 Yaokun Li1
Yaokun Li1 Xiangping Xu
Xiangping Xu Ming Deng
Ming Deng Xian Zou
Xian Zou Zhifeng Zhao
Zhifeng Zhao Sixiu Huang
Sixiu Huang Dewu Liu
Dewu Liu Guangbin Liu
Guangbin Liu- 1Guangdong Laboratory for Lingnan Modern Agriculture, College of Animal Science, South China Agricultural University, Guangzhou, China
- 2State Key Laboratory of Livestock and Poultry Breeding, Institute of Animal Science, Guangdong Academy of Agricultural Sciences, Guangzhou, China
The ovary is the most important reproductive organ in goats and directly affects the fecundity. Long non-coding RNAs (lncRNAs) are involved in the biological process of oocyte maturation. However, in the context of reproduction in goats, few studies have explored the regulation of lncRNAs. Therefore, we herein used the ovaries of high and low fecundity Leizhou black goats to identify differentially expressed lncRNAs (DElncRNAs) by high-throughput RNA sequencing; moreover, we analyzed the target genes of lncRNAs by functional annotation to explore the role of DElncRNAs in ovarian development. Twenty DElncRNAs were identified, of which six were significantly upregulated and 14 were significantly downregulated in high fecundity goats. Gene Ontology analyses suggested that MSTRG.3782 positively influences the expression of the corresponding gene API5, exerting regulative effects on the development of follicles, through which litter size might show variations. The target gene KRR1 of ENSCHIT00000001883 is significantly enriched in cell components, and ENSCHIT00000001883 may regulate cell growth and thus affect follicular development. Further, as per Kyoto Encyclopedia of Genes and Genomes pathway analyses, MSTRG.2938 was found to be significantly enriched, and we speculate that MSTRG.2938 could regulate ribosomal biogenesis in the pre-snoRNP complex as well as cell transformation in eukaryotes. Quantitative real-time PCR results were consistent with sequencing data. To conclude, our research results indicate that some lncRNAs play a key role in regulating follicle development and cell growth during goat’ s ovarian development.
Introduction
Litter size is influenced not only by nutrition levels and environment but also by inheritance (Cui et al., 2009). The ovary is the most important organ for the normal reproductive function of goats. It secretes estrogen to maintain sexual characteristics and cyclical reproductive activity; further, oocytes and ovulation have a major impact on the fertility of goats (Barnett et al., 2006; Zhao et al., 2015). Studies have shown that the ovulation rate of goats is linked to high productivity (Pramod et al., 2013). lncRNAs play a chief role in reproduction-related processes in animals, but very limited information is available on the functions of lncRNAs in goats. In particular, in the context of reproduction in goats, few studies have explored the regulation of lncRNAs (Xing et al., 2014). Long non-coding RNAs are non-coding RNA transcripts of >200 nucleotides in length; they have a complex structure and lack the ability to code proteins (Jarroux et al., 2017). They can regulate gene expression and protein function to perform biological functions. Studies have reported that lncRNAs can regulate reproductive processes, such as ovarian development and maturation in female animals (Li et al., 2015; Ling et al., 2017; Liu et al., 2020). Therefore, it is crucial to study their role by exploring the function of key target genes.
High-throughput RNA sequencing and functional analyses have been used to elucidate the reproductive function of lncRNAs that were identified to be differentially expressed between the ovaries of multiparous and uniparous Anhui white goats; TCONS_00136407, TCONS_00146968, and TCONS_00320849, for example, were suggested to participate in oocyte meiosis (Ling et al., 2017). Using the same method to study the function of differentially expressed lncRNAs (DElncRNAs) in Chuanzhong black goats, ENSCHIT00000005909 and ENSCHIT00000005910 were suggested to regulate the viability and proliferation of keratinocyte-derived cells by influencing IL1R2 (interleukin 1 receptor type II) thereby affecting ovarian function (Bouckenheimer et al., 2018). Leizhou black goat is a special local goat breed in southern China, which shows excellent adaptability to the living circumstance with high humidity and high temperature, and using high-throughput sequencing and bioinformatics analysis can help us to explore the novel functional DElncRNAs in the ovaries of goats.
Litter size is one of the most important economic traits in goat production, determining the benefit of farming enterprises. To provide a theoretical basis for goat breeding and improve the production efficiency of goat industry, it is vital to conduct in-depth research on the mechanisms regulating litter size. We herein screened DElncRNAs between the ovaries of high and low fecundity Leizhou black goats and predicted the target genes of DElncRNAs. In addition, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were used to analyze the function of target genes. Our results not only enrich the transcriptomic data of the goat ovary but also provide a theoretical basis for combining molecular breeding and conventional breeding technologies.
Materials and Methods
Ethics Statement
All study protocols were approved by the Ethics Committee for the Care and Use of Laboratory Animals at the South China Agricultural University (permit no.: SYXK-2014-0136). Further, all experiments were performed in accordance with the guidelines of the South China Agricultural University.
Animals and Sample Collection
Seven healthy female Leizhou black goats (age, 3.5–4.5 years) were divided into high and low fecundity groups. The litter size of high-fecundity group (n = 3) and low-fecundity group (n = 4) were more than one and only one, respectively. Meanwhile, all of the samples in this study were from goats with three parity records of litter size. The female goats were injected with 0.1 mg cloprostenol to induce estrus (Perera et al., 1978; Martemucci and D’Alessandro, 2011; Fierro et al., 2013). The goats were kept under observation to determine whether they were in heat (bleating, searching for the male goat, frequent urination, hyperemia, edema, contraction of the vulva, and vaginal mucus discharge). The basis of estrus was the female goat shaking their tail, standing, and accepting to mate with the male goat (Taylor, 1978; Mekuriaw et al., 2016). The ovaries were collected within 24 h of estrus. The selected goats were killed and dissected, and both whole ovaries from each goat were collected immediately. The intact ovaries were collected and washed with 75% alcohol thrice. Then they were soaked into phosphate buffered saline. After the collection of the ovary, the ligaments and attached tissues were trimmed off under surgical anatomy microscope, ovarian follicles were isolated from the ovary, and the isolated ovarian tissue was frozen in liquid nitrogen and stored at –80°C.
Total RNA Isolation, cDNA Library Construction, and Transcriptome Sequencing
After thoroughly grinding the ovarian tissue, total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA, United States). NanoDrop ND-2000 was used to measure RNA concentration (Thermo Science, Wilmington, DE, United States). RNA integrity was assessed by denaturing agarose gel electrophoresis. Further, the cDNA library was constructed using 3 μl of total RNA from each sample, and double-terminal sequencing was performed on the HiSeq X-TEN sequencing platform by Shanghai Parsons Biotech Co., Ltd.
Quality Control of Raw Sequences
We used Cutadapt to remove reads with an average quality score below Q20. The Q20 value referred to the error probability of 1% for the identified bases in the process of base recognition. The reference genome index (GCF_001704415.1_ARS1_genomic1) was established by Bowtie 2, and the filtered reads were compared with the reference genome using TopHat 2. If the mismatch between the reads and the reference genome sequence was within 2, we considered the alignment to be successful (Kim et al., 2013).
Assembly and Novel lncRNA Prediction
According to the TopHat 2 results, StringTie was used for transcript assembly, and candidate lncRNAs were then selected based on the splicing results and structural features of lncRNAs. The screening conditions to identify lncRNAs were as follows: (1) transcripts with low expression levels, low credible single exon transcripts, and exon numbers < 2 were filtered out and (2) transcripts < 200 bp in length were excluded (Trapnell et al., 2010; Cabili et al., 2011). Moreover, Coding-Non-Coding-Index v2 (Barnett et al., 2006), Coding Potential Calculator (0.9-r2; Sun et al., 2013), Pfam Scan v1.3 (Punta et al., 2012), and phylogenetic codon substitution frequency (v20121028; Lin et al., 2011) were used for coding potential analyses. Transcripts without coding potential comprised the candidate set of lncRNAs. lncRNA expression at the transcription level was analyzed with StringTie. DESeq was used to analyze the expression of lncRNAs; the screening conditions were | Log2FoldChange| > 1 and P < 0.05 (Love et al., 2014). The ggplot 2 software package was used to construct a volcano map of DElncRNAs, and the pheatmap software package was used to perform clustering according to the expression level of same lncRNAs in different samples and that of different lncRNAs in the same sample. Distance was calculated with the Euclidean method and clustering was performed using hierarchical agglomerative clustering (Wang et al., 2010).
Target Gene Prediction
To explore the functions of lncRNAs, we predicted the target genes of DElncRNAs. Because the reliability of the analysis results is not high when the sample number is small, the function of trans-regulation can not be predicted. We searched the genes 100 kb downstream and upstream of lncRNAs and analyzed their functions.
GO and KEGG Pathway Analyses for Target Genes of DElncRNAs
GO analysis was performed with the predicted target genes using DAVID2. Furthermore, we used the KEGG database to analyze the potential functions of these genes in pathways3 (Dennis et al., 2003; Han et al., 2012). A hypergeometric test was applied to discover the significant enrichment of GO terms and KEGG pathways so as to determine the main biological functions of differentially expressed genes (Wang et al., 2020; Huang et al., 2007). P < 0.05 indicated statistical significance.
Quantitative Real-Time PCR (qRT-PCR) for DElncRNAs
Total RNA (1 μg) was first reverse-transcribed using an RT Reagent Kit with gDNA Eraser (Takara, Dalian, China), according to manufacturer instructions. qRT-PCR was performed on a StepOnePlus Real-Time PCR System (Life Technologies, United StatesA), as per the standard protocol, using TB Green Fast qPCR Mix (Takara, Dalian, China). Primer Premier 5 used in primer design. Capra hircus β-actin served as the endogenous control for mRNA and lncRNA expression analyses.
Results
Sequencing Data Quality Control
The raw reads from the high and low fecundity groups were analyzed for quality control before further analyses. The Q30 value for each sample exceeded 93% (Table 1). Within the mapped reads, >85% of total reads were mapped to the reference genome without any mismatch (Table 2), indicating that the sequencing data was of high quality and suitable for subsequent analyses.
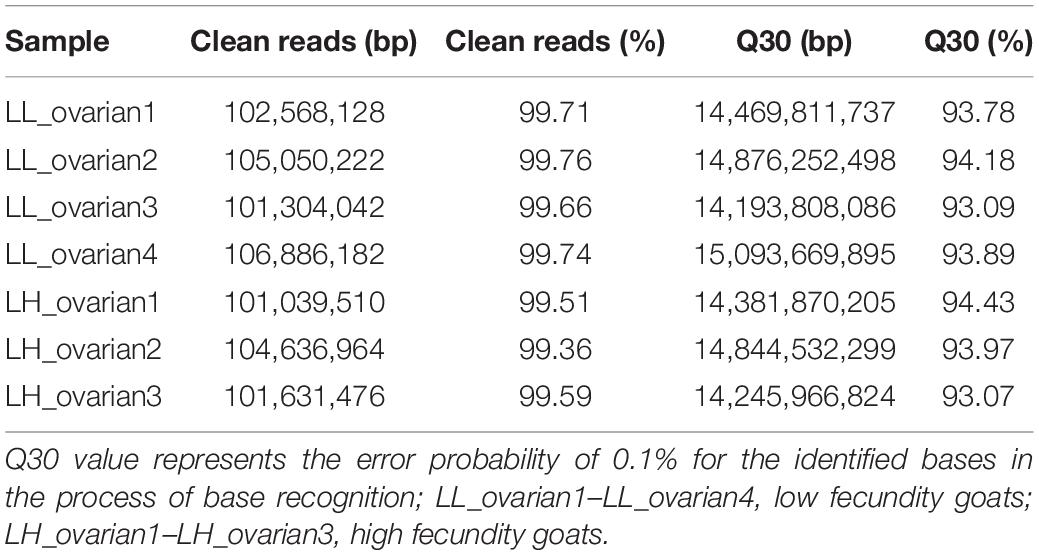
Table 1. Quality control of RNA-seq data.
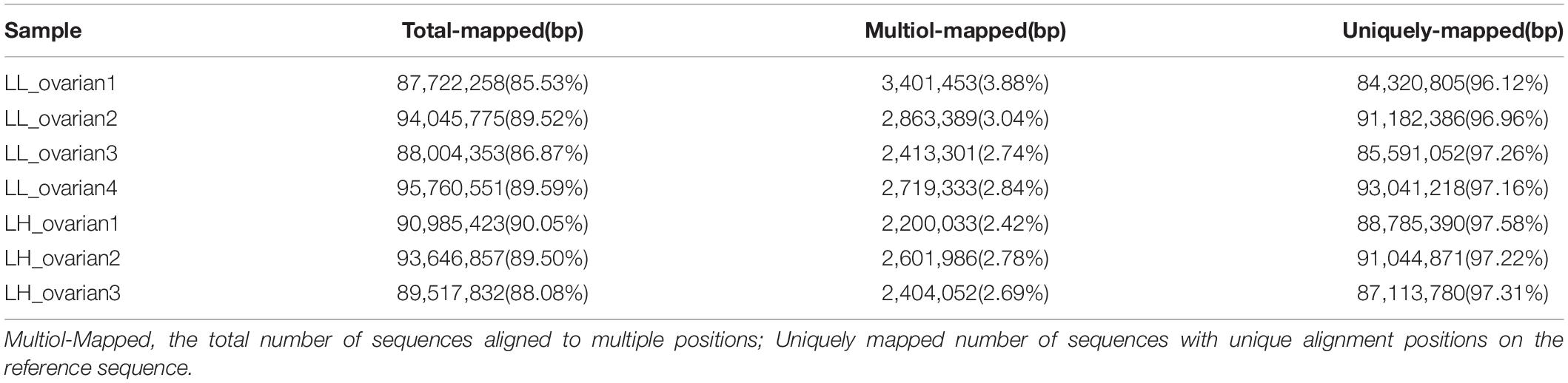
Table 2. Statistics of the mapping result.
Screening and Validation of DElncRNAs
Of 4,462 lncRNAs, 20 were differentially expressed between the high and low fecundity groups. Compared with the low fecundity group, six lncRNAs were upregulated and 14 were downregulated in the high fecundity group (P < 0.05; Figure 1A). From the heatmap analysis, the expression level of the same lncRNA in the same group was essentially the same, indicating that there was little difference between the samples in the same group. Four and three samples belonging to the low and high fecundity groups, respectively, were clustered together, indicating that lncRNA expression patterns in the groups were different (Figure 1B). Six DElncRNAs were randomly selected for qRT-PCR to verify the reliability of RNA sequencing data. qRT-PCR results were fundamentally consistent with sequencing results, confirming that the sequencing data had high reliability (Figure 1C).
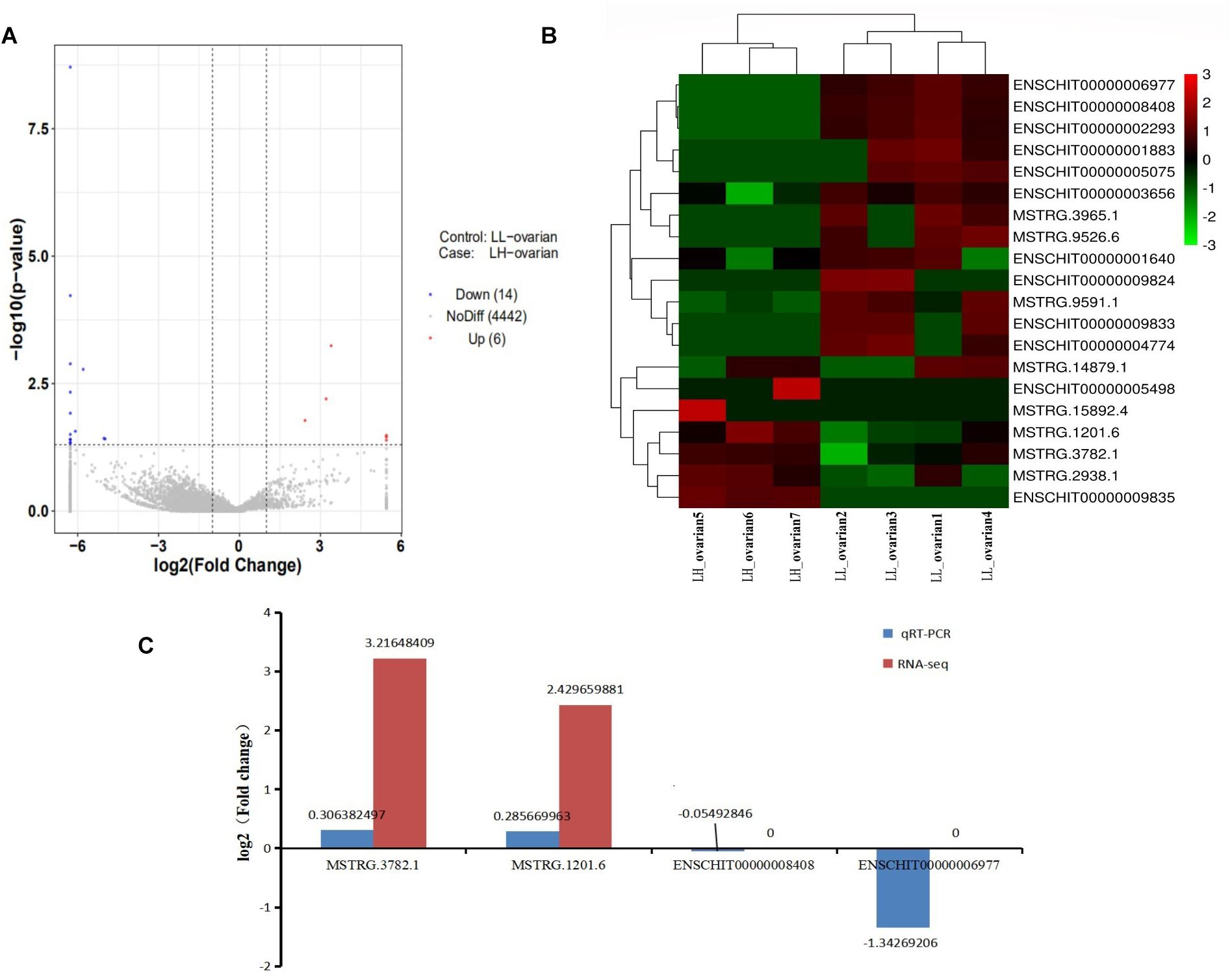
Figure 1. Analysis and validation of DElncRNAs in RNA-seq libraries. (A) Volcano map in analyzing DElncRNAs between high fecundity group and low fecundity group. The red plot represented up-regulated expression in high fecundity group; the blue plot represented down-regulated expression in high fecundity group. (B) Hierarchical clustering analysis of lncRNA expression profiles from libraries with 20 DElncRNAs. Data were expressed as FPKM. Red: relatively high expression; Green: relatively low expression. (C) qRT-PCR results pertaining to DElncRNAs were compared with RNA-seq data. Red: RNA-seq; blue: qRT-PCR.
GO Analyses for Target Genes of lncRNAs
GO Analyses for Target Genes of All lncRNAs
The 4,462 lncRNAs corresponded to 2,870 genes, of which DElncRNAs corresponded to 19 target genes. To explore the biological function of lncRNAs involved in regulating litter size, we performed GO and KEGG pathway analysis to identify the functions of target genes. GO analyses revealed diverse biological functions, such as positive regulation of transcription from RNA polymerase II promoter, patterning of blood vessels, and palate development, and multiple target genes were involved, such as CTNNB1 (encoding catenin beta-1), WNT5A, and EDN1 (endothelin-1). Transcription factor complex, nucleus, and integral component of plasma membrane were the top three terms significantly enriched in the cellular component, whereas transcriptional repressor activity, RNA polymerase II core promoter proximal region sequence-specific binding, sequence-specific DNA binding and RNA polymerase II core promoter proximal region sequence-specific DNA binding were the top three terms significantly enriched in the molecular function (P < 0.05; Table 3 and Figure 2A). ZNF536 and SALL1 (sal-like 1) are noted to be involved in these functions.
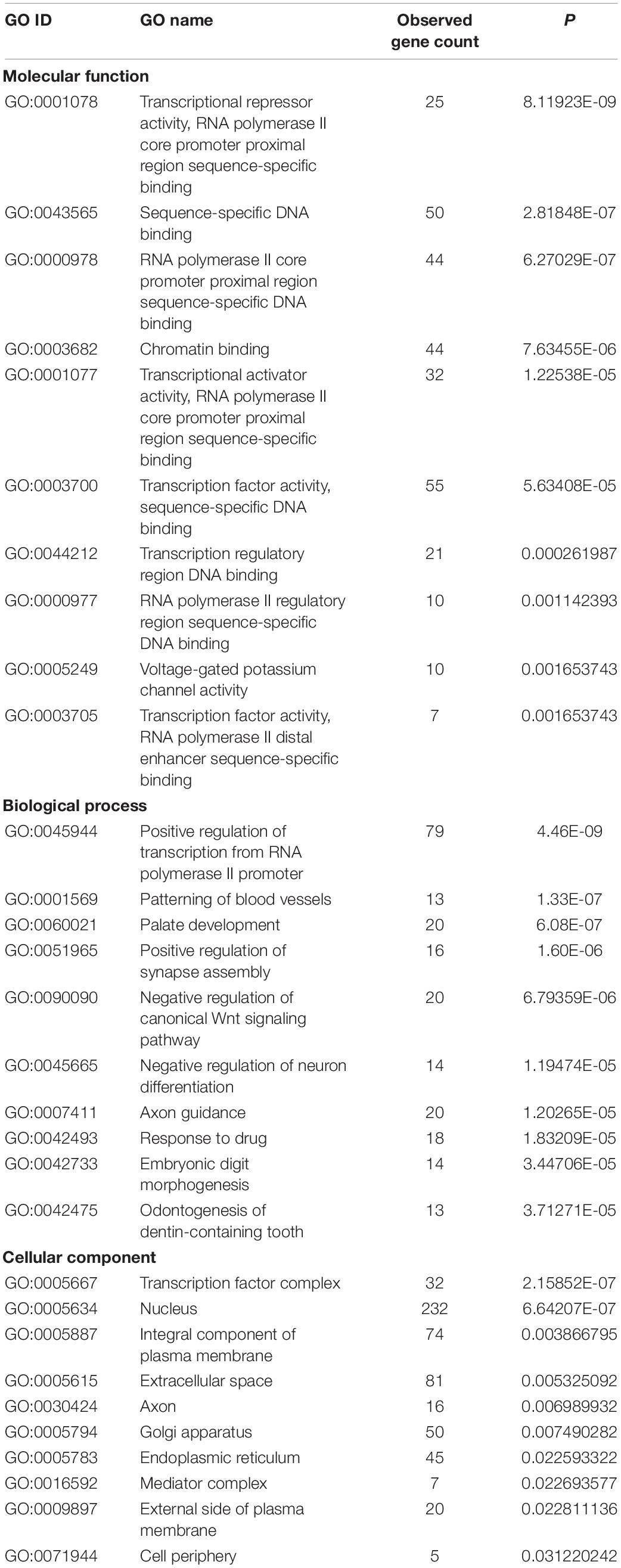
Table 3. Top 10 significantly enriched Gene Ontology (GO) terms of target genes of all long non-coding RNAs (lncRNAs).
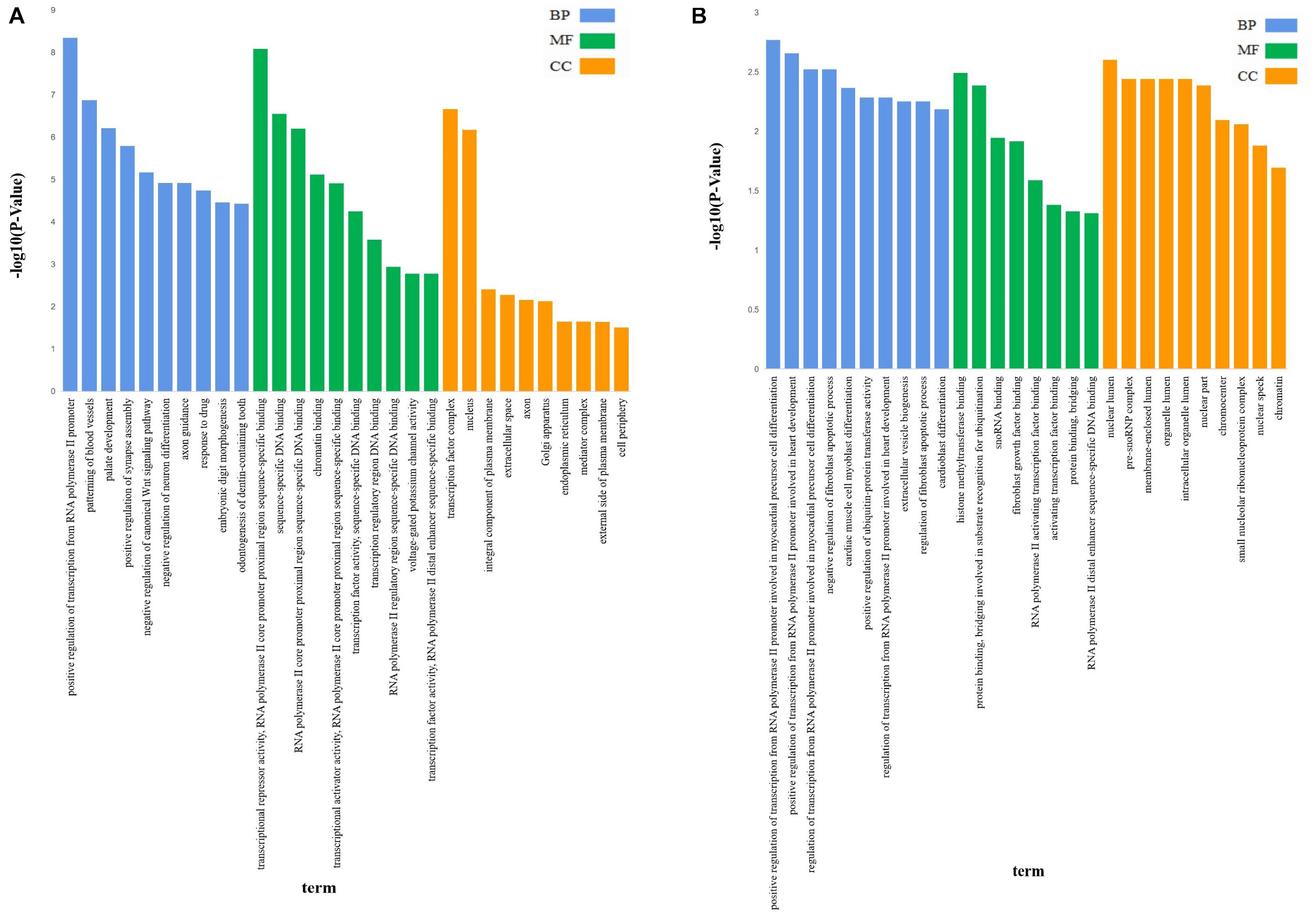
Figure 2. The GO analysis of target genes of lncRNAs. GO analysis of lncRNA-target genes according to biological process (BP), cell component (CC), and molecular function (MF); (A) The GO analysis of target genes of all lncRNAs, X-axis was P-value(-log10), Y-axis was the GO term. (B) The GO analysis of target genes of DElncRNAs, X-axis was P-value(-log10), Y-axis was the GO term.
GO Analyses for Target Genes of DElncRNAs
GO analysis revealed that 47 terms were significantly enriched between the high and low fecundity groups; the target genes involved were IER2 (immediate early response protein 2), TBXT, API5 (encoding apoptosis inhibitor 5), KRR1, ARRDC4 (arrestin domain containing 4), NOP56 (encoding nucleolar protein 56), and OIP5 (encoding OPA-interacting protein 5) (P < 0.05). The target gene API5 of MSTRG.3782 participated in 14 GO terms, including nuclear lumen, negative regulation of fibroblast apoptotic process, and regulation of fibroblast apoptotic process. The target gene NOP56 of MSTRG.2938 participated in 13 GO terms, including nuclear lumen, histone methyltransferase binding, and pre-snoRNP complex. Further, we classified the function of the target genes into the three major GO categories of biological process, cellular component, and molecular function. Positive regulation of transcription from RNA polymerase II promoter involved in myocardial precursor cell differentiation, positive regulation of transcription from RNA polymerase II promoter involved in heart development, regulation of transcription from RNA polymerase II promoter involved in myocardial precursor cell differentiation were the top three abundant terms in the biological process category (P < 0.05). In the cellular component category, nuclear lumen, pre-snoRNP complex, and membrane-enclosed lumen were the top three abundant terms, whereas in the molecular function category, histone methyltransferase binding, protein binding, bridging involved in substrate recognition for ubiquitination, and snoRNA binding were the top three abundant terms (P < 0.05; Table 4 and Figure 2B).
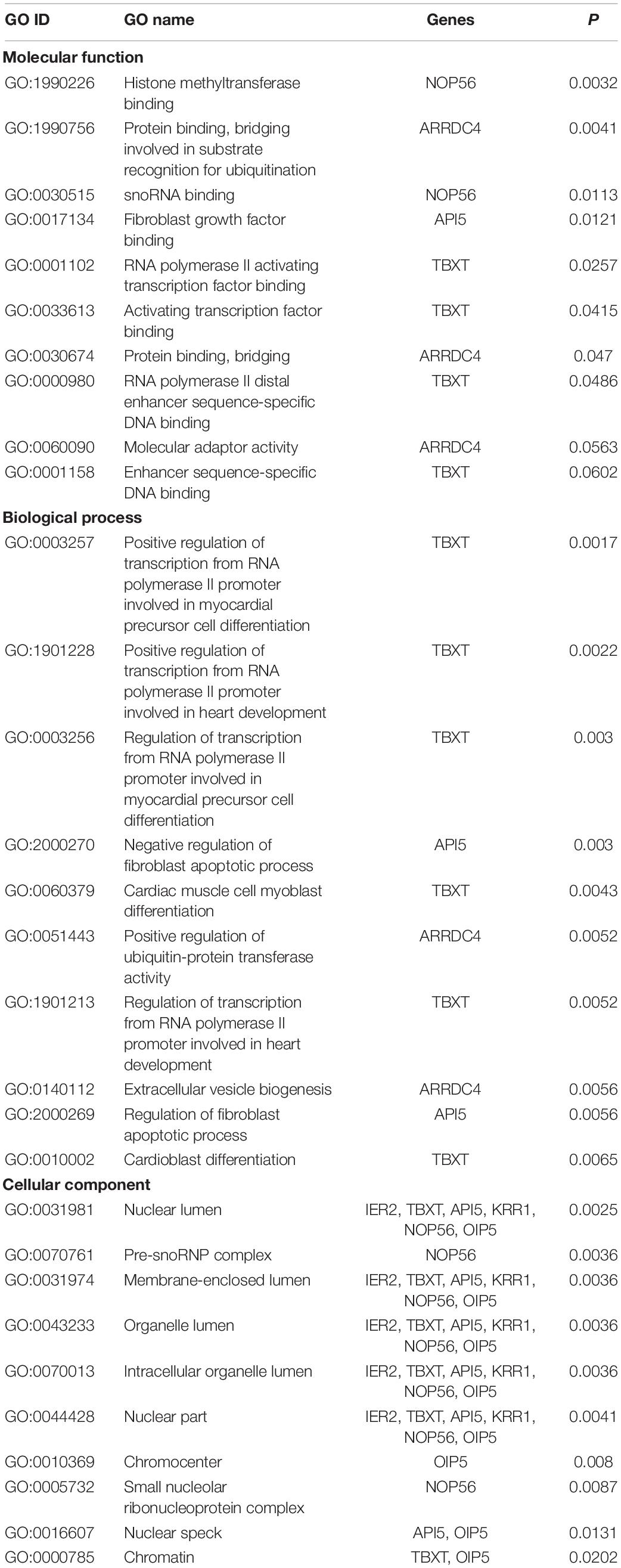
Table 4. Top 10 significantly enriched Gene Ontology (GO) terms of target genes of differentially expressed long non-coding RNAs (DElncRNAs).
KEGG Pathway Analyses for Target Genes of lncRNAs
KEGG Pathway Analyses for Target Genes of All lncRNAs
As per KEGG pathway analysis, 20 pathways were significantly enriched (P < 0.05). The top 10 pathways were primarily associated with transforming growth factor-beta (TGF-β), signaling pathways regulating pluripotency of stem cells, pathways in cancer, basal cell carcinoma, Wnt signaling pathway, HTLV-I infection, neuroactive ligand-receptor interaction, proteoglycans in cancer, Hippo signaling pathway, and transcriptional misregulation in cancer (Table 5 and Figure 3A). The target genes included CTNNB1, WNT5A, and TGF-β2, among others. The target gene WNT5A of ENSCHIG00000000774 was involved in seven signaling pathways, such as the Wnt signaling pathway, basal cell carcinoma, and HTLV-I infection.
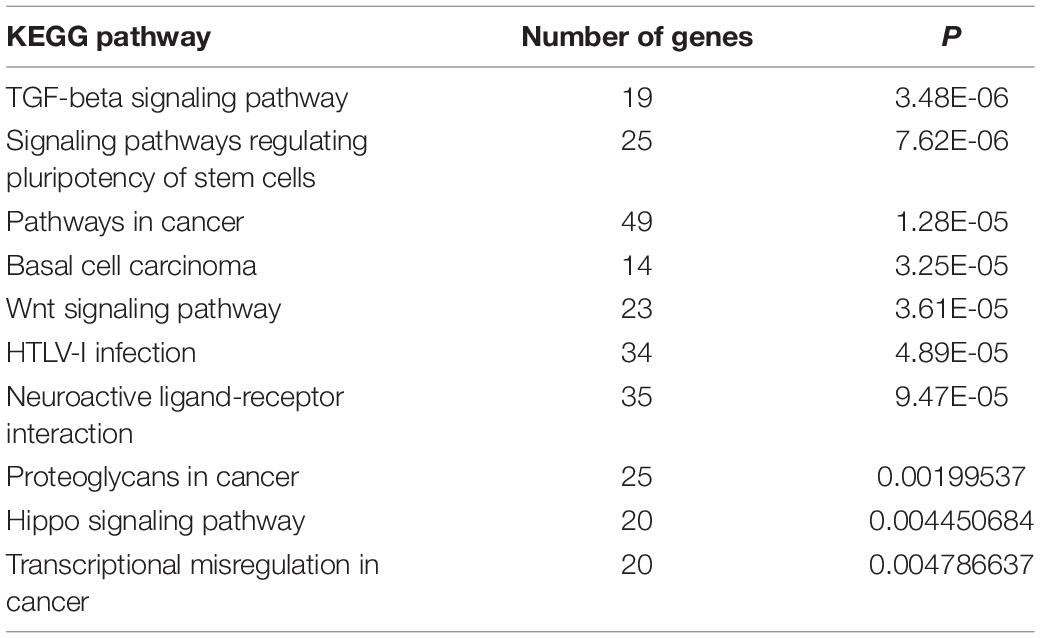
Table 5. Top 10 significantly enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of target genes of all long non-coding RNAs.
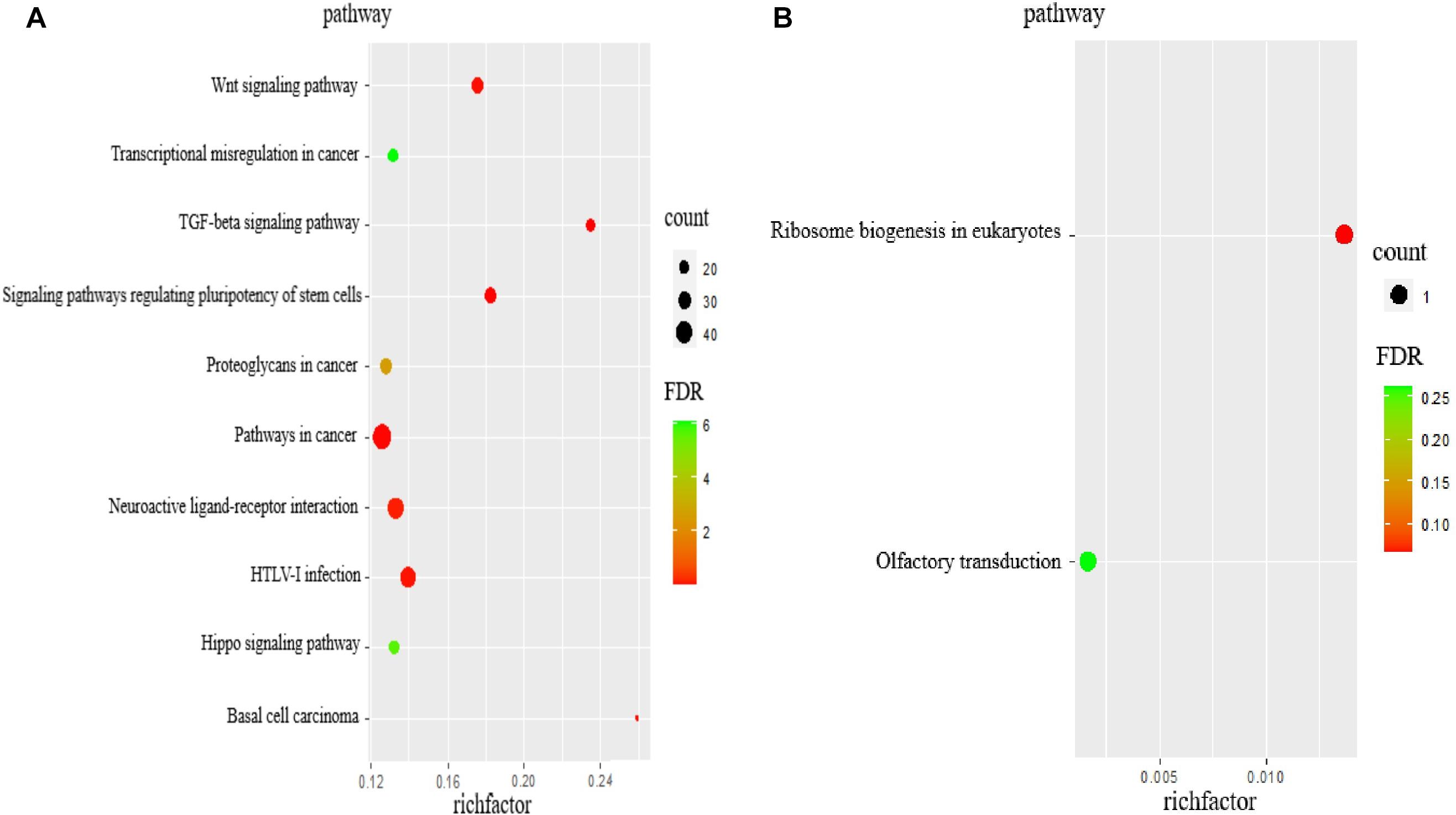
Figure 3. The KEGG analysis of target genes of lncRNAs. (A) KEGG pathway analyses of target genes of all lncRNAs. KEGG enrichment was measured by rich factor, FDR and the number of genes enriched on this pathway. (B) KEGG pathway analyses of target genes of DElncRNAs. KEGG enrichment was measured by rich factor, FDR and the number of genes enriched on this pathway.
KEGG Pathway Analyses for Target Genes of DElncRNAs
According to KEGG pathway enrichment analyses, the target genes involved ribosomal biogenesis in eukaryotes and olfactory transduction pathways, of which only the former showed significant enrichment (P < 0.05). The target gene NOP56 of MSTRG.2938 was involved in this pathway (Table 6 and Figure 3B).
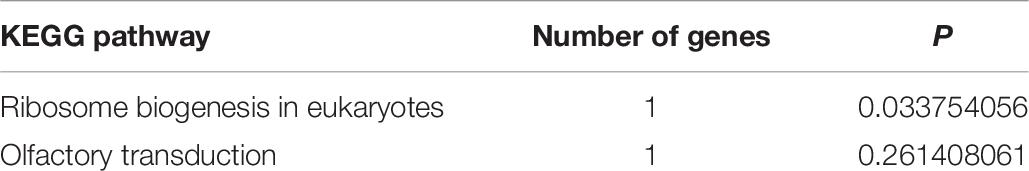
Table 6. Top significantly enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of target genes of differentially expressed long non-coding RNAs.
Discussion
The elucidation of mechanisms regulating litter size can provide a theoretical basis for breeding technologies in goats. Therefore, in this study, we used the ovaries of high and low fecundity Leizhou black goats to identify DElncRNAs by high-throughput RNA sequencing; moreover, we analyzed the target genes of lncRNAs to explore the role of DElncRNAs in ovarian development.
We herein identified enriched terms and signaling pathways; subsequently, we analyzed them as well as pertinent target genes involved in the regulation of reproduction. Fibroblasts are the main cellular component of loose connective tissue (Cai et al., 2012; Yeung et al., 2013). Carcinoma-associated fibroblasts evidently regulate the development of epithelial ovarian cancer by affecting the proliferation, apoptosis, migration, and invasive activity of ovarian cancer cells (Zhang et al., 2011). Fibroblast growth factor (FGFs) is involved in follicular development and follicular atresia (Costa et al., 2009; Miyoshi et al., 2010; Asgari et al., 2015; Coticchio et al., 2015). The expression of FGFs related to follicular development changes with the development of follicles. FGFs need to bind to distinct receptors to physiologically function (Basu et al., 2014). Apoptosis inhibitor 5, which is encoded by API5, is involved in regulating the cell cycle. Further, it promotes DNA synthesis and cell cycle G1/S transition, and regulates cell growth, proliferation, and apoptosis (Garcia-Jove Navarro et al., 2013). API5 plays an important role in the termination of diapause and early embryonic development of Artemia sinica (Zhang et al., 2017). Through GO analysis, we found that the target gene API5 of MSTRG.3782 was involved in the regulation of fibroblast apoptotic process and FGF binding. MSTRG.3782 was significantly upregulated in the high fecundity group. Accordingly, we hypothesized that lncRNA participates in cell growth, thereby affecting follicular development.
The nucleus is the main repository of genetic information in eukaryotic cells, and the site of DNA replication and transcription; it consequently controls genetic and metabolic activities (Lynch and Marinov, 2017). Chromosomes are the most important structures in the nucleus and carry hereditary information (Zetterström, 2008). A study found that OPA-interacting protein 5 (OIP5) was enriched in centrosomes during the G1 phase of the cell cycle and mediated the regulation of cell division (Naetar et al., 2007). In addition, OIP5 reportedly has a fundamental role in maintaining the structure and function of centrosomes/centromeres (Fujita et al., 2007). KRR1 encodes proteins present in early 90 S precursor particles of the small ribosomal subunit, and its locus has been implied to contribute to the development of polycystic ovary syndrome (Gromadka and Rytka, 2000; Zheng et al., 2014; Pau et al., 2017). In this study, we found that the target gene OIP5 of MSTRG.1201 and the target gene KRR1 of ENSCHIT00000001883 were significantly enriched in the cellular component of nuclear lumen, chromatin, organelle lumen, intracellular organelle lumen, among others. In the high fecundity group, MSTRG.1201 was significantly upregulated and ENSCHIT00000001883 was significantly downregulated. Therefore, we believe that MSTRG.1201 and ENSCHIT00000001883 affect follicular development by regulating cell division.
Mature snoRNP particles are composed of a series of small nucleolar RNA and core proteins. snoRNPs regulate the processing and modification of pre-rRNA and play an important role in ribosomal biogenesis (Richard and Kiss, 2006). Nucleolar protein 56 (encoded by Nop56) is involved in the synthesis of snoRNP as a core protein (Lykke-Andersen et al., 2018). Furthermore, an increase in the ribosome biosynthesis rate can promote the expression of the proto-oncogene C-myc and enhance the proliferative ability of cancer cells (Tomczak et al., 2015). C-myc encodes a transcription factor with a direct role in controlling translation (Ruggero, 2009). Nol5a/Nop56 may be a critical gene involved in Myc-mediated oncogenic transformation (Cowling et al., 2014). According to our GO and KEGG pathway analyses, MSTRG.2938 was significantly upregulated in the high fecundity group, and its target gene NOP56 was involved in ribosomal biogenesis in eukaryotes and the pre-snoRNP complex. We thus speculate that MSTRG.2938 regulates ribosomal biogenesis in the pre-snoRNP complex as well as cell transformation in eukaryotes. However, the specific mechanism of regulation of each lncRNA remains to be further investigated.
According to GO analyses, the target genes [Guanine Nucleotide Binding Protein, alpha 13(GNA13), Mothers against decapentaplegic homolog 2 (SMAD2), and Fibronectin Leucine Rich Transmembrane Protein 2 (FLRT2)] of all lncRNAs were mainly enriched in positive regulation of transcription from RNA polymerase II promoters, patterning of blood vessels, palate development, and positive regulation of synapse assembly. RNA polymerase II plays a pivotal role in the transcription of protein-encoding genes in all eukaryotic cells (Bernecky et al., 2016). GNA13 participates in regulating cell movement and developmental angiogenesis (Offermanns et al., 1997). Moreover, SMAD2 overexpression has been reported to repair secondary cleft palate by increasing apoptosis of medial edge epithelial cells in the TGF-β3 pathway (Miyazono et al., 2018). We thus report that these genes play a major role in maintaining the healthy growth of goats.
The Wnt signaling pathway and its downstream effectors not only regulate physiological processes such as cell growth and differentiation, cell migration, and genetic material stability but are also important for cancer progression, including for regulating tumor growth, cell senescence, and cell death. The Wnt/β-catenin signaling pathway is involved in various important processes, such as the regulation of embryo development, cell proliferation, and cell migration (Nusse and Clevers, 2017; Peng et al., 2017; Steinhart and Angers, 2018). β-Catenin is an essential structural component of cadherin-based adherens junctions and is a key component of Wnt/β-catenin signal transduction. Aberrant expression of CTNNB1 and WNT5A has been observed to affect cell proliferation and lead to cancer occurrence. A mutation in CTNNB1 is one of the many causes of β-catenin degradation (Kim et al., 2018). The Wnt/CTNNB1 pathway is a pivotal signaling pathway that regulates steroid production (Abedini et al., 2016). WNT5A is a highly evolved conservative non-classical Wnt ligand, which is required for normal ovarian follicle development (Abedini et al., 2016). WNT5A is differentially expressed during the development of mouse follicles, and it can significantly inhibit steroid production in atretic follicles (Lapointe and Boerboom, 2011; Abedini et al., 2016; Kumawat and Gosens, 2016). By blocking the function of FSH (follicle-stimulating hormone) and luteinizing protein, WNT5A can induce the down-regulation of CTNNB1 and cAMP-response element binding protein (CREB), thus affecting follicle development and gonadotropin reactivity (Abedini et al., 2015). We found that the target gene CTNNB1 of ENSCHIG00000000641 is one of the two signaling pathway members of Wnt and proteoglycans in cancer. ENSCHIG00000000641 was downregulated in the high fecundity group. Further, the target gene WNT5A of ENSCHIG00000000774 participated in the negative regulation of canonical Wnt signaling pathway terms and the Wnt signaling pathway. ENSCHIG00000000774 was also downregulated in the high fecundity group. Thus, we believe that ENSCHIG00000000641 and ENSCHIG00000000774 affect follicular development by regulating cell proliferation and steroid production, respectively.
The TGF-β superfamily participates in many physiological activities in mammals via autocrine and paracrine pathways, and TGF-β is mainly produced locally in the ovary. It has been reported that TGF-β1 can promote the growth of mice follicles. Both TGF-β and activin A have proliferative action and cytodifferentiative action on granulosa cells (Liu et al., 1999). As a member of the transforming growth factor β family, TGF-β2 also plays an important role in the growth and development of follicles. TGF-β2 is located in follicular membrane cells and luteal cells and regulates the production of inhibitors and activins in granulosa cells and luteal cells (Knight and Glister, 2003). Smad2/Smad3 are key molecules in the TGF-β/Smad signaling pathway that regulate ovarian growth and development and maintain ovarian function (Coutts et al., 2008; AlMegbel and Shuler, 2020). Smad2 and Smad3 can maintain normal fertility in women, and further support the Smad2/3 pathway in the ovary to participate in the regulation of signals produced by oocytes, which plays an important role in the coordination of ovulation (Li et al., 2008). In this study, we found that TGF-β2, TGF-βR2, and Smad2 participated in the TGF-β signaling pathway, and they were regulated by ENSCHIG00000000886, ENSCHIG00000000609, and ENSCHIG00000002761, respectively. We accordingly speculate that these lncRNAs regulate follicle development, but the specific mechanism needs to be further studied.
To conclude, we found that target genes of all lncRNAs were mainly involved in protein transcription and played a role in maintaining the healthy growth of animals. In addition, the TGF-β and Wnt signaling pathways were found to be related to reproduction in animals. Based on functional analyses of target genes of DElncRNAs, fibroblast apoptotic process, FGF binding, pre-snoRNP complex, and ribosomal biogenesis in eukaryotes were associated with reproduction in goats. Our data improves the current understanding of the transcriptome of goats and provides valuable information for functional genomics resources and biological studies; moreover, we believe that our results are of great significance for in-depth studies of candidate lncRNAs in breeding techniques.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI (accession: PRJNA728366).
Ethics Statement
The animal study was reviewed and approved by all study protocols were approved by the Ethics Committee for the Care and Use of Laboratory Animals at the South China Agricultural University (permit no. SYXK-2014-0136). Further, all experiments were performed in accordance with the guidelines of the South China Agricultural University.
Author Contributions
YL: conceptualization, methodology, writing-reviewing, and editing. XX: data curation, writing-original draft preparation, software, and validation. MD: conceptualization. DL: visualization and investigation. GL: supervision. XZ: investigation. ZZ: investigation. All authors contributed to the article and approved the submitted version.
Funding
The Modern Agricultural Industrial Technology System of Guangdong Province (2019KJ127), the Guangdong Provincial Promotion Project of Modern Seed Industry, Guangdong Provincial Promotion Project on Preservation and Utilization of Local Breed of Livestock and Poultry, and Natural Science Foundation of Guangdong Province (2019B1515210017), and Natural Science Foundation of Guangdong Province, Regulation of miR-128 and miR-450 on proliferation and apoptosis of goat follicular granulosa cells (2021A1515010636).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank WENS Company for helping us with performing experiments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.648158/full#supplementary-material
Supplementary File 1 | The DElncRNAs in the ovary of high fecundity goats and low fecundity goats. (XLSX)
Supplementary File 2 | lncRNA primers sequences used for the qRT-PCR. (XLSX)
Supplementary File 3 | Protein-coding genes detected 100-kb upstream and downstream of the DElncRNAs. (XLSX)
Supplementary File 4 | GO enrichment analysis of target genes co-located with the all lncRNAs. (XLSX)
Supplementary File 5 | GO enrichment analysis of target genes co-located with the DElncRNAs. (XLSX)
Supplementary File 6 | KEGG enrichment analysis of target genes co-located with the all lncRNAs. (XLSX)
Supplementary File 7 | KEGG enrichment analysis of target genes co-located with the DElncRNAs. (XLSX)
Supplementary File 8 | Sequences of the DElncRNAs. (DOC)
Footnotes
References
Abedini, A., Zamberlam, G., Boerboom, D., and Price, C. A. (2015). Non-canonical WNT5A is a potential regulator of granulosa cell function in cattle. Mol. Cell Endocrinol. 403, 39–45. doi: 10.1016/j.mce.2015.01.017
Abedini, A., Zamberlam, G., Lapointe, E., Tourigny, C., Boyer, A., Paquet, M., et al. (2016). WNT5a is required for normal ovarian follicle development and antagonizes gonadotropin responsiveness in granulosa cells by suppressing canonical WNT signaling. FASEB J. 30, 1534–1547. doi: 10.1096/fj.15-280313
AlMegbel, A. M., and Shuler, C. F. (2020). SMAD2 overexpression rescues the TGF-β3 null mutant mice cleft palate by increased apoptosis. Differ. Res. Biol. Diversity 111, 60–69. doi: 10.1016/j.diff.2019.10.001
Asgari, F., Valojerdi, M. R., Ebrahimi, B., and Fatehi, R. (2015). Three dimensional in vitro culture of preantral follicles following slow-freezing and vitrification of mouse ovarian tissue. Cryobiology 71, 529–536. doi: 10.1016/j.cryobiol.2015.11.001
Barnett, K. R., Schilling, C., Greenfeld, C. R., Tomic, D., and Flaws, J. A. (2006). Ovarian follicle development and transgenic mouse models. Hum. Reprod. Update 12, 537–555. doi: 10.1093/humupd/dml022
Basu, M., Mukhopadhyay, S., Chatterjee, U., and Roy, S. S. (2014). FGF16 promotes invasive behavior of SKOV-3 ovarian cancer cells through activation of mitogen-activated protein kinase (MAPK) signaling pathway. J. Biol. Chem. 289, 1415–1428.
Bernecky, C., Herzog, F., Baumeister, W., Plitzko, J. M., and Cramer, P. (2016). Structure of transcribing mammalian RNA polymerase II. Nature 529, 551–554. doi: 10.1038/nature16482
Bouckenheimer, J., Fauque, P., Lecellier, C. H., Bruno, C., Commes, T., Lemaître, J. M., et al. (2018). Differential long non-coding RNA expression profiles in human oocytes and cumulus cells. Sci. Rep. 8:2202. doi: 10.1038/s41598-018-20727-0
Cabili, M. N., Trapnell, C., Goff, L., Koziol, M., Tazon-Vega, B., Regev, A., et al. (2011). Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 25, 1915–1927. doi: 10.1101/gad.17446611
Cai, J., Tang, H., Xu, L., Wang, X., Yang, C., Ruan, S., et al. (2012). Fibroblasts in omentum activated by tumor cells promote ovarian cancer growth, adhesion and invasiveness. Carcinogenesis 33, 20–29. doi: 10.1093/carcin/bgr230
Costa, I., Teixeira, N., Ripamonte, P., Guerra, D., Price, C., Buratini, J., et al. (2009). Fgf1 mRNA in. (bovine)antral follicles and corpora lutea. Anim. Reprod. 6, 409–415.
Coticchio, G., Dal Canto, M., Mignini Renzini, M., Guglielmo, M. C., Brambillasca, F., Turchi, D., et al. (2015). Oocyte maturation: gamete-somatic cells interactions, meiotic resumption, cytoskeletal dynamics and cytoplasmic reorganization. Hum. Reprod. Update 21, 427–454. doi: 10.1093/humupd/dmv011
Coutts, S. M., Childs, A. J., Fulton, N., Collins, C., Bayne, R. A., McNeilly, A. S., et al. (2008). Activin signals via SMAD2/3 between germ and somatic cells in the human fetal ovary and regulates kit ligand expression. Dev. Biol. 314, 189–199. doi: 10.1016/j.ydbio.2007.11.026
Cowling, V. H., Turner, S. A., and Cole, M. D. (2014). Burkitt’s lymphoma-associated c-Myc mutations converge on a dramatically altered target gene response and implicate Nol5a/Nop56 in oncogenesis. Oncogene 33, 3519–3527. doi: 10.1038/onc.2013.338
Cui, H. X., Zhao, S. M., Cheng, M. L., Guo, L., Ye, R. Q., Liu, W. Q., et al. (2009). Cloning and expression levels of genes relating to the ovulation rate of the Yunling black goat. Biol. Reprod. 80, 219–226. doi: 10.1095/biolreprod.108.069021
Dennis, G. Jr., Sherman, B. T., Hosack, D. A., Yang, J., Gao, W., Lane, H. C., et al. (2003). DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 4:3.
Fierro, S., Gil, J., Viñoles, C., and Olivera-Muzante, J. (2013). The use of prostaglandins in controlling estrous cycle of the ewe: a review. Theriogenology 79, 399–408. doi: 10.1016/j.theriogenology.2012.10.022
Fujita, Y., Hayashi, T., Kiyomitsu, T., Toyoda, Y., Kokubu, A., Obuse, C., et al. (2007). Priming of centromere for CENP-A recruitment by human hMis18alpha, hMis18beta, and M18BP1. Dev. Cell 12, 17–30. doi: 10.1016/j.devcel.2006.11.002
Garcia-Jove Navarro, M., Basset, C., Arcondéguy, T., Touriol, C., Perez, G., Prats, H., et al. (2013). Api5 contributes to E2F1 control of the G1/S cell cycle phase transition. PLoS One 8:e71443. doi: 10.1371/journal.pone.0071443
Gromadka, R., and Rytka, J. (2000). The KRR1 gene encodes a protein required for 18S rRNA synthesis and 40S ribosomal subunit assembly in Saccharomyces cerevisiae. Acta Biochim. Pol. 47, 993–1005.
Han, L., Zhang, K., Shi, Z., Zhang, J., Zhu, J., Zhu, S., et al. (2012). LncRNA profile of glioblastoma reveals the potential role of lncRNAs in contributing to glioblastoma pathogenesis. Int. J. Oncol. 40, 2004–2012. doi: 10.3892/ijo.2012.1413
Huang, D. W., Sherman, B. T., Tan, Q., Kir, J., Liu, D., Bryant, D., et al. (2007). DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 35, W169–W175. doi: 10.1093/nar/gkm415
Jarroux, J., Morillon, A., and Pinskaya, M. (2017). History, discovery, and classification of lncRNAs. Adv. Exp. Med. Biol. 1008, 1–46. doi: 10.1007/978-981-10-5203-3_1
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S. L. (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14:R36. doi: 10.1186/gb-2013-14-4-r36
Kim, G., Kurnit, K. C., Djordjevic, B., Singh, C., Munsell, M. F., Wang, W. L., et al. (2018). Nuclear β-catenin localization and mutation of the CTNNB1 gene: a context-dependent association. Modern Pathol. 31, 1553–1559. doi: 10.1038/s41379-018-0080-0
Knight, P. G., and Glister, C. (2003). Local roles of TGF-beta superfamily members in the control of ovarian follicle development. Anim. Reprod. Sci. 78, 165–183. doi: 10.1016/s0378-4320(03)00089-7
Kumawat, K., and Gosens, R. (2016). WNT-5A: signaling and functions in health and disease. Cell. Mol. Life Sci. CMLS 73, 567–587. doi: 10.1007/s00018-015-2076-y
Lapointe, E., and Boerboom, D. (2011). WNT signaling and the regulation of ovarian steroidogenesis. Front. Biosci. 3:276–285.
Li, J., Cao, Y., Xu, X., Xiang, H., Zhang, Z., Chen, B., et al. (2015). Increased new lncRNA-mRNA gene pair levels in human cumulus cells correlate with oocyte maturation and embryo development. Reproductive Sci. 22, 1008–1014. doi: 10.1177/1933719115570911
Li, Q., Pangas, S. A., Jorgez, C. J., Graff, J. M., Weinstein, M., and Matzuk, M. M. (2008). Redundant roles of SMAD2 and SMAD3 in ovarian granulosa cells in vivo. Mol. Cell. Biol. 28, 7001–7011. doi: 10.1128/MCB.00732-08
Lin, M. F., Jungreis, I., and Kellis, M. (2011). PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 27, i275–i282. doi: 10.1093/bioinformatics/btr209
Ling, Y., Xu, L., Zhu, L., Sui, M., Zheng, Q., Li, W., et al. (2017). Identification and analysis of differentially expressed long non-coding RNAs between multiparous and uniparous goat (Capra hircus) ovaries. PLoS One 12:e0183163. doi: 10.1371/journal.pone.0183163
Liu, G., Liu, S., Xing, G., and Wang, F. (2020). lncRNA PVT1/MicroRNA-17-5p/PTEN axis regulates secretion of E2 and P4, proliferation, and apoptosis of ovarian granulosa cells in PCOS. Mol. Therapy. Nucleic Acids 20, 205–216. doi: 10.1016/j.omtn.2020.02.007
Liu, X., Andoh, K., Abe, Y., Kobayashi, J., Yamada, K., Mizunuma, H., et al. (1999). A comparative study on transforming growth factor-beta and activin A for preantral follicles from adult, immature, and diethylstilbestrol-primed immature mice. Endocrinology 140, 2480–2485. doi: 10.1210/endo.140.6.6827
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lykke-Andersen, S., Ardal, B. K., Hollensen, A. K., Damgaard, C. K., and Jensen, T. H. (2018). Box C/D snoRNP autoregulation by a cis-Acting snoRNA in the NOP56 Pre-mRNA. Mol. Cell. 72, 99–111.e5. doi: 10.1016/j.molcel.2018.08.017
Lynch, M., and Marinov, G. K. (2017). Membranes, energetics, and evolution across the prokaryote-eukaryote divide. eLife 6:e20437. doi: 10.7554/eLife.20437
Martemucci, G., and D’Alessandro, A. G. (2011). Induction/synchronization of oestrus and ovulation in dairy goats with different short term treatments and fixed time intrauterine or exocervical insemination system. Anim. Reprod. Sci. 126, 187–194. doi: 10.1016/j.anireprosci.2011.05.011
Mekuriaw, Z., Assefa, H., Tegegne, A., and Muluneh, D. (2016). Estrus response and fertility of Menz and crossbred ewes to single prostaglandin injection protocol. Trop. Animal Health Prod. 48, 53–57. doi: 10.1007/s11250-015-0919-z
Miyazono, K. I., Moriwaki, S., Ito, T., Kurisaki, A., Asashima, M., and Tanokura, M. (2018). Hydrophobic patches on SMAD2 and SMAD3 determine selective binding to cofactors. Sci. Signal. 11:eaao7227. doi: 10.1126/scisignal.aao7227
Miyoshi, T., Otsuka, F., Yamashita, M., Inagaki, K., Nakamura, E., Tsukamoto, N., et al. (2010). Functional relationship between fibroblast growth factor-8 and bone morphogenetic proteins in regulating steroidogenesis by rat granulosa cells. Mol. Cell. Endocrinol. 325, 84–92. doi: 10.1016/j.mce.2010.04.012
Naetar, N., Hutter, S., Dorner, D., Dechat, T., Korbei, B., Gotzmann, J., et al. (2007). LAP2alpha-binding protein LINT-25 is a novel chromatin-associated protein involved in cell cycle exit. J. Cell Sci. 120(Pt 5), 737–747. doi: 10.1242/jcs.03390
Nusse, R., and Clevers, H. (2017). Wnt/β-Catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999. doi: 10.1016/j.cell.2017.05.016
Offermanns, S., Mancino, V., Revel, J. P., and Simon, M. I. (1997). Vascular system defects and impaired cell chemokinesis as a result of Galpha13 deficiency. Science 275, 533–536. doi: 10.1126/science.275.5299.533
Pau, C. T., Mosbruger, T., Saxena, R., and Welt, C. K. (2017). Phenotype and tissue expression as a function of genetic risk in polycystic ovary syndrome. PLoS One 12:e0168870. doi: 10.1371/journal.pone.0168870
Peng, Y., Zhang, X., Feng, X., Fan, X., and Jin, Z. (2017). The crosstalk between microRNAs and the Wnt/β-catenin signaling pathway in cancer. Oncotarget 8, 14089–14106. doi: 10.18632/oncotarget.12923
Perera, B. M., Bongso, T. A., and Abeynaike, P. (1978). Oestrus synchronisation in goats using cloprostenol. Vet. Record 102:314. doi: 10.1136/vr.102.14.314-a
Pramod, R. K., Sharma, S. K., Singhi, A., Pan, S., and Mitra, A. (2013). Differential ovarian morphometry and follicular expression of BMP15, GDF9 and BMPR1B influence the prolificacy in goat. Reprod. Domest. Anim. 48, 803–809. doi: 10.1111/rda.12165
Punta, M., Coggill, P. C., Eberhardt, R. Y., Mistry, J., Tate, J., Boursnell, C., et al. (2012). The Pfam protein families database. Nucleic Acids Res. 40, D290–D301. doi: 10.1093/nar/gkr1065
Richard, P., and Kiss, T. (2006). Integrating snoRNP assembly with mRNA biogenesis. EMBO Rep. 7, 590–592. doi: 10.1038/sj.embor.7400715
Ruggero, D. (2009). The role of Myc-induced protein synthesis in cancer. Cancer Res. 69, 8839–8843. doi: 10.1158/0008-5472.CAN-09-1970
Steinhart, Z., and Angers, S. (2018). Wnt signaling in development and tissue homeostasis. Development 145:dev146589. doi: 10.1242/dev.146589
Sun, L., Luo, H., Bu, D., Zhao, G., Yu, K., Zhang, C., et al. (2013). Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 41:e166. doi: 10.1093/nar/gkt646
Tomczak, K., Czerwińska, P., and Wiznerowicz, M. (2015). The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemporary Oncol. 19, A68–A77. doi: 10.5114/wo.2014.47136
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
Wang, H., Nie, X., Li, X., Fang, Y., Wang, D., Wang, W., et al. (2020). Bioinformatics analysis and high-throughput sequencing to identify differentially expressed genes in nebulin gene (NEB) mutations mice. Med. Sci. Monitor 26:e922953. doi: 10.12659/MSM.922953
Wang, L., Feng, Z., Wang, X., Wang, X., and Zhang, X. (2010). DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138. doi: 10.1093/bioinformatics/btp612
Xing, Z., Lin, A., Li, C., Liang, K., Wang, S., Liu, Y., et al. (2014). lncRNA directs cooperative epigenetic regulation downstream of chemokine signals. Cell 159, 1110–1125. doi: 10.1016/j.cell.2014.10.013
Yeung, T. L., Leung, C. S., Wong, K. K., Samimi, G., Thompson, M. S., Liu, J., et al. (2013). TGF-β modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res. 73, 5016–5028. doi: 10.1158/0008-5472.CAN-13-0023
Zetterström, R. (2008). The discovery of the structure and function of chromosomes: the basis of cytogenetics. Acta Paediatrica 97, 673–676. doi: 10.1111/j.1651-2227.2008.00755.x
Zhang, S., Yao, F., Jing, T., Zhang, M., Zhao, W., Zou, X., et al. (2017). Cloning, expression pattern, and potential role of apoptosis inhibitor 5 in the termination of embryonic diapause and early embryo development of Artemia sinica. Gene 628, 170–179. doi: 10.1016/j.gene.2017.07.021
Zhang, Y., Tang, H., Cai, J., Zhang, T., Guo, J., Feng, D., et al. (2011). Ovarian cancer-associated fibroblasts contribute to epithelial ovarian carcinoma metastasis by promoting angiogenesis, lymphangiogenesis and tumor cell invasion. Cancer Lett. 303, 47–55. doi: 10.1016/j.canlet.2011.01.011
Zhao, Z. Q., Wang, L. J., Sun, X. W., Zhang, J. J., Zhao, Y. J., Na, R. S., et al. (2015). Transcriptome analysis of the Capra hircus ovary. PLoS One 10:e0121586. doi: 10.1371/journal.pone.0121586
Keywords: long non-coding RNA, litter size, goats, reproduction, fertility, high-throughput nucleotide sequencing
Citation: Li Y, Xu X, Deng M, Zou X, Zhao Z, Huang S, Liu D and Liu G (2021) Identification and Comparative Analysis of Long Non-coding RNAs in High- and Low-Fecundity Goat Ovaries During Estrus. Front. Genet. 12:648158. doi: 10.3389/fgene.2021.648158
Received: 31 December 2020; Accepted: 06 May 2021;
Published: 25 June 2021.
Edited by:
Rui Su, Inner Mongolia Agricultural University, ChinaReviewed by:
Ran Di, Institute of Animal Sciences, Chinese Academy of Agricultural Sciences, ChinaZhibin Ji, Shandong Agricultural University, China
Jiangjiang Zhu, Southwest Minzu University, China
Adeyemi Adenaike, Federal University of Agriculture, Abeokuta, Nigeria
Copyright © 2021 Li, Xu, Deng, Zou, Zhao, Huang, Liu and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dewu Liu, ZHdsaXVAc2NhdS5lZHUuY24=; Guangbin Liu, Z2JsaXVAc2NhdS5lZHUuY24=