 Jia-Xuan Liu
Jia-Xuan Liu Qiong Wu
Qiong Wu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
DATA REPORT article
Front. Genet. , 04 March 2021
Sec. Livestock Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.645401
This article is part of the Research Topic Omics-based Analysis on the Interaction Between Microbe and Agricultural Animals View all 17 articles
Several Gram-negative bacteria, including Actinobacillus pleuropneumoniae and Haemophilus parasuis, are responsible for respiratory diseases and cause huge economic losses to the swine industry worldwide. Lipopolysaccharide (LPS) is a cell outer membrane component of Gram-negative bacteria and serves as a major pro-inflammatory stimulus binding to pattern recognition receptor Toll-like receptor 4 (TLR4) (Ciesielska et al., 2020). LPS is ubiquitous in nature and exists in high concentrations in air pollution, soil, and organic dust. Inhalation of LPS is involved in the pathogenesis of lung inflammation (Kaelberer et al., 2020).
Alveolar macrophages (AMs) are the predominant immune cells located at the air-surface interface of alveoli. Resident AMs that arise during embryogenesis and recruited AMs that originate postnatally from circulating monocytes coexist in the inflamed lung. Once infection occurs, AMs move between alveoli to sense and phagocytose inhaled bacteria before they can induce harmful lung inflammation (Neupane et al., 2020). Meanwhile, the Gram-negative bacterial LPS binding to the TLR4 of AMs initiates multiple intracellular signaling pathways and induces the production of some pro-inflammatory cytokines, such as interleukin 1β (IL-1β) (Li et al., 2017). These pro-inflammatory cytokines induce superfluous neutrophil recruitment, leading to continuous lung inflammation and injury. The activation states of AMs are divided into classically activated (M1) and alternatively activated (M2). M1-type AMs generally induced by TLR signaling and interferon-gamma (IFN-γ) secrete pro-inflammatory cytokines, and M2-type AMs generally induced by interleukin-4 (IL-4) are anti-inflammatory and typically express the transforming growth factor-β (TGF-β) (Hussell and Bell, 2014). However, the gene reprogramming and polarization states of macrophages are also affected by stimulation intensity and tissue origin. A meta-analysis of in vitro differentiated macrophages showed that macrophages display distinguishing activation states even after early (2–4 h) or late (18–24 h) LPS infection (Chen et al., 2019). In M1-type AMs, increased levels of reactive oxygen species, such as hydrogen peroxide, superoxide, and hydroxyl, are implicated in DNA damage and membrane dysfunction (Riazanski et al., 2020). Therefore, the cellular antioxidant capacity of AMs is indispensable for controlling the homeostasis of intracellular oxidative stress and maintaining immune defense.
Selenium (Se) is considered as a functional element of thioredoxin reductase, glutathione peroxidase, and other Se-containing enzymes and protects against oxidative injury (Silvestrini et al., 2020). LPS infection impairs Se metabolism and leads to dysregulation of selenoprotein expression in the spleen, thymus, and lymph node of pigs (Sun et al., 2017). An animal study using a chicken model of Se deficiency has demonstrated the negative correlation between Se deficiency and inflammation-related gene expression in skeletal muscles (Wu et al., 2014). Se supplementation can attenuate inflammatory response and lung injury induced by a variety of stimuli, including virus (Liu et al., 2015), bacteria (Xu et al., 2020), and heavy metal (Ghorbel et al., 2017). It was also reported that supplementation of Se to macrophages ameliorates the pro-inflammatory response induced by LPS (Vunta et al., 2008). However, the potential molecular mechanism of the anti-inflammatory function of Se is still unclear. Transcriptome sequencing is proven to be a powerful tool to comprehensively view the immune response of porcine AMs (PAMs) to bacterial or viral infection (Kim et al., 2019; Park et al., 2020). In this study, we performed transcriptome sequencing to deepen the understanding of the mechanism of Se protecting PAMs against LPS infection.
The porcine lung alveolar macrophage cell line 3D4/31 (ATCC CRL-2844) was cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% heat-inactivated fetal calf serum, 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 1 mM of sodium pyruvate. Confluent cell monolayers were treated under three different conditions: (i) RPMI 1640 medium alone (CON group), (ii) LPS from Escherichia coli O111:B4 (1 μg/ml, 3 ml) infection alone (LPS group), and (iii) pretreatment with Se as sodium selenite containing 0.1 μM for 6 h followed by LPS infection (1 μg/ml) (SeL group).
At 12 h after LPS infection, total RNA was extracted, and the RNA integrity number was further assessed using an RNA 6000 Nano kit (Agilent Technologies, Santa Clara, CA, USA). PCR amplification was performed to obtain the final libraries. The constructed library was quantified and pooled in the flow cell. After cBot clustering, the RNA-seq libraries were sequenced using Illumina high-throughput sequencing Novaseq 6000 platform, with a paired-end read length of 150 base pairs (bp).
The clean reads were mapped to the pig reference genome Sscrofa11 using TopHat v2.1.1. The mapped reads were assembled into transcripts using StringTie v1.3.3b. The genes were annotated by BLAST based on the Cluster of Orthologous Groups of proteins (COG), Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases.
The expression of genes was calculated and normalized to fragments per kilobases per million reads (FPKM) using RSEM v1.3.1. Differentially Expressed Genes (DEGs) were identified using DESeq2 v1.24.0. The p-value was adjusted using Benjamini and Hochberg's (BH) approach for controlling the false discovery rate. Genes with an adjusted p-value < 0.05 and fold change (FC) > 1.5 were assigned as DEGs.
GO enrichment analysis based on Fisher's exact test was carried out to specify the potential roles of DEGs using Goatools v0.6.5. The p-value was adjusted by BH, and GO terms with adjusted p-value < 0.05 were considered significantly enriched. KEGG enrichment analysis was performed to evaluate significantly enriched signal transduction or metabolic pathways using KOBAS v2.1.1. A Venn diagram was generated using the R package Venndiagram. The protein–protein interaction (PPI) analysis of DEGs was based on the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database v11.0, and the minimum STRING score was set at 1,000. The interaction with a combined score >0.4 was considered to be significant. The protein network was visualized using NetworkX.
The raw transcriptome data have been deposited in the US National Center for Biotechnology Information Sequence Read Archive database under accession no. SRR13277478–SRR13277486.
A raw dataset consisting of 487.3 million reads (~73.6 Gbps) was yielded. After filtering low-quality reads, adaptor or ambiguous sequences, and removal of contamination, 51.9 (98.73%, the percentage of clean reads), 57.1 (98.81%), and 51.9 (98.77%) million clean reads from the CON groups, 52 (98.82%), 49.2 (98.88%), and 58.3 (99.18%) million clean reads from the LPS group, and 51.2 (99.21%), 56.3 (99.24%), and 54.7 (99.29%) million clean reads from the SeL group were retained. The average of Q30 of clean reads was >94.64%, indicating that the obtained clean reads were of high quality (Supplementary Table 1). The saturation curve of sequencing showed that the FPKM values of ~22.18% of genes from the CON, LPS, and SeL groups were expressed between 0.3 and 3.5, and that only a few of 6.53% of genes were highly expressed with an FPKM value >60. Most genes with medium or above expression level (i.e., the genes with FPKM value >3.5) were nearly saturated at 40% of the sequencing reads (ordinate value tended to 1), indicating that the sequencing quantity can cover most of the expressed genes (Supplementary Table 2; Supplementary Figure 1).
A total of 27,576 genes were found across all samples, including 25,880 (93.85%) annotated genes and 1,696 (6.15%) unannotated novel genes. Among 63,606 transcripts identified, there were 14,158 novel transcripts, including 538 transcripts with exonic overlap with reference on the opposite strand, 666 transcripts with transfrag falling entirely within a reference intron, 10,491 transcripts with potentially novel isoform: at least one splice junction was shared with a reference transcript, 2,058 unknown transcripts, and intergenic transcripts, and 405 transcripts with generic exonic overlap with a reference transcript (Supplementary Figure 2).
A total of 964 (57.72%) novel transcripts and 12,473 (90.04%) novel genes were successfully annotated by BLAST, with 437 transcripts and 7,437 genes in the GO database, 91 and 7,664 in the KEGG database, 331 and 11,080 in the COG database, 955 and 12,451 in the NR database, 366 and 11,199 in the Swiss-Prot database, and 192 and 10,015 in the Pfam database, respectively (Supplementary Table 3). According to GO analysis, catalytic activity (259 genes) in molecular function, membrane (190 genes) and membrane part (186 genes) in the cellular component, and cellular process (122 genes) in the biological process were the most enriched ontology terms (Supplementary Figure 3A). A total of 91 novel genes were classified into 110 KEGG pathways involving 32 KEGG functional categories, mainly functioning in signal transduction, endocrine system, immune system, digestive system, translation, and environmental adaptation (Supplementary Figure 3B). The COG analysis showed that 37 novel genes were assigned into 13 COG functional categories (Supplementary Figure 3C), mainly including “intracellular trafficking, secretion, and vesicular transport” (Class U; 12 genes), “posttranslational modification, protein turnover, chaperones” (Class O; 9 genes), and “chromatin structure and dynamics” (Class B; 8 genes).
In the CON relative to the LPS group (CON_LPS), a total of 223 DEGs, including 28 up-regulated and 195 down-regulated DEGs, were identified (Supplementary Figure 4A; Supplementary Table 4). The top 10 known up-regulated genes were RF00030, CTF1, CCDC103, STMN3, WIPI1, RELB, PHLDA1, FLT3, CHCHD10, EXOSC6, HSD17B10, OTUD1, PPP1R14B, YRDC, GMIP, SCLY, CEBPB, and SESN2. The top 10 down-regulated genes were VMAC, ECM2, TMOD1, C17orf78, IGSF6, COCH, NAALADL1, MILR1, GSDMC, and SV2A. Out of 58 identified DEGs in LPS relative to the SeL group (LPS_SeL), 25 DEGs were up-regulated, and 33 DEGs were down-regulated (Supplementary Figure 4B; Supplementary Table 5). Se treatment induced the expression of anti-apoptosis protein BCL-2 and antioxidant defense-related glutathione peroxidase 1 (GPX1) and selenoprotein H and P (SELENOP). Se acts as a rare amino acid selenocysteine through incorporation into selenoproteins. It was reported that Se supplementation protects against apoptosis induced by reactive oxygen species or toxic heavy metal lead in a BCL-2-dependent manner (Khera et al., 2017; Wang et al., 2018). In CON relative to the SeL group (CON_SeL), out of 252 identified DEGs, 27 DEGs were up-regulated, and 225 DEGs were down-regulated (Supplementary Figure 4C; Supplementary Table 6).
KEGG enrichment analysis of DEGs was performed. The DEGs in the CON_LPS group were enriched in the IL-17 signaling pathway, tumor necrosis factor (TNF) signaling pathway, cytokine–cytokine receptor interaction, lysine degradation, and graft-versus-host disease (Figure 1A). The “TGF-β signaling pathway” possessed the highest rich factor in the up-regulated DEGs in the CON_LPS group. TGF-β could skew LPS-stimulated M1-type macrophage polarization toward the M2 phenotype via the Akt/FoxO1 pathway and reduce inflammatory reactions in sepsis (Liu et al., 2019). In a previous study, IFN-γ at a concentration of 50 ng/ml and LPS at a concentration of 100 ng/ml classically induce M1 activation of PAMs, accompanied by enriched TNF pathway and down-regulated TGF-β signaling pathway (Liu et al., 2018). LPS stimulation intensity could significantly affect the gene expression profile and polarization state of macrophages. Compared with short exposure (2–4 h) to LPS, short exposure (18–24 h) to LPS increases the expression of M2-related genes, including the tyrosine protein kinase MER and arginase in macrophages (Chen et al., 2019). Further study is needed to explore the regulatory effect of a high concentration of LPS on the polarization state of PAMs. Among the down-regulated DEGs in the CON_LPS group, the enriched KEGG pathways were related to “graft-versus-host disease,” followed by “endocrine resistance” and “IL-17 signaling pathway,” and “cytokine–cytokine receptor interaction” had the most DEGs. Consistent with the transcriptome analysis of PAMs activated by LPS, down-regulated genes involving cytokine–cytokine receptor interaction suggested their important role in cellular activation (Liu et al., 2018). In contrary to this study, co-infection of Mycoplasma gallisepticum and E. coli leads to inflammatory damage of chicken lung involving the enriched IL-17 signaling pathway (Wu et al., 2019). Both genes encoding matrix metallopeptidase 9 (MMP9, Log2FC = −1.44) and CCAAT enhancer binding protein beta (CEBPβ, Log2FC = 0.60) were involved in the IL-17 signaling pathway and TNF signaling pathway (Supplementary Figure 5A). The DEGs in the LPS_SeL group were highly related to categories including protein digestion and absorption, AGE–RAGE signaling pathway in diabetic complications, TGF-β signaling pathway, thyroid hormone synthesis, relaxin signaling pathway, amoebiasis, glutathione metabolism, PI3K–Akt signaling pathway, and arachidonic acid metabolism (Figure 1A). The “tryptophan metabolism” and “phototransduction” pathway possessed the highest rich factor in the up-regulated and down-regulated DEGs in the LPS_SL group, respectively. LPS and IFN-γ-stimulated RAW264.7 macrophages cultured in tryptophan-deficient medium exhibit a significant reduction in iNOS expression involved in pathogen killing (Poormasjedi-Meibod et al., 2013). The expression of proteins involved in tryptophan metabolism indoleamine 2,3-dioxygenase and kynurenic acid is activated in pig bone marrow-derived macrophages infected with LPS (Kapetanovic et al., 2012). The metabolomic analysis also showed that LPS stimulation reprograms metabolomic profiling of the human M1-type AMs and induces tryptophan degradation in tryptophan metabolism (Fall et al., 2020).
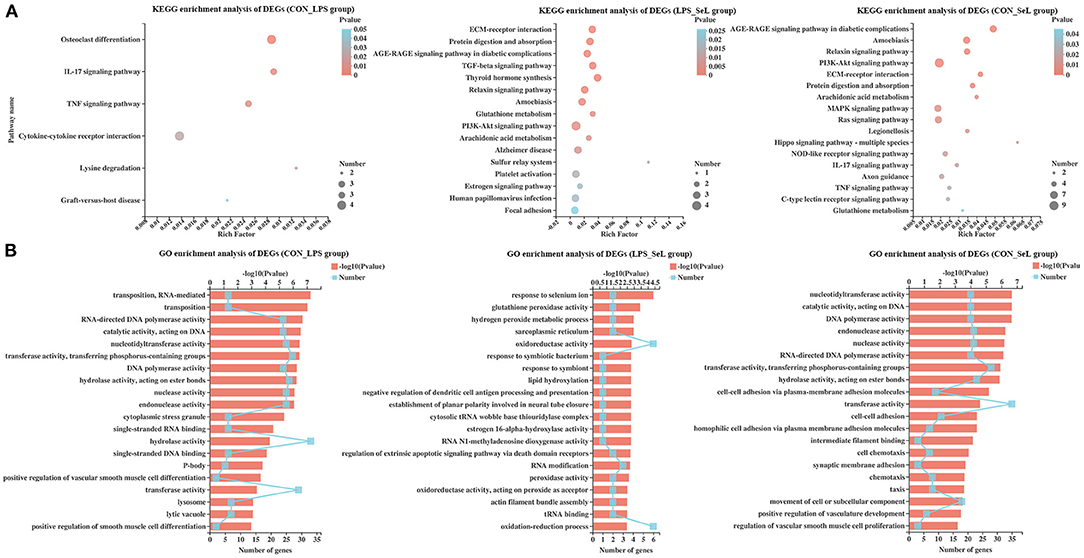
Figure 1. KEGG and GO enrichment analysis of DEGs. Enriched bubble chart showing the enrichment of the KEGG pathway in the CON_LPS, LPS_SeL, and CON-SeL groups (A). X-axis represented the enrichment ratio, and Y-axis represented the top 20 KEGG pathways. Number: bubble size represented the number of genes annotated to a KEGG pathway. Pvalue: color indicated the enriched p-value. Enriched bar chart showing the enrichment of the GO pathway in the CON_LPS, LPS_SeL, and CON-SeL groups (B). The length of the X-axis column represents the p-value. The value of the box on the fold line above X is the number of DEGs annotated to the GO terms.
Compared with the LPS challenge, Se treatment up-regulated the expression of GPX1 (Log2FC = 2.12) expression and down-regulated the expression of GPX2 (Log2FC = −0.72), which participated in thyroid hormone synthesis, glutathione metabolism, and arachidonic acid metabolism (Supplementary Figure 5B). The presence of prostaglandin E2 (PGE2), a main arachidonic acid derivative, is necessary to the LPS-induced production of the pro-inflammatory cytokine IL-1β (Zaslona et al., 2017). Up-regulated thrombospondin 1 (THBS1, Log2FC = 0.64) was involved in the TGF-β signaling pathway and PI3K–Akt signaling pathway. The enriched KEGG pathways for the CON_SeL group, out of 252 DEGs, were very similar to the CON_LPS group, but ranked differently: amoebiasis, relaxin signaling pathway, PI3K–Akt signaling pathway, extracellular matrix (ECM)–receptor interaction, protein digestion and absorption, arachidonic acid metabolism, MAPK signaling pathway, Ras signaling pathway, and Legionellosis, which had many genes in common, including up-regulated genes [GPX1, TLR4, nuclear factor kappa-B (NF-κB) subunit RELβ, chemokine C-X-C motif ligand 2 (CXCL2), platelet-derived growth factor subunit B (PDGFB), GRB2-associated binding protein 1 (GAB1), etc.] and down-regulated genes [phospholipase A2 group 1β (PLA2G1β), MMP9, muscle RAS (MRAS), ephrin A2 (EFNA2), GPX2, etc.] (Figure 1A; Supplementary Figure 5C). The main limitation of this study was that the Se alone treatment group was not included, which was limited to assess the effect of Se treatment on the gene expression profile of PAMs. Se supplementation was proven to attenuate the levels of oxidative stress and pro-inflammatory gene expression in macrophages (Vunta et al., 2008; Ghorbel et al., 2017). A comprehensive gene expression profile of Se-treated PAMs is needed in further study.
GO enrichment analysis was performed. Among the DEGs in the CON_LPS group, RNA-mediated transposition and transposition classified into biological process class occupied the strongest enrichment degree. The molecular function class was the most abundant function groups, mainly including some enzyme activity, such as polymerase activity, catalytic activity, nucleotidyltransferase activity, transferase activity, hydrolase activity, nuclease activity, and endonuclease activity. The cytoplasmic stress granule was the main type in cellular component (Figure 1B). The presence of CEBPB and CD28 was directly related to the positive regulation of IL-4 production (Supplementary Figure 6A). The DEGs in the LPS_SeL group were predicted to be involved in the response to Se ion, followed by a response to the symbiotic bacterium, regulation of extrinsic apoptotic signaling pathway via death domain receptors, and some oxidative stress-related functional terms, including hydrogen peroxide metabolic process, glutathione peroxidase activity, oxidation–reduction process, oxidoreductase activity, peroxidase activity, and oxidoreductase activity acting on peroxide as acceptor (Figure 1B). Up-regulated GPX1 (Log2FC = 2.12) and SELENOP (Log2FC = 0.61) were related to the response to Se ion (Supplementary Figure 6B). Cytochrome P450 1A1 (CYP1A1, Log2FC = 2.47), GPX1, GPX2 (Log2FC = −0.72), and hydroxysteroid 17-beta dehydrogenase 10 (HSD17B10, Log2FC = −0.69) were related to oxidative stress-related function. THBS1 (Log2FC = 0.64) was related to the regulation of extrinsic apoptotic signaling pathway via death domain receptors. The DEGs in the CON_SeL group were found to be involved in the significant enrichment of GO biological terms including enzyme activity and cell adhesion function (Figure 1B; Supplementary Figure 6C).
According to the Venn analysis, a total of 113, 34, and 138 genes were specifically expressed in the CON_LPS, LPS_SeL, and CON_SeL groups, respectively (Figure 2A). A total of 14 and 100 genes were shared by the LPS_SeL and CON_SeL groups and the CON_LPS and CON_SeL groups, respectively. Moreover, only 10 DEGs were shared by the CON_LPS and LPS_SeL groups. The KEGG enrichment analysis showed that the 10 DEGs shared by the CON_LPS and LPS_SeL groups were enriched in Alzheimer disease; valine, leucine, and isoleucine degradation; cardiac muscle contraction; TGF-β signaling pathway; and Jak–STAT signaling pathway (Figure 2B). Among the top 20 GO terms identified by enrichment analysis based on 10 DEGs, the first three topmost enriched were regulation of spongiotrophoblast cell proliferation, regulation of cell proliferation involving embryonic placenta development, and regulation of growth hormone activity (Figure 2C). The other GO terms were involved in mitochondrial RNA processing and modification.
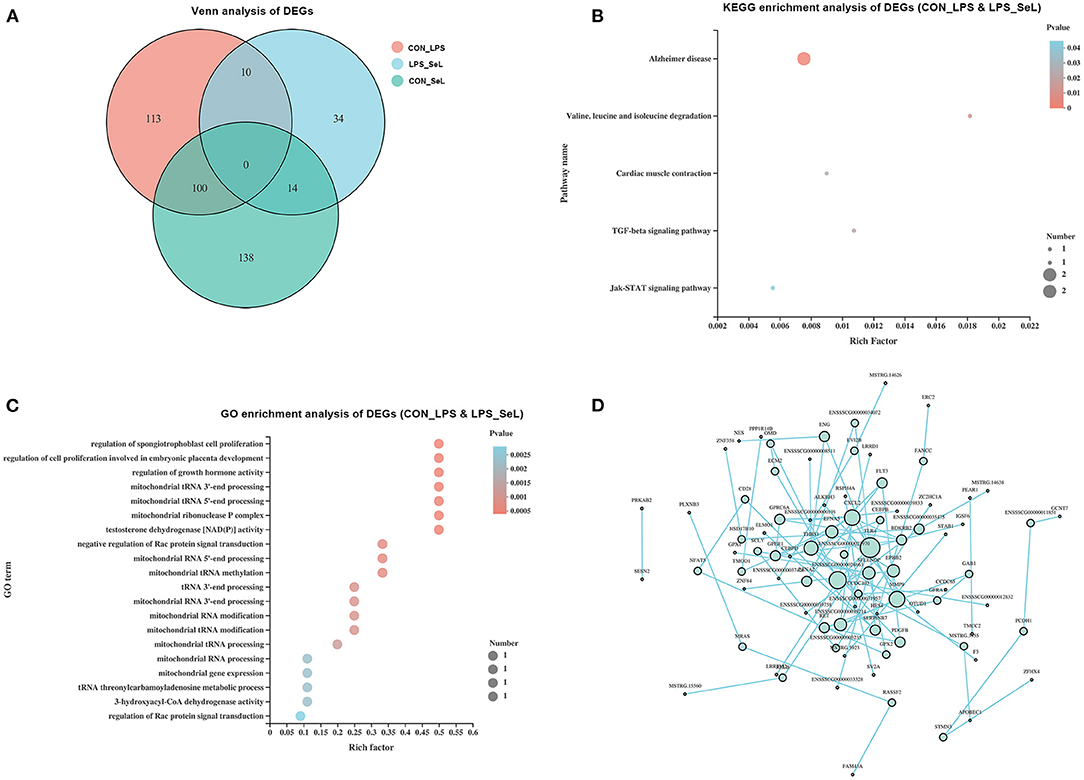
Figure 2. Functional annotation of DEGs affected by both Se treatment and LPS infection. Venn diagram presenting co-expressed and uniquely expressed DEGs in the CON_LPS, LPS_SeL, and CON_SeL groups (A). The KEGG (B) and GO (C) enrichment analysis of DEGs affected by both Se treatment and LPS infection was indicated. PPI was performed to generate interaction analysis (D). Node color from white to green represents the lowest to highest betweenness centrality. The size of each node corresponded to the number of connections (degree).
The main protein interaction cluster derived from 409 DEGs contained 84 nodes, each representing 1 protein and connected by 84 edges (Figure 2D). TLR4, CXCL2, MMP9, and THBS1, followed by SELENOP, had the highest scores for betweenness centrality, indicating that they accounted for many direct and indirect interactions within the network of PPI. TLR4, MMP9, CXCL2, and THBS1 had the highest number of direct connections (degree). TLR4 and CXCL2 represented the immune response of M1-type PAMs to LPS infection (Herrera-Uribe et al., 2020).
In this study, most DEGs in PAMs infected with LPS compared with the control group are enriched in the IL-17 signaling pathway, TNF signaling pathway, and cytokine–cytokine receptor interaction. LPS promotes the early activation of TLR4 and CXCL2. Se treatment enhances the antioxidant and anti-inflammatory responses to LPS through integrating GPX1, GPX2, SELENOP, CYP1A1, HSD17B10, and THBS1 genes. These findings provide an important view of the mechanism of Se protecting the host against infection. The study also suggests that dietary Se supply to pigs may help prevent respiratory infection.
The raw transcriptome data have been deposited in the US National Center for Biotechnology Information Sequence Read Archive database under the accession no. SRR13277478–SRR13277486.
QW and J-XL: conceived and designed the experiments and prepared the manuscript. J-XL, X-YC, PC, Y-DW, T-JS, ML, and R-YX: Performed the RNA extraction. QW, J-XL, X-YC, and PC: Performed the analysis of data. All authors contributed to the article and approved the submitted version.
This work was supported financially by the National Key R&D Program of China (Project no. 2017YFD0502200), Undergraduate Scientific Research Project of Cross Training Program of High Level Talents in Beijing Universities (Project no. PXM2020_014207_000009), Research Project for Youth Teachers of Beijing University of Agriculture (Project no. SXQN201904), General Projects of Beijing Municipal Education Commission Science and Technology Plan (Project no. KM202010020009), and Undergraduate Scientific Research Training Project of BUA (Project no. kx2019008).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.645401/full#supplementary-material
Supplementary Figure 1. Saturation curve of sequencing of each sample. Each color line represented the saturation curve of gene expression at different expression levels in the sample.
Supplementary Figure 2. Classification of new transcripts. New transcripts were classified according to the overlapping relationship between spliced transcripts and known transcripts. The percentage of new transcripts were shown.
Supplementary Figure 3. Annotation analysis of identified novel genes. The novel genes were annotated using based on GO (A), KEGG (B), and COG (C) databases.
Supplementary Figure 4. DEGs in PAMs in response to Se treatment or LPS infection. Volcano plots displaying DEGs in the CON_LPS (A), CON-SeL (B), and LPS_SeL (C) groups. The longitudinal dashed lines indicated an expression level of |FC| ≥ 1.5. The horizontal dashed lines indicated an expression level with a p-value < 0.05. Blue dots (up) represented significantly up-regulated genes; gray dots (down) represented significantly down-regulated genes; red dots (no significance) represented insignificantly DEGs.
Supplementary Figure 5. KEGGChord plot of top 20 ranked KEGG terms. Chords indicated a detailed relationship between the expression levels of DEGs (left semicircle perimeter) in the CON_LPS (A), LPS_SeL (B), and CON-SeL (C) groups and their enriched KEGG pathways (right semicircle perimeter). The genes were linked to their annotated KEGG terms via colored ribbons. Genes were ordered according to log2FC.
Supplementary Figure 6. GOChord plot of top 20 ranked GO terms. Chords indicated a detailed relationship between the expression levels of DEGs (left semicircle perimeter) in the CON_LPS (A), LPS_SeL (B), and CON-SeL (C) groups and their enriched KEGG pathways (right semicircle perimeter). The genes are linked to their annotated terms via colored ribbons.
Chen, H. J., Li Yim, A. Y. F., Griffith, G. R., de Jonge, W. J., Mannens, M. M. A. M., Ferrero, E., et al. (2019). Meta-analysis of in vitro-differentiated macrophages identifies transcriptomic signatures that classify disease macrophages in vivo. Front. Immunol. 10:2887. doi: 10.3389/fimmu.2019.02887
Ciesielska, A., Matyjek, M., and Kwiatkowska, K. (2020). TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell. Mol. Life Sci. 78, 1233–1261. doi: 10.1007/s00018-020-03656-y
Fall, F., Lamy, E., Brollo, M., Naline, E., Lenuzza, N., Thévenot, E., et al. (2020). Metabolic reprograming of LPS-stimulated human lung macrophages involves tryptophan metabolism and the aspartate-arginosuccinate shunt. PLoS ONE 15:e0230813. doi: 10.1371/journal.pone.0230813
Ghorbel, I., Elwej, A., Chaabane, M., Jamoussi, K., Mnif, H., Boudawara, T., et al. (2017). Selenium alleviates oxidative stress and lung damage induced by aluminum chloride in adult rats: biochemical and histological approach. Biol. Trace Elem. Res. 176, 181–191. doi: 10.1007/s12011-016-0818-9
Herrera-Uribe, J., Liu, H., Byrne, K. A., Bond, Z. F., Loving, C. L., and Tuggle, C.K. (2020). Changes in H3K27ac at gene regulatory regions in porcine alveolar macrophages following LPS or PolyIC exposure. Front. Genet. 11:817. doi: 10.3389/fgene.2020.00817
Hussell, T., and Bell, T. J. (2014). Alveolar macrophages: plasticity in a tissue-specific context. Nat. Rev. Immunol. 14, 81–93. doi: 10.1038/nri3600
Kaelberer, M., Caceres, A., and Jordt, S. E. (2020). Activation of a nerve injury transcriptional signature in airway-innervating sensory neurons after lipopolysaccharide-induced lung inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 318, L953–L964. doi: 10.1152/ajplung.00403.2019
Kapetanovic, R., Fairbairn, L., Beraldi, D., Sester, D. P., Archibald, A. L., Tuggle, C. K., et al. (2012). Pig bone marrow-derived macrophages resemble human macrophages in their response to bacterial lipopolysaccharide. J. Immunol. 188, 3382–3394. doi: 10.4049/jimmunol.1102649
Khera, A., Vanderlelie, J. J., Holland, O., and Perkins, A. V. (2017). Overexpression of endogenous anti-oxidants with selenium supplementation protects trophoblast cells from reactive oxygen species-induced apoptosis in a Bcl-2-dependent manner. Biol. Trace Elem. Res. 177, 394–403. doi: 10.1007/s12011-016-0870-5
Kim, S., Oh, M. W., Bin, P. W., and Yoo, H. S. (2019). Global gene networks in 3D4/31 porcine alveolar macrophages treated with antigenic epitopes of Actinobacillus pleuropneumoniae ApxIA, IIA, and IVA. Sci. Rep. 9:5269. doi: 10.1038/s41598-019-41748-3
Li, B., Fang, J., Zuo, Z., Yin, S., He, T., Yang, M., et al. (2017). Activation of porcine alveolar macrophages by Actinobacillus pleuropneumoniae lipopolysaccharide via the Toll-like Receptor 4/NF-κB-mediated pathway. Infect. Immun. 86, e00642–e00617. doi: 10.1128/IAI.00642-17
Liu, F., Qiu, H., Xue, M., Zhang, S., Zhang, X., Xu, J., et al. (2019). MSC-secreted TGF-β regulates lipopolysaccharide-stimulated macrophage M2-like polarization via the Akt/FoxO1 pathway. Stem Cell Res. Ther. 10:345. doi: 10.1186/s13287-019-1447-y
Liu, G., Yang, G., Guan, G. P., Zhang, Y. Z., Ren, W. K., Yin, J., et al. (2015). Effect of dietary selenium yeast supplementation on porcine circovirus type 2 (PCV2) infections in mice. PLoS ONE 10:e0115833. doi: 10.1371/journal.pone.0115833
Liu, Q., Zhang, Y. L., Hu, W., Hu, S. P., Zhang, Z., Cai, X. H., et al. (2018). Transcriptome of porcine alveolar macrophages activated by interferon-gamma and lipopolysaccharide. Biochem. Biophys. Res. Commun. 503, 2666–2672. doi: 10.1016/j.bbrc.2018.08.021
Neupane, A. S., Willson, M., Chojnacki, A. K., Vargas E Silva Castanheira, F., Morehouse, C., Carestia, A., et al. (2020). Patrolling alveolar macrophages conceal bacteria from the immune system to maintain homeostasis. Cell 183, 110–125. doi: 10.1016/j.cell.2020.08.020
Park, I. B., Choi, Y. C., Lee, K. T., and Chun, T. (2020). Transcriptome analysis of pig macrophages expressing porcine reproductive and respiratory syndrome virus non-structural protein 1. Vet. Immunol. Immunopathol. 231:110147. doi: 10.1016/j.vetimm.2020.110147
Poormasjedi-Meibod, M. S., Jalili, R. B., Hosseini-Tabatabaei, A., Hartwell, R., and Ghahary, A. (2013). Immuno-regulatory function of indoleamine 2,3 dioxygenase through modulation of innate immune responses. PLoS ONE 8:e71044. doi: 10.1371/journal.pone.0071044
Riazanski, V., Sui, Z., and Nelson, D. J. (2020). Kinetic separation of oxidative and non-oxidative metabolism in single phagosomes from alveolar macrophages: impact on bacterial killing. iScience 23:101759. doi: 10.1016/j.isci.2020.101759
Silvestrini, A., Mordente, A., Martino, G., Bruno, C., Vergani, E., Meucci, E., et al. (2020). The role of selenium in oxidative stress and in nonthyroidal illness syndrome (NTIS): an overview. Curr. Med. Chem. 27, 423–449. doi: 10.2174/0929867325666180201111159
Sun, L. H., Pi, D. A., Zhao, L., Wang, X. Y., Zhu, L. Y., Qi, D. S., et al. (2017). Response of selenium and selenogenome in immune tissues to LPS-induced inflammatory reactions in pigs. Biol. Trace Elem. Res. 177, 90–96. doi: 10.1007/s12011-016-0863-4
Vunta, H., Belda, B. J., Arner, R. J., Reddy, C. C., Heuvel, J. P. V., and Prabhu, K. S. (2008). Selenium attenuates pro-inflammatory gene expression in macrophages. Mol. Nutr. Food Res. 52, 1316–1323. doi: 10.1002/mnfr.200700346
Wang, X., An, Y., Jiao, W., Zhang, Z., Han, H., Gu, X., et al. (2018). Selenium protects against lead-induced apoptosis via endoplasmic reticulum stress in chicken kidneys. Biol. Trace Elem. Res. 182, 354–363. doi: 10.1007/s12011-017-1097-9
Wu, Q., Yao, H. D., Tan, S. R., Zhang, Z. W., Zhu, Y. H., and Xu, S. (2014). Possible correlation of selenoprotein W with inflammation factors in chicken skeletal muscles. Biol. Trace Elem. Res. 161, 167–172. doi: 10.1007/s12011-014-0092-7
Wu, Z. Y., Ding, L. J., Bao, J. X., Liu, Y. H., Zhang, Q. M., Wang, J., et al. (2019). Co-infection of Mycoplasma gallisepticum and Escherichia coli triggers inflammatory injury involving the IL-17 signaling pathway. Front. Microbiol. 10:2615. doi: 10.3389/fmicb.2019.02615
Xu, J. Y., Jia, W., Hu, C. X., Nie, M., Ming, J. J., Cheng, Q., et al. (2020). Selenium as a potential fungicide could protect oilseed rape leaves from Sclerotinia sclerotiorum infection. Environ. Pollut. 257:113495. doi: 10.1016/j.envpol.2019.113495
Keywords: transcriptome (RNA-seq), selenium, porcine alveolar macrophage, lipopolysaccharide, differentially expressed genes
Citation: Liu J-X, Chao X-Y, Chen P, Wang Y-D, Su T-J, Li M, Xu R-Y and Wu Q (2021) Transcriptome Analysis of Selenium-Treated Porcine Alveolar Macrophages Against Lipopolysaccharide Infection. Front. Genet. 12:645401. doi: 10.3389/fgene.2021.645401
Received: 23 December 2020; Accepted: 02 February 2021;
Published: 04 March 2021.
Edited by:
Jing Wang, Beijing Academy of Agriculture and Forestry Sciences, ChinaReviewed by:
Haoyu Liu, Uppsala University, SwedenCopyright © 2021 Liu, Chao, Chen, Wang, Su, Li, Xu and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiong Wu, d3VxaW9uZ2J1YUAxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.