 Marcela D. Mena1
Marcela D. Mena1 Angélica A. Moresco2
Angélica A. Moresco2 Sofía H. Vidal3
Sofía H. Vidal3 Diana Aguilar-Cortes1
Diana Aguilar-Cortes1 María G. Obregon2
María G. Obregon2 Adriana C. Fandiño3
Adriana C. Fandiño3 Juan M. Sendoya1
Juan M. Sendoya1 Andrea S. Llera1†
Andrea S. Llera1† Osvaldo L. Podhajcer1*†
Osvaldo L. Podhajcer1*†- 1Laboratorio de Terapia Molecular y Celular (Genocan), Fundación Instituto Leloir, CONICET, Buenos Aires, Argentina
- 2Servicio de Genética, Hospital de Pediatría Prof. Dr. Juan P. Garrahan, Buenos Aires, Argentina
- 3Servicio de Oftalmología, Hospital de Pediatría Prof. Dr. Juan P. Garrahan, Buenos Aires, Argentina
Purpose: To describe the clinical and molecular spectrum of Stargardt disease (STGD) in a cohort of Argentinean patients.
Methods: This retrospective study included 132 subjects comprising 95 probands clinically diagnosed with STGD and relatives from 16 of them. Targeted next-generation sequencing of the coding and splicing regions of ABCA4 and other phenocopying genes (ELOVL4, PROM1, and CNGB3) was performed in 97 STGD patients.
Results: We found two or more disease-causing variants in the ABCA4 gene in 69/95 (73%) probands, a single ABCA4 variant in 9/95 (9.5%) probands, and no ABCA4 variants in 17/95 (18%) probands. The final analysis identified 173 variants in ABCA4. Seventy-nine ABCA4 variants were unique, of which nine were novel. No significant findings were seen in the other evaluated genes.
Conclusion: This study describes the phenotypic and genetic features of STGD1 in an Argentinean cohort. The mutations p.(Gly1961Glu) and p.(Arg1129Leu) were the most frequent, representing almost 20% of the mutated alleles. We also expanded the ABCA4 mutational spectrum with nine novel disease-causing variants, of which eight might be associated with South American natives.
Introduction
Stargardt disease (STGD), initially described by Karl Stargardt in 1909, is the most common hereditary macular dystrophy with an estimated prevalence of 1 in 8,000 to 1 in 10,000 people (Blacharski, 1988). Mutations in the adenosine triphosphate (ATP)-binding cassette A4 gene (ABCA4) are responsible for the autosomal recessive form of the disease (STGD1, OMIM: 248200) (Allikmets et al., 1997). Most STGD1 patients typically present with bilateral central visual loss in early adolescence and young adulthood with a progressive vision loss during their life (Garces et al., 2018). Ophthalmic examination classically reveals bilateral atrophic changes in the macula due to the loss of photoreceptors and the retinal pigment epithelium (RPE) cells, the presence of yellowish flecks at the level of the RPE, and frequent dark or silent choroid due to the accumulation of lipofuscin in the outer retinal layers (Zernant et al., 2011; Fujinami et al., 2015; Molday, 2015). However, ABCA4-associated retinopathies are clinically variable and genetically heterogeneous. Although age-onset and disease progression varies widely depending on the combination of specific disease-causing alleles and modifiers, early onset is associated with a worse prognosis, while late-onset is associated with a milder phenotype (Fujinami et al., 2015; Zernant et al., 2017; Cremers et al., 2020).
Even though there is a high allelic heterogeneity in ABCA4 with more than 1200 disease-causing variants reported to date1, there are founder mutations associated with STGD1 in various racial and ethnic groups, especially in Europeans, African–Americans, and Asians (Cremers et al., 2020). Those mutations are scarcely found in the limited studies in Latin American STGD1 patients that showed additional founder mutations (Chacón-Camacho et al., 2013; Salles et al., 2018).
In the present study, we aimed to search for the first time the mutational spectrum in Argentinean Stargardt patients and to describe their clinical features. We also expanded the ABCA4 mutational spectrum with nine novel disease-causing variants, of which eight might be associated with native South Americans. Furthermore, we found two de novo variants in ABCA4, including a novel one, c.6214A>C. This study represents an advance in the accurate diagnosis and genetic counseling of STGD1 patients in our country; we also show that disease-causing ABCA4 variants prevalent in other Latin American countries such as Mexico and Brazil were seldom detected in our patients’ cohort.
Materials and Methods
Subjects and Clinical Evaluation
This retrospective study included 132 subjects comprising 82 probands and 16 families (16 probands and their relatives). Most subjects were from Argentina, except two from Uruguay. This study was conducted according to the Declaration of Helsinki. The protocol was approved by the Research and Ethics Committee of Hospital de Pediatría Prof. Dr. Juan P. Garrahan (HPG, protocol #892) and by the Bioethics Committee of Fundación Instituto Leloir (protocol #27). Written informed consent was obtained from all participants or legal guardians before enrollment in the study. Pre- and post-test counseling was offered at HPG for all participants. Self-referred ancestry and family history of retinal disorders were obtained from each participant. All the probands (n = 98) and two siblings suspected of STGD1 disease underwent a comprehensive ophthalmological evaluation between November 2015 and December 2017 at HPG. Clinical assessment included the best-corrected Snellen visual acuity (BCVA), converted to equivalent logMAR visual acuity, fundus examination, color fundus photography (RG), and autofluorescence imaging (AF) performed with a retinal fundus camera (Carl Zeiss Meditec, Germany). When feasible, fluorescein angiography (FA), electrophysiology tests (ERG), and optical coherence tomography (OCT) were evaluated. The fundus appearance was classified into four groups, and the AF pattern into three types (Fujinami et al., 2015). The age of onset and duration of the disease was assigned as described by Fujinami et al. (2015). Early onset of disease was defined as ≤ 17 years-old.
DNA Sample Collection and Sequencing
Six ml of peripheral blood were collected into EDTA tubes from all probands; saliva samples were collected using Oragene buccal swabs kit (DNA Genotek) from family members. The samples were transported following strict standard operating procedures from HPG to Genocan at Fundación Instituto Leloir. Genomic DNA was extracted using the QIAamp DNA Mini Kit (Qiagen) and quantified in a Nanodrop 2000 spectrophotometer (Thermo Scientific).
Next-generation sequencing (NGS) was performed in probands (n = 95) and two siblings, using a custom panel created with the Ion AmpliSeqTM Designer software (Thermo Fisher Scientific, Inc.). The panel allowed the analysis of coding exons and adjacent intronic sequences (±10 bp) of the ABCA4 (NM_000350.2), ELOVL4 (NM_022726.3), PROM1 (NM_006017.2), and CNGB3 (NM_019098.4) genes by sequencing of 130 amplicons distributed in 2 pools (pool 1: 66 amplicons; pool 2: 64 amplicons). The total panel size was 37.92 kb, with global coverage of 99.9%. DNA input was assessed with the Qubit fluorometer 2.0 (Thermo Fisher Scientific, Inc.) using the Qubit dsDNA HS (high-sensitivity) assay kit. Twenty nanograms of each sample (10 ng/pool) were used for library preparation with the Ion AmpliSeqTM Library Kit 2.0 (Thermo Fisher Scientific, Inc.). Targeted sequencing was performed using the Ion Hi-Q Sequencing Kit (Thermo Fisher Scientific, Inc.) on the Ion PGMTM platform (Ion Torrent; Thermo Fisher Scientific, Inc.). For segregation studies, family members’ samples were analyzed by Sanger sequencing to search for the variants detected in their respecting probands (Macrogen2).
Bioinformatics Analysis
Primary data analyses comprising raw signal conversion, base calling, read filtering, and alignments to the hg19/GRCh37 human reference genome were performed using the Torrent Suite software v. 4.0 (Ion Torrent; Thermo Fisher Scientific, Inc.). Coverage analysis plug-in (5.0.2.0 version) was used to verify overall amplicon coverage and check for strand bias. Following primary data analysis, all BAM files were uploaded on the Ion Reporter Server Software (Thermo Fisher Scientific, Inc.) for variant calling and annotation, resulting in a table of 30–50 variants per patient. Further, the targeted region’s coverage was manually inspected with the Integrative Genomics Viewer (IGV3) and Alamut Visual v.2.9 (Interactive Biosoftware, Rouen, France).
Analysis of the target regions revealed the systematic failure of four amplicons (coverage < 20x), corresponding to exons 21 and 29 of the ABCA4 gene, exon 3 of the ELOVL4 gene, and exon 4 of the PROM1 gene. Sanger sequencing was performed for these four exons and other regions identified with low coverage (<20x). Variants with minor allele frequency (MAF) > 1% reported in different population databases such as 1.000 Genomes Project4, dbSNP5 or the Genome Aggregation Database (gnomAD6) were initially filtered out. Causative ABCA4 variants with a high population frequency, such as the hypomorphic variant c.5603A > T p.(Asn1868Ile), were rescued by manually reviewing the filtered-out variants. Candidate variants were checked against ClinVar7 and Leiden Open Variation Database (LOVD; see footnote 1). Variants classified as benign or likely benign were excluded from the study. Variants not found in the databases mentioned above were subjected to an exhaustive PubMed8 literature search. The pathogenicity assessment was tested using the in silico prediction tools; Sorting Intolerant From Tolerant (SIFT9), Polymorphism Phenotyping v2 (PolyPhen-210), Mutation Taster11, Human Splicing Finder 3.012 and Combined Annotation Dependent Depletion (CADD13). Candidate variants were manually reviewed in the BAM file using Alamut Visual v.2.9 and validated using Sanger sequencing. Novel variants were classified using the terms “pathogenic,” “likely pathogenic,” “uncertain significance” (VUS), “likely benign,” and “benign,” according to the guideline of the American College of Medical Genetics and Genomics (ACMG) (Richards et al., 2015).
Results
Clinical Assessment of the Cohort
From the 100 patients recruited, including 98 probands and two siblings, three probands were excluded from the study after clinical reexamination (Supplementary Figure 1). The mean age at diagnosis of the remaining 95 probands was 18 years (range 3–48 years), and the average time from the age of onset to diagnosis was 5 years (range 0–44 years). NGS identified at least one ABCA4 mutant allele in 78/95 (82%) probands (45 female and 33 male). At the time of enrollment, the mean age of the 78 probands was 34 years old (range 10–65 years), with a mean age at onset of 14 years old (range 4–47 years). Sixty-three probands (81%) presented early onset disease with a mean duration of the disease of 21 years (range 1–52 years). The patients’ cohort showed symmetric grade 3a or higher, fundus abnormalities consistent with central retinal atrophy and vision loss in both eyes with a mean BCVA of 1.2 logMAR (Snellen 20/300) showing a range of 0.1–2.3 logMAR (Snellen 20/25 to hand movements) in both eyes (Supplementary Table 1). Frequently, the vision loss was associated with photophobia and, in fewer cases, with slow dark adaptation. Besides, the mean foveal thickness was 136.5 μm (range 43–251 μm) in both eyes, and over half of the probands showed dark choroid on FA images (Supplementary Table 1). Of note was the fact that only 16 out of 82 patients with at least one variant found in ABCA4, displayed a previous family history of STGD.
Variants Present in the ABCA4 Gene and Their Correlation With Clinical Features
We found two variants or more in ABCA4 in 69/95 (73%) probands (Supplementary Tables 2, 3); a single ABCA4 variant was found in 9/95 (9.5%) probands with STGD1 phenotype. A second variant could be found in four of these nine patients by Khan et al. (2020) (Figure 1 and Supplementary Tables 2, 4). Therefore, a total of 73/95 (77%) probands of our cohort ended up having two or more ABCA4 variants. In 17/95 (18%) probands, no variant was found in any of the sequenced genes.
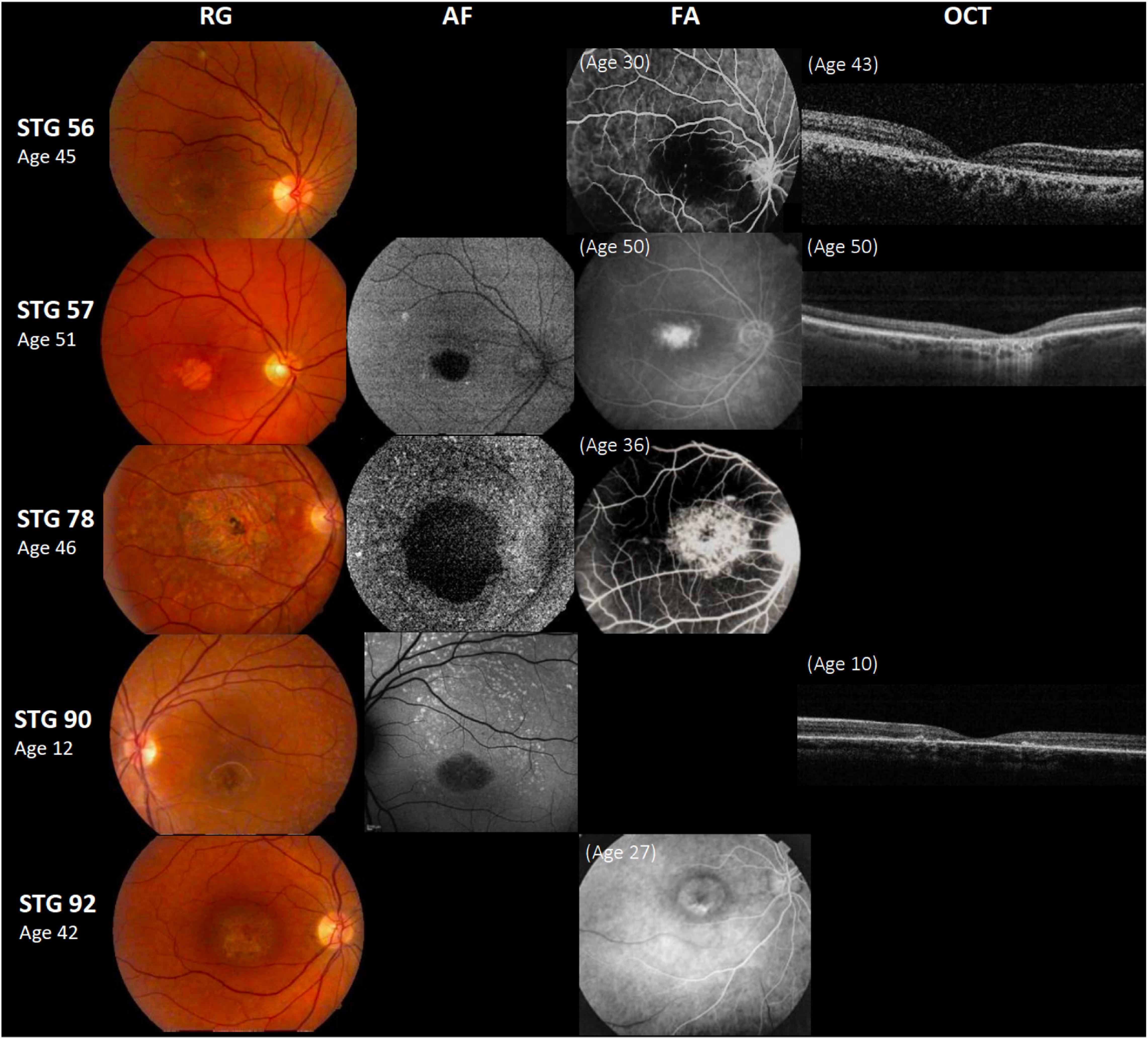
Figure 1. Clinical presentation in patients harboring a single ABCA4 variant. Patients STG 56, STG 57, STG 78, and STG 90 show central atrophy surrounded by macular and peripheral flecks (fundus grade: 3b), which are more visible on AF image of STG 78 and STG 90, showing widespread multiple foci of high and low AF signal with heterogeneous background (AF type 2). These patients demonstrate dark “silent” choroid on Fluorescein angiography images (FA) and macular hyperfluorescence consequence of retinal pigment epithelium and photoreceptors loss. Patient STG 92 present on FA image at the age of 27 years-old, hyperfluorescent annular lesion showing perifoveal atrophy without flecks (Bull’s eye maculopathy) according to fundus grade 3a, further in the course of the disease with 42 years-old, note an enlargement of macular atrophy on color fundus photograph (RG). The optical coherence tomographic images (OCT) present decreased retinal thickness with disruption of external layers.
The final analysis identified 173 variants in ABCA4 spread out from exon 3 to exon 48. Seventy-nine variants were unique, and nine have not been previously associated with STGD (Table 1). Among the 79 unique variants found, the most common type of variant was missense (51/79), followed by frameshift (11/79), splice-site (9/79), nonsense (7/79), and in-frame deletion (1/79). All the variants were heterozygous, except for two homozygous variants identified in proband STG 80.

Table 1. Assessment of pathogenicity of novel ABCA4 variants.
Of the 156 alleles mutated in unrelated patients, p.(Gly1961Glu) and p.(Arg1129Leu) showed an allelic frequency of 10.25% (16/156) and 9.7% (15/156), respectively (Supplementary Figure 2). Half of the patients (8/16) harboring p.(Gly1961Glu) presented early onset of the disease, and 9/16 had severe visual impairment, with a mean BCVA of 1 logMAR for the right eye (range 0.2–2.3) and 0.9 logMAR for the left eye (0.2–1.9) and mean duration time of the disease of 24 years (2–50 years). Most patients (11/14) harboring p.(Arg1129Leu) demonstrated severe compromise in visual acuity with a mean BCVA of 1.2 logMAR (range 0.5–1.9) and a mean age of onset of 14 years old (range 7-24 years). Proband STG 80 carried two homozygous mutations p.(Arg1129Leu) and p.(Thr2240Ala) and showed a typical STGD phenotype (Figure 2). As expected, we observed that proband STG 9 and her affected brother (STG 10) carried the same variants; likewise, the monozygotic twins (STG 17 and STG 18) carried the same ABCA4 variants; also, both pairs of siblings showed a concordant phenotype (Supplementary Table 2).
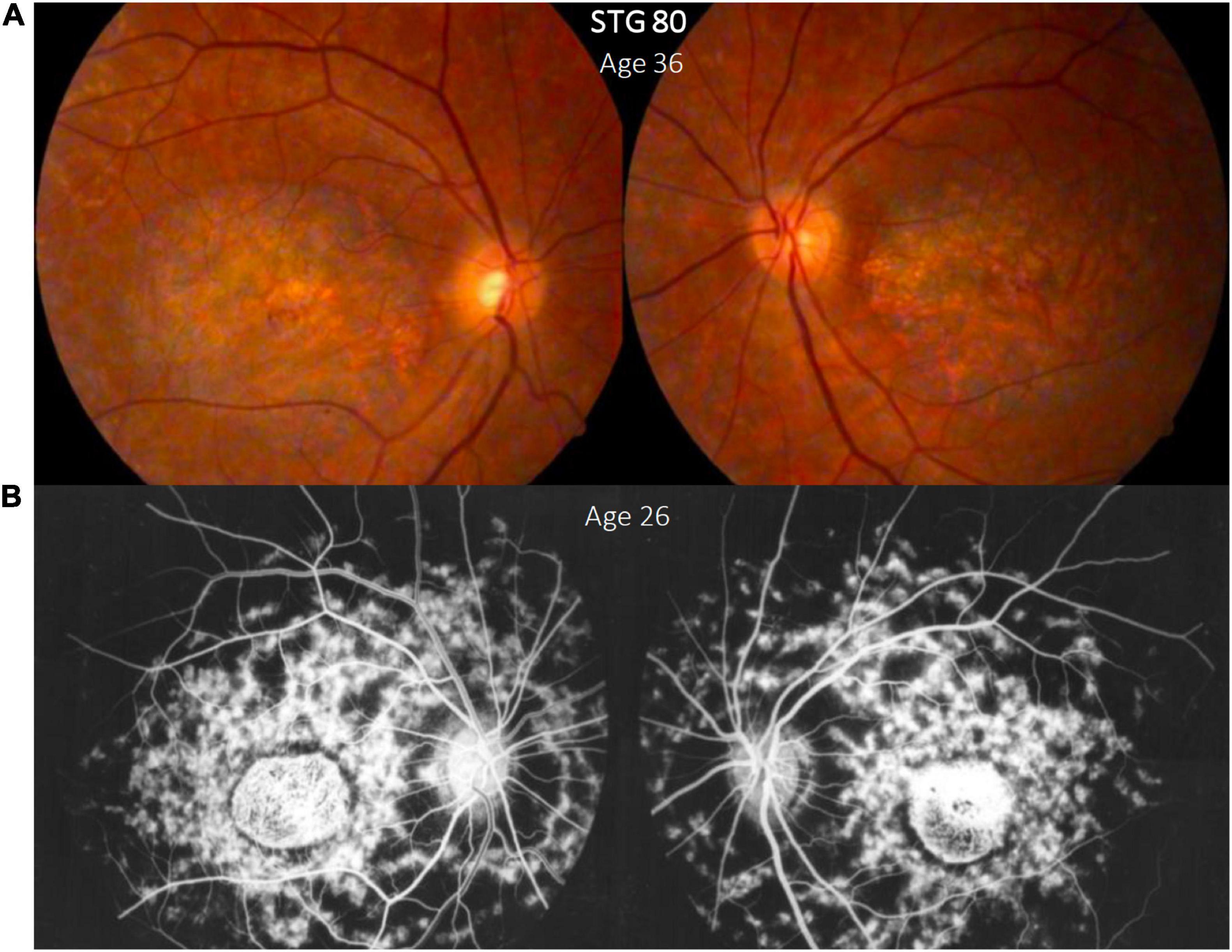
Figure 2. Clinical presentation in patient STG 80, (A) Color fundus photographs (RG) of both eyes with macular atrophy and peripheral flecks (fundus grade: 3b) (B) fluorescein angiography images (FA) clearly show dark “silent” choroid and central hyperfluorescence due to macular atrophy in this angiogram that was made years before our assessment. Note the enlargement of the diameter of the macular atrophy between the FA image brought by the patient (B) and the color RG (A) taken years later for this study.
We performed segregation analysis in 16 families, although the co-segregation was not complete in six families due to the absence of additional family members (five cases) or incomplete sequencing information (one case) (Supplementary Figure 1).
We found the hypomorphic c.5603A>T, p.(Asn1868Ile) variant with an allelic frequency of 7% (13/190) mainly in heterozygous state and combined mostly with two causal ABCA4 variants (Supplementary Tables 2, 3), except two probands who carried one (STG 2) or no variant (STG 41). STG 2 carried a heterozygous frameshift variant combined with c.5603A>T; upon segregation analysis, we found that both parents harbored c.5603A>T in heterozygous state.
Complex Alleles in ABCA4 and Their Clinical Correlates
We detected complex ABCA4 alleles in 13.7% (13/95) probands (Supplementary Table 3). Most of these probands carried variants previously identified as complex ABCA4 alleles in retinal dystrophy patients. All but one patient (STG 15) presented early onset, and their clinical records were associated with higher aggressive disease, severe visual loss, and early morphologic changes (Supplementary Table 3).
The frequent complex allele, c.[1622T>C;3113C>T], p.[Leu541Pro;Ala1038Val] was found in two probands (STG 15 and STG 42), that also carried p.(Gly1961Glu). This complex allele was associated with a complete ABCA4 loss of function and early onset, resembling truncating ABCA4 variants (Zhang et al., 2015; Cremers et al., 2020). However, STG 15 had a late-onset at 24 years old, legal blindness after 20 years of the disease, showed macular atrophy with flecks, and compromised OCT images, likely due to the mild mutation p.(Gly1961Glu) present in the second allele; on the other side, STG 42, had an early onset at 16 years-old; this patient, that was enrolled 2 years after disease onset, showed a moderate visual loss and fundus with foveal atrophy without flecks. It is likely that a long-time follow-up would clarify the severity of the disease and the clinical correlation with this specific genotype in both patients (Supplementary Table 3). Segregation analysis confirmed the presence of the complex ABCA4 alleles in relatives of STG 14, STG 15, and STG 42 (Supplementary Table 3 and Supplementary Figure 1).
STG 81 presented the complex allele c.688T>A(;)6718A>G. Of note, the variant c.6718A>G was not present in STG 81 family, even when the other two variants that were part of the genotype, c.688T>A and c.5714+5G>A were present (Supplementary Table 3). Therefore, we considered c.6718A>G as a de novo variant.
Five probands carried the hypomorphic variant c.5603A>T combined with the disease-causing variants c.5461-10T>C, 4469G>A p.(Cys1490Tyr), and c.2588G>C p.(Gly863Ala) (Supplementary Table 3). Segregation analysis in these five probands was not possible due to the lack of DNA samples of their family members. Other probands harboring c.5603A>T combined with two ABCA4 variants not included as complex alleles are also shown (Supplementary Table 2).
Novel Variants and Correlation With the Clinics
We found nine ABCA4 novel variants in eight unrelated patients with STGD clinical phenotypes (Figure 3); eight were considered as likely pathogenic/pathogenic, and one as VUS (Table 1). Four were missense, three frameshifts, one nonsense, and one splice-site variant (Figure 4). None but one variant (c.1698C>A) has been previously found in two alleles in the population database gnomAD (Table 1). Most of the probands (5/8) carried novel variants associated with a known ABCA4 mutation and exhibited clinical features of STGD1 with early onset (Supplementary Table 2).
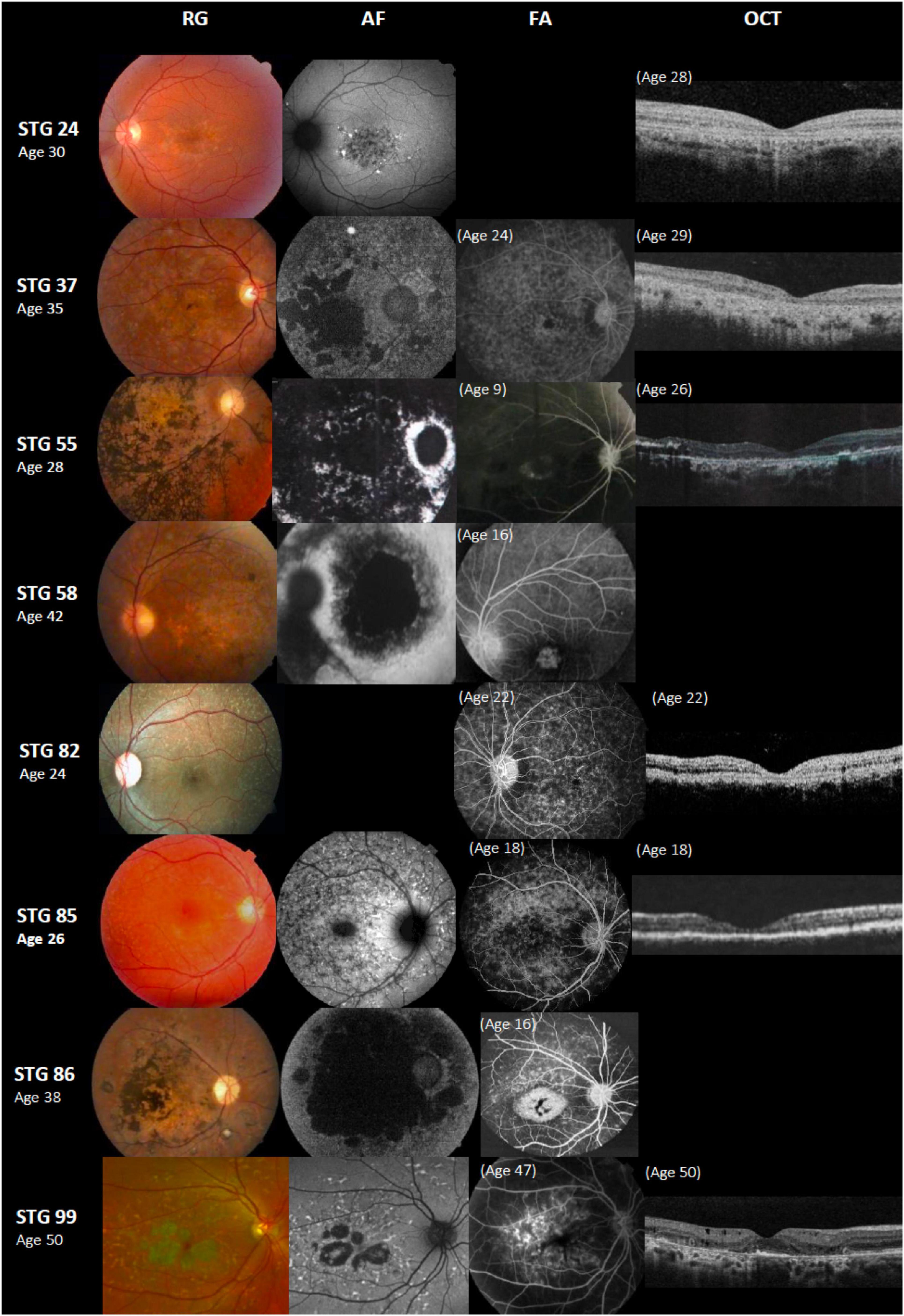
Figure 3. Color fundus photographs (RG), autofluorescence (AF), fluorescein angiography (FA), and optical coherence tomographic images (OCT) of eight patients with novel ABCA4 variants. All of the shown cases had early onset except STG 24, STG 82, and STG 99. Patients STG 24, STG 37, STG 55, STG 82, and STG 85 present central atrophy with macular and peripheral flecks (fundus grade: 3b) on RG images; AF images demonstrate low AF signal at the macula and posterior pole as black lesions corresponding to cells death of retinal pigment epithelium and photoreceptors. The available OCT images in these patients present severe decreased retinal thickness below 150 μm and absence of external retinal layers. Patient STG 55 presents on FA image at the age of 9 years-old, hyperfluorescent annular lesion showing perifoveal atrophy without flecks (Bull’s eye maculopathy). Further, in the course of the disease with 28 years-old, note on AF how retinal atrophy increases as multiple areas of low AF signal at the posterior pole (AF type 3) and on RG image, there is extensive chorioretinal atrophy with large bone spicule pigmentation. Patient STG 58 at the age of 16 years-old, showed hyperfluorescent foveal lesion compatible with central atrophy on the FA image, that 26 years later in RG and AF images showed multiple extensive atrophic changes of the retinal pigment epithelium extending beyond the vascular arcades (fundus grade:4, AF type 3). Patient STG 86 showed on FA image at the age of 16 years-old, hyperfluorescent central lesion as macular atrophy surrounded by hyperfluorescent flecks. RG and AF images performed 22 years later, and there is an enlargement of macular atrophy with pigmentation clearly seen on RG and multiple areas of low AF signal in the posterior pole (AF type 3). Patient STG 99 presents central atrophy with foveal sparing and peripheral flecks in the color image; AF demonstrates low central AF signal with foveal sparing surrounded by marked flecks corresponding to foci of high signal as lipofuscin deposits. This patient had late onset of the disease at the age of 47 years old with BCVA 0.1 logMAR (20/25 Snellen) in both eyes. OCT image showed parafoveal disruption of external retinal layers with chronic structural changes.
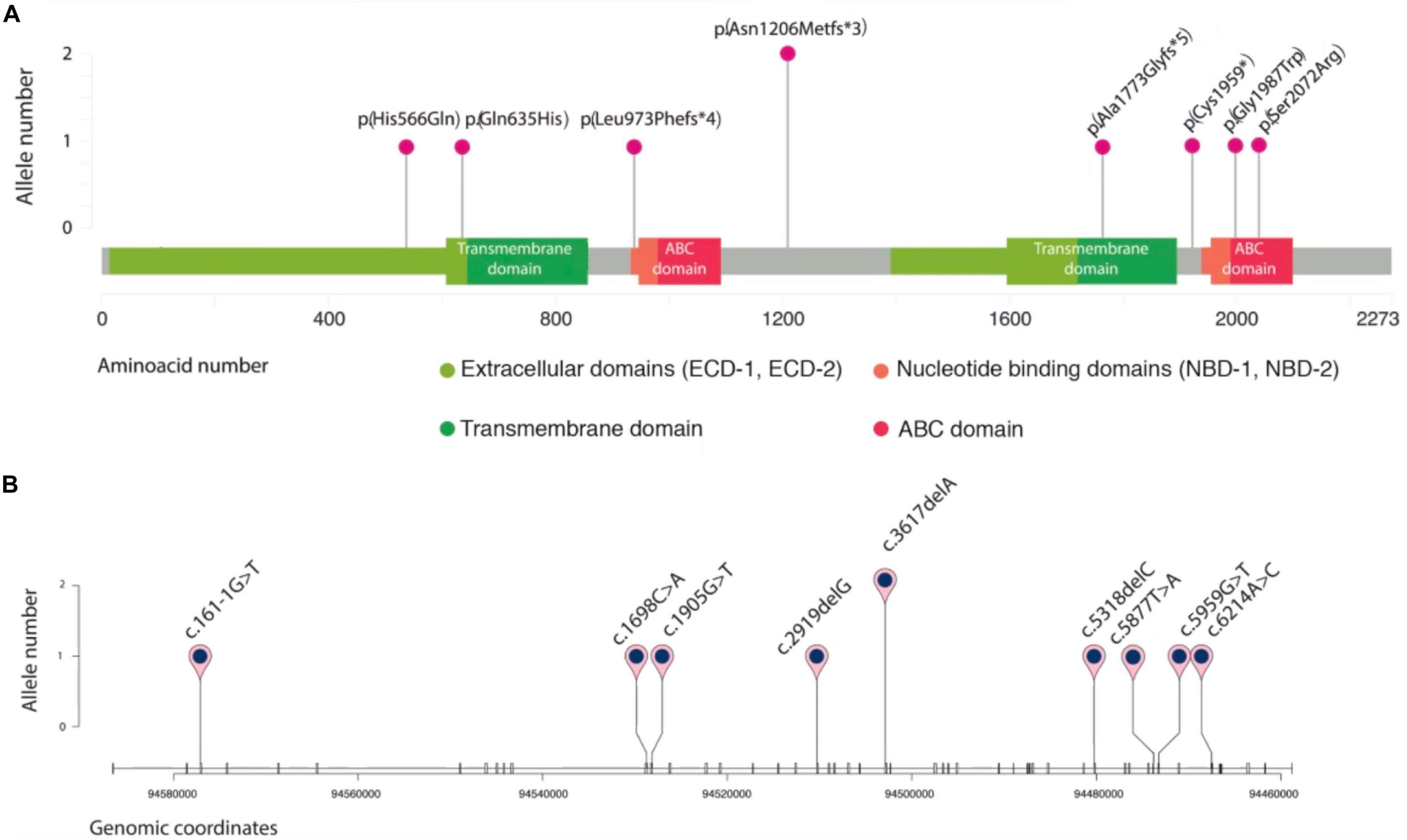
Figure 4. Schematic representation of novel ABCA4 variants found in eight unrelated patients. Plots are adapted from cBioportal’s MutationMapper and R package trackViewer. Variants are displayed in two contexts: (A): linear ABCA4 protein sequence (NP_000341.2), highlighting its typical A-subclass ABC transporter domains, and (B): ABCA4 gene sequence (NM_00350.2), where each tick represents an exon. Genomic coordinates correspond to hg19/GRCh37 human reference genome.
Interestingly, STG 86, a 38-year-old female, carried two novel frameshifts variants p.[(Ala1773Glyfs∗5);(Asn1206Metfs∗3) that were considered pathogenic. She presented early onset with a visual acuity markedly affected (1.9 logMAR) after 30 years of the disease and a typical STGD atrophic fundus appearance (Supplementary Table 2 and Figure 3). Segregation analyses showed that both unaffected parents were heterozygous carriers of each one of those frameshift variants (Supplementary Figure 1).
Patient STG 99, a 50-year-old female, carried two novel missense variants p.(His566Gln)(;)(Gln635His). This patient presented late-onset (47 years old) with preserved visual acuity [BCVA of 0.1 logMAR (20/25 Snellen)] combined with paracentral atrophy with foveal sparing, peripheral flecks, and dark choroid on AF images consistent with STGD features (Figure 3). We were unable to perform segregation analysis due to the lack of DNA samples of her family. Of note, a different variant in the same amino acid position p.(Gln635Lys) was reported in a patient with STGD1 (Rivera et al., 2000) and classified as likely pathogenic (ClinVar: VCV000099095.2).
Patient STG 85 carried three variants; two known variants and a novel one, c.5959G>T, that we could confirm as present in cis with c.5961_5964del, as they both appeared in the same read in the NGS BAM file. Segregation analysis showed that the unaffected mother was heterozygous carrier of c.5714+5G>A (Supplementary Table 3 and Figure 3).
Interestingly, in patient STG 55 (Figure 3), the novel variant c.6214A>C p.(Ser2072Arg) was absent in either of the parents; therefore, we considered it as a de novo variant.
Variants Present in PROM1, ELOVL4, and CNBG3 Genes
Sequencing of the PROM1 and ELOVL4 genes showed only variants classified as benign or likely benign in all probands. A novel heterozygous variant, c.1528C>T p.(Leu510Phe), was detected in the CNGB3 gene in proband STG 52. In silico analysis predicted p.(Leu510Phe) as deleterious on protein function [Polyphen-2 (score: 0.890), Mutation Taster (p-value: 0.989), and SIFT (score: 0.03)]. Patient STG 52 also carried c.1957C>T, p.(Arg653Cys) and c.6320G>A p.(Arg2107His) in the ABCA4 gene. This patient presented early onset disease with a slow progression in vision loss after 19 years of the disease (0,6 logMAR; 20/80 Snellen). Considering all evidence, p.(Leu510Phe) was classified as VUS.
Discussion
In the present study, we performed the first clinical and molecular screening of STGD in Argentinean patients using targeted next-generation sequencing. This study revealed a total of 173 variants identified in ABCA4, from which 79 were unique. More than half were detected only once. Among them, we found nine novel variants. The most common ABCA4 mutations were c.5882G>A p.(Gly1961Glu) and c.3386G>T p.(Arg1129Leu), representing almost 20% of the mutated alleles found in this representative cohort.
The mutation p.(Gly1961Glu) is the most common among STGD1 patients from different ethnic backgrounds and was reported with variable frequency (6.5–21%) (Ścieżyńska et al., 2016; Fujinami et al., 2018). Distinct clinical phenotypes were associated with patients harboring p.(Gly1961Glu) (Cella et al., 2009; Burke et al., 2012), including late-onset disease, Bull’s eye maculopathy, and the absence of dark choroid signs on FA and retinal flecks (Cella et al., 2009; Lee et al., 2017). Our patients harboring p.(Gly1961Glu) presented a mean age of onset disease of 18 years old, but only 1/16 probands exhibited Bull’s eye maculopathy. Furthermore, most of these probands presented flecks on RG, and dark choroid on FA, showing differences in the clinical phenotype of our STGD1 patients carrying this mutation compared to patients from other ethnic backgrounds (Cella et al., 2009; Lee et al., 2017).
The second more frequent pathogenic variant in our cohort, p.(Arg1129Leu), is considered a mild variant associated with later onset and foveal sparing (Del Pozo-Valero et al., 2020) that might enhance disease severity when combined with a “null mutation” (Riveiro-Alvarez et al., 2013). Interestingly, most of our patients harboring p.(Arg1129Leu) presented early onset (range 7–24 years) with a phenotype that included visual acuity already compromised at early stages, regardless of whether it was combined with a “null” or missense variant; this suggests once again that the ABCA4 variants alone might not predict neither onset nor severity. One of these patients was double homozygous for p.(Arg1129Leu) combined with p.(Thr2240Ala). A few rare cases of double homozygous mutations were described in STGD1 patients (Burke et al., 2012; Serapinas et al., 2013). Unfortunately, no segregation analysis could be performed on this patient. Further studies, such as copy number variation and haplotype analysis, should be performed to exclude uniparental disomy (UPD) as previously reported in STGD1 patients (Fingert et al., 2006; Riveiro-Alvarez et al., 2007; Khan et al., 2020).
We found the hypomorphic c.5603A>T p.(Asn1868Ile) variant with an allele frequency of ∼7%, three times higher than in the general Latin American population (MAF ∼2.28) (see footnote 6). This result is consistent with previous studies showing that c.5603A>T (p.Asn1868Ile) is present three to four times more frequently in ABCA4-associated retinopathy patients than in the general population (Aguirre-Lamban et al., 2011; Zernant et al., 2017). Besides, c.5603A>T was found in probands that carried the disease-causing alleles c.5461–10T>C, p.(Cys1490Tyr), and p.(Gly863Ala). The variant c.5603A>T was reported to be always in cis with these alleles, and it was shown to be required for the pathogenicity of p.(Gly863Ala) (Zernant et al., 2017; Midgley et al., 2020).
It was of note the near absence of the Latin American-exclusive mutations described in the Mexican and Brazilian populations. Indeed, the most prevalent mutation in the Mexican population p.(Ala1773Val) (Chacón-Camacho et al., 2013) was utterly absent in our cohort; moreover, p.(Arg602Trp), the most prevalent mutation in Brazilian patients (Salles et al., 2018) was only found in one allele (0.6%). Besides, we found that only ∼14% of our probands harbored complex alleles in ABCA4 compared to 30% reported in Brazilian patients (Salles et al., 2017). These findings can be explained by Argentina’s differential ancestral admixture compared to Brazil and Mexico (Muzzio et al., 2018). In that sense, p.(Arg1129Leu), a founder mutation in Spain (Valverde et al., 2006) considered rare in many populations, was found in 9.62% (15/156) alleles in our cohort. Also, two probands (STG14 and STG 27) carried the same set of three ABCA4 variants that were reported in patients from a Spanish cohort (Riveiro-Alvarez et al., 2013). Further rare variants such as p.(Leu2140Gln); p.(Ser84Thrfs∗16) and p.(Leu464Glufs∗2) reported either as novel variants or only found in Italian cohorts (Simonelli et al., 2000; Fumagalli et al., 2001; Passerini et al., 2010; Testa et al., 2012) were identified in our cohort. In fact, patients harboring these mutations self-referred with admixture ancestry, mostly from Spain and Italy, consistent with the immigration history of Argentina.
Founder mutations in Danish and Chinese patients such as p.(Asn965Ser) (Rosenberg et al., 2007; Jiang et al., 2016; Cremers et al., 2020) or p.[Gly863Ala,Gly863del] in Western/Northern Europe (Maugeri et al., 1999, 2002) were detected at low frequency in our cohort. Further, the most frequent complex allele p.[Leu541Pro; Ala1038Val] described as a founder mutation in Germany (Rivera et al., 2000) also reported in high-frequency in patients from Eastern Europe (Ścieżyńska et al., 2016; Zolnikova et al., 2017; Tracewska et al., 2019) was found in 1.3% of our mutated alleles.
Eight unrelated patients carried nine novel variants. Two probands carried two novel variants each, while in the other cases, the novel variant was accompanied by known disease-causing ABCA4 variants. Interestingly, two unrelated patients carried the same novel frameshift variant, p.(Asn1206Metfs∗3). All the probands with novel variants showed typical STGD1 phenotype, including the proband with two novel missenses, who showed late-onset and foveal sparing, apparently presenting the milder end of the clinical spectrum STGD1 (van Huet et al., 2014). Thus, it is tempting to hypothesize that eight of these novel variants might be associated with Native Americans from South America.
A study performed on 27 probands of our cohort with a single or no disease-causing ABCA4 variants using single-molecule molecular inversion probes (smMIPs) led to the identification of the second coding/splice site ABCA4 variant in three probands and two deep intronic variants in one proband (Khan et al., 2020); the three coding/splice site variants were confirmed in our NGS BAM files; however, they were missed in the analysis using Ion reporter variant caller software. These results indicate the necessity to combine and normalize the output of various variant calling tools to improve the sensitivity and precision to identify real variants (Sandmann et al., 2017).
Overall, a total of six probands could not be genetically solved, including five probands with a single ABCA4 variant and a proband (STG 2) harboring a frameshift variant and the hypomorphic variant c.5603A>T in heterozygous state. This latter variant is considered a mild conditional allele, causal of STGD1 when it is present in trans with a loss-of-function variant (Zernant et al., 2017). In this regard, the segregation analysis within the STG 2 family could not establish the variants’ configuration (cis/trans) because both parents were heterozygous carriers of the c.5603A>T variant. Further studies involving haplotype analysis should be performed to establish the phase of both variants. No other ABCA4 disease-causing variant was found in this proband when tested by Khan et al. (2020).
The missing ABCA4 disease-causing alleles in these five probands could result from changes that affect ABCA4 transcription or heterozygous deletions (<400 bp), insertions, or inversions (>40 bp), not detected by smMIPs-based sequencing (Khan et al., 2020). Although all our patients were diagnosed clinically with STGD, the overlapping of phenotypes between different macular dystrophies and several stages of ABCA4-associated retinopathy could result in a misdiagnosis of STGD with mutations in phenocopying genes, such as PRPH2, ROM1, CRX, or RPGR (Cremers et al., 2020). Other genomic techniques, including whole-exome sequencing, might eventually help to determine the molecular diagnosis of the remaining unsolved patients.
This study represents the first integrative study of retinopathy in Argentina. We are aware of the limitations of the study, in particular, the segregation studies due to the fact that families were difficult to approach and, in some cases, were reluctant to be included. Despite that, we can conclude that p.(Gly1961Glu) and p.(Arg1129Leu) were the most frequent mutations in our cohort, consistent with a Southern European ancestry’s broad representation in Argentina population’s admixture. We also expanded the ABCA4 mutational spectrum with nine novel disease-causing variants; eight of them might be associated with native South Americans. Interestingly, there were phenotypic differences in patients of this cohort harboring mutations already described in the literature compared to other cohorts. These findings, along with the lack of concordance with variants seen in other Latin American populations, reinforce the need for genotype-phenotype studies in large cohorts and diverse populations.
Data Availability Statement
The datasets presented in this study can be found in the online repository ABCA4 LOVD (https://databases.lovd.nl/shared/genes/ABCA4). The names of the repository/repositories and accession number(s) can be found below https://databases.lovd.nl/shared/variants?search_owned_by_=%3D%22Marcela%20Mena%22.
Ethics Statement
This protocol was approved by the Research and Ethics Committee of Hospital de Pediatría Prof. Dr. Juan P. Garrahan (HPG, protocol #892) and by the Bioethics Committee of Fundación Instituto Leloir (protocol #27). Written informed consent to participate in this study was provided by the participants or legal guardians.
Author Contributions
MM, AL, and OP contributed to conception and design of the study. AM and MO recruited the patients and performed the genetic counseling. SV and AF performed the clinical examination. MM and JS performed the NGS sequencing. JS, DA-C, and MM performed the Sanger sequencing. MM analyzed and interpreted the sequencing data, wrote the original draft of the manuscript. SV and AM wrote sections of the manuscript. MM, AL, and OP wrote, reviewed, and edited the manuscript. All the authors have read and agreed to the submitted version of the manuscript.
Funding
Financial support was provided by the Ministry of Science, Technology and Productive Innovation (www.argentina.gob.ar/ciencia); Stargardt APNES Argentina (www.stargardt.com.ar) and Federación Argentina de Enfermedades Poco Frecuentes (Fadepof) (www.fadepof.org.ar).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank all the participants in this study. Our gratitude goes to the patients and their families, to Stargardt-APNES Argentina, in particular to Marcela Ciccioli and FADEPOF, without whom this research could not have been performed. We would also like to thank Dr. Guillermo Chantada for his administrative support, to Dr. Luis Diaz Gonzalez for collaborating in ophthalmological exams, to Dr. Daniel Hidalgo for referring patients, and to Josefina Borda, Julieta Viglino, and Daniel Scheifer for their technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.646058/full#supplementary-material
Footnotes
- ^ https://www.lovd.nl
- ^ http://www.macrogen.com/
- ^ www.broadinstitute.org/igv
- ^ https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/
- ^ https://www.ncbi.nlm.nih.gov/snp/
- ^ http://gnomad.broadinstitute.org/
- ^ https://www.ncbi.nlm.nih.gov/clinvar/
- ^ https://pubmed.ncbi.nlm.nih.gov/
- ^ http://sift.jcvi.org/
- ^ http://genetics.bwh.harvard.edu/pph2/
- ^ http://www.mutationtaster.org/
- ^ http://www.umd.be/HSF/
- ^ https://cadd.gs.washington.edu/
References
Aguirre-Lamban, J., González-Aguilera, J. J., Riveiro-Alvarez, R., Cantalapiedra, D., Avila-Fernandez, A., Villaverde-Montero, C., et al. (2011). Further associations between mutations and polymorphisms in the ABCA4 Gene: clinical implication of allelic variants and their role as protector/risk factors. Invest. Opthalmol. Vis. Sci. 52:6206. doi: 10.1167/iovs.10-5743
Allikmets, R., Singh, N., Sun, H., Shroyer, N. F., Hutchinson, A., Chidambaram, A., et al. (1997). A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 15, 236–246. doi: 10.1038/ng0397-236
Blacharski, P. (1988). “Fundu flavimaculatus In,” in Retinal Dystrophies and Degenerations, ed. D. A. Newsome (New York, NY: Raven Press).
Burke, T. R., Fishman, G. A., Zernant, J., Schubert, C., Tsang, S. H., Smith, R. T., et al. (2012). Retinal phenotypes in patients homozygous for the g1961e mutation in the ABCA4 gene. Invest. Ophthalmol. Vis. Sci. 53:4458. doi: 10.1167/iovs.11-9166
Cella, W., Greenstein, V. C., Zernant-Rajang, J., Smith, T. R., Barile, G., Allikmets, R., et al. (2009). G1961E mutant allele in the Stargardt disease gene ABCA4 causes bull’s eye maculopathy. Exp. Eye Res. 89, 16–24. doi: 10.1016/j.exer.2009.02.001
Chacón-Camacho, O. F., Granillo-Alvarez, M., Ayala-Ramírez, R., and Zenteno, J. C. (2013). ABCA4 mutational spectrum in mexican patients with Stargardt disease: identification of 12 novel mutations and evidence of a founder effect for the common p.A1773V mutation. Exp. Eye Res. 109, 77–82. doi: 10.1016/j.exer.2013.02.006
Cremers, F. P. M., Lee, W., Collin, R. W. J., and Allikmets, R. (2020). Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Progress in Retinal and Eye Research 79, 100861. doi: 10.1016/j.preteyeres.2020.100861
Del Pozo-Valero, M., Riveiro-Alvarez, R., Blanco-Kelly, F., Aguirre-Lamban, J., Martin-Merida, I., Iancu, I.-F., et al. (2020). Genotype–phenotype correlations in a spanish cohort of 506 families with biallelic abca4 pathogenic variants. Am. J. Ophthalmol. 219, 195–204. doi: 10.1016/j.ajo.2020.06.027
Fingert, J. H., Eliason, D. A., Phillips, N. C., Lotery, A. J., Sheffield, V. C., and Stone, E. M. (2006). Case of Stargardt disease caused by uniparental isodisomy. Arch. Ophthalmol. 124, 744–745. doi: 10.1001/archopht.124.5.744
Fujinami, K., Strauss, R. W., Chiang, J. P.-W., Audo, I. S., Bernstein, P. S., Birch, D. G., et al. (2018). Detailed genetic characteristics of an international large cohort of patients with Stargardt disease: ProgStar study report 8. Br. J. Ophthalmol. 103, 390–397. doi: 10.1136/bjophthalmol-2018-312064
Fujinami, K., Zernant, J., Chana, R. K., Wright, G. A., Tsunoda, K., Ozawa, Y., et al. (2015). Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology 122, 326–334. doi: 10.1016/j.ophtha.2014.08.012
Fumagalli, A., Ferrari, M., Soriani, N., Gessi, A., Foglieni, B., Martina, E., et al. (2001). Mutational scanning of the ABCR gene with double-gradient denaturing-gradient gel electrophoresis (DG-DGGE) in italian Stargardt disease patients. Hum. Genet. 109, 326–338. doi: 10.1007/s004390100583
Garces, F., Jiang, K., Molday, L. L., Stöhr, H., Weber, B. H., Lyons, C. J., et al. (2018). Correlating the expression and functional activity of ABCA4 disease variants with the phenotype of patients with Stargardt disease. Invest. Ophthalmol. Vis. Sci. 59, 2305–2315. doi: 10.1167/iovs.17-23364
Jiang, F., Pan, Z., Xu, K., Tian, L., Xie, Y., Zhang, X., et al. (2016). Screening of ABCA4 gene in a chinese cohort with Stargardt disease or cone-rod dystrophy with a report on 85 novel mutations. Invest. Opthalmol. Vis. Sci. 57:145. doi: 10.1167/iovs.15-18190
Khan, M., Cornelis, S. S., Pozo-Valero, M. D., Whelan, L., Runhart, E. H., Mishra, K., et al. (2020). Resolving the dark matter of ABCA4 for 1054 Stargardt disease probands through integrated genomics and transcriptomics. Genet. Med. 22, 1235–1246. doi: 10.1038/s41436-020-0787-4
Lee, W., Schuerch, K., Zernant, J., Collison, F. T., Bearelly, S., Fishman, G. A., et al. (2017). Genotypic spectrum and phenotype correlations of ABCA4-associated disease in patients of south Asian descent. Eur. J. Hum. Genet. 25, 735–743. doi: 10.1038/ejhg.2017.13
Maugeri, A., Flothmann, K., Hemmrich, N., Ingvast, S., Jorge, P., Paloma, E., et al. (2002). The ABCA4 2588G>C Stargardt mutation: single origin and increasing frequency from South-West to North-East Europe. Eur. J. Hum. Genet. 10, 197–203. doi: 10.1038/sj.ejhg.5200784
Maugeri, A., van Driel, M. A., van de Pol, D. J., Klevering, B. J., van Haren, F. J., Tijmes, N., et al. (1999). The 2588G–>C mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease. Am. J. Hum. Genet. 64, 1024–1035. doi: 10.1086/302323
Midgley, N., Roberts, L., Rebello, G., and Ramesar, R. (2020). The impact of the c.5603A>T hypomorphic variant on founder mutation screening of ABCA4 for Stargardt disease in South Africa. Mol. Vis. 26, 613–622.
Molday, R. S. (2015). Insights into the molecular properties of ABCA4 and its role in the visual cycle and Stargardt disease. Prog. Mol. Biol. Trans. Sci. 134, 415–431. doi: 10.1016/bs.pmbts.2015.06.008
Muzzio, M., Motti, J. M. B., Paz Sepulveda, P. B., Yee, M., Cooke, T., Santos, M. R., et al. (2018). Population structure in Argentina. PLoS One 13:e0196325. doi: 10.1371/journal.pone.0196325
Passerini, I., Sodi, A., Giambene, B., Mariottini, A., Menchini, U., and Torricelli, F. (2010). Novel mutations in the ABCR gene in Italian patients with Stargardt disease. Eye 24, 158–164. doi: 10.1038/eye.2009.35
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Riveiro-Alvarez, R., Lopez-Martinez, M.-A., Zernant, J., Aguirre-Lamban, J., Cantalapiedra, D., Avila-Fernandez, A., et al. (2013). Outcome of ABCA4 disease-associated alleles in autosomal recessive retinal dystrophies: retrospective analysis in 420 Spanish families. Ophthalmology 120, 2332–2337. doi: 10.1016/j.ophtha.2013.04.002
Riveiro-Alvarez, R., Valverde, D., Lorda-Sanchez, I., Trujillo-Tiebas, M. J., Cantalapiedra, D., Vallespin, E., et al. (2007). Partial paternal uniparental disomy (UPD) of chromosome 1 in a patient with Stargardt disease. Mol. Vis. 13, 96–101.
Rivera, A., White, K., Stöhr, H., Steiner, K., Hemmrich, N., Grimm, T., et al. (2000). A comprehensive survey of sequence variation in the ABCA4 (ABCR) Gene in Stargardt disease and age-related macular degeneration. Am. J. Hum. Genet. 67, 800–813. doi: 10.1086/303090
Rosenberg, T., Klie, F., Garred, P., and Schwartz, M. (2007). N965S is a common ABCA4 variant in Stargardt-related retinopathies in the Danish population. Mol. Vis. 13, 1962–1969.
Salles, M. V., Motta, F. L., Dias da Silva, E., Varela, P., Costa, K. A., Filippelli-Silva, R., et al. (2017). Novel complex ABCA4 alleles in brazilian patients with stargardt disease: genotype–phenotype correlation. Invest. Ophthalmol. Vis. Sci. 58, 5723–5730. doi: 10.1167/iovs.17-22398
Salles, M. V., Motta, F. L., Martin, R., Filippelli-Silva, R., Dias da Silva, E., Varela, P., et al. (2018). Variants in the ABCA4 gene in a Brazilian population with Stargardt disease. Mol. Vis. 24, 546–559.
Sandmann, S., de Graaf, A. O., Karimi, M., van der Reijden, B. A., Hellström-Lindberg, E., Jansen, J. H., et al. (2017). Evaluating variant calling tools for non-matched next-generation sequencing data. Sci. Rep. 7:43169. doi: 10.1038/srep43169
Ścieżyńska, A., Oziȩbło, D., Ambroziak, A. M., Korwin, M., Szulborski, K., Krawczyński, M., et al. (2016). Next-generation sequencing of ABCA4 : High frequency of complex alleles and novel mutations in patients with retinal dystrophies from central Europe. Exp. Eye Res. 145, 93–99. doi: 10.1016/j.exer.2015.11.011
Serapinas, D., Obrikytë, V., and Sakalauskas, R. (2013). Stargardt disease caused by a rare combination of double homozygous mutations. Medicina 49:60. doi: 10.3390/medicina49080060
Simonelli, F., Testa, F., Giuseppė de Crecchio, Rinaldi, E., Hutchinson, A., Atkinson, A., et al. (2000). New ABCR mutations and clinical phenotype in italian patients with stargardt disease. Invest. Ophthalmol. Vis. Sci. 41, 892–897.
Testa, F., Rossi, S., Sodi, A., Passerini, I., Di Iorio, V., Corte, M. D., et al. (2012). Correlation between photoreceptor layer integrity and visual function in patients with stargardt disease: implications for gene therapy. Invest. Opthalmol. Vis. Sci. 53:4409. doi: 10.1167/iovs.11-8201
Tracewska, A. M., Kocyła-Karczmarewicz, B., Rafalska, A., Murawska, J., Jakubaszko-Jablonska, J., Rydzanicz, M., et al. (2019). Genetic spectrum of ABCA4-associated retinal degeneration in poland. Genes 10:959. doi: 10.3390/genes10120959
Valverde, D., Riveiro-Alvarez, R., Bernal, S., Jaakson, K., Baiget, M., Navarro, R., et al. (2006). Microarray-based mutation analysis of the ABCA4 gene in spanish patients with Stargardt disease: evidence of a prevalent mutated allele. Mol. Vis. 12, 902–908.
van Huet, R. A. C., Bax, N. M., Westeneng-Van Haaften, S. C., Muhamad, M., Zonneveld-Vrieling, M. N., Hoefsloot, L. H., et al. (2014). Foveal sparing in Stargardt disease. Invest. Ophthalmol. Vis. Sci. 55:7467. doi: 10.1167/iovs.13-13825
Zernant, J., Lee, W., Collison, F. T., Fishman, G. A., Sergeev, Y. V., Schuerch, K., et al. (2017). Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J. Med. Genet. 54, 404–412. doi: 10.1136/jmedgenet-2017-104540
Zernant, J., Schubert, C., Im, K. M., Burke, T., Brown, C. M., Fishman, G. A., et al. (2011). Analysis of the ABCA4 gene by next-generation sequencing. Invest. Opthalmol. Vis. Sci. 52:8479. doi: 10.1167/iovs.11-8182
Zhang, N., Tsybovsky, Y., Kolesnikov, A. V., Rozanowska, M., Swider, M., Schwartz, S. B., et al. (2015). Protein misfolding and the pathogenesis of ABCA4-associated retinal degenerations. Hum. Mol. Genet. 24, 3220–3237. doi: 10.1093/hmg/ddv073
Keywords: Stargardt disease, next-generation sequencing – NGS, Latin America, mutation, ABCA4 gene
Citation: Mena MD, Moresco AA, Vidal SH, Aguilar-Cortes D, Obregon MG, Fandiño AC, Sendoya JM, Llera AS and Podhajcer OL (2021) Clinical and Genetic Spectrum of Stargardt Disease in Argentinean Patients. Front. Genet. 12:646058. doi: 10.3389/fgene.2021.646058
Received: 24 December 2020; Accepted: 05 March 2021;
Published: 26 March 2021.
Edited by:
Nelson L. S. Tang, The Chinese University of Hong Kong, ChinaReviewed by:
Yin-Hsiu Chien, National Taiwan University Hospital, TaiwanAna Westenberger, University of Lübeck, Germany
Vladimir V. Strelnikov, Federal State Budgetary Scientific Institution Research Centre for Medical Genetics (RCMG), Russia
Copyright © 2021 Mena, Moresco, Vidal, Aguilar-Cortes, Obregon, Fandiño, Sendoya, Llera and Podhajcer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Osvaldo L. Podhajcer, b3BvZGhhamNlckBsZWxvaXIub3JnLmFy
†These authors have contributed equally to this work