 Xun Wang
Xun Wang Yue Zhong
Yue Zhong Mao Ding2*
Mao Ding2*
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 15 February 2021
Sec. Computational Genomics
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.638330
This article is part of the Research TopicArtificial Intelligence in Bioinformatics and Drug Repurposing: Methods and ApplicationsView all 13 articles
Alzheimer's disease (AD) is a common neurodegenerative dementia in the elderly. Although there is no effective drug to treat AD, proteins associated with AD have been discovered in related studies. One of the proteins is mitochondrial fusion protein 2 (Mfn2), and its regulation presumably be related to AD. However, there is no specific drug for Mfn2 regulation. In this study, a three-tunnel deep neural network (3-Tunnel DNN) model is constructed and trained on the extended Davis dataset. In the prediction of drug-target binding affinity values, the accuracy of the model is up to 88.82% and the loss value is 0.172. By ranking the binding affinity values of 1,063 approved drugs and small molecular compounds in the DrugBank database, the top 15 drug molecules are recommended by the 3-Tunnel DNN model. After removing molecular weight <200 and topical drugs, a total of 11 drug molecules are selected for literature mining. The results show that six drugs have effect on AD, which are reported in references. Meanwhile, molecular docking experiments are implemented on the 11 drugs. The results show that all of the 11 drug molecules could dock with Mfn2 successfully, and 5 of them have great binding effect.
Alzheimer's disease (AD) is a destructive nervous system disease, which is characterized by a progressive dementia. The incidence of AD accounts for 50–70% of the total number of senile dementias. It mostly occurs in middle or late life, and the psychological skills, cognitive function, and physiological function of the patients have gradually lost (McKhann et al., 1984; Navarro et al., 2020). With the increasing aging of the population, AD has become an important world problem to be solved. However, the pathogenesis of AD is still unclear. The cascade hypothesis of amyloid β protein (Aβ) is the most concerned. The hypothesis holds that the formation of senile plaques by a large amount of Aβ in the brain is related to cognitive dysfunction and pathological changes of AD (Lin Zhang et al., 2020). Abnormal deposition of Aβ is considered to be the vital pathogenesis of AD. And Aβ in cerebrospinal fluid has been included as a diagnostic marker of AD (Jia and Wei, 2018; Cui et al., 2020). A variety of AD-targeted drugs are difficult to be used in clinical practice because of poor efficacy or side effects in phase III clinical trials. More scholars focus on controlling the progression of mild cognitive impairment. Consequently, the regulation mechanism of Aβ production and clearance has become an important research direction (Cui et al., 2020).
Mitochondria is the main site of cellular aerobic respiration. And mitochondrial dysfunction has effect on the production and toxicity of Aβ. Mitochondrial dynamics and mitochondrial dysfunction caused by abnormal mitochondrial autophagy play an important role in the pathogenesis of AD. The mitochondrial fusion protein 2 (Mfn2) is a dynamic protein expressed in the outer membrane of mitochondria. Mfn2 not only participates in mitochondrial fusion but also affects cell metabolism by regulating cell apoptosis, mitochondrial autophagy, and other biological processes. At present, Mfn2 has been proved to be closely related to the occurrence of many kinds of common diseases. Although some specific mechanisms are still unclear, Mfn2 is expected to become a new therapeutic target for some diseases (Li et al., 2020). In addition, Mfn2 involved in the regulation of protein homeostasis and pathogenesis of AD has become a research hotspot.
The approved drugs are designed and developed based on the concept of single target. Therefore, no drug has been specifically developed for Mfn2 regulation till now. It takes 10–15 years to implement the de novo drug design. In order to reduce the cost of drug development and the risk in the process of drug research, it has become an important strategy to repurpose the approved drugs and explore their new functions. And deep learning methods provide powerful technical supports in computing the drug-target interactions (DTIs). The prediction of DTIs is the focus of drug design and the key step of drug repositioning. However, it is obviously not accurate to divide the drug–target pairs (DT pairs) into effective and ineffective in the classification method. Therefore, more attention has been paid to the regression method, which directly predicts the binding affinity values of drug–target (DT) pairs with dissociation constant (Kd).
The DeepDTA model (Ozturk et al., 2018) considers the sequence information of drug molecules and proteins in the prediction of binding affinity values. Convolutional neural network is used in the research. It is considered to be the state-of-the-art model of predicting DTA (Huang et al., 2020). However, the model fluctuates greatly when training for many times. The GraphDTA model (Nguyen et al., 2020) uses graph convolution neural network to represent the features of drug molecules. Although its loss value is tiny, the calculation cost is too high. Recurrent neural networks (RNNs) such as gated recurrent units (GRU) (Cho et al., 2014) and long short-term memory units (LSTM) (Hochreiter and Schmidhuber, 1997) are widely used to capture temporal dependence in sequence-based data such as time series and text (Chuang et al., 2020). Extending on the use of a single RNN, the ensemble of RNNs with CNNs is a common hybrid architecture in recent applications that seeks to combine the ability of RNN in analyzing sequential data and CNN on extracting local features (Cao et al., 2020). Nonetheless, in the representation of drug molecules, the results are not better than that of CNN. The latest DeepGS model (Lin et al., 2020) inputs the sequence information and two-dimensional structure information of drug molecules as well as the protein sequence information into the model for prediction. It also has the problem of higher calculation cost. Moreover, information redundancy is inevitable as drug molecules are encoded twice by different encoding strategies. In addition, DeepPurpose (Huang et al., 2020) provides a toolkit that integrates a variety of encoding methods of drug molecules and protein amino acid sequences. Two kinds of encoding methods are selected to input the model to predict the binding affinity values of DT pairs. The toolkit provides great convenience for future research.
In this study, we implement an approach that considers the binding affinity information and negative samples of DT pairs to reposition regulatory drugs Mfn2 as candidate medications of AD. First, a three–tunnel deep neural network (3-Tunnel-DNN) model is constructed and trained on the expanded Davis dataset using drug–protein binding affinity information. The three tunnels are protein sequences, drug molecules of positive samples, and negative samples. The accuracy of the 3-Tunnel-DNN model is 0.8882 and the loss value is 0.172 in the test set. Finally, the well-trained model is used to reposition 1,063 drugs/compounds from the DrugBank database to Mfn2 regulatory. A total of 15 drugs are recommended for Mfn2 regulation by ranking the binding affinity values of drugs/compounds from the database with Mfn2. After removing three molecules with molecular weight <200 and a topical drug, a total of 11 drug molecules are selected for literature mining and molecular docking experiments.
Davis dataset contains the selective analysis of kinase protein family and related inhibitors and their respective Kd values, and it includes 30,056 binding affinity values of 442 proteins and 68 compounds (Ozturk et al., 2018; Davis et al., 2020). Negative samples are expected to be considered in our model. Davis dataset is widely used as training set in the field of drug-target binding affinity prediction, such as DeepDTA (Ozturk et al., 2018), DeepGS (Lin et al., 2020), GraphDTA (Nguyen et al., 2020), etc. Therefore, binding affinity values of DT pairs in Davis dataset are applied as training set in the 3-Tunnel DNN model as well. Besides, information of negative samples is added into Davis dataset to extend the dataset.
In the original Davis dataset, binding affinity data of DT pairs are measured by Kd values. It ranges from 0 to 10,000. The extended Davis dataset consists of four files, which are SMILES sequences file of compounds, FASTA sequences file of proteins, binding affinity values file of DT pairs, and SMILES sequences file of negative samples. The original Davis dataset consists of the first three files. The first and second files contain the sequence information needed in the model training process. The third file, in particular, is a 68 × 442 dimensional digital matrix [i.e., M(68 × 442)], in which each number [m(i, j)] represents the Kd value of the i-th compound and the j-th protein. The fourth file is a matrix [i.e., NM(68 × 442)] composed of SMILES sequences of negative samples. Each element [nm(i, j)] represents the SMILES sequence of the negative sample of the i-th compound and the j-th protein. In the research, the Kd value of 50 is taken as the boundary between positive and negative samples. It means that for each protein, the compounds with binding affinity value ≤ 50 are positive samples, and the compounds with >50 are negative samples. The extended Davis dataset is given in Supplementary Tables 1–4.
In fact, we compare the recommendation of Zeng et al. that the boundary of positive and negative samples is set as 10 (Zeng et al., 2019) with our boundary of 50 as well. The results show that the Kd value of 50 as the boundary performs better. The results are shown in Table 1.

Table 1. Comparison list of consistency index (CI) and mean square error (MSE) of different boundaries on test set.
In the training process, the Kd values converted into log space (pKd) are used as the actual binding affinity values for easier calculation of regression. The explanation is similar to Equation (1) (Huang et al., 2020).
The DrugBank database contains common compounds (amino acids, polypeptides, choline, etc.), approved drugs (azithromycin, etc.), and approved small molecular compounds (5-fluorouridine, etc.). The first 1,063 drugs/compounds in the DrugBank database are used here as potential candidates for repositioning regulatory Mfn2. Particularly, SMILES sequences of drugs are used for calculation. Mfn2 sequence of human protein (Mfn2_Human) is used as the target protein for repositioning. Mfn2_Human in form of FASTA sequence from the UniProt database is used for binding affinity calculations. The usage of data is shown in Figure 1.
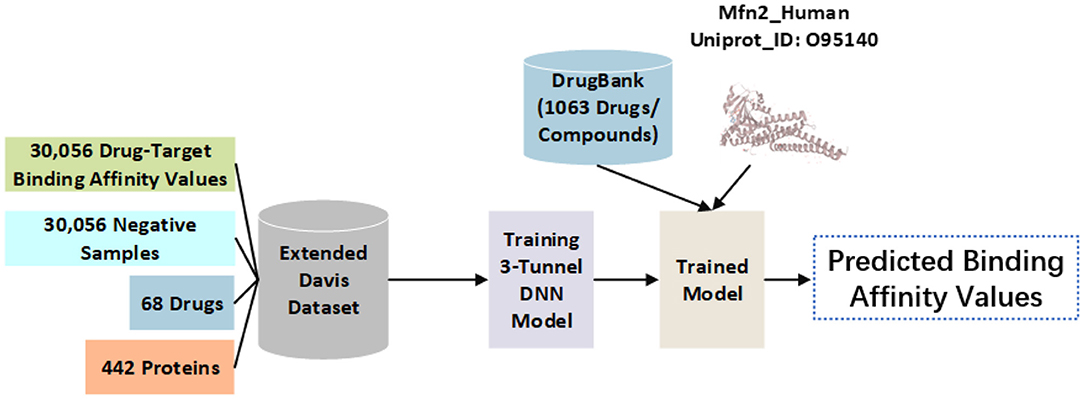
Figure 1. The use of data in the model. Binding affinity values of DT pairs in the extended Davis dataset are used for training set. The 1,063 drugs/compounds from the DrugBank database and Mfn2_Human from the UniProt database are used for external test set. Binding affinity values among drugs and Mfn2_Human are predicted using the well-trained model to recommend repositioning regulatory drugs to Mfn2_Human.
Extended-connectivity fingerprints (ECFPs) are a novel class of topological fingerprints for molecular characterization, which is a 1,024-length bits vector (Rogers and Hahn, 2010). In the study, n=2 (i.e., ECFP_2) is chosen as the circular radius that encodes the substructure of drug molecules. RDKit (Bento et al., 2020) is used to generate fingerprints of molecules. A multi-layer perceptron (MLP) (Chuang et al., 2020) is then applied on the binary fingerprint vector (Huang et al., 2020). In the 3-Tunnel DNN model, MLP is constructed as a four-layer neural network that the number of neurons is 1,024, 256, 64, and 256, respectively, to extract feature representations of drug molecules.
For proteins, there are 25 unique characters in protein FASTA sequence in Davis dataset (Ozturk et al., 2018). In our model, the symbol “?” is filled in the beginning of each sequence (Huang et al., 2020). Therefore, there are 26 unique characters in FASTA sequences. Each character is mapper into a unique integer, and the FASTA sequences are transformed into one-dimensional vectors. After that, the vector is extended into square data structure, in form of binary matrix with one-hot encoding strategy. The maximum length of FASTA sequences is set as 1,000 (Ozturk et al., 2018), so the matrix size of FASTA sequences is “1,000 × 26.” In particular, if the length of FASTA sequence is <1,000, the matrix is filled with 0. The matrix is input into the convolutional neural network (CNN), which consists of three layers of one-dimensional convolutional network and a global maximum pooling layer. The convolutional kernel is 32 × 1, 32 × 2, and 32 × 3, respectively (Ozturk et al., 2018; Huang et al., 2020). The activation function is Rectified Linear Unit (ReLU) (Nair and Hinton, 2010). And then, the representation vector of the features is generated.
The feature extraction methods are shown in Figure 2B.
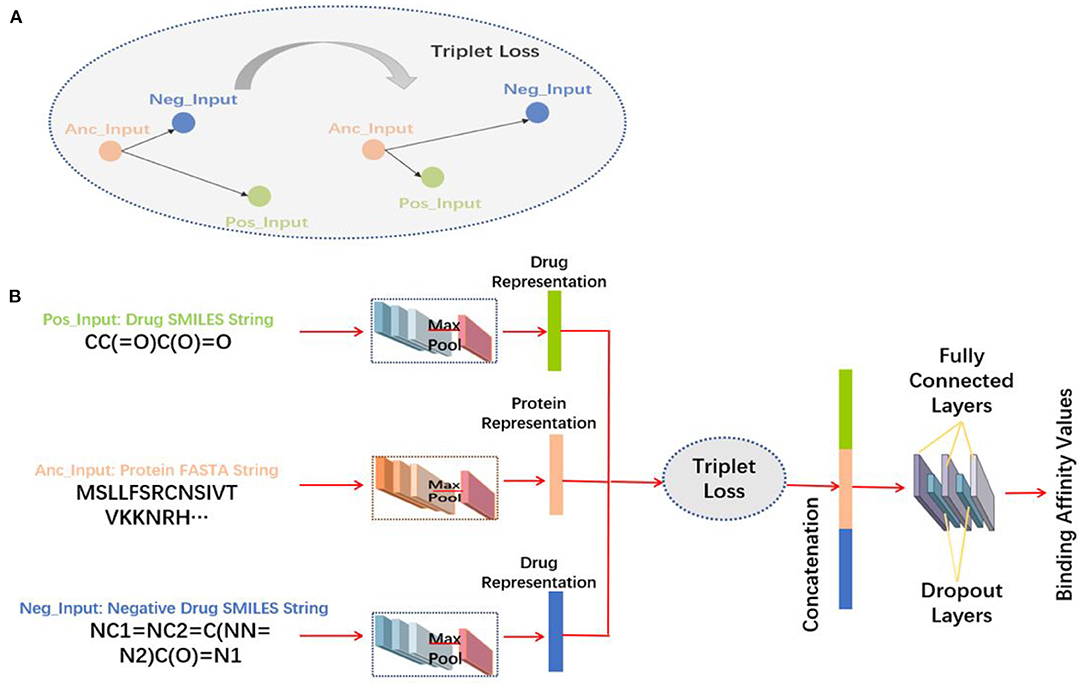
Figure 2. Structure diagram of the 3-Tunnel DNN model. (A) Diagram of triplet loss and (B) flowchart of the 3-Tunnel DNN model.
Information of negative samples is expected to be considered in the process of model training. Therefore, on the basis of two tunnels of drug molecules and proteins amino acid sequences, the third tunnel is added to process the data of negative samples to accurately reposition the drugs. The amino acid sequence of the protein is set as the anchor input (Anc_Input), the positive samples of the extended Davis dataset are taken as positive input (Pos_Input), and negative samples are set as negative input (Neg_Input). In the process of learning feature representations, triplet loss (Davis et al., 2020) is used to minimize the distance between Anc_Input and Pos_Input, and maximize the distance between Anc_Input and Neg_Input. The triplet loss is explained in Equation (2).
where represents the feature representation of the ith protein amino acid sequence, represents the feature representation of the ith positive sample, while represents the feature representation of the ith negative sample. In addition, means the square of the Euclidean distance between the vectors of the ith Anc_Input and Pos_Input. Similarly, means the square of the Euclidean distance between the vectors of the ith Anc_Input and Neg_Input. And M is a hyperparameter, which is set to 1 in the manuscript.
The three tunnels are used to process the FASTA sequences of proteins, the SMILES sequences of positive samples, and negative samples of drug molecules, respectively. The triplet loss (Schroff et al., 2015) is used here to obtain more accurate feature representations by maximizing the distance between proteins and negative samples and minimizing the distance between proteins and negative samples (Figure 2A). And then, three feature representations are concatenated together and input into the fully connected layers to make nonlinear changes to these extracted feature representations. In particular, the first two fully connected layers are followed by a dropout layer, respectively, which randomly “delete” hidden neurons to prevent over fitting, and finally map to the output space. The output of the model is the predicted binding affinity values of DT pairs. The 3-Tunnel DNN model is based on the MLP_CNN model (MLP for drugs encoding, CNN for proteins encoding) in DeepPurpose toolkit (Lin et al., 2020), and its topologic structure is shown in Figure 2B.
The 30,056 DT pairs from the extended Davis dataset are taken as training set, which are divided into three subsets in the ratio of 7:1:2 (Huang et al., 2020). It means that 70% of the data are used for training, 10% for validation, and 20% for testing. We use 256 small batch data to update the weights of neural networks. The number of epochs of the 3-Tunnel DNN model is 100, as well Adam optimization algorithm (with learning rate of 10−4) is applied to optimize the model.
After the well-trained 3-Tunnel DNN model is saved, 1,063 SMILES sequences of drugs/compounds from the DrugBank database and Mfn2_human protein sequence in the form of FASTA sequence from the UniProt database are input into the well-trained model. These predicted values are ranked to get the drug recommendation list. After removing the molecules with molecular weight <200 and topical drugs, a total of 11 drug molecules are recommended to regulate Mfn2. Literature mining and molecular docking experiments are implemented to verify the effectiveness of these molecules.
A total of 11 structures of recommended drug molecules and Mfn2 are analyzed by molecular docking experiments. The X-ray crystal structure of Mfn2 (PDB code: 6JFK, Resolution: 2.00 Å) is downloaded from RCSB Protein Data Bank (http://www.rcsb.org) in PDB format, and the first conformation is chosen as the receptor structure. The three-dimensional structures of recommended molecules are downloaded from the DrugBank database (https://www.drugbank.ca) in PDB format as well. UCSF Chimera software (Pettersen et al., 2004) is used to prepare receptor protein binding sites, establish the three-dimensional structure of molecules, and minimize the energy.
Before the formal docking experiments, it is necessary to prepare the documents of receptor protein, binding sites, protein surface, and drug molecules. For the receptor protein, all structures of ligands and hydrogens are deleted first. Dock Prep module is used to supplement the parameters of the receptor protein. Hydrogens, AMBER ff14SB force field, and AM1-BCC charges (Jakalian et al., 2000, 2010) of receptor and ligand are added, respectively. After that, the results are saved to a file in mol2 format. Then all hydrogens are deleted again and saved in PDB format. For the binding sites in the receptor, the same operation is implemented and the result is saved in the format of mol2. The DMS tool in UCSF Chimera (Pettersen et al., 2004) is used to generate the surface of the receptor using a probe atom with a 1.4 Å radius, which is saved as the file in the format of dms. Similarly, for drug molecules, hydrogenation, and charging operations are performed, and the results are saved as a file in the format of mol2 as well.
For each binding site, the Sphgen module is used to generate a spherical collection around the active site. The grid file is generated by the Grid module, which is used for grid-based energy assessment. Then, the semi-flexible docking is implemented with the program of Dock6.8 (Lang et al., 2009; Mukherjee et al., 2010), and 1,000 different orientations are generated. In particular, some drug molecules (e.g., adinazolam, pyrimethamine, and carbamazepine) are implemented rigid molecular docking to evaluate whether the receptor can accommodate the conformation. After that, the van der Waals force and electrostatic interaction between the ligand and the binding site are obtained, and the grid scores are also calculated. Finally, the best conformation is obtained by using cluster analysis (RMSD threshold is 2.0 Å) in semi-flexible docking, and in rigid docking, only one conformation is obtained.
In the training process, consistency index (CI) (Pahikkala et al., 2014) is used to evaluate the training performance, and mean square error (MSE) (Kansal et al., 2019) is used as the loss function to measure the error of each epoch.
We compare the performance of boundary with the K_d value of 10 (Zeng et al., 2019) and 50 as positive and negative samples. The performance of CNN_CNN model (CNN for drugs encoding, CNN for proteins encoding) and the 3-Tunnel DNN model at different boundaries are compared primarily. The results are shown in Table 1 and Figure 3. The results show that the K_d value of 50 as the boundary of positive and negative samples makes the model perform better. And our 3-Tunnel DNN model performs better than CNN_CNN model.
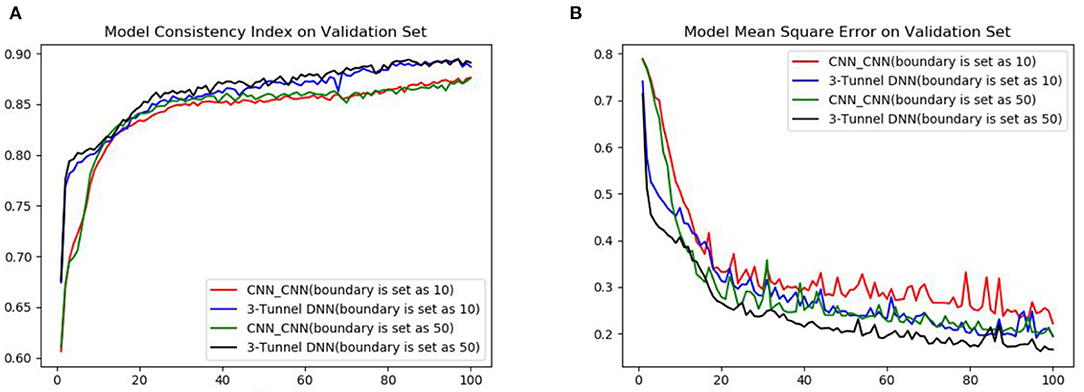
Figure 3. Comparison diagram of different boundaries in CNN_CNN model and MLP_CNN model. (A) Diagram of the accuracy on validation set and (B) diagram of the loss on validation set.
The 3-Tunnel DNN model is compared with the performance with the state-of-the-art model at present, such as DeepDTA (Ozturk et al., 2018), GraphDTA (Nguyen et al., 2020), and the latest DeepGS (Lin et al., 2020) model. At the same time, the original CNN_CNN model (Huang et al., 2020) and other models obtained in the DeepPurpose toolkit using our extended Davis dataset, such as CNN_CNN, CNN+LSTM_CNN (CNN+LSTM for drugs encoding, CNN for proteins encoding), and CNN+GRU_CNN (CNN+GRU for drugs encoding, CNN for proteins encoding) model, are also compared with the 3-Tunnel DNN model. The performances of these models are shown in Table 2.
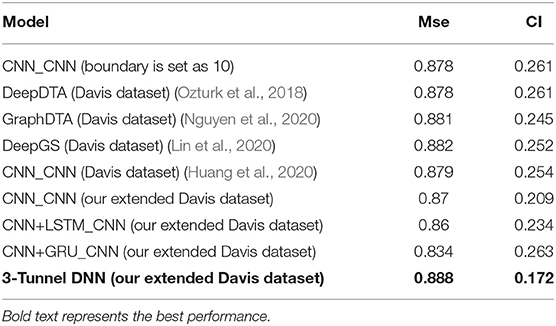
Table 2. Comparison list of consistency index (CI) and mean square error (MSE) of different models on test set.
According to results of the 3-Tunnel DNN model, the value of CI on test set is 0.888 and that of MSE is 0.172, which performs best among these models. The CI value of the 3-Tunnel DNN model is improved by 0.6% compared with DeepGS model (Lin et al., 2020), that is, with best accuracy. And the MSE value is improved by 7.3% compared with GraphDTA model (Nguyen et al., 2020), that is, with minimum loss. In addition, we also find that the MSE value of models using the extended Davis dataset as the training set is significantly smaller than models trained on the original Davis dataset, except for CNN+GRU_CNN model.
For repositioning regulatory drugs to Mfn2, the well-trained 3-Tunnel DNN model is used to calculate the binding affinity value of each pair of potential drug-Mfn2 pairs. The SMILES of potential drugs are the 1,063 approved drugs in the DrugBank database, and Mfn2 is the amino acid sequence of human in the form of FASTA sequence from the UniProt database. According to the ranking of predicted values, the 11 drugs recommended by 3-Tunnel DNN model are shown in Table 3 after removing drugs with molecular weight <200 (niacin, ethionamide, and acetohydroxamic acid) and an anesthetic (dyclonine). In particular, although dyclonine is reported to be effective for targets of AD in the reference (Zhang et al., 2019), it is still removed from the results because of topical drug.
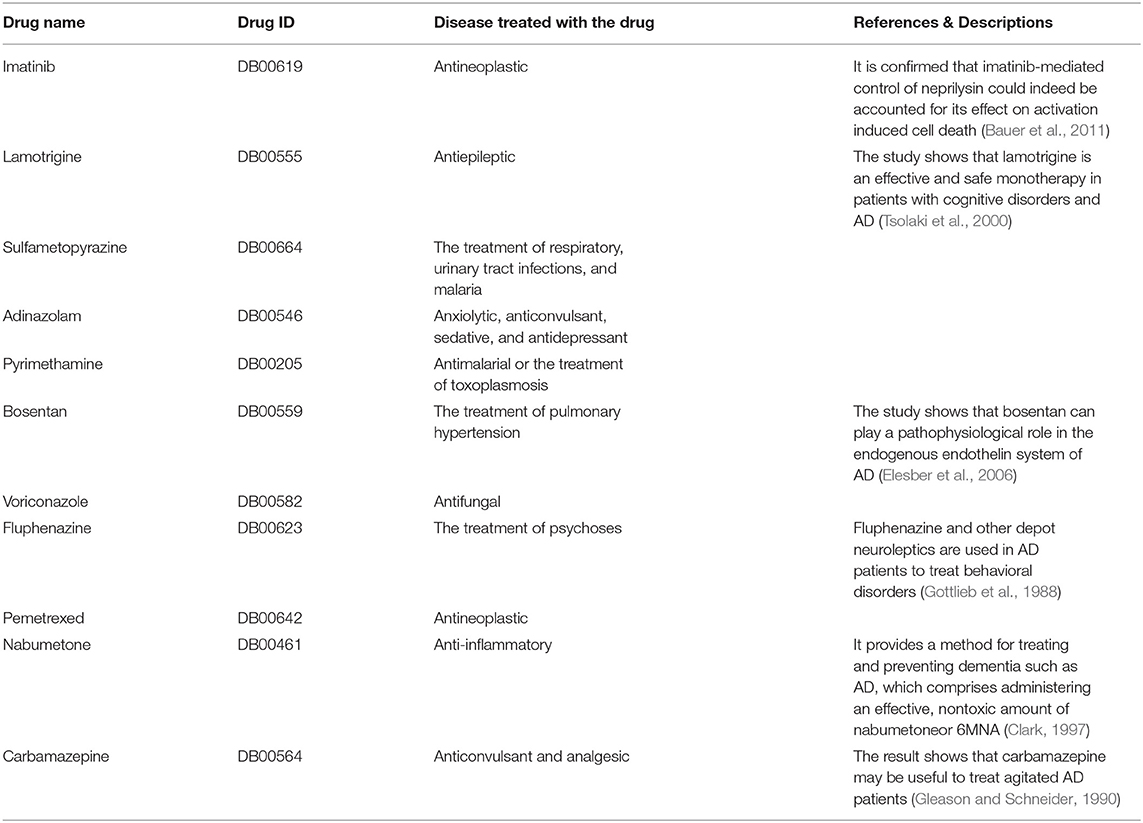
Table 3. Recommended drugs by the three-tunnel deep neural network (3-Tunnel DNN) model.
We search the keywords “Alzheimer; Drug name” and find that 6 drugs have supported references. Names, DrugBank ID, original functions, and description of supported references of these drugs are shown in Table 3.
It is reported that neuroleptic medication appears to have modest efficacy in controlling behavioral symptoms in dementia patients (Lemke, 1995). And the three drugs (adinazolam, fluphenazine, and carbamazepine) are related to mental illness. In addition, anti-tumor drugs, anti-epilepsy drugs, anti-infection drugs, and drugs for the treatment of hypertension are included in the recommended list.
Dock6.8 program (Lang et al., 2009; Mukherjee et al., 2010) is used to predict the binding patterns of 11 drug molecules in Mfn2. The value of Grid_Score is used to evaluate the molecular docking results, which represents the sum of van der Waals force and electrostatic interaction. The negative value of Grid_Score indicates that the drug molecule is bound to the target, while the positive value indicates no binding. And the smaller the Grid_Score, the stronger binding of drug molecules to Mfn2. Generally, the value of Grid_Score >−40 kcal/mol indicates poor binding, the value between −40 and −50 kcal/mol indicates medium binding, and the value < −50 kcal/mol indicates great binding (Liu et al., 2018). The Grid_Score values of each drug molecule binding to Mfn2 are shown in Table 4.
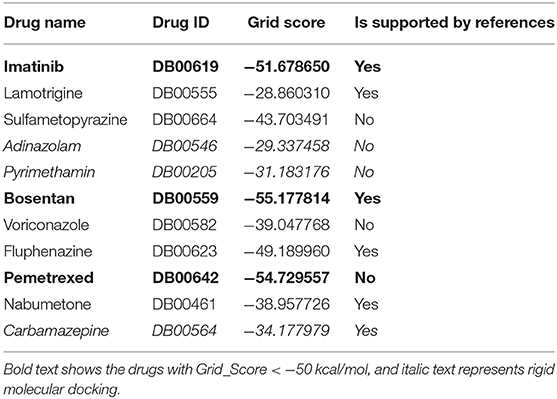
Table 4. Results of molecular docking.
According to the results of molecular docking, the binding effect of bosentan and Mfn2 is the best, and it is supported by the reference (Elesber et al., 2006) as well. In addition, the Grid_Score of imatinib and pemetrexed are < −50 kcal/mol, and they also have strong binding with Mfn2. Pemetrexed is not supported by any reference, but its binding capacity to Mfn2 is slightly lower than bosentan. And sulfametopyrazine and fluphenazine have medium binding with Mfn2. However, lamotrigine, voriconazole, and nabumetone have poor binding with Mfn2 in the experiments of semi-flexible docking. Lamotrigine, in particular, has the lowest score among these drug molecules, although it is supported by reference (Tsolaki et al., 2000). In addition, the Grid_Scores of adinazolam, pyrimethamine, and carbamazepine are not satisfactory. We speculate that it is caused by the rigid molecular docking. Since atomic bonds cannot rotate, there is only one conformation in the rigid docking.
In this study, we construct a 3-Tunnel DNN model based on the original drug-target binding affinity prediction model to consider the influence of negative samples. The binding affinity values of DT pairs are trained on the extended Davis dataset (i.e., the positive and negative samples are divided by the K_d value of 50 on the original Davis dataset). Then, the binding affinity values of 1,063 drug molecules with Mfn2 protein are predicted using a well-trained deep learning model. Literature mining and molecular docking experiments are implemented on the recommended 11 molecules. The values of accuracy and loss of the model are obviously better than the existing models, especially the loss value is 0.172. Six of the 11 molecules have been reported by other researchers. The results of molecular docking show that all of the 11 drug molecules can dock with Mfn2 successfully. And five drug molecules have medium or strong binding force. In particular, bosentan has the best performance of molecular docking, which is also supported by the reference (Elesber et al., 2006). In addition, pemetrexed and imatinib are prospect drugs as well. Specially, pemetrexed has not been used in the treatment of AD, and its molecular docking result is just tiny lower than bosentan. In the following work, we will evaluate the pharmacology and toxicology of pemetrexed, and in vitro experiments could be prepared to verify its effectiveness.
Although we find that positive and negative samples in Davis dataset with the K_d value of 50 as the boundary is better than 10, its specific value is still worthy to study. In addition, other datasets (such as KIBA and BindingDB) are expected to be extended and implemented in the model in our future work. And, it is a promising research to add gene information (Chen et al., 2013; Zhang et al., 2013; Shi et al., 2014; Tan et al., 2014) into the drug–target relationships and explore the relationships between genes (Li et al., 2015, 2016; Shi et al., 2015) and drugs. Furthermore, the training speed and accuracy of the feature representations of drug molecules extracted by molecular fingerprint is obviously better than that of SMILES sequences. For further research, a better feature extraction method for protein characteristics is expected to be obtained. And spiking neural P systems (Song et al., 2013, 2015a,b; Song and Pan, 2015) is also considered to be implemented in the future.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
XW conceived and designed the experiments. YZ performed the experiments and wrote the code. MD analyzed the data. XW and YZ drafted the work and revised it. All authors contributed to the article and approved the submitted version.
This work was supported by National Natural Science Foundation of China (Grant Nos. 61873280, 61873281, 61672033, 61672248, 61972416), Taishan Scholarship (tsqn201812029), Natural Science Foundation of Shandong Province (No. 2019GGX101067, ZR2019MF012), Fundamental Research Funds for the Central Universities (18CX02152A, 19CX05003A-6), and Foundation of Science and Technology Development of Jinan (201907116).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.638330/full#supplementary-material
Supplementary Tables 1–4. The extended Davis dataset.
Bauer, C., Pardossi-Piquard, R., Dunys, J., Roy, M., and Checler, F. (2011). γ-secretase-mediated regulation of neprilysin: influence of cell density and aging and modulation by imatinib. J. Alzheimer's Dis. 27, 511–520. doi: 10.3233/JAD-2011-110746
Bento, A. P., Hersey, A., Felix, E., Landrum, G., Gaulton, A., Atkinson, F., et al. (2020). An open source chemical structure curation pipeline using RDkit. J. Cheminform. 12:51. doi: 10.1186/s13321-020-00456-1
Cao, Y., Geddes, T. A., Yang, J. Y. H., and Yang, P. (2020). Ensemble deep learning in bioinformatics. Nat. Mach. Intell. 2, 1–9. doi: 10.1038/s42256-020-0217-y
Chen, Z., Song, T., Huang, Y., and Shi, X. (2013). Solving vertex cover problem using DNA tile assembly model. J. Appl. Math. 407816, 2541–2565. doi: 10.1155/2013/407816
Cho, K., van Merrienboer, B., Gulcehre, C., Fethi Bougares, D. B., Schwenk, H., and Bengio, Y. (2014). “Learning phrase representations using RNN encoder-decoder for statistical machine translation,” in Conference on Empirical Methods in Natural Language Processing (EMNLP 2014) (Doha). doi: 10.3115/v1/D14-1179
Chuang, K. V., Gunsalus, L. M., and Keiser, M. J. (2020). Learning molecular representations for medicinal chemistry. J. Med. Chem. 63, 8705–8722. doi: 10.1021/acs.jmedchem.0c00385
Clark, M. S. G. (1997). Use of nabumetone or 6-methoxynaphthyl acetic acid for the treatment of dementia. US5695774 A.
Cui, Y., Wang, Y., and Wang, P. (2020). Regulation of amyloidogenesis and clearance of β-amyloid in Alzheimer's disease. Chinese J. Biochem. Mol. Biol. 1–7. doi: 10.13865/j.cnki.cjbmb.2020.11.1384
Davis, M. I., Hunt, J. P., Herrgard, S., Ciceri, P., Wodicka, L. M., Pallares, G., et al. (2020). Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051. doi: 10.1038/nbt.1990
Elesber, A. A., Bonetti, P. O., Julie E Woodruma, X. Z., Lerman, L. O., Pallares, G., et al. (2006). Bosentan preserves endothelial function in mice overexpressing app. Neurobiol. Aging 27, 446–450. doi: 10.1016/j.neurobiolaging.2005.02.012
Gleason, R. P., and Schneider, L. S. (1990). Carbamazepine treatment of agitation in Alzheimer's outpatients refractory to neuroleptics. J. Clin. Psychiatry 51, 115–118.
Gottlieb, G. L., McAllisfer, T. W., and Gur, R. C. (1988). Depot neuroleptics in the treatment of bekavioral disbrders in patients with Alzheimer's disease. J. Am. Geriatr. Soc. 36, 619–621. doi: 10.1111/j.1532-5415.1988.tb06157.x
Hochreiter, S., and Schmidhuber, J. (1997). Long short-term memory. Neural Comput. 9, 1735–1780. doi: 10.1162/neco.1997.9.8.1735
Huang, K., Fu, T., Xiao, C., Glass, L. M., and Sun, J. (2020). Deeppurpose: a deep learning library for drug-target interaction prediction and applications to repurposing and screening. Bioinformatics 1–3. doi: 10.1093/bioinformatics/btaa1005
Jakalian, A., Bush, B. L., Jack, D. B., and Bayly, C. I. (2000). Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 21, 132–146. doi: 10.1002/(SICI)1096-987X(20000130)21:2<132::AID-JCC5>3.0.CO;2-P
Jakalian, A., Jack, D. B., and Bayly, C. I. (2010). Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 23, 1623–1641. doi: 10.1002/jcc.10128
Jia, J., and Wei, C. (2018). 2018 chinese guidelines for the diagnosis and treatment of dementia and cognitive impairment, guidelines for the diagnosis and treatment of Alzheimer's disease. Chinese J. Med. 13, 971–977. doi: 10.3760/cma.j.issn.0376-2491.2018.13.004
Kansal, S., Bansod, P. P., and Kunar, A. (2019). Prediction of instantaneous heart rate using adaptive algorithms. Int. J. Adapt. Innov. Syst. 2, 267–281. doi: 10.1504/IJAIS.2019.108397
Lang, P. T., Brozell, S. R., Mukherjee, S., Pettersen, E. F., Meng, E. C., Thomas, V., et al. (2009). Dock 6: combining techniques to model RNA-small molecule complexes. RNA 15, 1219–1230. doi: 10.1261/rna.1563609
Lemke, M. R. (1995). Effect of carbamazepine on agitation in Alzheimer's inpatients refractory to neurolepticss. J. Clin. Psychiatry 56, 354–357.
Li, H., Lei, L., and Han, S. (2020). Role of mitofusin 2 in major diseases. Biol. Chem. Eng. 6, 160–162.
Li, X., Song, T., Chen, Z., Shi, X., Chen, C., and Zhang, Z. (2016). A universal fast colorimetric method for DNA signal detection with DNA strand displacement and gold nanoparticles. J. Nanomater. 16, 464–469. doi: 10.1155/2015/407184
Li, X., Wang, X., Song, T., Lu, W., Chen, Z., and Shi, X. (2015). Novel computational method to reduce leaky reaction in DNA strand displacement. J. Analyt. Methods Chem. 2015:675827. doi: 10.1155/2015/675827
Lin Zhang, D. F., Zhao, D., Wang, Y., and Liu, C. (2020). Study on uptake of Puerarin by SH-SY5Y cells and improvement of aβ1-42 induced cell damage. Chinese Arch. Tradit. Chinese Med, 1–7. Available online at: http://kns.cnki.net/kcms/detail/21.1546.R.20201106.1612.028.html
Lin, X., Zhao, K., Xiao, T., Quan, Z., Wang, Z., and Yu, P. S. (2020). “Deepgs: Deep representation learning of graphs and sequences for drug-target binding affinity prediction,” in The 24th European Conference on Artificial Intelligence (ECAI 2020), (Santiago de Compostela).
Liu, T., Liu, M., Chen, F., Chen, F., Tian, Y., and Huang, Q. (2018). A small-molecule compound has anti-influenza A virus activity by acting as a 'pb2 inhibitor'. Mol. Pharm. 15, 4110–4120. doi: 10.1021/acs.molpharmaceut.8b00531
McKhann, G., Drachman, D., Folstein, M., Katzman, R., Price, D., and Stadlan, E. M. (1984). Clinical diagnosis of Alzheimer's disease: Report of the NINCDS-ADRDA work group under the auspices of department of health and human services task force on Alzheimer's disease. Nat. Neurol. 34, 939–944. doi: 10.1212/WNL.34.7.939
Mukherjee, S., Balius, T. E., and Rizzo, R. C. (2010). Docking validation resources: protein family and ligand flexibility experiments. J. Chem. Inform. Model. 50, 1986–2000. doi: 10.1021/ci1001982
Nair, V., and Hinton, G. E. (2010). “Rectified linear units improve restricted boltzmann machines,” in Proceedings of the 27th International Conference on International Conference on Machine Learning (Haifa), 807–814.
Navarro, J. F., Croteau, D. L., Jurek, A., Andrusivova, Z., Yang, B., Wang, Y., et al. (2020). Associated with dysregulated mitochondrial functions and stress signaling in Alzheimer disease. iScience 23:101556. doi: 10.1016/j.isci.2020.101556
Nguyen, T., Le, H., and Venkatesh, S. (2020). Graphdta: prediction of drug-target binding affinity using graph convolutional networks. Bioinformatics btaa921. doi: 10.1093/bioinformatics/btaa921
Ozturk, H., Ozkirimli, E., and Ozgur, A. (2018). Deepdta: Deep drug-target binding affinity prediction. Bioinformatics 34, 821–829. doi: 10.1093/bioinformatics/bty593
Pahikkala, T., Airola, A., Sami Pietilä, S. S., Shakyawar, S., Szwajda, A., Tang, J., et al. (2014). Toward more realistic drug-target interaction predictions. Brief. Bioinformatics 16, 325–337. doi: 10.1093/bib/bbu010
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Rogers, D., and Hahn, M. (2010). Extended-connectivity fingerprints. J. Chem. Inform. Model. 50, 742–754. doi: 10.1021/ci100050t
Schroff, F., Kalenichenko, D., and Philbin, J. (2015). “Facenet: A unified embedding for face recognition and clustering,” in 2015 IEEE Conference on Computer Vision and Pattern Recognition (CVPR) (Boston, MA), 815–823. doi: 10.1109/CVPR.2015.7298682
Shi, X., Chen, C., Li, X., Song, T., Chen, Z., Zhang, Z., et al. (2015). Size controllable DNA nanoribbons assembled from three types of reusable brick single-strand DNA tiles. Soft Matter 11, 8484–8492. doi: 10.1039/C5SM00796H
Shi, X., Wang, Z., Deng, C., Song, T., Pan, L., and Chen, Z. (2014). A novel bio-sensor based on DNA strand displacement. PLoS ONE 9:e108856. doi: 10.1371/journal.pone.0108856
Song, T., and Pan, L. (2015). Spiking neural p systems with rules on synapses working in maximum spikes consumption strategy. IEEE Trans. Nanobiosci. 14, 38–44. doi: 10.1109/TNB.2014.2367506
Song, T., Pan, L., and Paun, G. (2013). Asynchronous spiking neural P systems with local synchronization. Inform. Sci. 219, 197–207. doi: 10.1016/j.ins.2012.07.023
Song, T., Xu, J., and Pan, L. (2015a). On the universality and non-universality of spiking neural P systems with rules on synapses. IEEE Trans. Nanobiosci. 14, 960–966. doi: 10.1109/TNB.2015.2503603
Song, T., Zou, Q., Zeng, X., and Liu, X. (2015b). Asynchronous spiking neural p systems with rules on synapses. Neurocomputing 151, 1439–1445. doi: 10.1016/j.neucom.2014.10.044
Tan, G., Song, T., and Chen, Z. (2014). Spiking neural p systems with anti-spikes and without annihilating priority as number acceptors. J. Syst. Eng. Electron. 25, 464–469. doi: 10.1109/JSEE.2014.00053
Tsolaki, M., Kourtis, A., Divanoglou, D., Bostanzopoulou, M., and Kazis, A. (2000). Monotherapy with lamotrigine in patients with Alzheimer's disease and seizures. Am. J. Alzheimer's Dis. Other Dement. 15, 74–79. doi: 10.1177/153331750001500209
Zeng, X., Zhu, S., Liu, X., Zhou, Y., and Cheng, F. (2019). deepdr: A network-based deep learning approach to in silico drug repositioning. Bioinformatics 35, 5191–5198. doi: 10.1093/bioinformatics/btz418
Zhang, B., Pang, X., Jia, H., Wang, Z., Liu, A., and Du, G. (2019). Repositioning drug discovery for Alzheimer's disease based on global marketed drug data. Acta Pharm. Sin. 54, 1214–1224. doi: 10.16438/j.0513-4870.2019-0165
Keywords: Alzheimer's disease, drug repositioning, prediction of binding affinity values, three-tunnel deep neural network, molecular docking
Citation: Wang X, Zhong Y and Ding M (2021) Repositioning Drugs to the Mitochondrial Fusion Protein 2 by Three-Tunnel Deep Neural Network for Alzheimer's Disease. Front. Genet. 12:638330. doi: 10.3389/fgene.2021.638330
Received: 06 December 2020; Accepted: 08 January 2021;
Published: 15 February 2021.
Edited by:
Quan Zou, University of Electronic Science and Technology of China, ChinaReviewed by:
Pan Zheng, University of Canterbury, New ZealandCopyright © 2021 Wang, Zhong and Ding. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xun Wang, d2FuZ3N5dW5AdXBjLmVkdS5jbg==; Mao Ding, MTgyNjQxODEzMTJAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.