholog. Hence, these analyses revealed that most of the IGs identified in the mouse genome remained with this genetic structure in other species thus supporting their high conservation across vertebrate genomes.
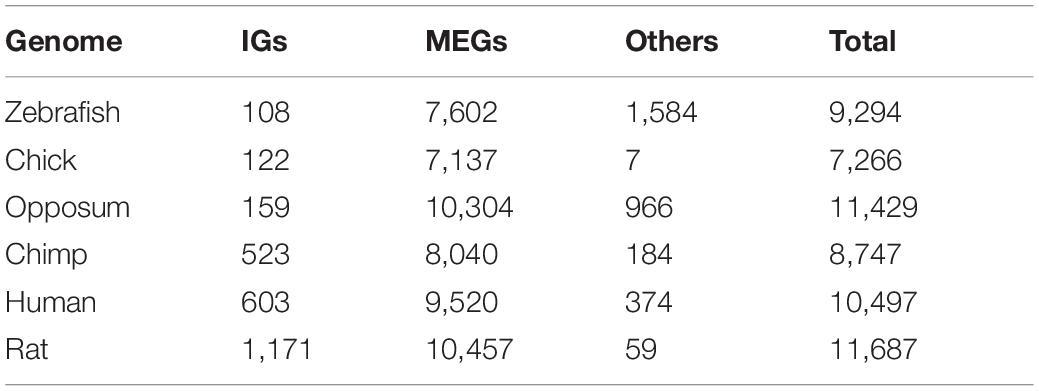
Table 2. Summary of mouse MEG orthologs in selected genomes.
From the previous analysis we clusterized IGs and MEGs into five age-groups named by the taxonomic category that includes all the species of each group and thus represents the most recent common ancestor (MRCA) of each ortholog as inferred from the extant species analyzed. These groups were: Vertebrata, Tetrapoda, Theria, Eutheria, and Muridae (Figure 9). From the reconstruction of the evolutionary history of the mouse IGs, our results revealed that their conservation is more marked in the Muridae as it contains the largest number of orthologs common to its members (Figure 9A) followed in abundance by Vertebrata. This indicates that a large number of IGs are sufficiently old to have orthologs in all the vertebrates analyzed, and that the clades that include the closest relatives to mice have increasing IG ortholog abundance. In contrast, the highest conservation of MEGs in gene numbers is among Vertebrata thereby revealing a much older age than that of Muridae IGs (Figure 9B). For both IG and MEG orthologs, the number of paralog-related genes increases with gene age consistently with the rate of duplication of the edges of each clade (Figure 9). Moreover, a significant number of in-paralogous genes in the zebrafish genome, generated via duplication after speciation, have an ortholog in the mouse genome.
With the purpose of determining whether there was a differential functional enrichment of IGs according to their evolutionary age, we analyzed the enrichment of molecular pathway GO terms in both IGs and MEGs. In agreement with a specialized role of IGs, our results show that IG and MEG orthologs are involved in different biological pathways although some shared pathways were detected as well (Figure 10). Conserved IG proteins with the MRCA among Vertebrata are histones highly enriched in negative regulation of megakaryocyte differentiation (–log10, –20.82). Other orthologs conserved to this group are linked in a lower level to thermogenesis, basal cell carcinoma, positive regulation of protein kinase A signaling, ribosomal large subunit assembly, wound healing, and vascular process in the circulatory system, platelet aggregation and development process such as cell-fate specification, negative regulation of animal organ morphogenesis, pituitary gland development, and regulation of bicellular tight junction assembly (–log10, –9.33 > −2.81). For Theria we found G alpha signaling events (–log10, –8.86), while in the Eutheria group peptidyl-serine phosphorylation of STAT protein, and chromatin silencing were enriched (–log10, –6.47;–5.69). Noticeably, the most recent genes which belong to the Muridae group are exclusively enriched in intracellular sequestering of iron, complement receptor-mediated signaling pathway, and histone deubiquitination (–log10, –8.64 > −3.63).
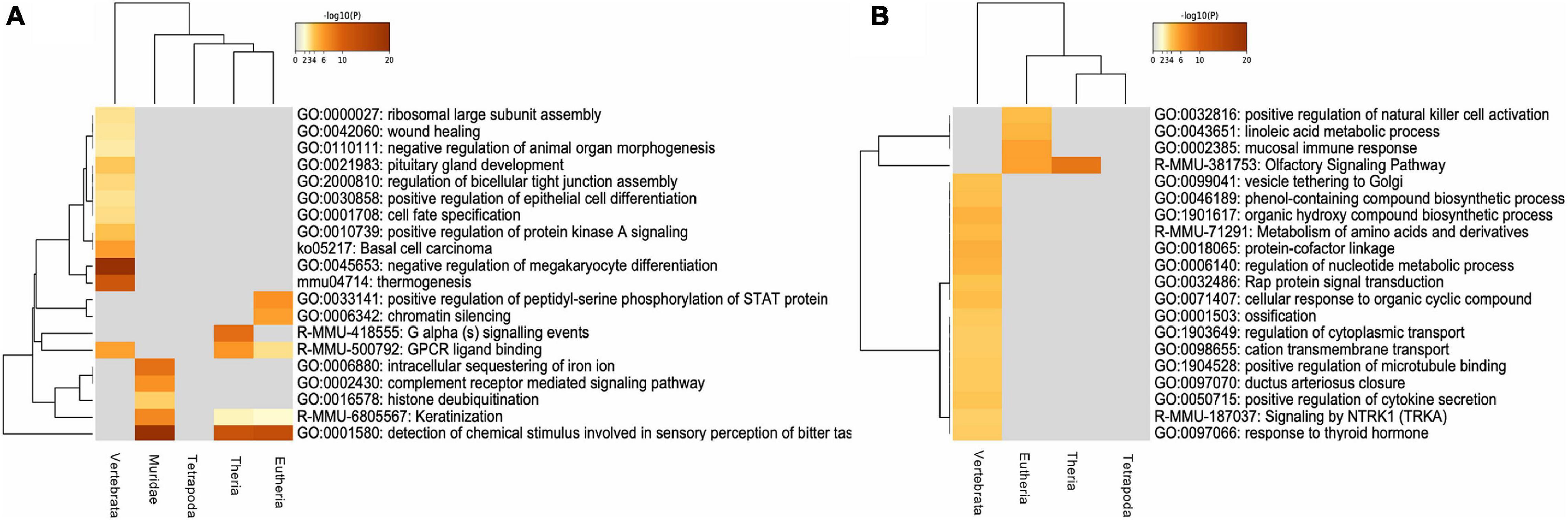
Figure 10. Functional enrichment of conserved mouse IG and MEG orthologs. (A) Gene ontology enrichment and pathways for mouse IG conserved as IG in the selected species, orthologs are clustered regarding their “age” group. (B) Gene ontology enrichment and pathways for a sample of 1,116 mouse MEGs conserved as MEG in the selected species, orthologs are clustered regarding their “age” group. The color key from yellow to brown indicates high to low p-values, respectively.
Our analysis also identified IG proteins with enriched pathways shared among the various age groups. Detection of chemical stimulus involved in sensory perception of bitter taste, and keratinization are GO terms shared among Muridae (–log10, –22.91;–7.07), Eutheria (–log10, –13.36;–2.10) and Theria (–log10, –12.37;–2.39). Meanwhile, GPCR ligand binding is shared among Vertebrata (–log10, –5.59), Theria (–log10, –6.11), and Eutheria (–log10, –3.05) (Figure 10A).
Then we focused on determining the conservation of the biological role of IG orthologs among the different genomes. GO terms that are highly enriched in the seven genomes analyzed were detection of chemical stimulus involved in sensory perception of smell, organic substance metabolism, DNA packaging, signaling, multicellular organismal process, cell communication, transport, and localization, while GO molecular function terms enriched are olfactory receptor activity, Wnt protein binding, odorant binding, protein binding, catalytic, and molecular transducer activity.
The mechanism of alternative splicing is a pivotal contributor to the diversity of proteins and the functional complexity of eukaryotic genomes. Intron-containing genes are capable of generating multiple protein isoforms by this process by which exons can be removed, lengthened, or shortened (Sakharkar et al., 2005b). In contrast, protein coding genes lacking introns produce a single peptide of predictable sequence which may undergo postranslational fine tuning. The availability of detailed annotation of sequenced genomes for many organisms contributes toward a better understanding of their structure which has been shaped by flexible evolutionary pressure (Bult et al., 2019). Studying the evolutionary dynamics of exon-intron patterns at the genomic level is likely to shed light on their role in genome structure and gene architecture.
To further our insight into the structure and the evolution of mouse IGs, we examined their function, differential expression in the developing brain, the signatures for post-translational modifications of their encoded proteins, their potential as modulators of multiprotein complexes, as well as their evolutionary dynamics in comparison to their orthologs in other vertebrates.
Our work revealed that, in accordance with previous studies, IGs and MEGs appear to specialize in different functions which is supported by their enrichment in distinct biological pathways and differential abundance of post-translational modifications. As an additional indication of this specialization, IGs that are up-regulated during the development of the mouse telencephalon, are associated with specific developmental programs in this structure and display a functional enrichment profile that differs from that of up-regulated MEGs. Moreover, mouse IGs, some of which fit the criteria to be defined as regulatory microproteins, appear to be of more recent origin than MEGs in vertebrates. Consistent with this notion, about half of IGs do not appear to have orthologs in other genomes thus suggesting a relevant role in mouse evolution. The synteny of the β-protocadherins, however, points out to a mammalian conserved function of these IGs although some species-specific changes in this gene cluster were observed for the various species analyzed.
In the mouse genome, coding genes are predominantly of MEG type11 (80% of a total of 22,481). However, a considerable number of IGs are present in this genome, and the conservation of this fraction has been reported for other mammalian genomes (Sakharkar et al., 2006).
Our comparative analysis of the types of proteins encoded by IGs and MEGs revealed that these two populations have very divergent functional profiles. The most abundant types of proteins found among the former were the chromatin components histones and centromere proteins, transmembrane proteins of the G-protein coupled receptor family 1 and cadherins, and transcription factors containing BTB, forkhead, and HLH domains. In contrast, among MEGs, the most abundant proteins were those containing zinc finger, Pkinase (Protein-kinase), and PH (Pleckstrin homology) domains. These observations are consistent with previous findings that IGs are highly enriched in GPCRs, and seven transmembrane domain proteins and reveal a functional divergence between IGs and MEGs likely to be the result of differential evolutionary constraints (Sakharkar et al., 2006).
Among the transmembrane IG proteins, vomeronasal and taste receptors stand out as the most abundant. These proteins play a highly relevant role in chemoreception which is the most salient means of the interaction of Muridae with their environment, with conspecifics, with potential prey or predators. These findings suggest that some olfaction and taste receptors are required to be constantly transcribed in an efficient and rapid process, which may be a factor that favors their overrepresentation among IGs. Moreover, the taste receptor cells of vertebrates are continually renewed throughout the organism’s life which suggests a high demand for the housekeeping expression of these genes.
A considerable enrichment was also observed among IGs of BTB, Forkhead, HLH, HMG-Box transcription factors, and the chromatin components core histones H2AC, CENP-TC, and the histone Linker cluster. Thus, this indicates that IGs are playing also an important role in packaging, transcription and chromatin assembly.
Altogether, these results suggest that intronless genes have specialized roles, and a strong link to gene expression regulation and chromatin structure. Overall, our results suggest that IG proteins have specialized, prevalent, and unique biological roles.
Embryonic development relies on the complex interplay of fundamental cellular processes, including proliferation, differentiation, and apoptosis. Regulation of these events is essential for the establishment of structures and organ development. The formation of the telencephalic architecture results from the interaction of the signaling centers located on the edges of the pallium. In this process, the Wnt signaling pathway plays an essential role in the dorsomedial pattern, where signals from the cortical hem direct the morphogenesis of the hippocampus, the corpus callosum, and the generation of migratory Cajal-Retzius cells. From the lateral pallium, the anti-hem signals, EGF, FGF, Frizzled, and Sfrp determine the development of the olfactory cortex.
In a previous study, we highlighted the up-regulation of Wnt signaling genes of the canonical pathway in the early stages of the developing telencephalon in mice (Muley et al., 2020). The main receptors of the Wnt/beta-catenin signaling pathway are Frizzled domain proteins (Fzd), a family of seven-transmembrane G-protein coupled receptors that also possess a large extracellular cysteine-rich domain.
In this work, we found Fzd7, Fzd8, and Fzd10 among the IG transmembrane proteins that were developmentally regulated in the embryonic telencephalon, as well as a group of 11 IGs of the protocadherin β-cluster. Previous studies have described Fzd8 as an essential receptor of the Wnt pathway implicated in brain development and size (Boyd et al., 2015). This gene is also highly expressed in two human cancer cell lines, indicating that it may play a role in tumorigenesis (Li et al., 2017; Murillo-Garzón et al., 2018). Fzd10 functions in the canonical Wnt/beta-catenin signaling pathway which may be involved in signal transduction during tissue morphogenesis (Wang et al., 2005). In keeping with this, protocadherins have also been described as regulators in the Wnt signaling pathway (Mah and Weiner, 2017). More specifically, protocadherins of the β-cluster, along with those of the α and γ clusters, act cooperatively in mice in olfactory-axon targeting, in the formation of diverse neural circuits, and in neuronal survival (Hasegawa et al., 2016, 2017). These functions, however, correspond to developmental stages that occur later than the one addressed in this study. Hence, our striking finding that half of the 22 β-protocadherins are up-regulated along with Wnt receptors during the development of the telencephalon, suggest that this group of mostly IGs has a differential function thus far unknown related to Wnt signaling at this early stage. Consistent with this idea, the Wnt binding molecular function GO term is one of the most conserved among IGs in the genomes analyzed in this study.
Our expression analysis additionally revealed up-regulation of Olig1, Bhlhe22, Bhlhe23, Pou3f1, Pou3f2, Pou3f4, Foxq1, and Neurog1. Notably, this represents the up-regulation of three of the four members of the Pou3 class of transcription factors present in mouse. The Pou genes encode a broad family of 6 classes (Pou1f–Pou6f) which are involved in developmental processes, mainly cell fate determination and differentiation (Tantin, 2013). Among those, the four members of the Pou3f class are preferentially expressed in ectodermal derivatives such as the developing mammalian nervous system (Bally-Cuif and Hammerschmidt, 2003). The human Pou3f3 is an intronless gene also named Brain-1, which is a well-known transcription factor involved in the development of the central nervous system and its variant alleles have been associated with intellectual disability and language neurodevelopmental disorders (Snijders Blok et al., 2019). Furthermore, an important role of Neurog1 is as a promoter of proliferation or neuronal differentiation, while Olig1 is involved in the generation and maturation of specific neural cells during the development of the spinal cord (Qi et al., 2016; Song et al., 2017). Bhlhe22 and Bhlhe23 in turn, are among those that were up-regulated the most in mice during telencephalon development. In humans, Bhlhe22 has been identified as a highly methylated gene in endometrial cancer with potential epigenetic biomarkers in cervical scrapings (Liew et al., 2019), while Bhlhe23 has been linked to mammalian retinal development (Woods et al., 2018).
Finally, among the histone group, H2bc21 (Hist2h2be), H2bu2 (Hist3h2ba), H2aw (Hist3h2a) were also up-regulated. The H2b histone family members are responsible for the chromosomal fiber nucleosome structure in eukaryotes. H2bc21/Hist2h2be has been described in mouse as expressed in olfactory epithelium, while H2bu2 (Hist3h2ba) in neocortex and lens of camera type-eye, and H2aw (Hist3h2a) in retina12. In humans, Hist2h2be is a hub gene related to poor prognosis in rhabdomyosarcoma tumors in pediatric patients (Li et al., 2019).
Summarizing, IGs appear to play crucial roles in the mouse telencephalon involved in gliogenesis, eye, and sensory organ development, canonical Wnt signaling, nucleosome organization, and have molecular regulatory roles. Therefore, in accordance with the functional assignment, our expression analysis supports that IGs play a critical role during mammalian brain development.
In agreement with the link of IGs to chromatin structure found in this and previous works, we also identified unique and highly represented PTM signatures in the histone protein category. Proteins encoded by mouse IGs have enriched signatures for histone H2A and H2B, characteristic of key core histones involved in chromatin structure in eukaryotic cells, as well as linker histone H1/H5 and the CENB-type HTH. In addition to the identification of exclusive signatures, we compared potential regulatory mechanisms of IG and MEG PTMs. Although the variability of PTMs is high, these modifications are typically very specific and, altogether, 300 types are known to occur in proteins (Witze et al., 2007). Among all PTMs, we found that the most abundant (Phosphorylation and Acetylation) are differentially represented among IG and MEG proteins.
As it could be expected from the observed enrichment in chromatin remodeling protein domains, our results show that proteins encoded by IGs undergo specific PTMs for histones such as crotonylation, methylation, sumoylation, citrullination, and sumoylation. However, our results suggest that IG-encoded histones have high specificity for Lysine-crotonylation, which is a recently identified post-translational modification associated with active promoters to directly stimulate transcription. Moreover, PTMs with changes in the physicochemical properties of amino acids like citrullination and amidation, are a characteristic feature highly enriched in IG proteins.
When we assessed the potential role of IGs as microproteins we found proteins with strong potential to be modulators of multi-protein complexes. The targets or microproteins are mostly transcription factors that bind DNA as dimers. In this study, we found potential miPs encoded by intronless genes that are bHLH transcription factors with a regulatory role during critical events such as neural development and function. For example, Ferd3l, an evolutionarily conserved bHLH protein, is expressed in the developing central nervous system and functions as a transcriptional inhibitor. Other examples are bHLHe23, a transcriptional regulator in the pancreas and brain that marks the dimesencephalic boundary (Bramblett et al., 2002), Bhlha9 a regulator of apical ectodermal ridge formation during limb development (Kataoka et al., 2018), Msg1 which is predominantly expressed in nascent mesoderm, the heart tube, limb bud, and sclerotome during mouse embryogenesis (Dunwoodie et al., 1998), and Ascl5 member of the ASCL family of proneural transcription factors that control the development of the nervous system, particularly neuroblast cell fate determination (Guillemot et al., 1993). Moreover, its potential role in tumorigenesis has been described with up-regulation in lung cancer and down-regulation in brain tumors such as glioblastoma, anaplastic oligoastrocytoma, anaplastic oligodendroglioma, and oligodendroglioma (Wang et al., 2017). Additionally, consistent with the potential role in the development of IG-encoded miPs, we identified members of the H1 linker histone group that fit the criteria to be classified as miPs. These histone proteins belong to a complex family with distinct specificity for tissues, developmental stages, and organisms in which they are expressed (Izzo et al., 2008).
According to earlier studies that reported the high conservation of mouse IG orthologs among other eukaryotic genomes, our analysis across seven species belonging to three classes of vertebrates revealed that the most numerous orthologs in each species were also IGs. This high rate of genetic structure conservation has been previously associated with their essential role in cell housekeeping functions, particularly those functionally pivotal proteins involved in molecular and biological roles such as transcription, translation, energy metabolism, amino-acid biosynthesis, and binding, which must be highly conserved (Sakharkar et al., 2006). Individually, eutherians (rat, chimp, human) are the species with the most orthologs. Moreover, Muridae, the clade that includes the mouse and rat, has the largest number of conserved orthologs and this number decreases as the clades gradually include the more distantly related species. Moreover, a distinctly large number was also found common to all species analyzed (Vertebrata), which is consistent with previous findings that identified functional and evolutionary conservation of eukaryotic IGs with highly distant genomes such as bacteria (Sakharkar et al., 2006). The higher abundance of conserved IG orthologs among species more closely related to mouse and lower in groups including more distant species, could be the result of the gradual loss of IG orthologs during the divergence of the diverse vertebrate branches. This abundance, however, could also be due to an increased rate of IG generation among mammals. Evidence supporting the latter possibility comes from our finding that in stark contrast, an increase of conserved MEG orthologs among species more closely related to mouse was not observed.
The clade Vertebrata contains the older IGs that are involved in diverse functions, among which nucleosome structure stands out. Histone IGs are conserved among all species, with some losses in opossum and chick. As histones are basic proteins known to be conserved across eukaryotes, it is not surprising that they are found to be some of the oldest IGs. In some cases, histones are conserved in the genome as clusters, and in some others, they appear to have been generated in a specific lineage, due to multiple gene duplications. Muridae, in contrast to Vertebrata, contains the more recent IGs which are involved in keratinization, GPCRs, and chemodetection by GPCRs. We can conclude that Muridae IGs are younger and have more specific functions, whereas IGs conserved in all vertebrates are more ancient and have more general or basic functions, as expected due to the high conservation of sequence and function among vertebrates.
Gene duplication is an important mechanism for the acquisition of new genes, frequently providing specialized or new gene functions (Magadum et al., 2013). Known mechanisms of gene duplication include retroposition, tandem duplication, and genome duplication (Pan and Zhang, 2008). Our analysis shows that the vast majority (48%) of one exon genes in the mouse genome are a consequence of retroposition. Moreover, regarding the duplication events, we found clear examples of IG tandem repeat cluster organization. For example, the syntenic conservation of the tandem cluster of β-protocadherins and their neural tissue-specific expression suggest that some aspects of the nervous system characteristic of mammals could be associated with the duplication of intronless genes, such as olfactory-axon targeting, the formation of neural circuits, neuronal survival, or neurite self-avoidance during development (Dennis et al., 2016; Hasegawa et al., 2017; Brasch et al., 2019). Similar to the single exon β-cluster of protocadherins, we also observed that IG histones in the mouse genome are present as tandem families with a tendency to cluster in their chromosome organization. An example of this is the H2A histone family member L1J, a family of ten IG members in the mouse X chromosome, with only one ortholog (H2AL1RP) in human and one (H2A-beta) in opossum. Almost all of the tandem repeat genes have parallel transcription orientation, which means they are encoded on the same strand.
The disrupted gene structure of most eukaryotic genes has led to a long-lasting debate regarding the origin of introns. The “exon theory of genes” also known as “introns-early,” proposed the presence of introns in prokaryotic primordial genes (Gilbert, 1987; Roy and Gilbert, 2006), while in the “insertional theory of introns” or “introns-late theory,” introns are a eukaryotic innovation (Doolittle and Stoltzfus, 1993; Stoltzfus, 1994; Stoltzfus et al., 1994; Mattick, 1994; Logsdon, 1998). Recent genomic evidence supports a view that combines aspects of both theories but still placing the invasion of eukaryotic genes by introns at the emergence of eukaryotic cells (Koonin, 2007). In accordance with this combined view, the comparison of mouse intron-bearing and intron-lacking IG orthologs among the analyzed organisms, suggests that IGs are more recent than MEGs. This is also consistent with findings that have revealed that intron-exon gene structure is highly stable among vertebrates and that individual intron losses outnumber intron gains in diverse vertebrate lineages (Roy et al., 2003; Coulombe-Huntington and Majewski, 2007; Loh et al., 2008; Venkatesh et al., 2014). This evolutionary stability also suggests that the observed increase in IG abundance in Muridae is due to gene duplication of IGs rather than intron loss of MEG orthologs. Our findings, however, also revealed some interesting gene superfamilies of a few members each, containing only IGs which were restricted to one or two species. These IGs are also likely to have been generated recently in the branches leading to the species analyzed herein but the mechanism involved remains to be studied further.
The present study aimed to identify the conservation of the role of intronless genes in mammals and other vertebrate genomes. A comprehensive understanding of their biological function is essential to compare and contrast their evolution with that of intron-containing genes. Hence, we studied the complex regulatory role of intronless genes and their conservation in cellular environments using computational functional assignment, gene expression analysis, and evolutionary reconstruction. First, we determined that the functions associated with IGs are very different from those associated with MEGs. Expression analysis of the developing telencephalon also revealed specific up-regulation of IGs that encode genes involved in Wnt signaling, bHLH and Pou transcription factors, as well as chromatin proteins. Among Wnt signaling-related proteins, it was striking to detect up-regulation of half of all protocadherins of the β-cluster. Moreover, some IG transcription factors meet the criteria to be considered microproteins and thus appear to have modulatory properties of protein complex formation. Overall, our results highlight a role for IGs as essential modulators of diverse biological processes as pivotal as cortical development, chemosensory functions, chromatin condensation, and gene silencing. In fact, specific modifications of IG proteins indicate that their regulatory roles extend to the post-translational level. Notably, some of the IGs highlighted in this study also have potential clinical relevance in humans. For example, Fzd8 and pcdhs which are associated to Wnt signaling, an evolutionarily conserved regulatory pathway related to cell fate determination and proliferation during development, have also been identified as part of a key mechanism in cancer biology. Other IGs discussed in this study and linked with cancer and neurodevelopmental disorders were Pou3f3, bHLHE22, ASCL5, and Hist2h2be.
Furthermore, the analysis of the evolutionary patterns of IGs revealed a large fraction of genes that appear to be of more recent generation as compared to the older and more conserved MEGs. Overall, this analysis reveals specific functions of IGs that distinguish them from MEGs and therefore strengthen the notion suggested by previous observations that these two groups are under differential evolutionary constraints.
Publicly available datasets were analyzed in this study. This data can be found here: https://data.mendeley.com/datasets/rdt5757cbw/1.
KA-P: project design, performed data collection, manuscript writing, proofreading, carried out bioinformatic analyses, prepared figures, and their interpretation. JR-R: expertise in bioinformatic analysis methods, performed evolutionary reconstruction, prepared figures. GH-O: literature search, bioinformatic analyses, and prepared figures. DV: writing, bioinformatic analysis, and prepared figures. ED-V: writing, bioinformatic analysis, and prepared figures. AG-G: expertise in data analysis methods for the API-REST and data collection, prepared figures. VM: proofreading, performed DEG analysis, prepared figures. AV-E: supervised the study, provided advice on the research strategy, and participated in manuscript writing. MH-R: co-director of the study and project development, performed bioinformatic analysis and interpretation, writing, and proofreading. All authors contributed to the article and approved the submitted version.
This project was supported by research funding provided by CONACYT grants QRO-2018-01-01-88344, 314869, 254206, 267749, and 315802 as well as DGAPA IN229620. KA-P received financial support from the DGAPA program for a postdoctoral fellowship at the INB UNAM and is a current holder of support from CONACyT (CVU: 227919).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
KA-P acknowledges the CABANA program for training in bioinformatics. For technical support we thank Luis Alberto Aguilar Bautista, Alejandro de León Cuevas, Carlos Sair Flores Bautista, and Jair García of the Laboratorio Nacional de Visualización Científica Avanzada (LAVIS). Critical comments and suggestions to this project development were received from Roddy Jorquera, Carolina Gonzalez, Michael Jerzioski, and Carlos Lozano Flores.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.654256/full#supplementary-material
Supplementary Code | https://github.com/GEmilioHO/intronless_genes.
Supplementary Tool | https://gitlab.com/jarr.tecn/revolutionh-tl.
Supplementary Figure 1 | Prevalence of intronless protein-coding genes among single-exon genes in the mouse genome. All mouse genes having one exon are classified regarding their gene biotype, proportion of protein-coding intronless genes (IGs) is highlighted in pink.
Supplementary Figure 2 | Enrichment of SUPERFAMILY assignments of mouse IG and MEG proteins. (A) Enriched scop families in IG proteins: The scop families with the largest gene ratios are plotted in order of gene ratio. The size of the dots represents the number of genes in the significant background list associated with the scop family, while the color of the dots represents the adjusted p-values, (B) Enriched scop families in MEG proteins: The scop families with the largest gene ratios are plotted in order of gene ratio. The size of the dots represents the number of genes in the significant background list associated with the scop family, while the color of the dots represents the p-adjusted values.
Amigo, J. D., Opazo, J. C., Jorquera, R., Wichmann, I. A., Garcia-Bloj, B. A., Alarcon, M. A., et al. (2018). The reprimo gene family: a novel gene lineage in gastric cancer with tumor suppressive properties. Int. J. Mol. Sci. 19:1862. doi: 10.3390/ijms19071862
Bally-Cuif, L., and Hammerschmidt, M. (2003). Induction and patterning of neuronal development, and its connection to cell cycle control. Curr. Opin. Neurobiol. 13, 16–25. doi: 10.1016/s0959-4388(03)00015-1
Boyd, J. L., Skove, S. L., Rouanet, J. P., Pilaz, L. J., Bepler, T., Gordân, R., et al. (2015). Human-chimpanzee differences in a FZD8 enhancer alter cell-cycle dynamics in the developing neocortex. Curr. Biol. 25, 772–779. doi: 10.1016/j.cub.2015.01.041
Bramblett, D. E., Copeland, N. G., Jenkins, N. A., and Tsai, M. J. (2002). BHLHB4 Is a BHLH transcriptional regulator in pancreas and brain that marks the dimesencephalic boundary. Genomics 79, 402–412. doi: 10.1006/geno.2002.6708
Brasch, J., Goodman, K. M., Noble, A. J., Micah, R., Seetha, M., Fabiana, B., et al. (2019). Visualization of clustered protocadherin neuronal self-recognition complexes”. Nature 569, 280–283. doi: 10.1038/s41586-019-1089-3
Bult, C. J., Blake, J. A., Smith, C. L., Kadin, J. A., and Richardson, J. E. (2019). Mouse Genome Database (MGD) 2019. Nucleic Acids Res. 47, D801–D806.
Coulombe-Huntington, J., and Majewski, J. (2007). Characterization of intron loss events in mammals. Genome Res. 17, 23–32. doi: 10.1101/gr.5703406
de Klein, N., Magnani, E., Banf, M., and Rhee, S. Y. (2015). MicroProtein Prediction Program (MiP3): a software for predicting microproteins and their target transcription factors. Int. J. Genomics 2015:734147.
de Souza, S. J. (2003). “The emergence of a synthetic theory of intron evolution,” in Origin and Evolution of New Gene Functions, ed. M. Long (Dordrecht: Springer), 117–121. doi: 10.1007/978-94-010-0229-5_2
Dennis, D., Picketts, D., Slack, R. S., and Schuurmans, C. (2016). Forebrain neurogenesis: from embryo to adult. Trends Dev. Biol. 9:77.
Doolittle, W. F., and Stoltzfus, A. (1993). Genes-in-pieces revisited. Nature 361:403. doi: 10.1038/361403a0
Dunwoodie, S. L., Rodriguez, T. A., and Beddington, R. S. (1998). Msg1 and Mrg1, founding members of a gene family, show distinct patterns of gene expression during mouse embryogenesis. Mech. Dev. 72, 27–40. doi: 10.1016/s0925-4773(98)00011-2
Eguen, T., Straub, D., Graeff, M., and Wenkel, S. (2015). MicroProteins: small size–big impact. Trends Plant Sci. 20, 477–482. doi: 10.1016/j.tplants.2015.05.011
Fedorova, L., and Fedorov, A. (2003). “Introns in gene evolution,” in Origin and Evolution of New Gene Functions, ed. M. Long (Berlin: Springer), 123–131. doi: 10.1007/978-94-010-0229-5_3
Fong, N., Kim, H., Zhou, Y., Ji, X., Qiu, J., Saldi, T., et al. (2014). Pre-MRNA splicing is facilitated by an optimal RNA polymerase II elongation rate. Genes Dev. 28, 2663–2676. doi: 10.1101/gad.252106.114
Gentles, A. J., and Karlin, S. (1999). Why are human G-Protein-coupled receptors predominantly intronless? Trends Genet. 15, 47–49. doi: 10.1016/s0168-9525(98)01648-5
Grzybowska, E. A. (2012). Human intronless genes: functional groups, associated diseases, evolution, and MRNA processing in absence of splicing. Biochem. Biophys. Res. Commun. 424, 1–6. doi: 10.1016/j.bbrc.2012.06.092
Guillemot, F., Lo, L. C., Johnson, J. E., Auerbach, A., Anderson, D. J., and Joyner, A. L. (1993). Mammalian achaete-scute homolog 1 is required for the early development of olfactory and autonomic neurons. Cell 75, 463–476. doi: 10.1016/0092-8674(93)90381-y
Hasegawa, S., Kobayashi, H., Kumagai, M., Nishimaru, H., Tarusawa, E., Kanda, H., et al. (2017). Clustered protocadherins are required for building functional neural circuits. Front. Mol. Neurosci. 10:114. doi: 10.3389/fnmol.2017.00114
Hasegawa, S., Kumagai, M., Hagihara, M., Nishimaru, H., Hirano, K., Kaneko, R., et al. (2016). Distinct and cooperative functions for the protocadherin-α,-β and-γ clusters in neuronal survival and axon targeting. Front. Mol. Neurosci. 9:155. doi: 10.3389/fnmol.2016.00155
Hellmuth, M., Hernandez-Rosales, M., Huber, K. T., Moulton, V., Stadler, P. F., and Wieseke, N. (2013). Orthology relations, symbolic ultrametrics, and cographs. J. Math. Biol. 66, 399–420. doi: 10.1007/s00285-012-0525-x
Hernandez-Rosales, M., Hellmuth, M., Wieseke, N., Huber, K. T., Moulton, V., and Stadler, P. F. (2012). From event-labeled gene trees to species trees. BMC Bioinformatics 13:S6.
Hung, M. S., Lin, Y. C., Mao, J. H., Kim, I. J., Xu, Z., Yang, C. T., et al. (2010). Functional polymorphism of the CK2α intronless gene plays oncogenic roles in lung cancer. PLoS One 5:e11418. doi: 10.1371/journal.pone.0011418
Izzo, A., Kamieniarz, K., and Schneider, R. (2008). The histone H1 family: specific members, specific functions? Biol. Chem. 389, 333–343. doi: 10.1515/bc.2008.037
Jorquera, R., González, C., Clausen, P., Petersen, B., and Holmes, D. S. (2018). Improved ontology for eukaryotic single-exon coding sequences in biological databases. Database 2018, 1–6. doi: 10.1002/9783527678679.dg08413
Jorquera, R., Ortiz, R., Ossandon, F., Cardenas, J. P., Sepulveda, R., Gonzalez, C., et al. (2016). SinEx DB: a database for single exon coding sequences in mammalian genomes. Database 2016:baw095. doi: 10.1093/database/baw095
Kaessmann, H., Vinckenbosch, N., and Long, M. (2009). RNA-based gene duplication: mechanistic and evolutionary insights. Nat. Rev. Genet. 10, 19–31. doi: 10.1038/nrg2487
Kataoka, K., Matsushima, T., Ito, Y., Sato, T., Yokoyama, S., and Asahara, H. (2018). Bhlha9 regulates apical ectodermal ridge formation during limb development. J. Bone Miner. Metab. 36, 64–72. doi: 10.1007/s00774-017-0820-0
Koonin, E. V. (2007). The biological big bang model for the major transitions in evolution. Biol. Direct 2:21. doi: 10.1186/1745-6150-2-21
Lechner, M., Findeiß, S., Steiner, L., Marz, M., Stadler, P. F., and Prohaska, S. J. (2011). Proteinortho: detection of (Co-) orthologs in large-scale analysis. BMC Bioinformatics 12:124.
Lechner, M., Hernandez-Rosales, M., Doerr, D., Wieseke, N., Thévenin, A., Stoye, J., et al. (2014). Orthology detection combining clustering and synteny for very large datasets. PLoS One 9:e105015. doi: 10.1371/journal.pone.0105015
Li, Q., Ye, L., Zhang, X., Wang, M., Lin, C., Huang, S., et al. (2017). FZD8, a Target of P53, promotes bone metastasis in prostate cancer by activating canonical Wnt/β-Catenin signaling. Cancer Lett. 402, 166–176. doi: 10.1016/j.canlet.2017.05.029
Li, Q., Zhang, L., Jiang, J., Zhang, Y., Wang, X., Zhang, Q., et al. (2019). CDK1 and CCNB1 as potential diagnostic markers of rhabdomyosarcoma: validation following bioinformatics analysis. BMC Med. Genomics 12:198.
Liew, P. L., Huang, R. L., Wu, T. I., Liao, C. C., Chen, C. W., Su, P. H., et al. (2019). Combined genetic mutations and DNA-methylated genes as biomarkers for endometrial cancer detection from cervical scrapings. Clin. Epigenetics 11:170.
Liu, X. Y., Fan, Y. C., Gao, S., Zhao, J., Chen, L. Y., Li, F., et al. (2017). Methylation of SOX1 and VIM promoters in serum as potential biomarkers for hepatocellular carcinoma. Neoplasma 64, 745–753. doi: 10.4149/neo_2017_513
Logsdon, J. M. Jr. (1998). The recent origins of spliceosomal introns revisited. Curr. Opin. Genet. Dev. 8, 637–648. doi: 10.1016/s0959-437x(98)80031-2
Loh, Y. H., Brenner, S., and Venkatesh, B. (2008). Investigation of Loss and gain of introns in the compact genomes of pufferfishes (Fugu and Tetraodon). Mol. Biol. Evol. 25, 526–535. doi: 10.1093/molbev/msm278
Louhichi, A., Fourati, A., and Rebaï, A. (2011). IGD: a resource for intronless genes in the human genome. Gene 488, 35–40. doi: 10.1016/j.gene.2011.08.013
Magadum, S., Banerjee, U., Murugan, P., Gangapur, D., and Ravikesavan, R. (2013). Gene duplication as a major force in evolution. J. Genet. 92, 155–161. doi: 10.1007/s12041-013-0212-8
Mah, K. M., and Weiner, R. A. (2017). Regulation of Wnt signaling by protocadherins. Semin. Cell Dev. Biol. 69, 158–171. doi: 10.1016/j.semcdb.2017.07.043
Mattick, J. S. (1994). Introns: evolution and function. Curr. Opin. Genet. Dev. 4, 823–831. doi: 10.1016/0959-437x(94)90066-3
Muley, V. Y., López-Victorio, C. J., Ayala-Sumuano, J. T., González-Gallardo, A., González-Santos, L., Lozano-Flores, C., et al. (2020). Conserved and divergent expression dynamics during early patterning of the telencephalon in mouse and chick embryos. Prog. Neurobiol. 186:101735. doi: 10.1016/j.pneurobio.2019.101735
Murillo-Garzón, V., Gorroño-Etxebarria, I., Åkerfelt, M., Puustinen, M. C., Sistonen, L., Nees, M., et al. (2018). Frizzled-8 integrates Wnt-11 and transforming growth factor-β signaling in prostate cancer. Nat. Commun. 9:1747.
Noonan, J. P., Grimwood, J., Schmutz, J., Dickson, M., and Myers, R. M. (2004). Gene conversion and the evolution of protocadherin gene cluster diversity. Genome Res. 14, 354–366. doi: 10.1101/gr.2133704
Ohki, R., Nemoto, J., Murasawa, H., Oda, E., Inazawa, J., Tanaka, N., et al. (2000). Reprimo, a new candidate mediator of the P53-mediated cell cycle arrest at the G2 phase. J. Biol. Chem. 275, 22627–22630. doi: 10.1074/jbc.c000235200
Pan, D., and Zhang, L. (2008). Tandemly arrayed genes in vertebrate genomes. Comp. Funct. Genomics 2008:545269.
Qi, Q., Zhang, Y., Shen, L., Wang, R., Zhou, J., Lü, H., et al. (2016). Olig1 Expression pattern in neural cells during rat spinal cord development. Neuropsychiatr. Dis. Treat. 12, 909–916. doi: 10.2147/ndt.s99257
Roy, S. W., and Gilbert, W. (2006). The evolution of spliceosomal introns: patterns, puzzles and progress. Nat. Rev. Genet. 7, 211–221. doi: 10.1038/nrg1807
Roy, S. W., Fedorov, A., and Gilbert, W. (2003). Large-Scale comparison of intron positions in mammalian genes shows intron loss but no gain. Proc. Natl. Acad. Sci. U.S.A. 100, 7158–7162. doi: 10.1073/pnas.1232297100
Sakharkar, K. R., Sakharkar, M. K., Culiat, C. T., Chow, V. T., and Pervaiz, S. (2006). Functional and evolutionary analyses on expressed intronless genes in the mouse genome. FEBS Lett. 580, 1472–1478. doi: 10.1016/j.febslet.2006.01.070
Sakharkar, M. K., Chow, V. T., Ghosh, K., Chaturvedi, I., Lee, P. C., Bagavathi, S. P., et al. (2005a). Computational prediction of SEG (Single Exon Gene) function in humans. Front. Biosci. 10:1382–1395. doi: 10.2741/1627
Sakharkar, M. K., Kangueane, P., Petrov, D. A., Kolaskar, A. S., and Subbiah, S. (2002). “SEGE: a databasse on ‘intron less/single exonic’ genes from eukaryotes. Bioinformatics 18, 1266–1267. doi: 10.1093/bioinformatics/18.9.1266
Sakharkar, M. K., Perumal, B. S., Lim, Y. P., Chern, L. P., Yu, Y., and Kangueane, P. (2005b). Alternatively spliced human genes by exon skipping–a database (ASHESdb). In Silico Biol. 5, 221–225.
Snijders Blok, L., Kleefstra, T., Venselaar, H., Maas, S., Kroes, H. Y., Lachmeijer, A. M. A., et al. (2019). De novo variants disturbing the transactivation capacity of POU3F3 cause a characteristic neurodevelopmental disorder. Am. J. Hum. Genet. 105, 403–412. doi: 10.1016/j.ajhg.2019.06.007
Song, Z., Jadali, A., Fritzsch, B., and Kwan, K. Y. (2017). NEUROG1 regulates CDK2 to promote proliferation in otic progenitors. Stem Cell Rep. 9, 1516–1529. doi: 10.1016/j.stemcr.2017.09.011
Staudt, A. C., and Wenkel, S. (2011). Regulation of protein function by ‘MicroProteins.’. EMBO Rep. 12, 35–42. doi: 10.1038/embor.2010.196
Stoltzfus, A., Spencer, D. F., Zuker, M., Logsdon, J. M., and Doolittle, W. F. (1994). Testing the exon theory of genes: the evidence from protein structure. Science 265, 202–207. doi: 10.1126/science.8023140
Straub, D., and Wenkel, S. (2017). Cross-species genome wide identification of evolutionary conserved microproteins. Genome Biol. Evol. 9, 777–789. doi: 10.1093/gbe/evx041
Sunahara, R. K., Niznik, H. B., Weiner, D. M., Stormann, T. M., Brann, M. R., Kennedy, J. L., et al. (1990). Human dopamine D 1 receptor encoded by an intronless gene on chromosome 5. Nature 347, 80–83. doi: 10.1038/347080a0
Supek, F., Bošnjak, M., Škunca, N., and Šmuc, T. (2011). REVIGO Summarizes and visualizes long lists of gene ontology terms. PLoS One 6:e21800. doi: 10.1371/journal.pone.0021800
Tantin, D. (2013). Oct transcription factors in development and stem cells: insights and mechanisms. Development 140, 2857–2866. doi: 10.1242/dev.095927
Tedder, M., Corneil, D., Habib, M., and Paul, C. (2008). “Simpler linear-time modular decomposition via recursive factorizing permutations,” in International Colloquium on Automata, Languages, and Programming, eds L. Aceto, I. Damgård, L. A. Goldberg, M. M. Halldórsson, A. Ingólfsdóttir, and I. Walukiewicz (Berlin: Springer), 634–645. doi: 10.1007/978-3-540-70575-8_52
Tine, M., Kuhl, H., Beck, A., Bargelloni, L., and Reinhardt, R. (2011). Comparative Analysis of intronless genes in teleost fish genomes: insights into their evolution and molecular function. Mar. Genomics 4, 109–119. doi: 10.1016/j.margen.2011.03.004
Uversky, V. N. (2015). Functional roles of transiently and intrinsically disordered regions within proteins. FEBS J. 282, 1182–1189. doi: 10.1111/febs.13202
Venkatesh, B., Lee, A. P., Ravi, V., Maurya, A. K., Lian, M. M., Swann, J. B., et al. (2014). Elephant shark genome provides unique insights into gnathostome evolution. Nature 505, 174–179. doi: 10.1038/nature12826
Wang, C. Y., Shahi, P., Huang, J. T., Phan, N. N., Sun, Z., Lin, Y. C., et al. (2017). Systematic analysis of the achaete-scute complex-like gene signature in clinical cancer patients. Mol. Clin. Oncol. 6, 7–18. doi: 10.3892/mco.2016.1094
Wang, Z., Shu, W., Lu, M. M., and Morrisey, E. E. (2005). Wnt7b activates canonical signaling in epithelial and vascular smooth muscle cells through interactions with Fzd1, Fzd10, and LRP5. Mol. Cell. Biol. 25, 5022–5030. doi: 10.1128/mcb.25.12.5022-5030.2005
Witze, E. S., Old, W. M., Resing, K. A., and Ahn, N. G. (2007). Mapping protein post-translational modifications with mass spectrometry. Nat. Methods 4, 798–806.
Woods, S. M., Mountjoy, E., Muir, D., Ross, S. E., and Atan, D. (2018). A comparative analysis of rod bipolar cell transcriptomes identifies novel genes implicated in night vision. Sci. Rep. 8:5506.
Yan, H., Zhang, W., Lin, Y., Dong, Q., Peng, X., Jiang, H., et al. (2014). Different evolutionary patterns among intronless genes in maize genome. Biochem. Biophys. Res. Commun. 449, 146–150. doi: 10.1016/j.bbrc.2014.05.008
Yu, G., Wang, L. G., Han, Y., and He, Q. Y. (2012). ClusterProfiler: an R package for comparing biological themes among gene clusters. Omics 16, 284–287. doi: 10.1089/omi.2011.0118
Yu, W. P., Rajasegaran, V., Yew, K., Loh, W. L., Tay, B. H., Amemiya, C. T., et al. (2008). Elephant shark sequence reveals unique insights into the evolutionary history of vertebrate genes: a comparative analysis of the protocadherin cluster. Proc. Natl. Acad. Sci. U.S.A. 105, 3819–3824. doi: 10.1073/pnas.0800398105
Keywords: intronless genes, exon-intron architecture, embryonic telencephalon, protocadherins, histones, transcription factors, microproteins, evolutionary histories
Citation: Aviña-Padilla K, Ramírez-Rafael JA, Herrera-Oropeza GE, Muley VY, Valdivia DI, Díaz-Valenzuela E, García-García A, Varela-Echavarría A and Hernández-Rosales M (2021) Evolutionary Perspective and Expression Analysis of Intronless Genes Highlight the Conservation of Their Regulatory Role. Front. Genet. 12:654256. doi: 10.3389/fgene.2021.654256
Received: 15 January 2021; Accepted: 01 June 2021;
Published: 09 July 2021.
Edited by:
Katja Nowick, Freie Universität Berlin, GermanyReviewed by:
Jarosław Bryk, University of Huddersfield, United KingdomCopyright © 2021 Aviña-Padilla, Ramírez-Rafael, Herrera-Oropeza, Muley, Valdivia, Díaz-Valenzuela, García-García, Varela-Echavarría and Hernández-Rosales. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alfredo Varela-Echavarría, YXZhcmVsYUB1bmFtLm14; Maribel Hernández-Rosales, bWFyaWJlbC5ockBjaW52ZXN0YXYubXg=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.