
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 07 May 2021
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.643452
 Yuqi Shen1†
Yuqi Shen1† Shi Shu1,2†
Shi Shu1,2† Yaqiong Ren1†Weibo Xia3Jianhua Chen2
Yaqiong Ren1†Weibo Xia3Jianhua Chen2 Liling Dong2Haijun Ge1
Liling Dong2Haijun Ge1 Shiqi Fan1
Shiqi Fan1 Lei Shi1,4*
Lei Shi1,4* Bin Peng2*
Bin Peng2* Xue Zhang1,4
Xue Zhang1,4Primary familial brain calcification (PFBC, OMIM#213600), also known as Fahr's disease, is characterized by bilateral and symmetric brain calcification in the basal ganglia (globus pallidus, caudate nucleus, and putamen), thalamus, subcortical white matter, and cerebellum. PFBC can be caused by loss-of-function mutations in any of the six known causative genes. The most common clinical manifestations include movement disorders, cognitive impairment, and neuropsychiatric signs that gradually emerge in middle-aged patients. To broaden the PFBC mutation spectrum, we examined nine members of a family with PFBC and two sporadic cases from clinical departments, and sequenced all PFBC-causative genes in the index case. Two novel frameshift mutations in SLC20A2 [NM_001257180.2; c.806delC, p.(Pro269Glnfs*49) and c.1154delG, p.(Ser385Ilefs*70)] and one novel splice donor site mutation (NM_002608.4, c.456+1G>C, r.436_456del) in PDGFB were identified in the patient cohort. c.806delC co-segregated with brain calcification and led to SLC20A2 haploinsufficiency among the affected family members. The c.456+1G>C mutation in PDGFB resulted in aberrant mRNA splicing, thereby forming mature transcripts containing an in-frame 21 base pair (bp) deletion, which might create a stably truncated protein [p.(Val146_Gln152del)] and exert a dominant negative effect on wild-type PDGFB. All three mutations were located in highly conserved regions among multiple species and predicted to be pathogenic, as evaluated by at least eight common genetic variation scoring systems. This study identified three novel mutations in SLC20A2 and PDGFB, which broadened and enriched the PFBC mutation spectrum.
Primary familial brain calcification (PFBC, OMIM #213600) is a rare neurodegenerative disorder characterized by vascular calcification affecting multiple brain regions, particularly the basal ganglia, thalamus, subcortical white matter, and cerebellum (Nicolas et al., 2013a, 2015; Tadic et al., 2015; Batla et al., 2017). Brain calcification first appears in the globus pallidus, caudate nucleus, and putamen, and progressively affects the thalamus, hypothalamus, subcortical white matter, cerebral cortex, and dentate gyrus of the cerebellum (Kimura et al., 2016; Paucar et al., 2016, 2017). Clinical symptoms include cognitive impairment, psychiatric symptoms, and movement disorders (Nicolas et al., 2013a, 2015; Grangeon et al., 2019). Most patients have no obvious neurological manifestations before their middle age; however, ~60–70% of patients may exhibit progressive motor coordination dysfunction and neuropsychiatric signs, such as tremor paralysis, dystonia, ataxia, dementia, aphasia, mental confusion, and chronic headache, after the age of 40 years (Donzuso et al., 2019; Grangeon et al., 2019; Westenberger et al., 2019).
Genetically, loss-of-function mutations in SLC20A2 cause familial and sporadic cases of PFBC (Hsu et al., 2013; Lemos et al., 2015). SLC20A2 mutations can impair the inward transport of phosphate through loss-of-function (Wang et al., 2012), dominant negative effects (Kimura et al., 2016; Larsen et al., 2017), or haploinsufficiency mechanisms (Zhang et al., 2013). In addition, PIT-1/SLC20A1 (Inden et al., 2016) and XPR1 (Giovannini et al., 2013; Legati et al., 2015) are important for regulating phosphate homeostasis in the brain. PIT-2 and PIT-1 proteins belong to the type III sodium-dependent phosphate co-transporter family that mediates phosphate influx (Li et al., 2006; Crouthamel et al., 2013), whereas XPR1 is the only known transporter for phosphate efflux (Legati et al., 2015). PDGFRB (Nicolas et al., 2013b), PDGFB (Keller et al., 2013), MYORG (Yao et al., 2018), and JAM2 (Cen et al., 2020; Schottlaender et al., 2020) are four known PFBC-causative genes, all of which may help maintain the structural integrity of the neurovascular unit (NVU) and regulate the permeability of the blood-brain barrier (BBB) (Westenberger et al., 2019; Zarb et al., 2019a).
To date, SLC20A2 and PDGFB are most frequently involved in familial cases of autosomal dominant (AD) PFBC (Batla et al., 2017; Donzuso et al., 2019), whereas the presence of bi-allelic mutations in MYORG is the major cause of recessive PFBC (Bauer et al., 2019). This study aimed to identify novel pathogenic mutations in six known PFBC-causative genes and to provide new insights into the clinical diagnosis of PFBC.
One Chinese family and two Chinese sporadic cases of PFBC were recruited from the Peking Union Medical College Hospital (PUMCH). The eligibility criteria for patient enrollment were: (1) bilateral and symmetrical basal ganglia calcification; and (2) PFBC-related neurological symptoms. Exclusion criteria were: (1) individuals with blood biochemical disorders related to calcium, phosphate, alkaline phosphatase (ALP), or parathyroid hormone (PTH) metabolism; (2) traumatic brain injuries; (3) parasitic or viral infections; and (4) physiological and senile calcification. Written informed consent was obtained from each participant, and ethical approval was obtained from the Institutional Ethics Committee of Peking Union Medical College, Chinese Academy of Medical Sciences (CAMS&PUMC). Subsequently, the recruited patients underwent systemic physical, neurological, and blood biochemical examinations. Brain computed tomography (CT) or magnetic resonance imaging (MRI) scans were routinely adopted as part of the diagnostic workup for evidence of brain calcification and other intracranial abnormalities. All clinical examinations and diagnoses were carefully evaluated and revised by the relevant experts.
A three-generation HB-PFBC family was recruited from the Department of Endocrinology, PUMCH (Figure 1A). The regions of brain calcification were revealed using intracranial CT scanning, and the high radiopacity and density areas inside the brain parenchyma represented calcification. Members (I-1, II-1, II-3, and III-1) of the HB-PFBC family, who underwent CT scanning with the imaging authorized to researchers, showed symmetric and bilateral calcification in the caudate nuclei, globus pallidus, and putamen regions, whereas no retina/lens calcification, microphthalmia, or cataracts were observed (Figure 1B). The molecular and clinical characteristics of the patients are summarized in Table 1. The old father (I-1) showed prominent and severe calcification in the cerebellar hemisphere and vermis, as well as in the hippocampus, which is seldom calcified in PFBC patients. His neurological symptoms included parkinsonism, cerebral infarction, dysarthria, gait rigidity, and ataxia. The proband II-1 reported suffering from chronic and repetitive dizziness for 2 years. Biochemical tests showed that only total thyroxine (TT4) levels were slightly lower than normal; however, PTH levels were much higher than those in the reference (Supplementary Table 1). Mutational analysis of the four known AD PFBC-causative genes revealed the c.806delC mutation in SLC20A2 of proband II-1 (Figure 1C) that co-segregated with brain calcification in this family (Supplementary Figure 1). The c.806delC mutation is located in highly conserved regions of exon 7 of SLC20A2 (NM_001257180.2) in multiple species (Figure 1C), theoretically resulting in a prematurely terminated mRNA transcript, which might activate a surveillance mechanism called nonsense-mediated mRNA decay (NMD) (Khajavi et al., 2006), or probably create a C-terminal truncated PIT-2 protein [p.(Pro269Glnfs*49)] with impaired phosphate transport function. We evaluated the impact of the c.806delC mutation on SLC20A2 mRNA expression and found a 40–65% relative level in heterozygous carriers compared with controls, confirming that SLC20A2 haploinsufficiency causes brain calcification in the HB-PFBC family members (Figure 1D). Furthermore, SLC20A1, which is considered to play an essential role in inorganic phosphate-induced cardiovascular calcification (Li et al., 2006), showed no compensation effect for the c.806delC mutation in SLC20A2 (Figure 1D).
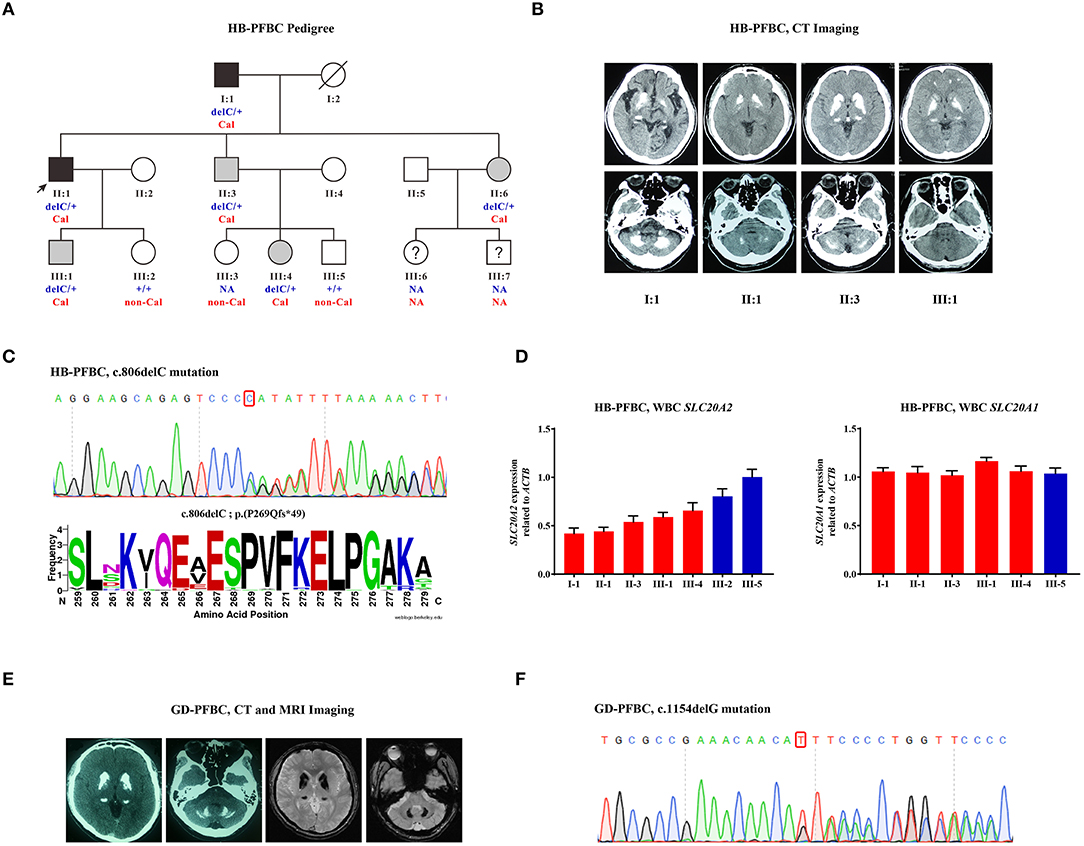
Figure 1. (A) HB-PFBC pedigree. Filled symbols represent family members affected with brain calcification, including both symptomatic (black) and asymptomatic (gray) cases. The black arrow indicates the proband and question marks indicate individuals whose blood samples and brain imaging were both not available. delC: c.806delC; +: wild-type allele; Cal, calcification; non-Cal, no calcification; NA, not available. (B) Brain computed tomography (CT) images of affected individuals in the HB-PFBC family. High radiopacity and density areas represent brain calcification. (C) The c.806delC mutation of SLC20A2 in the HB-PFBC proband and frequency diagram of protein conservation analysis for mutated Pro269 site. (D) SLC20A2 and SLC20A1 mRNA expression in the peripheral leukocytes of the HB-PFBC family members. Red bar indicates the patient, and blue indicates normal people. (E) Brain CT and MRI axial T2*GRE images of the GD-PFBC patient. (F) The c.1154delG mutation of SLC20A2 in the GD-PFBC patient.
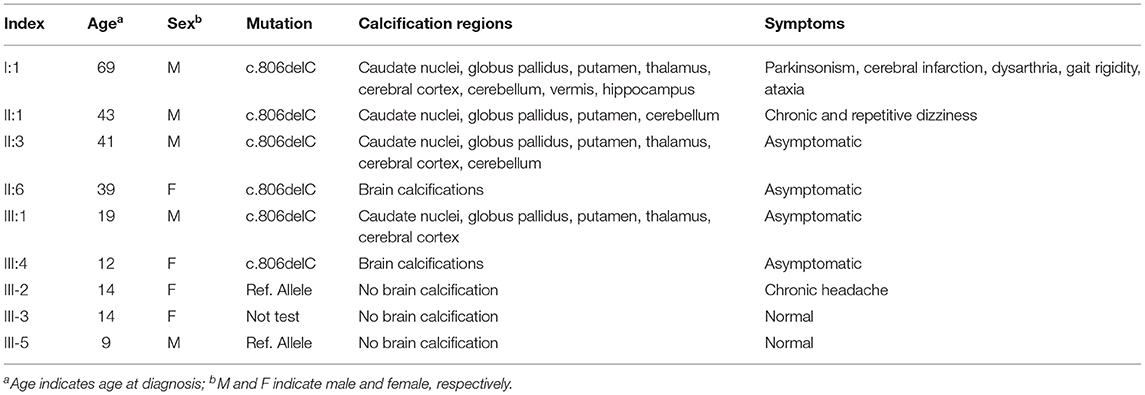
Table 1. Molecular and clinical findings in the HB-PFBC family.
A case of a patient with sporadic PFBC, named GD-PFBC, was recruited from the Department of Neurology, PUMCH. The patient was a 45-year-old man who had been suffering from involuntary movement of his left limbs and bradykinesia for 3 years. No headache, dizziness, nausea, vomiting, convulsion, language or speech problems, or consciousness disorders were present. A family history was also referred to. Systemic physical and neurological examinations revealed no abnormalities. Serum calcium, phosphate, and PTH concentrations and thyroid function were normal. The total serum cholesterol was normal; however, triglycerides (TG: 4.78 mmol/L, normal: 0.56–1.70 mmol/L), high-density lipoproteins (HDL-C: 0.75 mmol/L, normal: 1.16–1.42 mmol/L), low-density lipoproteins (LDL-C: 2.56 mmol/L, normal: 2.70–4.10 mmol/L), and apolipoprotein A-I (apoA-I: 0.90 g/L, normal: 1.20–1.60 g/L) were all out of reference, implicating dyslipidemia. Serum homocysteine (HCY: 36.9 μmol/L, normal: 4.0–17.0 μmol/L) and blood lactic acid (2.50 mmol/L, normal: 0.50–2.20 mmol/L) levels were higher than normal values. Abdominal ultrasonography showed mild fatty lesions in the liver and prostatic calcification. Brain CT imaging revealed bilateral symmetric calcification in the caudate nuclei, lentiform nuclei, thalami, and dentate nuclei of the cerebellum (Figure 1E). Brain MRI axial T2*GRE also showed bilateral symmetric hypointense signal changes in the aforementioned brain regions (Figure 1E). The patient was eventually diagnosed with Fahr's disease with hypertriglyceridemia and hyperhomocysteinemia. We identified a novel c.1154delG mutation at exon 8 of SLC20A2 (NM_001257180.2) (Figure 1F), in an evolutionarily conserved region, and predicted the formation of an NMD-directed degraded transcript or a truncated protein [p.(Ser385Ilefs*70)] in this patient.
The HLJ-PFBC patient in this case was a 46-year-old woman who complained of chronic headache, nausea, and slow writing in the 3 years prior to examination. Her physical examination revealed slightly elevated blood pressure (130/100 mmHg), and biochemical examination showed normal levels of serum calcium, phosphate, magnesium, calcitonin, and PTH. However, cranial CT revealed bilateral and symmetrical calcification in the basal ganglia, thalamus, and cerebellum (Figure 2A). A splice donor mutation (c.456+1G>C), located at the initiation of intron 4 of PDGFB (NM_002608.4), was suspected to be the causative mutation in this patient (Figure 2B). Mutational effects on mRNA splicing evaluated via cDNA-PCR and TA-cloning sequencing revealed that the c.456+1G>C mutation resulted in an in-frame deletion of 21 bp of nucleotides in vivo (Figure 2C). The relatively lower numbers of mutant TA clones (mt/wt clones = 22/44) and lower heights of cDNA sequencing peaks indicated that the newly deleted transcript was either undergoing the NMD process, the compensatory effect of the wild-type allele, or both, in the peripheral blood. Quantitative real-time PCR using SYBR Green dye and TaqMan probes revealed that the overall expression of PDGFB mRNA was increased by 2.47-fold when compared with age- and sex-matched controls, while the expression of the wild-type allele was increased by more than 3-fold (1.55 vs. 0.50%) (Figure 2D). Mutant PDGFB protein might be more stable than wild-type PDGFB and may induce the compensatory expression of wild-type protein in the HEK293T cell line (Figure 2E). The c.456+1G>C mutation effect on the PDGFB mRNA splicing process is illustrated in Figure 2F.
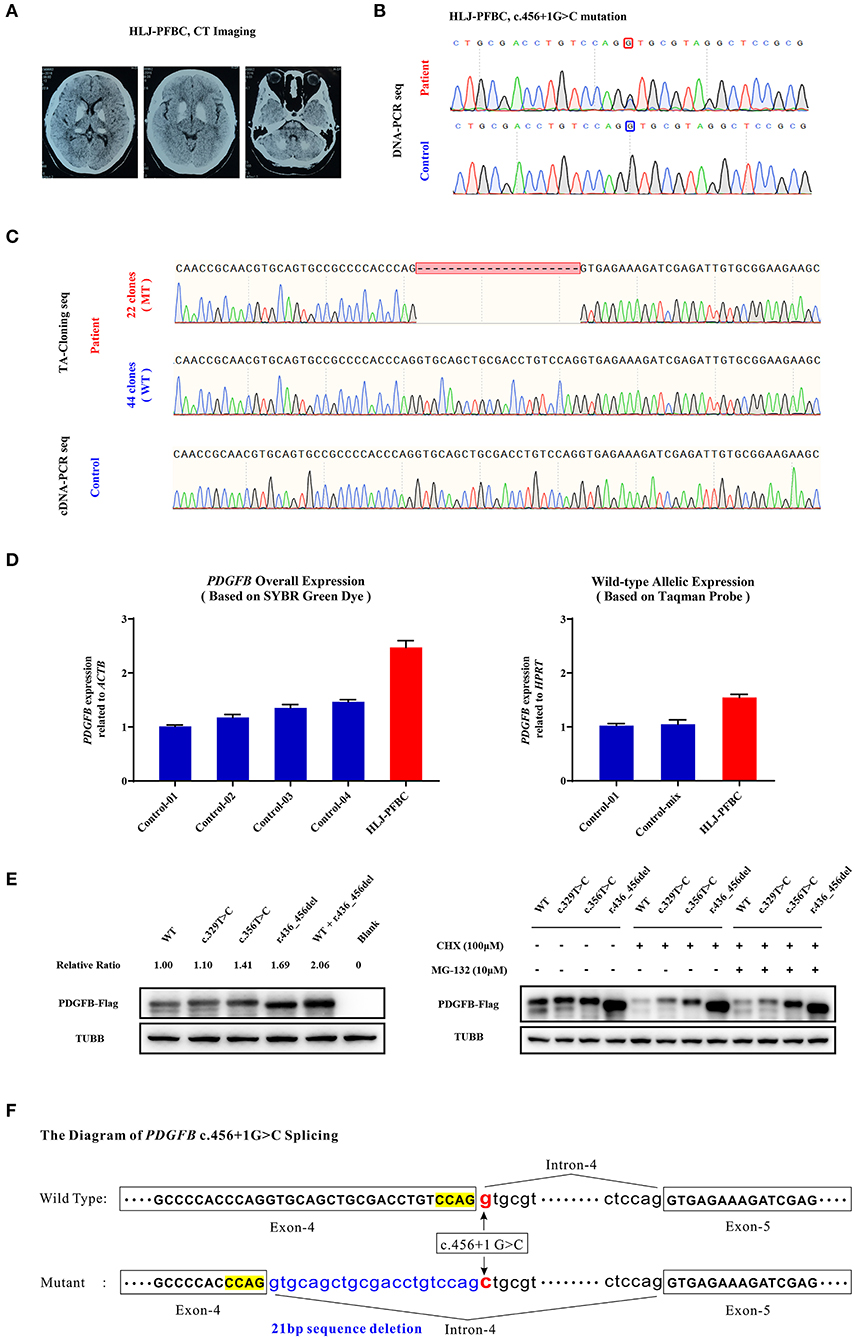
Figure 2. (A) Brain CT images of the HLJ-PFBC patient. (B) The PDGFB c.456+1G>C mutation in the HLJ-PFBC patient and normal control. (C) The impact of the PDGFB c.456+1G>C mutation on its mRNA splicing revealed by TA-cloning sequencing and cDNA-PCR sequencing in the HLJ-PFBC patient and normal control. (D) The overall and wild-type allele-specific expression of PDGFB in the peripheral leukocytes of the HLJ-PFBC patient (red) and normal controls revealed by SYBR Green dye- and TaqMan probe-based assays. Control-mix indicated a mixture of cDNA templates from six other normal controls. (E) Fusion protein expression of the PDGFB-Flag construct in the HEK293T cell line. The c.329T>C and c.356T>C of PDGFB are two previously reported PFBC-causative mutations. CHX (cycloheximide), a protein synthesis inhibitor; MG132, a ubiquitin-proteasome inhibitor. (F) The sketch of the mRNA splicing process of PDGFB c.456+1G>C.
Loss-of-function mutations in SLC20A2 are the major genetic causes of familial and sporadic PFBC (Hsu et al., 2013; Lemos et al., 2015). Most patients are asymptomatic before the age of 40 years. Notably, among members of the HB-PFBC family, SLC20A2 mRNA levels of III-2 (no calcification and no c.806delC mutation) were reduced to 80% of that of III-5 normal controls (Figure 1D). III-2 also presented with short and thick fingers and toes, and reported chronic headache and arthritis of the fingers. Biochemical results of proband II-1 in the HB-PFBC family showed slightly lower TT4 levels but significantly higher PTH levels than the reference range (Supplementary Table 1). Generally, TT4 decreases along with serum thyroid-binding globulin due to the intake of diazepam, testosterone, glucocorticoids, or other drugs. PTH, fibroblast growth factor-23 (FGF-23), and 1,25-dihydroxyvitamin D (1,25(OH)2D) are the main phosphate regulators in human physiology. PTH can increase renal phosphate excretion by reducing the expression of sodium-dependent phosphate co-transporters NaPi-IIa and NaPi-IIc in the proximal renal tubules (Bergwitz and Juppner, 2010). Elevated PTH is commonly regarded as a sub-clinical stage of hyperparathyroidism (OMIM#145000) or pseudo-hypoparathyroidism (OMIM#103580). Hypoparathyroidism, another clinical condition that can also lead to intracranial calcification, is usually caused by inadvertent removal of or accidental injury to the parathyroid gland during neck-related surgery, or by autoimmune disorders (Mannstadt et al., 2017; Gafni and Collins, 2019). Pseudo-hypoparathyroidism results from resistance to the biological effects of PTH in the peripheral organs. Both diseases share common biochemical features, such as hypocalcemia and hyperphosphatemia (Mantovani et al., 2018). Environmental factors and other genetic or epigenetic modifiers may also influence SLC20A2 expression, leading to recurrent headaches or repetitive dizziness.
The GD-PFBC patient showed moderate to severe hypertriglyceridemia and hyperhomocysteinemia, both of which are traditional risk factors for cardiovascular diseases (CVDs). Genetic variations and other secondary acquired factors could affect serum or plasma TG, HDL-C, LDL-C, and apoA-I levels, thereby modifying the risk of CVDs (Miller et al., 2011; Nordestgaard and Tybjaerg-Hansen, 2011; Nordestgaard, 2016; Ference et al., 2017; Rosenson et al., 2018). Elevated HCY levels are generally considered to be independent and strong risk factors for the incidence and progression of coronary artery calcification (Kullo et al., 2006; Karger et al., 2020), aortic calcification (Hirose et al., 2001; Karger et al., 2020), intracranial arterial calcification, and cerebral atherosclerosis (Kim et al., 2016). Hyperhomocysteinemia can induce vascular smooth muscle cell osteogenic differentiation and calcification, and increase endothelial cell apoptosis and vascular inflammation, all of which are adverse and pathogenic cardiovascular events (Hofmann et al., 2001; Van Campenhout et al., 2009; Fang et al., 2015; Zhu et al., 2019). High plasma homocysteine could also decrease HDL-C levels by enhancing its clearance and inhibiting apoA-I protein synthesis (Liao et al., 2006), which is in accordance with the lower HDL-C levels in patients with GD-PFBC. In addition, hyperhomocysteinemia might independently result in multifocal calcifications in the brain and coronary arteries, as revealed by brain and coronary CT scans in human patients (Nah and Kim, 2010). Homocysteine metabolism is largely dependent on the folate and methionine-homocysteine cycles (McCully, 1996; Welch and Loscalzo, 1998; Hankey and Eikelboom, 1999). Genetic variations in methylenetetrahydrofolate reductase encoded by the MTHFR gene could lead to variations in HCY concentrations at the individual and population levels (Frosst et al., 1995; Klerk et al., 2002; Selzer et al., 2003). In the GD-PFBC case, no common hypomorphic alleles were detected in four key enzymes closely associated with folate and methionine-homocysteine metabolism (MTHFR, 677C>T, 1298A>C; MTR, 2756A>G; MTRR, 66A>G; CBS, 844 ins68) (Kluijtmans et al., 2003; Moll and Varga, 2015). Treatment of this patient with vitamin B supplements (vitamin B6, B12, and folic acid) might alleviate hyperhomocysteinemia and improve clinical outcomes. However, dyslipidemia and hypertriglyceridemia, which are closely associated with critical cardiovascular events, such as mitral annular and aortic valve calcification and coronary artery calcification (Greif et al., 2013; Thanassoulis et al., 2013; Afshar et al., 2017; Zheng et al., 2019), also deserve special attention in this patient. We could not perform an informative analysis in the GD-PFBC patient due to the non-availability of RNA samples. However, we evaluated the pathogenicity of the c.1154delG variation in SLC20A2 (NM_001257180.2) in ten function-predicting scoring systems (ACMG, DDIG-in, FATHMM-Indel, MutPred-LOF, PROVEAN, SIFT-Indel, CADD/CADD-Splice, CAPICE/GAVIN, MutationTaster, and VEST/VEST-Indel), three software programs providing conservation scores (GERP++, phyloP100way, and phastCons100way), and four variation databases (gnomAD, ExAC, dbSNP, and 1000 Genomes Project), most of which supported the deleterious effect of this variation (Table 2).
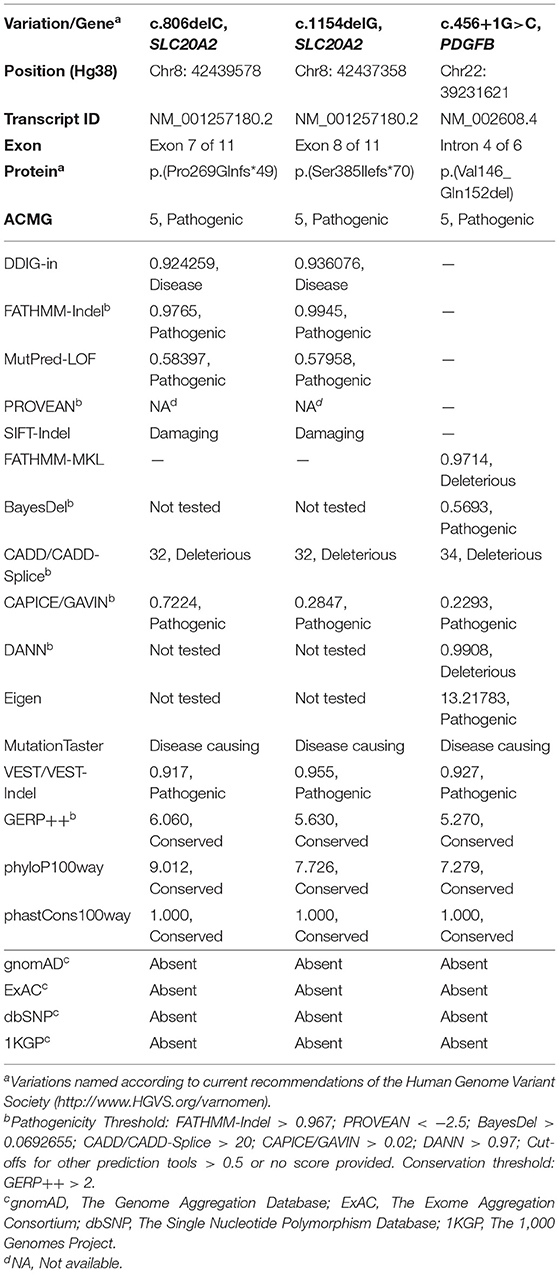
Table 2. Computational evidence for the pathogenicity of three novel variations.
The HLJ-PFBC patient, harboring the c.456+1G>C mutation of PDGFB, presented with nausea and higher blood pressure, which has rarely been reported previously. It seemed that her nausea was the result of elevated blood pressure because no other risk factors were found for this symptom. The PDGFB gene encodes platelet-derived growth factor beta, which is functionally activated when forming PDGF-BB homodimers or PDGF-AB heterodimers with PDGFA (Betsholtz and Keller, 2014). Loss-of-function mutations in PDGFB could affect its synthesis, maturation, and dimerization, resulting in impairment of the PDGFB-PDGFRB pathway and dysfunction of the BBB, eventually leading to PFBC (Vanlandewijck et al., 2015). We confirmed that the c.456+1G>C mutation removed the canonical 5-prime splice donor site and resulted in aberrant mRNA splicing, creating an in-frame deleted transcript. A similar splice donor site mutation, c.456+1G>A, was reported in a nuclear family, and predicted to lead to exon 4 (NM_002608.4) skipping and introduction of a frameshift version of PDGFB. However, both patients in the nuclear family had a severe migraine, a history of depression, and calcification in the basal ganglia, thalamus (only mother affected), cerebral cortex (only proband affected), and subcortical white matter (Ramos et al., 2018). Another splice acceptor site mutation in the same intron (c.457-1G>T) may lead to exon 5 (NM_002608.4) skipping and frameshift protein in carriers, leading to chronic headache and intellectual disability (Sekine et al., 2019). These results suggest that different substitutions on the same splicing unit (GT-AG in DNA code) could result in distinct molecular events and disease phenotypes. Of note, another three splice site mutations in PDGFB (c.64-3C>G, c.160+2T>A, and c.602-1G>T) also resulted in aberrant splicing processes and frameshift outcomes (Nicolas et al., 2015; Koyama et al., 2017; Sekine et al., 2019). In our study, the c.456+1G>C variant was functionally deleterious, as revealed by the comprehensive bioinformatic analyses (Table 2), and could activate the upstream cryptic splice donor site residing in exon 4 and create a novel mature transcript with an in-frame 21 bp deletion (NM_002608.4, r.436_456del). This novel transcript and its translated protein might be more stable than and promote the compensatory expression of the wild-type mRNA and protein in the peripheral blood and HEK293T cell line. The mutant PDGFB protein was resistant to degradation, perhaps via the lysosomal pathway, but not the ubiquitin-proteasome system, as revealed by the combinational post-treatment HEK293T cell line with cycloheximide and MG132 (Ostman et al., 1992). These results suggest that the mutant PDGFB protein might impair the PDGFB-PDGFRB pathway by disrupting disulfide bond formation and affecting its dimerization with wild-type PDGFB protein, thus producing a dominant negative effect (Shim et al., 2010). However, the exact and real molecular and functional effects of the c.456+1G>C mutation in the brain of patients with PFBC remain unclear.
Patients with PFBC can exhibit peripheral calcification, presenting as skin microangiopathy (Biancheri et al., 2016; Nicolas et al., 2017). Intracranial senile calcification is a common neuroimaging sign in healthy individuals. During the human lifespan, the choroid plexus, pineal gland, and habenular nuclei tend to accumulate physiologic calcium and phosphate, which is likely due to organ functional decline or insufficient hormone concentration (Grech et al., 2012). In addition, brain calcification load and location show significant inter-individual differences in PFBC patients and mouse models (Zarb et al., 2019b).
SLC20A2 gene haploinsufficiency is a likely pathogenic mechanism of brain calcification; half dosage of SLC20A2 expression cannot maintain the phosphate transport demand in the brain (Baker et al., 2014; Fujioka et al., 2015; Guo et al., 2019; Mu et al., 2019). Deletions adjacent to the regulatory element in the SLC20A2 coding region may also cause PFBC (Pasanen et al., 2017; Cassinari et al., 2020). SLC20A2 expression might be related to the severity of brain calcification to some extent, such as the genetic dosage effect observed in patients harboring MYORG mutations (Chen et al., 2019b, 2020; Grangeon et al., 2019). Some studies have also suggested a dominant negative function of SLC20A2 genetic variations (Kimura et al., 2016; Larsen et al., 2017), with bi-allelic pathogenic mutations in SLC20A2, resulting in more severe brain calcification (de Oliveira et al., 2013; Chen et al., 2019a). Phosphoric acid transport activities were significantly maintained in the presence of the c.680C>T mutation in SLC20A2. However, those harboring this mutation revealed severe and bilateral basal ganglia calcification (Nishii et al., 2019), suggesting that this mutation might exert a dominant negative effect, as seen in SLC20A2 variants encoding D28N, H502A, and E575K (Larsen et al., 2017). The effect of SLC20A2 dosage on PFBC remains controversial, and environmental factors and genetic or epigenetic modifiers need to be taken into consideration. PFBC-causative mutations in PDGFB might lead to complete loss of PDGFB function either through abolished protein synthesis or defective stimulation of PDGFRB and its downstream pathways (Vanlandewijck et al., 2015). However, some discrepancies were observed, such as when Pdgfrbredeye/redeye mice, which showed nearly complete reduction of PDGFB-PDGFRB signaling, did not develop brain calcification (Vanlandewijck et al., 2015). The relationship between impaired PDGFB-PDGFRB signaling and brain calcification requires further examination.
As of the end of January 2021, according to the Human Gene Mutation Database (HGMD, www.hgmd.cf.ac.uk/ac/index.php), 142 mutations in SLC20A2 have been identified, including 75 missense and non-sense mutations, 15 splice-site mutations, 32 small deletions, 5 small insertions and duplications, and 15 gross mutations; 24 mutations were identified in PDGFB, including 19 missense and non-sense mutations, three splice-site mutations, and two gross deletions. We summarized and compiled the other four pathogenic mutations in PDGFB from the literature (Sekine et al., 2019). In the present study, we identified two novel frameshift mutations in SLC20A2 (c.806delC and c.1154delG) and one splice donor site mutation in PDGFB (c.456+1G>C), which have broadened and enriched the SLC20A2 and PDGFB mutation spectrum (Supplementary Figure 2).
All datasets generated for this study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by the Institutional Ethics Committee of Peking Union Medical College, Chinese Academy of Medical Sciences (CAMS& PUMC). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
LS, BP, and XZ conceived and designed the study. All experiments and statistical analyses were conducted by LS, YR, YS, and HG. Three novel mutations were identified by LS and YS. Clinical samples and professional medical guidance were provided by SS, WX, JC, and LD. LS, SF, and YR cooperatively prepared the original manuscript. LS, YR, SF, and XZ independently checked and revised the grammar, syntax, and logical errors. All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This research was financially supported by the National Key Research and Development Program of China (Grant number: 2016YFC0905100), the CAMS Innovation Fund for Medical Sciences (CIFMS) (Grant number: 2016-I2M-1-002) at the Chinese Academy of Medical Sciences and Peking Union Medical College (CAMS&PUMC), the Beijing Municipal Science and Technology Commission (Grant number: Z151100003915078), the Fundamental Research Funds for the Central Universities (Grant number: 2016ZX310177), and the National Key R&D Program of China (Grant number: 2020YFA0804000).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Special thanks to Prof. Jingyu Liu and collaborators at Huazhong University of Science and Technology (HUST) for collaboratively identifying the first PFBC-causative gene. We also sincerely thank all the patients and their family members for their cooperation and support in this study. Prof. Jianqiang Tan of Liuzhou Maternal and Child Health Hospital and Prof. Lianqing Wang of Zibo Central Hospital also made great efforts for sample collection and experimental guidelines.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.643452/full#supplementary-material
Afshar, M., Luk, K., Do, R., Dufresne, L., Owens, D. S., Harris, T. B., et al. (2017). Association of triglyceride-related genetic variants with mitral annular calcification. J. Am. Coll. Cardiol. 69, 2941–2948. doi: 10.1016/j.jacc.2017.04.051
Baker, M., Strongosky, A. J., Sanchez-Contreras, M. Y., Yang, S., Ferguson, W., Calne, D. B., et al. (2014). SLC20A2 and THAP1 deletion in familial basal ganglia calcification with dystonia. Neurogenetics 15, 23–30. doi: 10.1007/s10048-013-0378-5
Batla, A., Tai, X. Y., Schottlaender, L., Erro, R., Balint, B., and Bhatia, K. P. (2017). Deconstructing Fahr's disease/syndrome of brain calcification in the era of new genes. Parkinsonism Relat. Disord. 37, 1–10. doi: 10.1016/j.parkreldis.2016.12.024
Bauer, M., Rahat, D., Zisman, E., Tabach, Y., Lossos, A., Meiner, V., et al. (2019). MYORG mutations: a major cause of recessive primary familial brain calcification. Curr. Neurol. Neurosci. Rep. 19:70. doi: 10.1007/s11910-019-0986-z
Bergwitz, C., and Juppner, H. (2010). Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu. Rev. Med. 61, 91–104. doi: 10.1146/annurev.med.051308.111339
Betsholtz, C., and Keller, A. (2014). PDGF, pericytes and the pathogenesis of idiopathic basal ganglia calcification (IBGC). Brain Pathol. 24, 387–395. doi: 10.1111/bpa.12158
Biancheri, R., Severino, M., Robbiano, A., Iacomino, M., Del Sette, M., Minetti, C., et al. (2016). White matter involvement in a family with a novel PDGFB mutation. Neurol. Genet. 2:e77. doi: 10.1212/NXG.0000000000000077
Cassinari, K., Rovelet-Lecrux, A., Tury, S., Quenez, O., Richard, A. C., Charbonnier, C., et al. (2020). Haploinsufficiency of the primary familial brain calcification gene SLC20A2 mediated by disruption of a regulatory element. Mov. Disord. 35, 1336–1345. doi: 10.1002/mds.28090
Cen, Z., Chen, Y., Chen, S., Wang, H., Yang, D., Zhang, H., et al. (2020). Biallelic loss-of-function mutations in JAM2 cause primary familial brain calcification. Brain 143, 491–502. doi: 10.1093/brain/awz392
Chen, S., Cen, Z., Fu, F., Chen, Y., Chen, X., Yang, D., et al. (2019a). Underestimated disease prevalence and severe phenotypes in patients with biallelic variants: a cohort study of primary familial brain calcification from China. Parkinsonism Relat. Disord. 64, 211–219. doi: 10.1016/j.parkreldis.2019.04.009
Chen, Y., Cen, Z., Chen, X., Wang, H., Chen, S., Yang, D., et al. (2020). MYORG mutation heterozygosity is associated with brain calcification. Mov. Disord. 35, 679–686. doi: 10.1002/mds.27973
Chen, Y., Fu, F., Chen, S., Cen, Z., Tang, H., Huang, J., et al. (2019b). Evaluation of MYORG mutations as a novel cause of primary familial brain calcification. Mov. Disord. 34, 291–297. doi: 10.1002/mds.27582
Crouthamel, M. H., Lau, W. L., Leaf, E. M., Chavkin, N. W., Wallingford, M. C., Peterson, D. F., et al. (2013). Sodium-dependent phosphate cotransporters and phosphate-induced calcification of vascular smooth muscle cells: redundant roles for PiT-1 and PiT-2. Arterioscler Thromb. Vasc. Biol. 33, 2625–2632. doi: 10.1161/ATVBAHA.113.302249
de Oliveira, M. F., Steinberg, S. S., and de Oliveira, J. R. (2013). The challenging interpretation of genetic and neuroimaging features in basal ganglia calcification. Gen. Hosp. Psychiatry 35, 210–211. doi: 10.1016/j.genhosppsych.2012.11.008
Donzuso, G., Mostile, G., Nicoletti, A., and Zappia, M. (2019). Basal ganglia calcifications (Fahr's syndrome): related conditions and clinical features. Neurol. Sci. 40, 2251–2263. doi: 10.1007/s10072-019-03998-x
Fang, K., Chen, Z., Liu, M., Peng, J., and Wu, P. (2015). Apoptosis and calcification of vascular endothelial cell under hyperhomocysteinemia. Med. Oncol. 32, 403. doi: 10.1007/s12032-014-0403-z
Ference, B. A., Ginsberg, H. N., Graham, I., Ray, K. K., Packard, C. J., Bruckert, E., et al. (2017). Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. evidence from genetic, epidemiologic, and clinical studies. a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 38, 2459–2472. doi: 10.1093/eurheartj/ehx144
Frosst, P., Blom, H. J., Milos, R., Goyette, P., Sheppard, C. A., Matthews, R. G., et al. (1995). A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat. Genet. 10, 111–113. doi: 10.1038/ng0595-111
Fujioka, S., Strongosky, A. J., Hassan, A., Rademakers, R., Dickson, D. W., and Wszolek, Z. K. (2015). Clinical presentation of a patient with SLC20A2 and THAP1 deletions: differential diagnosis of oromandibular dystonia. Parkinsonism Relat. Disord. 21, 329–331. doi: 10.1016/j.parkreldis.2014.12.024
Gafni, R. I., and Collins, M. T. (2019). Hypoparathyroidism. N. Engl. J. Med. 380, 1738–1747. doi: 10.1056/NEJMcp1800213
Giovannini, D., Touhami, J., Charnet, P., Sitbon, M., and Battini, J. L. (2013). Inorganic phosphate export by the retrovirus receptor XPR1 in metazoans. Cell Rep. 3, 1866–1873. doi: 10.1016/j.celrep.2013.05.035
Grangeon, L., Wallon, D., Charbonnier, C., Quenez, O., Richard, A. C., Rousseau, S., et al. (2019). Biallelic MYORG mutation carriers exhibit primary brain calcification with a distinct phenotype. Brain 142, 1573–1586. doi: 10.1093/brain/awz095
Grech, R., Grech, S., and Mizzi, A. (2012). Intracranial calcifications. a pictorial review. Neuroradiol. J. 25, 427–451. doi: 10.1177/197140091202500406
Greif, M., Arnoldt, T., von Ziegler, F., Ruemmler, J., Becker, C., Wakili, R., et al. (2013). Lipoprotein (a) is independently correlated with coronary artery calcification. Eur. J. Intern. Med. 24, 75–79. doi: 10.1016/j.ejim.2012.08.014
Guo, X. X., Su, H. Z., Zou, X. H., Lai, L. L., Lu, Y. Q., Wang, C., et al. (2019). Identification of SLC20A2 deletions in patients with primary familial brain calcification. Clin. Genet. 96, 53–60. doi: 10.1111/cge.13540
Hankey, G. J., and Eikelboom, J. W. (1999). Homocysteine and vascular disease. Lancet 354, 407–413. doi: 10.1016/S0140-6736(98)11058-9
Hirose, N., Arai, Y., Ishii, T., Tushima, M., and Li, J. (2001). Association of mild hyperhomocysteinemia with aortic calcification in hypercholesterolemic patients. J. Atheroscler. Thromb. 8, 91–94. doi: 10.5551/jat1994.8.91
Hofmann, M. A., Lalla, E., Lu, Y., Gleason, M. R., Wolf, B. M., Tanji, N., et al. (2001). Hyperhomocysteinemia enhances vascular inflammation and accelerates atherosclerosis in a murine model. J. Clin. Invest. 107, 675–683. doi: 10.1172/JCI10588
Hsu, S. C., Sears, R. L., Lemos, R. R., Quintans, B., Huang, A., Spiteri, E., et al. (2013). Mutations in SLC20A2 are a major cause of familial idiopathic basal ganglia calcification. Neurogenetics 14, 11–22. doi: 10.1007/s10048-012-0349-2
Inden, M., Iriyama, M., Zennami, M., Sekine, S. I., Hara, A., Yamada, M., et al. (2016). The type III transporters (PiT-1 and PiT-2) are the major sodium-dependent phosphate transporters in the mice and human brains. Brain Res. 1637, 128–136. doi: 10.1016/j.brainres.2016.02.032
Karger, A. B., Steffen, B. T., Nomura, S. O., Guan, W., Garg, P. K., Szklo, M., et al. (2020). Association between homocysteine and vascular calcification incidence, prevalence, and progression in the MESA Cohort. J. Am. Heart Assoc. 9:e013934. doi: 10.1161/JAHA.119.013934
Keller, A., Westenberger, A., Sobrido, M. J., Garcia-Murias, M., Domingo, A., Sears, R. L., et al. (2013). Mutations in the gene encoding PDGF-B cause brain calcifications in humans and mice. Nat. Genet. 45, 1077–1082. doi: 10.1038/ng.2723
Khajavi, M., Inoue, K., and Lupski, J. R. (2006). Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur. J. Hum. Genet. 14, 1074–1081. doi: 10.1038/sj.ejhg.5201649
Kim, J. M., Park, K. Y., Shin, D. W., Park, M. S., and Kwon, O. S. (2016). Relation of serum homocysteine levels to cerebral artery calcification and atherosclerosis. Atherosclerosis 254, 200–204. doi: 10.1016/j.atherosclerosis.2016.10.023
Kimura, T., Miura, T., Aoki, K., Saito, S., Hondo, H., Konno, T., et al. (2016). Familial idiopathic basal ganglia calcification: histopathologic features of an autopsied patient with an SLC20A2 mutation. Neuropathology 36, 365–371. doi: 10.1111/neup.12280
Klerk, M., Verhoef, P., Clarke, R., Blom, H. J., Kok, F. J., Schouten, E. G., et al. (2002). MTHFR 677C–>T polymorphism and risk of coronary heart disease: a meta-analysis. JAMA 288, 2023–2031. doi: 10.1001/jama.288.16.2023
Kluijtmans, L. A., Young, I. S., Boreham, C. A., Murray, L., McMaster, D., McNulty, H., et al. (2003). Genetic and nutritional factors contributing to hyperhomocysteinemia in young adults. Blood 101, 2483–2488. doi: 10.1182/blood.V101.7.2483
Koyama, S., Sato, H., Kobayashi, R., Kawakatsu, S., Kurimura, M., Wada, M., et al. (2017). Clinical and radiological diversity in genetically confirmed primary familial brain calcification. Sci. Rep. 7:12046. doi: 10.1038/s41598-017-11595-1
Kullo, I. J., Li, G., Bielak, L. F., Bailey, K. R., Sheedy, P. F. 2nd, Peyser, P. A., et al. (2006). Association of plasma homocysteine with coronary artery calcification in different categories of coronary heart disease risk. Mayo. Clin. Proc. 81, 177–182. doi: 10.4065/81.2.177
Larsen, F. T., Jensen, N., Autzen, J. K., Kongsfelt, I. B., and Pedersen, L. (2017). Primary brain calcification causal PiT2 transport-knockout variants can exert dominant negative effects on wild-type PiT2 transport function in mammalian cells. J. Mol. Neurosci. 61, 215–220. doi: 10.1007/s12031-016-0868-7
Legati, A., Giovannini, D., Nicolas, G., Lopez-Sanchez, U., Quintans, B., Oliveira, J. R., et al. (2015). Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export. Nat. Genet. 47, 579–581. doi: 10.1038/ng.3289
Lemos, R. R., Ramos, E. M., Legati, A., Nicolas, G., Jenkinson, E. M., Livingston, J. H., et al. (2015). Update and mutational analysis of SLC20A2: a major cause of primary familial brain calcification. Hum. Mutat. 36, 489–495. doi: 10.1002/humu.22778
Li, X., Yang, H. Y., and Giachelli, C. M. (2006). Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ. Res. 98, 905–912. doi: 10.1161/01.RES.0000216409.20863.e7
Liao, D., Tan, H., Hui, R., Li, Z., Jiang, X., Gaubatz, J., et al. (2006). Hyperhomocysteinemia decreases circulating high-density lipoprotein by inhibiting apolipoprotein A-I Protein synthesis and enhancing HDL cholesterol clearance. Circ. Res. 99, 598–606. doi: 10.1161/01.RES.0000242559.42077.22
Mannstadt, M., Bilezikian, J. P., Thakker, R. V., Hannan, F. M., Clarke, B. L., Rejnmark, L., et al. (2017). Hypoparathyroidism. Nat. Rev. Dis. Primers 3:17055. doi: 10.1038/nrdp.2017.55
Mantovani, G., Bastepe, M., Monk, D., de Sanctis, L., Thiele, S., Usardi, A., et al. (2018). Diagnosis and management of pseudohypoparathyroidism and related disorders: first international Consensus Statement. Nat. Rev. Endocrinol. 14, 476–500. doi: 10.1038/s41574-018-0042-0
McCully, K. S. (1996). Homocysteine and vascular disease. Nat. Med. 2, 386–389. doi: 10.1038/nm0496-386
Miller, M., Stone, N. J., Ballantyne, C., Bittner, V., Criqui, M. H., Ginsberg, H. N., et al. (2011). Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation 123, 2292–2333. doi: 10.1161/CIR.0b013e3182160726
Moll, S., and Varga, E. A. (2015). Homocysteine and MTHFR mutations. Circulation 132:e6–9. doi: 10.1161/CIRCULATIONAHA.114.013311
Mu, W., Tochen, L., Bertsch, C., Singer, H. S., and Baranano, K. W. (2019). Intracranial calcifications and dystonia associated with a novel deletion of chromosome 8p11.2 encompassing SLC20A2 and THAP1. BMJ Case Rep. 12:e228782. doi: 10.1136/bcr-2018-228782
Nah, H. W., and Kim, J. S. (2010). Premature intracranial arterial calcification in a patient with hyperhomocysteinemia. Neurology 75:2252. doi: 10.1212/WNL.0b013e31820203ef
Nicolas, G., Charbonnier, C., de Lemos, R. R., Richard, A. C., Guillin, O., Wallon, D., et al. (2015). Brain calcification process and phenotypes according to age and sex: lessons from SLC20A2, PDGFB, and PDGFRB mutation carriers. Am. J. Med. Genet. B Neuropsychiatr. Genet. 168, 586–594. doi: 10.1002/ajmg.b.32336
Nicolas, G., Marguet, F., Laquerriere, A., Mendes de Oliveira, J. R., and Hannequin, D. (2017). Microangiopathy in primary familial brain calcification: evidence from skin biopsies. Neurol. Genet. 3:e134. doi: 10.1212/NXG.0000000000000134
Nicolas, G., Pottier, C., Charbonnier, C., Guyant-Marechal, L., Le Ber, I., Pariente, J., et al. (2013a). Phenotypic spectrum of probable and genetically-confirmed idiopathic basal ganglia calcification. Brain 136, 3395–3407. doi: 10.1093/brain/awt255
Nicolas, G., Pottier, C., Maltete, D., Coutant, S., Rovelet-Lecrux, A., Legallic, S., et al. (2013b). Mutation of the PDGFRB gene as a cause of idiopathic basal ganglia calcification. Neurology 80, 181–187. doi: 10.1212/WNL.0b013e31827ccf34
Nishii, K., Shimogawa, R., Kurita, H., Inden, M., Kobayashi, M., Toyoshima, I., et al. (2019). Partial reduced Pi transport function of PiT-2 might not be sufficient to induce brain calcification of idiopathic basal ganglia calcification. Sci. Rep. 9:17288. doi: 10.1038/s41598-019-53401-0
Nordestgaard, B. G. (2016). Triglyceride-rich lipoproteins and atherosclerotic cardiovascular disease: new insights from epidemiology, genetics, and biology. Circ. Res. 118, 547–563. doi: 10.1161/CIRCRESAHA.115.306249
Nordestgaard, B. G., and Tybjaerg-Hansen, A. (2011). Genetic determinants of LDL, lipoprotein(a), triglyceride-rich lipoproteins and HDL: concordance and discordance with cardiovascular disease risk. Curr. Opin. Lipidol. 22, 113–122. doi: 10.1097/MOL.0b013e32834477d2
Ostman, A., Thyberg, J., Westermark, B., and Heldin, C. H. (1992). PDGF-AA and PDGF-BB biosynthesis: proprotein processing in the Golgi complex and lysosomal degradation of PDGF-BB retained intracellularly. J. Cell Biol. 118, 509–519. doi: 10.1083/jcb.118.3.509
Pasanen, P., Makinen, J., Myllykangas, L., Guerreiro, R., Bras, J., Valori, M., et al. (2017). Primary familial brain calcification linked to deletion of 5' noncoding region of SLC20A2. Acta Neurol. Scand. 136, 59–63. doi: 10.1111/ane.12697
Paucar, M., Almqvist, H., Jelic, V., Hagman, G., Jorneskog, G., Holmin, S., et al. (2017). A SLC20A2 gene mutation carrier displaying ataxia and increased levels of cerebrospinal fluid phosphate. J. Neurol. Sci. 375, 245–247. doi: 10.1016/j.jns.2017.02.007
Paucar, M., Almqvist, H., Saeed, A., Bergendal, G., Ygge, J., Holmin, S., et al. (2016). Progressive brain calcifications and signs in a family with the L9R mutation in the PDGFB gene. Neurol. Genet. 2:e84. doi: 10.1212/NXG.0000000000000084
Ramos, E. M., Carecchio, M., Lemos, R., Ferreira, J., Legati, A., Sears, R. L., et al. (2018). Primary brain calcification: an international study reporting novel variants and associated phenotypes. Eur. J. Hum. Genet. 26, 1462–1477. doi: 10.1038/s41431-018-0185-4
Rosenson, R. S., Brewer, H. B. Jr., Barter, P. J., Bjorkegren, J. L. M., Chapman, M. J., Gaudet, D., et al. (2018). HDL and atherosclerotic cardiovascular disease: genetic insights into complex biology. Nat. Rev. Cardiol. 15, 9–19. doi: 10.1038/nrcardio.2017.115
Schottlaender, L. V., Abeti, R., Jaunmuktane, Z., Macmillan, C., Chelban, V., O'Callaghan, B., et al. (2020). Bi-allelic JAM2 variants lead to early-onset recessive primary familial brain calcification. Am. J. Hum. Genet. 106, 412–421. doi: 10.1016/j.ajhg.2020.02.007
Sekine, S. I., Kaneko, M., Tanaka, M., Ninomiya, Y., Kurita, H., Inden, M., et al. (2019). Functional evaluation of PDGFB-variants in idiopathic basal ganglia calcification, using patient-derived iPS cells. Sci. Rep. 9:5698. doi: 10.1038/s41598-019-42115-y
Selzer, R. R., Rosenblatt, D. S., Laxova, R., and Hogan, K. (2003). Adverse effect of nitrous oxide in a child with 5,10-methylenetetrahydrofolate reductase deficiency. N. Engl. J. Med. 349, 45–50. doi: 10.1056/NEJMoa021867
Shim, A. H., Liu, H., Focia, P. J., Chen, X., Lin, P. C., and He, X. (2010). Structures of a platelet-derived growth factor/propeptide complex and a platelet-derived growth factor/receptor complex. Proc. Natl. Acad. Sci. U.S.A. 107, 11307–11312. doi: 10.1073/pnas.1000806107
Tadic, V., Westenberger, A., Domingo, A., Alvarez-Fischer, D., Klein, C., and Kasten, M. (2015). Primary familial brain calcification with known gene mutations: a systematic review and challenges of phenotypic characterization. JAMA Neurol. 72, 460–467. doi: 10.1001/jamaneurol.2014.3889
Thanassoulis, G., Campbell, C. Y., Owens, D. S., Smith, J. G., Smith, A. V., Peloso, G. M., et al. (2013). Genetic associations with valvular calcification and aortic stenosis. N. Engl. J. Med. 368, 503–512. doi: 10.1056/NEJMoa1109034
Van Campenhout, A., Moran, C. S., Parr, A., Clancy, P., Rush, C., Jakubowski, H., et al. (2009). Role of homocysteine in aortic calcification and osteogenic cell differentiation. Atherosclerosis 202, 557–566. doi: 10.1016/j.atherosclerosis.2008.05.031
Vanlandewijck, M., Lebouvier, T., Andaloussi Mae, M., Nahar, K., Hornemann, S., Kenkel, D., et al. (2015). Functional characterization of germline mutations in PDGFB and PDGFRB in primary familial brain calcification. PLoS ONE 10:e0143407. doi: 10.1371/journal.pone.0143407
Wang, C., Li, Y., Shi, L., Ren, J., Patti, M., Wang, T., et al. (2012). Mutations in SLC20A2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nat. Genet. 44, 254–256. doi: 10.1038/ng.1077
Welch, G. N., and Loscalzo, J. (1998). Homocysteine and atherothrombosis. N. Engl. J. Med. 338, 1042–1050. doi: 10.1056/NEJM199804093381507
Westenberger, A., Balck, A., and Klein, C. (2019). Primary familial brain calcifications: genetic and clinical update. Curr. Opin. Neurol. 32, 571–578. doi: 10.1097/WCO.0000000000000712
Yao, X. P., Cheng, X., Wang, C., Zhao, M., Guo, X. X., Su, H. Z., et al. (2018). Biallelic mutations in MYORG cause autosomal recessive primary familial brain calcification. Neuron 98, 1116–1123 e1115. doi: 10.1016/j.neuron.2018.05.037
Zarb, Y., Franzoso, F. D., and Keller, A. (2019a). Pericytes in primary familial brain calcification. Adv. Exp. Med. Biol. 1147, 247–264. doi: 10.1007/978-3-030-16908-4_11
Zarb, Y., Weber-Stadlbauer, U., Kirschenbaum, D., Kindler, D. R., Richetto, J., Keller, D., et al. (2019b). Ossified blood vessels in primary familial brain calcification elicit a neurotoxic astrocyte response. Brain 142, 885–902. doi: 10.1093/brain/awz032
Zhang, Y., Guo, X., and Wu, A. (2013). Association between a novel mutation in SLC20A2 and familial idiopathic basal ganglia calcification. PLoS ONE 8:e57060. doi: 10.1371/journal.pone.0057060
Zheng, K. H., Tsimikas, S., Pawade, T., Kroon, J., Jenkins, W. S. A., Doris, M. K., et al. (2019). Lipoprotein(a) and oxidized phospholipids promote valve calcification in patients with aortic stenosis. J. Am. Coll. Cardiol. 73, 2150–2162. doi: 10.1016/j.jacc.2019.01.070
Keywords: primary familial brain calcification, PFBC, SLC20A2, PDGFB, mutation
Citation: Shen Y, Shu S, Ren Y, Xia W, Chen J, Dong L, Ge H, Fan S, Shi L, Peng B and Zhang X (2021) Case Report: Two Novel Frameshift Mutations in SLC20A2 and One Novel Splice Donor Mutation in PDGFB Associated With Primary Familial Brain Calcification. Front. Genet. 12:643452. doi: 10.3389/fgene.2021.643452
Received: 21 December 2020; Accepted: 08 April 2021;
Published: 07 May 2021.
Edited by:
Nelson L. S. Tang, The Chinese University of Hong Kong, ChinaReviewed by:
Ana Westenberger, University of Lübeck, GermanyCopyright © 2021 Shen, Shu, Ren, Xia, Chen, Dong, Ge, Fan, Shi, Peng and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Shi, c2hpbGVpXzAzMjhAaWJtcy5wdW1jLmVkdS5jbg==; Bin Peng, cGVuZ2JpbjNAaG90bWFpbC5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.