 Suzanne Gonzalez
Suzanne Gonzalez- Department of Psychiatry and Behavioral Health, Department of Pharmacology, Penn State College of Medicine, Hershey, PA, United States
It has been postulated that mitochondrial dysfunction has a significant role in the underlying pathophysiology of bipolar disorder (BD). Mitochondrial functioning plays an important role in regulating synaptic transmission, brain function, and cognition. Neuronal activity is energy dependent and neurons are particularly sensitive to changes in bioenergetic fluctuations, suggesting that mitochondria regulate fundamental aspects of brain function. Vigorous evidence supports the role of mitochondrial dysfunction in the etiology of BD, including dysregulated oxidative phosphorylation, general decrease of energy, altered brain bioenergetics, co-morbidity with mitochondrial disorders, and association with genetic variants in mitochondrial DNA (mtDNA) or nuclear-encoded mitochondrial genes. Despite these advances, the underlying etiology of mitochondrial dysfunction in BD is unclear. A plausible evolutionary explanation is that mitochondrial-nuclear (mitonuclear) incompatibility leads to a desynchronization of machinery required for efficient electron transport and cellular energy production. Approximately 1,200 genes, encoded from both nuclear and mitochondrial genomes, are essential for mitochondrial function. Studies suggest that mitochondrial and nuclear genomes co-evolve, and the coordinated expression of these interacting gene products are essential for optimal organism function. Incompatibilities between mtDNA and nuclear-encoded mitochondrial genes results in inefficiency in electron flow down the respiratory chain, differential oxidative phosphorylation efficiency, increased release of free radicals, altered intracellular Ca2+ signaling, and reduction of catalytic sites and ATP production. This review explores the role of mitonuclear incompatibility in BD susceptibility and resilience against environmental stressors.
Introduction
Mitochondria are multifunctional organelles found in large numbers in almost all human cells. They are highly effective in producing more than 90% of the cellular energy needed by the body in the form of ATP (Wallace, 2005). Efficient mitochondrial functioning depends on synchronized gene expression and protein interactions transcribed from both the mitochondrial (mtDNA) and nuclear (nDNA) genomes. This synchronization between genomes is essential for energy production needed to sustain life and organ function in addition to regulating a wide range of cellular pathways including homeostatic regulation of ionic gradients, cell signaling, and cellular stress response and survival (Mattson et al., 2008). The mitochondria genome encodes 13 proteins of the electron transport chain (ETC), 22 transfer RNAs, and 2 ribosomal RNAs (Anderson et al., 1981). Other mitochondrial proteins are encoded in the two copies nDNA. Individual mitochondrion typically include 2–10 mtDNA copies, while the number of mitochondria per cell varies up to 1,000 (Robin and Wong, 1988) depending on energy demands, oxidative stress and pathological conditions (Clay Montier et al., 2009).
Mitochondrial function is particularly important in neurons due to the high bioenergetic demands of neuronal activities (Chan, 2006; Mattson et al., 2008). Mitochondrial functioning plays an important role in regulating synaptic transmission, brain function, and cognition (Picard and McEwen, 2014). Neuronal activity is energy-dependent, and neurons are particularly sensitive to changes in bioenergetic fluctuations, suggesting that mitochondria regulate fundamental aspects of brain function. Mitochondrial deficits including energy production, oxidative stress, oxidation-reduction (redox) imbalance, and Ca2+ metabolism, have been implicated in the etiology of psychiatric diseases (Manji et al., 2012; Park and Park, 2012; Srivastava et al., 2018). It has long been postulated that disruption of mitochondrial function contributes to the etiology of bipolar disorder (BD). Kato and Takahashi (1996) and Kato et al. (1997a, b) demonstrated leukocytes and autopsied brains from BD individuals exhibited increased levels of the 4,977-bp mtDNA deletion, as well as abnormal brain energy metabolism in BD including decreased intracellular pH (Kato et al., 1993, 1998; Hamakawa et al., 2004), decreased phosphocreatine (Kato et al., 1992, 1994, 1995), and enhanced response of phosphocreatine in lithium-resistant BD (Murashita et al., 2000). These findings led to the mitochondrial dysfunction hypothesis of BD in 2000, theorizing that “mtDNA mutations as well as common variations may confer a risk of BD by affecting intracellular calcium signaling systems” (Kato and Kato, 2000). Since then, mitochondrial abnormalities associated with bipolar disorders have been extensively reviewed at the structural, molecular and functional levels (Kato and Kato, 2000; Kato, 2005, 2006, 2008, 2017; Stork and Renshaw, 2005; Quiroz et al., 2008; Shao et al., 2008; Clay et al., 2011; Konradi et al., 2012; Kim et al., 2015; Duong et al., 2016; Machado et al., 2016; Scaini et al., 2016; Cikankova et al., 2017; Morris et al., 2017; Andreazza et al., 2018; Holper et al., 2018; Srivastava et al., 2018), as well potential therapeutic strategies (Kato, 2007; Wang, 2007; Quiroz et al., 2008; Nierenberg et al., 2013; de Sousa et al., 2014a; Callaly et al., 2015; Cikankova et al., 2017; Pereira et al., 2018).
While there is mounting evidence supporting the mitochondrial dysfunction hypothesis of BD, recent BD GWAS have failed to show significant association with mtDNA and/or nDNA mitochondria related genes (Prata et al., 2019; Stahl et al., 2019). The closest BD risk allele to a mitochondrial core gene is rs74446114, located 63.5 kb downstream of MRPL33, which encodes Mitochondrial Ribosomal Protein L33 (Stahl et al., 2019). A greater understanding of the core evolutionary processes driving mitonuclear and environmental interactions will provide key insights into the complex etiology underlying mitochondrial dysfunction in BD.
Co-Evolution and Co-Adaptation of Mitonuclear Genomes
Mitochondria are double-membrane structures with their own maternally inherited, circular genome (16,568 bp) consisting of transcription, translation, and protein assembly systems conserved from its endosymbiotic origin (Anderson et al., 1981; Gray et al., 1999). In the course of evolution, mitochondria became dependent on the nuclear genome for nuclear-encoded factors necessary for its integrity, replication and expression (Gray et al., 1999). While 13 mitochondrial proteins are encoded by mtDNA, approximately 1,158 mitochondrial proteins are encoded by nDNA and imported into the mitochondrion (Poyton and McEwen, 1996; Calvo et al., 2016). Studies suggest that mitochondrial and nuclear genomes co-evolve to ensure the co-adaptation and coordinated expression of interacting proteins vital for mitochondrial biosynthesis and organism survival (Cosmides and Tooby, 1981; Bar-Yaacov et al., 2012).
Mitonuclear allelic interactions are essential to maintain evolutionary co-adaptation. Nuclear genes coding for mitochondrial-related proteins that co-function with mtDNA gene products, must co-evolve with arising deleterious mtDNA variants (Hill, 2016). If a mutation arises in mtDNA, the nuclear genome needs to adapt (i.e., nuclear mutation needs to arise to off-set the mtDNA mutation) in order to restore the synergistic interactions between these two genomes. MtDNA are more susceptible to SNPs, indels, and structural changes compared to their nDNA counterparts, as it has a higher mutation rate, lacks recombination, and has an inefficient DNA repair mechanism. As deleterious mutations reach a frequency threshold, the host fitness becomes compromised. As these deleterious mtDNA mutations spread through a population, other mutations accumulate to restore the host fitness by either directly compensating the effect of the mtDNA mutation in the nDNA or by inducing weak compensatory mutations in both genomes to try to restore fitness through selection (see Chou and Leu, 2015 for review). This mitonuclear co-evolution results in differential accumulation of population-specific mitonuclear mutations over time. While some studies suggested that most mtDNA variants in populations are selectively neutral to prevent removal by selective pressure (Gregorius and Ross, 1984; Wallace et al., 1999), the question remains whether this selective neutrality is dependent on the ancestral nuclear background.
Mitonuclear genomes are under the regulation of retrograde and anterograde “cross-talk” signals to modulate mitochondrial function (Figure 1), most often originating from the mitochondria to induce specific nuclear responses to regulate mitochondrial structure and function (Quiros et al., 2016). Mitochondria generate retrograde response signals to the nucleus to modulate nDNA gene expression in response to mtDNA mutations and to maintain cellular function, metabolism, and adaptation to environmental stressors such as diet, exercise, and temperature (Quiros et al., 2016). Alternatively, anterograde regulation sends signals from the nucleus to the mitochondria in order to regulate mitochondrial activity and promote mitochondrial biogenesis through cell growth and survival. Anterograde signaling can modulate bioenergetics based on cellular demands via mitochondrial fusion/fission to regulate mitochondrial copy number, morphology, mitophagy, and movement (Chen and Chan, 2009; Chan, 2012; Quiros et al., 2016). Mutations in any of these essential components may disrupt cross-talk between mitonuclear genomes, affecting mitochondrial stability and function, and ultimately contributing to diseases (Poyton and McEwen, 1996).
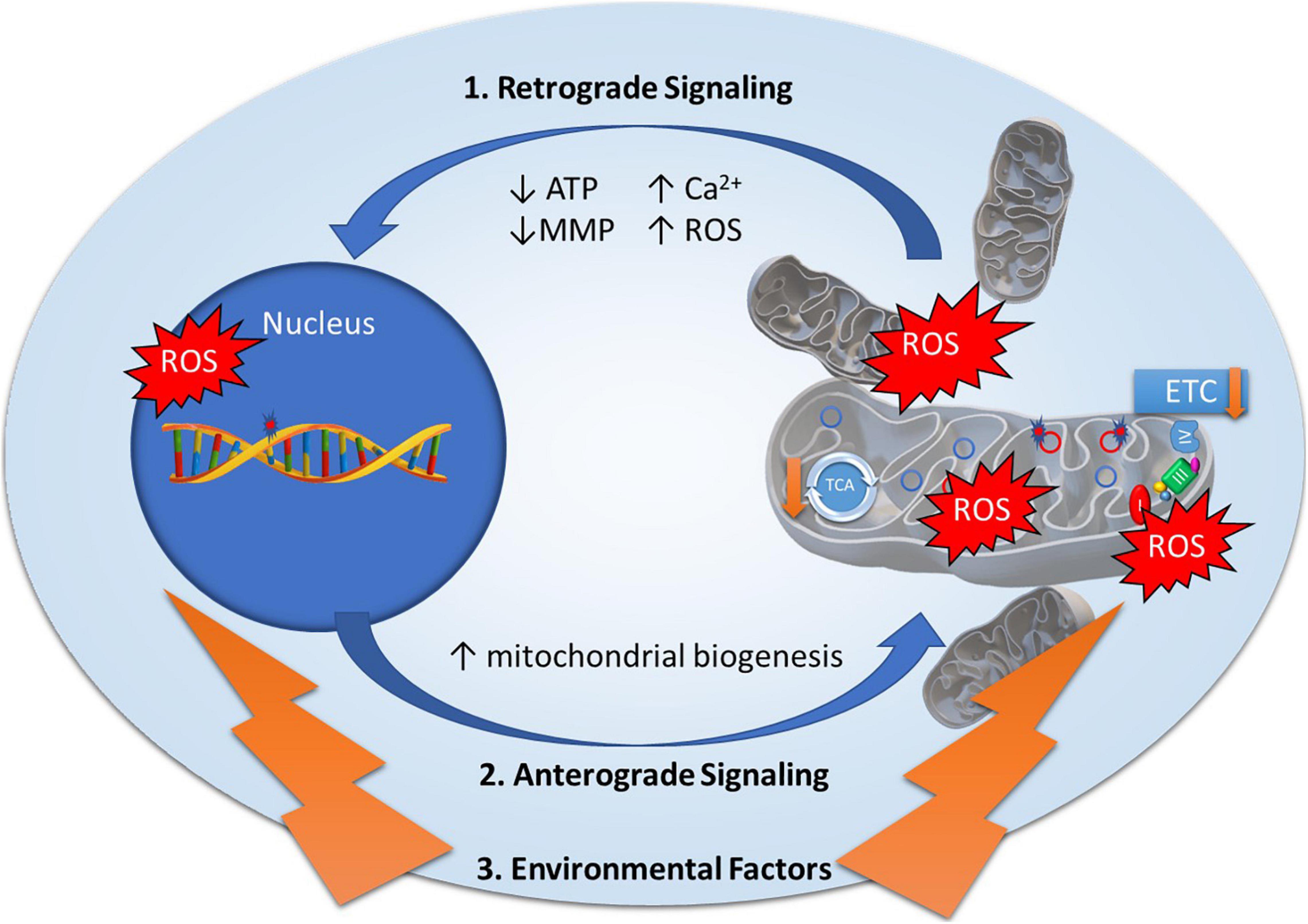
Figure 1. Mitonuclear communication in bipolar disorder. Mitochondrial dysfunction in bipolar disorder has been attributed to genetic variants in mitochondrial DNA and nuclear-encoded mitochondrial genes as well as decreased electron transport chain (ETC) and tricarboxylic acid (TCA) cycle activity. Mitochondrial function is under the regulation of retrograde and anterograde signals and adaptive responses to environmental factors. 1. Retrograde signaling (mitochondria to nucleus) activated by mitochondrial stress signals including Adenosine Triphosphate (ATP), Reactive Oxygen Species (ROS), Mitochondrial Membrane Potential (MMP), and calcium levels (Ca2+) to regulate nuclear gene expression to restore mitochondrial homeostasis. 2. Anterograde signaling (nucleus to mitochondria) are nucleus-controlled regulation of gene expression to modulate mitochondrial activity, mitochondrial biogenesis, and mitochondrial fusion/fission. 3. Environmental factors such as diet, exercise, and temperature elicit adaptive responses to modulate mitochondrial stress signals and expression of nuclear encoded mitochondrial related genes.
It has been suggested that population admixture may introduce mitonuclear discordance, in which mitochondrial and nuclear genomes originate from differentiated ancestries. Chou and Leu (2015) argued that if co-evolution in mtDNA and nDNA commonly occur in diverse populations, the balance between deleterious mtDNA mutations and compensatory nDNA variants may be dependent on their nuclear ancestral background, which may lead to genetic incompatibility between populations (Chou and Leu, 2015; Rand and Mossman, 2020). Individuals who have ancestors from different continental populations may have certain mtDNA genetic variants from one continental population, but might not have the corresponding adaptive mutation in the nuclear genome from the same continental population. Zaidi and Makova (2019) recently reported a significant increase in the local genetic ancestry at mitochondrial related genes in nDNA toward the corresponding mtDNA ancestry in African American and Native American admixed populations, providing empirical evidence that selective pressure helps restore mitonuclear compatibility in human admixed populations.
The evidence for differential lifetime prevalence of BD based on race/ethnicity is conflicting (Grant et al., 2005; Substance Abuse and Mental Health Services Administration, 2020) and specific prevalence of BD in admixed individuals are lacking. However, results from the 2019 National Survey on Drug Use and Health reports that the prevalence of any mental illness (AMI) and severe mental illness (SMI) is highest among adults reporting two or more races (AMI = 31.7% compared to 22.2% White adults; SMI = 9.3% compared to 5.7% White adults) (Substance Abuse and Mental Health Services Administration, 2020), suggesting admixed individuals are at higher risk for SMI, including BD. SMI is also higher in American Indian/Alaska Natives (6.7%) compared to White adults (Substance Abuse and Mental Health Services Administration, 2020). Lifetime mood disorders are significantly more prevalent among the American Indian/Alaska Natives population compared to Whites, with American Indian/Alaska Natives men showing greater trend toward BDI and women toward BDII (Brave Heart et al., 2016). American Indian/Alaska Natives admixed with two or more races reported a higher lifetime prevalence of diagnosed depressive disorder, more days of poor mental health, and more frequent reporting of mental distress compared to both American Indian/Alaska Natives-single race and White-single race groups. These results remained significant after adjustment for sociodemographic covariates (Asdigian et al., 2018), suggesting that increased prevalence mood disorders and related clinical characteristics reported in American Indian/Alaska Natives may be caused in part by increased population admixture.
While current evidence of mitonuclear discordance stems from crosses of various model organisms (Ballard, 2005; Kurbalija Novicic et al., 2015; Mossman et al., 2016b; Loewen and Ganetzky, 2018), human cytoplasmic hybrids (cybrids) (Esteves et al., 2008; Lin et al., 2012; Silva et al., 2013; Kenney et al., 2014a; Nashine et al., 2016; Shrivastava and Subbiah, 2016; Zhou et al., 2017) and conplastic animals (Yu et al., 2009; Weiss et al., 2012; Kumarasamy et al., 2013; Houstek et al., 2014; Latorre-Pellicer et al., 2016) studies are increasingly being used to demonstrate the mitonuclear effects of mtDNA variants in different nuclear ancestral backgrounds. Cybrids and conplastic animals are constructed using identical nuclei and external mitochondria with different mtDNA haplogroups in order to investigate resulting bioenergetic and physiological effects. These models also allow for investigations into environment-specific modulation of intraindividual mitonuclear interactions (Dowling et al., 2007; Soh et al., 2007; Zhu et al., 2014; Mossman et al., 2017; Rand et al., 2018; Dong et al., 2019).
Oxidative Phosphorylation Dysregulation in Bipolar Disorder
Mitochondria generate cellular energy through the OXPHOS system via the ETC, consisting of five multiprotein complexes (complex I–V) within and across the inner mitochondrial membrane. Thirteen ETC proteins are encoded by mtDNA genes, while > 1,000 proteins that make up the mitochondria are encoded by genes in the nuclear genome (Mattson et al., 2008; Calvo et al., 2016). OXPHOS involves respiration, an oxidative exergonic pathway, which leads to the phosphorylation of ADP into ATP. OXPHOS dysregulation has been previously reported in BD (Konradi et al., 2004; Iwamoto et al., 2005; Sun et al., 2006; Beech et al., 2010). Konradi et al. (2004) reported a significant decrease in mRNA expression levels of mitochondrial genes in the hippocampus of BD subjects, including genes regulating OXPHOS and proteasomal degradation. Iwamoto et al. (2005) found that 15.1% of 676 mitochondria-related genes were differentially expressed in BD. However, they concluded the reported global downregulation of mitochondrial genes was possibly a consequence of pharmaceutical effects, as BD medication-naive patients exhibited an up-regulation of a subset of 27 mitochondrial-related genes, including COX15, UQCRC2, ETFDH, and NDUFS1 involved in mitochondrial respiratory chain (Iwamoto et al., 2005). Using a modified Gene Set Enrichment Analysis (GSEA) computational screen to estimate collaborative candidate pathways co-expressed with OXPHOS dysregulation related to BD pathogenesis, Sawai et al. (2015) suggested that aberrant mitochondrial OXPHOS may induce dysregulation in the ubiquitin-proteasome pathway. Another study reported susceptibility to bioenergetic alterations within distinct brain regions in BD. They reported significant reductions in mitochondrial function in the PFC of patients with BD and increase in function in the hypothalamus and cerebellum, suggesting that mitochondrial dysfunction in brain specific regions may lead to increased oxidative stress, thus triggering compensatory mechanisms to offset oxidative damage of mtDNA (Bodenstein et al., 2019).
While several studies have reported associations of BD with various mtDNA variants and mitochondrial haplogroups (Kato et al., 2001; Munakata et al., 2004; Kazuno et al., 2009; Rollins et al., 2009; Frye et al., 2017), no mtDNA variants have reached GWAS thresholds of significance. In addition, mitonuclear co-adaptation could result in spurious associations of disease with mtDNA variants due to population stratification (Hagen et al., 2018). Several researchers have postulated that investigations focused on mitonuclear epistatic interactions may reveal additional genes associated with BD risk. Ryu et al. (2018) reported mitonuclear epistatic allelic interactions with nominal evidence for BD risk and/or early-onset BD based on European American GWAS data from the GAIN BD study. They reported nominal evidence of epistatic associations involving mitochondrial encoded ETC complex I genes mt-CytB and mt-ND4 with BD risk and risk of early onset BD. Mitonuclear epistatic allelic interactions associated with BD risk were reported for mt-CytB and MGAM, mt-CytB and AK5, and mt-ND4 and MGAM. For early onset BD risk among BD patients, the top association signals were detected in mt-CytB and CTNNA2, mt-CytB and IL34, and mt-ND4 and IL34 interactions (Ryu et al., 2018). Schulmann et al. (2019) reported on the association between the risk of SZ and mitonuclear allelic interactions in an Irish cohort. They reported 34 mitonuclear epistatic allelic interactions with significant joint effects between 10 mitochondrial and 21 nuclear genes, where six of the mitochondrial genes encoded ETC complex I components (Schulmann et al., 2019).
Several cybrid studies have utilized mtDNA-naive ρ0 human cells fused with platelets resulting in cell lines with identical nuclei and different mtDNA haplogroups in order to evaluate mtDNA variant and haplotype effects on mitochondrial energy production (Kenney et al., 2014a, b; Zhou et al., 2017). Zhou et al. (2017) reported several mtDNA SNPs associated with differential endogenous mitochondrial respiratory activity and uncoupled mitochondrial respiration. Different East Asian haplogroups exhibited differential effects on mtDNA copy number (mtDNAcn), transcription efficiency, complex III expression, and coupled and uncoupled mitochondrial respiration (Zhou et al., 2017). Kenney et al. compared OXPHOS related gene expression of mtDNA-encoded respiratory complex genes between West Asian and European mitotype cybrids (J and H haplotypes, respectively). West Asian mitotype cybrids exhibited lower OXPHOS utilization compared to European mitotype cybrids. They also compared cybrids with African, European, and West Asian mitotypes haplogroups (L, H, and J haplotypes, respectively). Haplogroups exhibited unique bioenergetic profiles with differential effects on mtDNAcn, OXPHOS gene expression, ATP turnover, and spare respiratory capacity. African mitotype cybrids displayed more efficient OXPHOS system, as evidenced by higher expression levels of mtDNA-encoded respiratory complex genes, decreased ATP turnover rates and lower reactive oxygen species (ROS) production (Kenney et al., 2014b). Cybrids with European mitotype had elevated ATP production and decreased lactate levels compared to West Asian mitotypes cybrids (Kenney et al., 2013). West Asian mitotypes cybrids exhibited altered bioenergetic profiles compared with European mitotype cybrids, including significantly decreased expression in mtDNA encoded respiratory genes encoded by mtDNA. In addition, West Asian and European mitotype cybrids had significantly altered expression of eight nuclear genes involved in inflammatory response and apoptosis (Kenney et al., 2013, 2014a). While these studies proposed that effects are dependent on mitochondrial haplotypes, the underlying ancestries of the ρ0 cells used in these studies must also be taken into consideration. For example, Zhou et al. (2017) utilized 143B ρ0 cells which are derived from an individual with an admixed nuclear genetic ancestral background consisting of South European (59%), South Asian (37%), and South East Asian (4%) genomic ancestries (Dutil et al., 2019) and compared the effects of differing East Asian mtDNA genetic backgrounds. Differences in OXPHOS and mitochondrial bioenergetics may reflect disruptions in co-adaptation of population specific mitonuclear interactions rather than effects of mitochondrial variants or haplotypes alone.
While the use of human cybrids are commonly used to study mitochondrial dysfunction in neurodegenerative disorders such as Alzheimer’s (Onyango et al., 2006; Yu et al., 2016, 2017; Weidling et al., 2020), and Parkinson’s disease (Keeney et al., 2009; Cronin-Furman et al., 2013; Reeve et al., 2015; Pignataro et al., 2017), there is a paucity of cybrid research in BD and other psychiatric disorders. Munakata et al. (2004) reported that the m.3644T > C variant in the mt-ND1 gene was associated with BD and 3644C cybrids exhibited mitochondrial dysfunction including decreases in mitochondrial membrane potential (MMP) and complex I activity compared to 3644T cybrids. Verma et al. (2016) determined that cybrids developed by fusing platelets from ADHD patients with SH-SY5Y ρ0-cells resulted in altered mitochondrial bioenergetics including significant decreases in mitochondrial respiration, ETC complex V activity, MMP, and increases in oxidative stress. A recent study reported potential therapeutic benefits of mitochondrial transplantation in SZ. Robicsek et al. (2018) demonstrated that the transfer of healthy mitochondria into SZ-iPSC differentiated neurons and rat SZ-models increased mtDNAcn and improved cellular respiration, MMP, mitochondrial distribution, and neuronal differentiation.
Compelling evidence in animal models suggests that population admixture with differing mtDNA can intensify oxidative stress and DNA damage, affect fitness, and bioenergetic activity due to mitonuclear discordance, and these effects can be modulated by environmental factors including temperature, diet, and exercise. In copepod (Tigriopus californicus) populations, Barreto and Burton (2013) studied 28 hybrid strains derived from 12 parental lines. Hybrids displayed a significant decrease in fitness and enhanced oxidative stress compared to the parental strains. In addition, admixed populations that showed evidence of decreased fecundity exhibited enhanced oxidative stress compared to those with no discernable change in fitness. Hybrid strains with high levels of mitonuclear discordance exhibited significant increases oxidative DNA damage, while hybrid lines with low mitonuclear discordance showed no significant difference from parental lines (Barreto and Burton, 2013).
Baris et al. (2016) reported persuasive evidence that natural selection to variable thermal environments maintains mitonuclear interactions regulating OXPHOS metabolism in admixed populations of mummichog fish (Fundulus heteroclitus) with northern or southern mitochondrial haplotypes. Effects of acute temperature change were dependent on northern and southern mitochondrial haplotypes. While fish with the southern mt-haplotype had acute effects in OXPHOS metabolism at low and high acclimation temperatures, little to no acute temperature effect was reported with fish with the northern mt-haplotype. Significant differential responses to acute temperature change were dependent on mitochondrial haplotypes at high acclimation temperatures for ADP stimulated respiration, mitochondrial uncoupling, and complex I and II respiration (Baris et al., 2016). Mummichog fish with a high level of mitonuclear discordance with northern mt-haplotype exhibited enhanced OXPHOS efficiency while those with a southern mt-haplotype showed decreased OXPHOS efficiency (Baris et al., 2017).
Environmental factors have also been shown to modulate mitonuclear responses in Drosophila. Hoekstra et al. (2013) crossed D. simulans with D. melanogaster to create four distinct mitonuclear genotype strains by combining two mtDNA alleles with two nuclear alleles previously shown to exhibit epistatic mitonuclear interactions (Montooth et al., 2010). One of the mitonuclear genotype strains resulted in delayed development, decreased fitness, height reduction, and inefficient bioenergetics that were directly proportional to temperature (Hoekstra et al., 2013). Montooth et al. (2019) also demonstrated that mitonuclear incompatibility in Drosophila adversely affects fertility in males developed at warmer temperatures and these deleterious effects could be partly reversed by diet. Dowling et al. (2007) reported significant mitonuclear interactions contributed to adult female fitness in a D. melanogaster population, in which main effects could not be attributed to mitotype or nuclear background alone. They also reported that minor variations in environment including temperature, humidity, and diet contributed to mitonuclear effects on relative fitness (Dowling et al., 2007). Soh et al. (2007) studied the effects of dietary intake on normal and extended longevity strains as well as introgressed mitonuclear hybrid strains in Drosophila. The extended longevity strains exhibited lower levels of ROS, enhanced mitochondrial bioenergetic efficiency, heightened antioxidant response, and reduced oxidative damage (Arking et al., 2002). Nutrition levels resulted in differential longevity dependent on mitonuclear combinations. Hybrids with the more efficient “extended longevity” mitochondria exhibited enhanced longevity in males but decreased longevity females under non-starvation nutritional diets (Soh et al., 2007). A study utilizing admixed D. simulans strains with differing mitotypes from mitonuclear compatible control strains also concluded that compensation for mitonuclear discordance may be temperature-dependent (Pichaud et al., 2013).
Investigations involving introgressive hybridized seed beetles (Callosobruchus maculatus), also investigated mitonuclear and environmental effects on fitness. Three mitochondrial haplotypes were backcrossed with nuclear genetic backgrounds from Brazil, California, and Yemen. The three mitotypes displayed significant differences in relative fitness. Strains with concordant mitonuclear genomes exhibited increased relative fitness compared to strains with differing mitonuclear genomes. At colder temperatures, Brazilian and Yemen mitotypes introgressed with California nuclear genomes showed opposite effects in fitness, again suggesting that temperature contributes to mitonuclear effects on relative fitness (Immonen et al., 2020).
Kumarasamy et al. (2013) constructed conplastic rat strains using two common rat models for cardiovascular disease, in which the mtDNA of the Dahl salt-sensitive (S) rat was exchanged with the mtDNA of the spontaneously hypertensive rat (SHR) and vice versa. The S.SHRmt and SHR.Smt conplastic strains displayed significant increases in mtDNAcn, decreases in mitochondrial ROS levels, and increases in aerobic fitness and survival compared to the S rat. The S.SHRmt conplastic strain also showed significant increases in mtDNAcn compared to the S rat (Kumarasamy et al., 2013). In toto, these results indicate that cellular bioenergetics, mtDNAcn, transcription efficiency, fitness, and survival are greatly affected by mitonuclear interactions modulated by environmental factors.
Altered Mitochondrial Calcium Regulation in Bipolar Disorder
Mitochondria play a critical role in intracellular Ca2+ homeostasis and Ca2+-mediated signaling processes (Mattson et al., 2008). Mitochondrial dysregulation is more pronounced in neurons, as the brain is the largest source of energy consumption due to the high bioenergetic demands of neuronal functions, and can lead to an imbalance in calcium homeostasis (Chan, 2006; Mattson et al., 2008). Mitochondria regulate rapid changes in intracellular Ca2+ dynamics. Increases in intracellular Ca2+ and uptake into the mitochondria matrix results in altered mitochondrial membrane permeability, ETC efficiency, and Ca2+-mediated signaling processes modulating gene expression, leading to dysregulation of several neuronal systems including neurotransmitter release, regulation of neuronal action potentials, dendritic developmental and remodeling, synaptic plasticity, regulation of gene expression, and apoptosis (Greer and Greenberg, 2008; Feissner et al., 2009; Peng and Jou, 2010; Trevelyan et al., 2010; Srivastava et al., 2018), and can alter neuronal function through the production ROS (Sena and Chandel, 2012).
Altered mitochondrial calcium regulation may contribute to the pathophysiology of neuropsychiatric disorders including BD (Kato et al., 2003; Kazuno et al., 2006; Kubota et al., 2006; Kato, 2007, 2008). Mertens et al. (2015) reported direct correlations between mitochondria defects, altered Ca2+ signaling, and hyperexcitability of neurons in BD. Hippocampal neurons differentiated from iPSCs from BD lithium responders and non-responders exhibited enhanced mitochondrial function as evidence by higher MMP and upregulated mitochondrial gene expression compared to control neurons. Ca2+ signaling pathways were altered in BD neurons compared to controls. Based on patch-clamp recording and somatic Ca2+ imaging in BD neurons, increased mitochondrial activity resulted in neuronal hyperexcitability, which was reduced by lithium in neurons from BD lithium responders, but not in BD non-responders (Mertens et al., 2015).
Kazuno et al. (2006) examined the phenotypic effect of mtDNA using calcium indicator cybrids 143B.TK–ρ0206 that stably expresses two ratiometric fluorescent proteins. Using these cybrids, they identified two mtDNA variants which altered mitochondrial pH and calcium concentration. In fluorescent images of cybrids, mitochondria exhibited increased calcium concentrations and pH in the mitochondrial matrix compared to cytosolic levels, suggesting m.10398A in the mt-ND3 gene of complex I and m.8701A in the ATPase6 gene of complex V alter mitochondrial pH and intracellular calcium homeostasis. Cytosolic calcium response to histamine stimulation also differed between cybrid cells carrying the m.10398A and m.8701A variants (Kazuno et al., 2006). The m.10398A mutation has been reported to be associated with BD (McMahon et al., 2000; Kato, 2001; Kato et al., 2001; Washizuka et al., 2003), as well as valproate (Kazuno et al., 2008) and lithium response (Washizuka et al., 2003), which suggests that these variants may contribute to the etiology of BD.
Mitochondrial Dna Copy Number in Bipolar Disorder
Alterations in mtDNAcn signify changes in mitochondrial bioenergetics, energy demands, and compensatory effects (Picard et al., 2018). Several studies have found altered mtDNAcn in BD patients compared to controls. Significantly lower mtDNAcn have been observed in BD leukocytes (Chang et al., 2014; Yamaki et al., 2018; Wang et al., 2020), postmortem hippocampus (Fries et al., 2019), and left frontopolar cortex (Tsujii et al., 2019). Chung et al. (2020) reported lower mtDNAcn in BDI patients compared to higher mtDNAcn in BDII patients compared to controls. However, Yamaki et al. (2018) reported that both BDI and BDII patients had significantly decreased mtDNAcn versus controls. In contrast, de Sousa et al. failed to find significant differences in mtDNAcn in BD subjects compared to controls or before and after lithium treatment. They did, however, report a trend for decreased mtDNAcn in BDI compared to controls and BDII (de Sousa et al., 2014b). Wang et al. (2018) reported a significant reduction in leukocyte relative mtDNAcn in BD patients with manic and depressive symptoms compared to controls. There were no significant changes in mtDNAcn between euthymic BD patients and controls. In addition, they found that mtDNAcn was negatively correlated with total number of manic episodes (Wang et al., 2018). While higher levels of mtDNAcn in BD have also been reported (Fries et al., 2017), a meta-analysis for previous BD-mtDNAcn studies (Chang et al., 2014; de Sousa et al., 2014b; Fries et al., 2017; Wang et al., 2018; Yamaki et al., 2018) did not find any significant association with mtDNAcn, although mtDNAcn was significantly decreased in BD patients in an Asian-specific meta-analysis (Yamaki et al., 2018). A post-mortem study of suicide completers reported higher mtDNAcn in peripheral blood and decreased mtDNAcn in post-mortem PFC samples from suicide victims versus controls (Otsuka et al., 2017).
Zaidi and Makova reported that mtDNAcn was negatively correlated with mitonuclear discordance across mtDNA haplogroups of different geographic origins, consistent with mitonuclear incompatibility in admixed individuals. The authors also reported enrichment of ancestry at nuclear-encoded mitochondrial genes in African Americans and Puerto Ricans toward the corresponding ancestry of mtDNA haplogroups, suggesting a compensatory method of selection in restoring mitonuclear compatibility (Zaidi and Makova, 2019).
Human cybrid lines also show variation in mtDNAcn. Cybrids containing mtDNA variants m.249del, m.13708A, m.13928C, and m.16304C resulted in a decrease in mtDNAcn, while cybrids encompassing variants m.489C, m.8701G, m.10398G, and m.10400T resulted in increased mtDNAcn. The majority of these SNPs are diagnostic of a specific mtDNA haplogroup in East Asia mtDNAcn (Zhou et al., 2017). Variants m.13708A and m.13928C displayed decreased mtDNAcn compared to other SNPs, and have been previously associated with increased risk of disease (Yu et al., 2008; Korkiamaki et al., 2013), while m.489C, m.10398G, and m.10400T exhibit increased mtDNAcn and have been reported to have protective effects against disease (Darvishi et al., 2007).
BD environmental stressors have also been associated with alterations mtDNAcn. Lower mtDNAcn have been reported in response to maternal psychosocial stress, pollution, poor dietary (Ma et al., 2020) and inactive lifestyles, and tobacco use (Brunst et al., 2017; Wong et al., 2017; Kaali et al., 2018; Revesz et al., 2018; Breton et al., 2019; Wu et al., 2019; Duan et al., 2020; Hu et al., 2020; Vyas et al., 2020). In contrast, prior evidence indicates an increase in mtDNAcn in relation to history of early life stress and childhood trauma (Cai et al., 2015; Tyrka et al., 2016; Ridout et al., 2019, 2020). Discrepancies in the literature pertaining to mtDNAcn alterations in both BD and environmental modulators, specifically stressful life events, may be explained in part by tissue-specific alterations. Baek et al. (2019) investigated the effects of chronic immobilization stress in C57BL/6 male mice on mtDNAcn in 12 tissues. While they reported that chronic stress resulted in an increase in mtDNAcn in leukocytes, mtDNAcn was significantly decreased in prefrontal cortex, suggesting that mtDNAcn variability is tissue-specific (Baek et al., 2019).
Discussion
While mtDNA variants can contribute to disease in humans (Taylor and Turnbull, 2005; Tuppen et al., 2010) and mtDNA haplogroups have been reported to associate with various disorders including psychiatric (Magri et al., 2007; Kazuno et al., 2009; Rollins et al., 2009), cardiovascular (Benn et al., 2008; Veronese et al., 2019), and metabolic diseases (Chinnery et al., 2007; Tavira et al., 2014; Mitchell et al., 2017; Zhao et al., 2019), these studies fail to encompass the complexity surrounding mitonuclear epistasis and environment interactions (Mossman et al., 2016a). Mitonuclear genetic variation embodies extraordinarily complex epistatic, pleiotropic and GxE interactions that function in non-additive ways to modify organismal fitness and survival. It has been argued that models optimized to study mitonuclear and environmental interactions are needed to better understand the underlying genetic architecture of complex phenotypes and to evaluate the nature of the polygenic models governed by mitonuclear cross-talk (Friedman and Nunnari, 2014; Chou and Leu, 2015; Quiros et al., 2016; Rand and Mossman, 2020).
This review postulates that disruptions in the co-evolution of the mtDNA and nuclear genomes, in addition to environmental factors, leads to mitochondrial impairment and increased risk of BD. Identification of the evolutionary mechanisms and genetic etiology underlying mitochondrial dysfunction is important in order to better understand the pathophysiology of the disease, to identify novel biomarkers of BD, to develop medications for treatment of BD, and to improve diagnostic predictive testing in diverse populations. Future BD research utilizing human cybrid lines, conplastic animals, and admixed human populations offer promising advancements in the field.
Experiments with human cybrids have shown that mtDNA representing populations from different geographic origins have differential OXPHOS efficiency and may play a role in susceptibilities to diseases (Lin et al., 2012; Kenney et al., 2014a, b). Incompatibilities between mtDNA and nuclear-encoded mitochondrial genes results in inefficient electron flow down the respiratory chain leading to increased release of free radicals, and a reduction of catalytic sites and ATP production (Lane, 2011; Meiklejohn et al., 2013). Mitochondrial haplogroups accumulate population-specific mtDNA variants which represent ancestral origins. Human cybrids studies using identical nuclei and common BD haplogroups can be used to investigate and potentially identify deleterious effects of mtDNA variants underlying bioenergetic deficiencies, altered OXPHOS metabolism, and deficient Ca2+ homeostasis when they are introduced into a new nuclear ancestral background lacking population specific nDNA suppressors.
Conplastic animal models can also help elucidate how mtDNA variation influences organismal pathophysiology and mitochondrial functioning. Previous studies investigating physiological and phenotypic variability influenced by mitochondria have reported that different mtDNA haplotypes can influence mitochondrial functioning, cellular processes, and behaviors that have been previously implicated in BD. These include the regulation of OXPHOS enzyme levels, reduced cellular ATP levels, pathological morphology of mitochondria, ROS generation, impaired glucose tolerance, reduced glucose-induced insulin secretion, and increased anxiety-related behavior, resulting in significant differences in health and longevity between various conplastic strains (Yu et al., 2009; Weiss et al., 2012; Houstek et al., 2014; Latorre-Pellicer et al., 2016).
Admixed populations, such as Hispanic and African Americans, also provide a unique opportunity to examine the role of mitonuclear discordance in BD, as they are genetically heterogeneous with varying proportions of European, African, and Native American ancestries (Bertoni et al., 2003). Therefore, mitonuclear incompatibility in these populations represents a possible disruption of 20,000–150,000 years of co-evolution between mitonuclear genomes. The increased genetic diversity in human admixed populations due to divergent underlying ancestral populations of mtDNA and nDNA may lead to increased collection and sharing of GWAS, transcriptome, and mtDNA sequence data from under-represented minority populations.
Research methods designed to capitalize on mitonuclear interactions and detect incompatibilities as outlined in this review can be applied to other psychiatric disorders, as mitochondrial dysfunction is well documented in schizophrenia (Goncalves et al., 2015; Hjelm et al., 2015; Monpays et al., 2016; Ben-Shachar, 2017; Ni and Chung, 2020; Roberts, 2020), depression (Bansal and Kuhad, 2016; Allen et al., 2018; Caruso et al., 2019; Forester et al., 2019), autism (Rossignol and Frye, 2012; Goh et al., 2014; Varga et al., 2018; Bennuri et al., 2019; Citrigno et al., 2020; Frye, 2020; Gevezova et al., 2020), and anxiety (Chakravarty et al., 2013; Khalifeh et al., 2016; Babenko et al., 2018). The studies outlined in this review suggest that mitochondrial dysfunction underlying BD and other psychiatric disorders with genetic overlap may be the consequence of mitonuclear incompatibility, and may be enhanced in admixed populations with mitonuclear genomes originating from different ancestral populations (Chou and Leu, 2015).
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work was supported by the Penn State College of Medicine, Hershey, PA.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Allen, J., Romay-Tallon, R., Brymer, K. J., Caruncho, H. J., and Kalynchuk, L. E. (2018). Mitochondria and mood: mitochondrial dysfunction as a key player in the manifestation of depression. Front. Neurosci. 12:386. doi: 10.3389/fnins.2018.00386
Anderson, S., Bankier, A. T., Barrell, B. G., de Bruijn, M. H., Coulson, A. R., Drouin, J., et al. (1981). Sequence and organization of the human mitochondrial genome. Nature 290, 457–465. doi: 10.1038/290457a0
Andreazza, A. C., Duong, A., and Young, L. T. (2018). Bipolar disorder as a mitochondrial disease. Biol. Psychiatry 83, 720–721. doi: 10.1016/j.biopsych.2017.09.018
Arking, R., Buck, S., Novoseltev, V. N., Hwangbo, D. S., and Lane, M. (2002). Genomic plasticity, energy allocations, and the extended longevity phenotypes of Drosophila. Ageing Res. Rev. 1, 209–228. doi: 10.1016/s1568-1637(01)00010-1
Asdigian, N. L., Bear, U. R., Beals, J., Manson, S. M., and Kaufman, C. E. (2018). Mental health burden in a national sample of American Indian and Alaska Native adults: differences between multiple-race and single-race subgroups. Soc. Psychiatry Psychiatr. Epidemiol. 53, 521–530. doi: 10.1007/s00127-018-1494-1
Babenko, V. N., Smagin, D. A., Galyamina, A. G., Kovalenko, I. L., and Kudryavtseva, N. N. (2018). Altered Slc25 family gene expression as markers of mitochondrial dysfunction in brain regions under experimental mixed anxiety/depression-like disorder. BMC Neurosci. 19:79. doi: 10.1186/s12868-018-0480-6
Baek, J. H., Son, H., Jeong, Y. H., Park, S. W., and Kim, H. J. (2019). Chronological aging standard curves of telomere length and mitochondrial DNA copy number in twelve tissues of C57BL/6 male mouse. Cells 8:247. doi: 10.3390/cells8030247
Ballard, J. W. (2005). Drosophila simulans as a novel model for studying mitochondrial metabolism and aging. Exp. Gerontol. 40, 763–773. doi: 10.1016/j.exger.2005.07.014
Bansal, Y., and Kuhad, A. (2016). Mitochondrial dysfunction in depression. Curr. Neuropharmacol. 14, 610–618. doi: 10.2174/1570159x14666160229114755
Baris, T. Z., Blier, P. U., Pichaud, N., Crawford, D. L., and Oleksiak, M. F. (2016). Gene by environmental interactions affecting oxidative phosphorylation and thermal sensitivity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 311, R157–R165. doi: 10.1152/ajpregu.00008.2016
Baris, T. Z., Wagner, D. N., Dayan, D. I., Du, X., Blier, P. U., Pichaud, N., et al. (2017). Evolved genetic and phenotypic differences due to mitochondrial-nuclear interactions. PLoS Genet. 13:e1006517. doi: 10.1371/journal.pgen.1006517
Barreto, F. S., and Burton, R. S. (2013). Elevated oxidative damage is correlated with reduced fitness in interpopulation hybrids of a marine copepod. Proc. Biol. Sci. 280:20131521. doi: 10.1098/rspb.2013.1521
Bar-Yaacov, D., Blumberg, A., and Mishmar, D. (2012). Mitochondrial-nuclear co-evolution and its effects on OXPHOS activity and regulation. Biochim. Biophys. Acta 1819, 1107–1111. doi: 10.1016/j.bbagrm.2011.10.008
Beech, R. D., Lowthert, L., Leffert, J. J., Mason, P. N., Taylor, M. M., Umlauf, S., et al. (2010). Increased peripheral blood expression of electron transport chain genes in bipolar depression. Bipolar Disord. 12, 813–824. doi: 10.1111/j.1399-5618.2010.00882.x
Benn, M., Schwartz, M., Nordestgaard, B. G., and Tybjaerg-Hansen, A. (2008). Mitochondrial haplogroups: ischemic cardiovascular disease, other diseases, mortality, and longevity in the general population. Circulation 117, 2492–2501. doi: 10.1161/CIRCULATIONAHA.107.756809
Bennuri, S. C., Rose, S., and Frye, R. E. (2019). Mitochondrial dysfunction is inducible in lymphoblastoid cell lines from children with autism and may involve the TORC1 pathway. Front. Psychiatry 10:269. doi: 10.3389/fpsyt.2019.00269
Ben-Shachar, D. (2017). Mitochondrial multifaceted dysfunction in schizophrenia; complex I as a possible pathological target. Schizophr. Res. 187, 3–10. doi: 10.1016/j.schres.2016.10.022
Bertoni, B., Budowle, B., Sans, M., Barton, S. A., and Chakraborty, R. (2003). Admixture in hispanics: distribution of ancestral population contributions in the continental United States. Hum. Biol. 75, 1–11.
Bodenstein, D. F., Kim, H. K., Brown, N. C., Navaid, B., Young, L. T., and Andreazza, A. C. (2019). Mitochondrial DNA content and oxidation in bipolar disorder and its role across brain regions. NPJ Schizophr. 5:21. doi: 10.1038/s41537-019-0089-5
Brave Heart, M. Y., Lewis-Fernandez, R., Beals, J., Hasin, D. S., Sugaya, L., Wang, S., et al. (2016). Psychiatric disorders and mental health treatment in American Indians and alaska natives: results of the national epidemiologic survey on alcohol and related conditions. Soc. Psychiatry Psychiatr. Epidemiol. 51, 1033–1046. doi: 10.1007/s00127-016-1225-4
Breton, C. V., Song, A. Y., Xiao, J., Kim, S. J., Mehta, H. H., Wan, J., et al. (2019). Effects of air pollution on mitochondrial function, mitochondrial DNA methylation, and mitochondrial peptide expression. Mitochondrion 46, 22–29. doi: 10.1016/j.mito.2019.04.001
Brunst, K. J., Sanchez Guerra, M., Gennings, C., Hacker, M., Jara, C., Bosquet Enlow, M., et al. (2017). Maternal lifetime stress and prenatal psychological functioning and decreased placental mitochondrial DNA copy number in the PRISM study. Am. J. Epidemiol. 186, 1227–1236. doi: 10.1093/aje/kwx183
Cai, N., Chang, S., Li, Y., Li, Q., Hu, J., Liang, J., et al. (2015). Molecular signatures of major depression. Curr. Biol. 25, 1146–1156. doi: 10.1016/j.cub.2015.03.008
Callaly, E., Walder, K., Morris, G., Maes, M., Debnath, M., and Berk, M. (2015). Mitochondrial dysfunction in the pathophysiology of bipolar disorder: effects of pharmacotherapy. Mini. Rev. Med. Chem. 15, 355–365.
Calvo, S. E., Clauser, K. R., and Mootha, V. K. (2016). MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 44, D1251–D1257. doi: 10.1093/nar/gkv1003
Caruso, G., Benatti, C., Blom, J. M. C., Caraci, F., and Tascedda, F. (2019). The many faces of mitochondrial dysfunction in depression: from pathology to treatment. Front. Pharmacol. 10:995. doi: 10.3389/fphar.2019.00995
Chakravarty, S., Reddy, B. R., Sudhakar, S. R., Saxena, S., Das, T., Meghah, V., et al. (2013). Chronic unpredictable stress (CUS)-induced anxiety and related mood disorders in a zebrafish model: altered brain proteome profile implicates mitochondrial dysfunction. PLoS One 8:e63302. doi: 10.1371/journal.pone.0063302
Chan, D. C. (2006). Mitochondria: dynamic organelles in disease, aging, and development. Cell 125, 1241–1252. doi: 10.1016/j.cell.2006.06.010
Chan, D. C. (2012). Fusion and fission: interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 46, 265–287. doi: 10.1146/annurev-genet-110410-132529
Chang, C. C., Jou, S. H., Lin, T. T., and Liu, C. S. (2014). Mitochondrial DNA variation and increased oxidative damage in euthymic patients with bipolar disorder. Psychiatry Clin. Neurosci. 68, 551–557. doi: 10.1111/pcn.12163
Chen, H., and Chan, D. C. (2009). Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum. Mol. Genet. 18, R169–R176. doi: 10.1093/hmg/ddp326
Chinnery, P. F., Mowbray, C., Patel, S. K., Elson, J. L., Sampson, M., Hitman, G. A., et al. (2007). Mitochondrial DNA haplogroups and type 2 diabetes: a study of 897 cases and 1010 controls. J. Med. Genet. 44:e80. doi: 10.1136/jmg.2007.048876
Chou, J. Y., and Leu, J. Y. (2015). The red queen in mitochondria: cyto-nuclear co-evolution, hybrid breakdown and human disease. Front. Genet. 6:187. doi: 10.3389/fgene.2015.00187
Chung, J. K., Ahn, Y. M., Kim, S. A., and Joo, E. J. (2020). Differences in mitochondrial DNA copy number between patients with bipolar I and II disorders. J. Psychiatr. Res. doi: 10.1016/j.jpsychires.2020.11.016 Online ahead of print
Cikankova, T., Sigitova, E., Zverova, M., Fisar, Z., Raboch, J., and Hroudova, J. (2017). Mitochondrial dysfunctions in bipolar disorder: effect of the disease and pharmacotherapy. CNS Neurol. Disord. Drug Targets 16, 176–186. doi: 10.2174/1871527315666161213110518
Citrigno, L., Muglia, M., Qualtieri, A., Spadafora, P., Cavalcanti, F., Pioggia, G., et al. (2020). The mitochondrial dysfunction hypothesis in autism spectrum disorders: current status and future perspectives. Int. J. Mol. Sci. 21:5785. doi: 10.3390/ijms21165785
Clay, H. B., Sillivan, S., and Konradi, C. (2011). Mitochondrial dysfunction and pathology in bipolar disorder and schizophrenia. Int. J. Dev. Neurosci. 29, 311–324. doi: 10.1016/j.ijdevneu.2010.08.007
Clay Montier, L. L., Deng, J. J., and Bai, Y. (2009). Number matters: control of mammalian mitochondrial DNA copy number. J. Genet. Genomics 36, 125–131. doi: 10.1016/S1673-8527(08)60099-5
Cosmides, L. M., and Tooby, J. (1981). Cytoplasmic inheritance and intragenomic conflict. J. Theor. Biol. 89, 83–129. doi: 10.1016/0022-5193(81)90181-8
Cronin-Furman, E. N., Borland, M. K., Bergquist, K. E., Bennett, J. P. Jr., and Trimmer, P. A. (2013). Mitochondrial quality, dynamics and functional capacity in Parkinson’s disease cybrid cell lines selected for Lewy body expression. Mol. Neurodegener. 8:6. doi: 10.1186/1750-1326-8-6
Darvishi, K., Sharma, S., Bhat, A. K., Rai, E., and Bamezai, R. N. (2007). Mitochondrial DNA G10398A polymorphism imparts maternal Haplogroup N a risk for breast and esophageal cancer. Cancer Lett. 249, 249–255. doi: 10.1016/j.canlet.2006.09.005
de Sousa, R. T., Machado-Vieira, R., Zarate, C. A. Jr., and Manji, H. K. (2014a). Targeting mitochondrially mediated plasticity to develop improved therapeutics for bipolar disorder. Expert Opin. Ther. Targets 18, 1131–1147. doi: 10.1517/14728222.2014.940893
de Sousa, R. T., Uno, M., Zanetti, M. V., Shinjo, S. M., Busatto, G. F., Gattaz, W. F., et al. (2014b). Leukocyte mitochondrial DNA copy number in bipolar disorder. Prog. Neuropsychopharmacol. Biol. Psychiatry 48, 32–35. doi: 10.1016/j.pnpbp.2013.09.002
Dong, W., Dobler, R., Dowling, D. K., and Moussian, B. (2019). The cuticle inward barrier in Drosophila melanogaster is shaped by mitochondrial and nuclear genotypes and a sex-specific effect of diet. PeerJ 7:e7802. doi: 10.7717/peerj.7802
Dowling, D. K., Friberg, U., Hailer, F., and Arnqvist, G. (2007). Intergenomic epistasis for fitness: within-population interactions between cytoplasmic and nuclear genes in Drosophila melanogaster. Genetics 175, 235–244. doi: 10.1534/genetics.105.052050
Duan, X., Yang, Y., Zhang, H., Liu, B., Wei, W., Wang, L., et al. (2020). Polycyclic aromatic hydrocarbon exposure, miRNA genetic variations, and associated leukocyte mitochondrial DNA copy number: a cross-sectional study in China. Chemosphere 246:125773. doi: 10.1016/j.chemosphere.2019.125773
Duong, A., Che, Y., Ceylan, D., Pinguelo, A., Andreazza, A. C., Trevor Young, L., et al. (2016). Regulators of mitochondrial complex I activity: a review of literature and evaluation in postmortem prefrontal cortex from patients with bipolar disorder. Psychiatry Res. 236, 148–157. doi: 10.1016/j.psychres.2015.12.015
Dutil, J., Chen, Z., Monteiro, A. N., Teer, J. K., and Eschrich, S. A. (2019). An interactive resource to probe genetic diversity and estimated ancestry in cancer cell lines. Cancer Res. 79, 1263–1273. doi: 10.1158/0008-5472.CAN-18-2747
Esteves, A. R., Domingues, A. F., Ferreira, I. L., Januario, C., Swerdlow, R. H., Oliveira, C. R., et al. (2008). Mitochondrial function in Parkinson’s disease cybrids containing an nt2 neuron-like nuclear background. Mitochondrion 8, 219–228. doi: 10.1016/j.mito.2008.03.004
Feissner, R. F., Skalska, J., Gaum, W. E., and Sheu, S. S. (2009). Crosstalk signaling between mitochondrial Ca2+ and ROS. Front. Biosci. 14:1197–1218. doi: 10.2741/3303
Forester, B. P., Mellen, E., and Cohen, B. M. (2019). Establishing a link between mitochondrial dysfunction and late-life depression. Am. J. Geriatr. Psychiatry 27, 972–974. doi: 10.1016/j.jagp.2019.04.013
Friedman, J. R., and Nunnari, J. (2014). Mitochondrial form and function. Nature 505, 335–343. doi: 10.1038/nature12985
Fries, G. R., Bauer, I. E., Scaini, G., Valvassori, S. S., Walss-Bass, C., Soares, J. C., et al. (2019). Accelerated hippocampal biological aging in bipolar disorder. Bipolar Disord. 22, 498–507. doi: 10.1111/bdi.12876
Fries, G. R., Bauer, I. E., Scaini, G., Wu, M. J., Kazimi, I. F., Valvassori, S. S., et al. (2017). Accelerated epigenetic aging and mitochondrial DNA copy number in bipolar disorder. Transl. Psychiatry 7:1283. doi: 10.1038/s41398-017-0048-8
Frye, M. A., Ryu, E., Nassan, M., Jenkins, G. D., Andreazza, A. C., Evans, J. M., et al. (2017). Mitochondrial DNA sequence data reveals association of haplogroup U with psychosis in bipolar disorder. J. Psychiatr. Res. 84, 221–226. doi: 10.1016/j.jpsychires.2016.09.027
Frye, R. E. (2020). Mitochondrial dysfunction in autism spectrum disorder: unique abnormalities and targeted treatments. Semin. Pediatr. Neurol. 35:100829. doi: 10.1016/j.spen.2020.100829
Gevezova, M., Sarafian, V., Anderson, G., and Maes, M. (2020). Inflammation and mitochondrial dysfunction in autism spectrum disorder. CNS Neurol. Disord. Drug Targets 19, 320–333. doi: 10.2174/1871527319666200628015039
Goh, S., Dong, Z., Zhang, Y., DiMauro, S., and Peterson, B. S. (2014). Mitochondrial dysfunction as a neurobiological subtype of autism spectrum disorder: evidence from brain imaging. JAMA Psychiatry 71, 665–671. doi: 10.1001/jamapsychiatry.2014.179
Goncalves, V. F., Andreazza, A. C., and Kennedy, J. L. (2015). Mitochondrial dysfunction in schizophrenia: an evolutionary perspective. Hum. Genet. 134, 13–21. doi: 10.1007/s00439-014-1491-8
Grant, B. F., Stinson, F. S., Hasin, D. S., Dawson, D. A., Chou, S. P., Ruan, W. J., et al. (2005). Prevalence, correlates, and comorbidity of bipolar I disorder and axis I and II disorders: results from the national epidemiologic survey on alcohol and related conditions. J. Clin. Psychiatry 66, 1205–1215.
Gray, M. W., Burger, G., and Lang, B. F. (1999). Mitochondrial evolution. Science 283, 1476–1481. doi: 10.1126/science.283.5407.1476
Greer, P. L., and Greenberg, M. E. (2008). From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron 59, 846–860. doi: 10.1016/j.neuron.2008.09.002
Gregorius, H. R., and Ross, M. D. (1984). Selection with gene-cytoplasm interactions. I. Maintenance of cytoplasm polymorphisms. Genetics 107, 165–178.
Hagen, C. M., Goncalves, V. F., Hedley, P. L., Bybjerg-Grauholm, J., Baekvad-Hansen, M., Hansen, C. S., et al. (2018). Schizophrenia-associated mt-DNA SNPs exhibit highly variable haplogroup affiliation and nuclear ancestry: Bi-genomic dependence raises major concerns for link to disease. PLoS One 13:e0208828. doi: 10.1371/journal.pone.0208828
Hamakawa, H., Murashita, J., Yamada, N., Inubushi, T., Kato, N., and Kato, T. (2004). Reduced intracellular pH in the basal ganglia and whole brain measured by 31P-MRS in bipolar disorder. Psychiatry Clin. Neurosci. 58, 82–88. doi: 10.1111/j.1440-1819.2004.01197.x
Hill, G. E. (2016). Mitonuclear coevolution as the genesis of speciation and the mitochondrial DNA barcode gap. Ecol. Evol. 6, 5831–5842. doi: 10.1002/ece3.2338
Hjelm, B. E., Rollins, B., Mamdani, F., Lauterborn, J. C., Kirov, G., Lynch, G., et al. (2015). Evidence of mitochondrial dysfunction within the complex genetic etiology of schizophrenia. Mol. Neuropsychiatry 1, 201–219. doi: 10.1159/000441252
Hoekstra, L. A., Siddiq, M. A., and Montooth, K. L. (2013). Pleiotropic effects of a mitochondrial-nuclear incompatibility depend upon the accelerating effect of temperature in Drosophila. Genetics 195, 1129–1139. doi: 10.1534/genetics.113.154914
Holper, L., Ben-Shachar, D., and Mann, J. J. (2018). Multivariate meta-analyses of mitochondrial complex I and IV in major depressive disorder, bipolar disorder, schizophrenia, Alzheimer disease, and Parkinson disease. Neuropsychopharmacology 44, 837–849. doi: 10.1038/s41386-018-0090-0
Houstek, J., Vrbacky, M., Hejzlarova, K., Zidek, V., Landa, V., Silhavy, J., et al. (2014). Effects of mtDNA in SHR-mtF344 versus SHR conplastic strains on reduced OXPHOS enzyme levels, insulin resistance, cardiac hypertrophy, and systolic dysfunction. Physiol. Genomics 46, 671–678. doi: 10.1152/physiolgenomics.00069.2014
Hu, C., Sheng, X., Li, Y., Xia, W., Zhang, B., Chen, X., et al. (2020). Effects of prenatal exposure to particulate air pollution on newborn mitochondrial DNA copy number. Chemosphere 253:126592. doi: 10.1016/j.chemosphere.2020.126592
Immonen, E., Berger, D., Sayadi, A., Liljestrand-Ronn, J., and Arnqvist, G. (2020). An experimental test of temperature-dependent selection on mitochondrial haplotypes in Callosobruchus maculatus seed beetles. Ecol. Evol. 10, 11387–11398. doi: 10.1002/ece3.6775
Iwamoto, K., Bundo, M., and Kato, T. (2005). Altered expression of mitochondria-related genes in postmortem brains of patients with bipolar disorder or schizophrenia, as revealed by large-scale DNA microarray analysis. Hum. Mol. Genet. 14, 241–253. doi: 10.1093/hmg/ddi022
Kaali, S., Jack, D., Delimini, R., Hu, L., Burkart, K., Opoku-Mensah, J., et al. (2018). Prenatal household air pollution alters cord blood mononuclear cell mitochondrial DNA Copy number: sex-specific associations. Int. J. Environ. Res. Public Health 16:26. doi: 10.3390/ijerph16010026
Kato, T. (2005). Mitochondrial dysfunction in bipolar disorder: from 31P-magnetic resonance spectroscopic findings to their molecular mechanisms. Int. Rev. Neurobiol. 63, 21–40. doi: 10.1016/S0074-7742(05)63002-4
Kato, T. (2006). The role of mitochondrial dysfunction in bipolar disorder. Drug News Perspect. 19, 597–602. doi: 10.1358/dnp.2006.19.10.1068006
Kato, T. (2007). Mitochondrial dysfunction as the molecular basis of bipolar disorder: therapeutic implications. CNS Drugs 21, 1–11. doi: 10.2165/00023210-200721010-00001
Kato, T. (2008). Role of mitochondrial DNA in calcium signaling abnormality in bipolar disorder. Cell Calcium 44, 92–102. doi: 10.1016/j.ceca.2007.11.005
Kato, T. (2017). Neurobiological basis of bipolar disorder: mitochondrial dysfunction hypothesis and beyond. Schizophr. Res. 187, 62–66. doi: 10.1016/j.schres.2016.10.037
Kato, T., Ishiwata, M., Mori, K., Washizuka, S., Tajima, O., Akiyama, T., et al. (2003). Mechanisms of altered Ca2+ signalling in transformed lymphoblastoid cells from patients with bipolar disorder. Int. J. Neuropsychopharmacol. 6, 379–389. doi: 10.1017/S1461145703003717
Kato, T., and Kato, N. (2000). Mitochondrial dysfunction in bipolar disorder. Bipolar Disord. 2(3 Pt 1), 180–190.
Kato, T., Kunugi, H., Nanko, S., and Kato, N. (2001). Mitochondrial DNA polymorphisms in bipolar disorder. J. Affect. Disord. 62, 151–164. doi: 10.1016/s0165-0327(99)00173-1
Kato, T., Murashita, J., Kamiya, A., Shioiri, T., Kato, N., and Inubushi, T. (1998). Decreased brain intracellular pH measured by 31P-MRS in bipolar disorder: a confirmation in drug-free patients and correlation with white matter hyperintensity. Eur. Arch. Psychiatry Clin. Neurosci. 248, 301–306. doi: 10.1007/s004060050054
Kato, T., Shioiri, T., Inubushi, T., and Takahashi, S. (1993). Brain lithium concentrations measured with lithium-7 magnetic resonance spectroscopy in patients with affective disorders: relationship to erythrocyte and serum concentrations. Biol. Psychiatry 33, 147–152. doi: 10.1016/0006-3223(93)90133-x
Kato, T., Shioiri, T., Murashita, J., Hamakawa, H., Takahashi, Y., Inubushi, T., et al. (1995). Lateralized abnormality of high energy phosphate metabolism in the frontal lobes of patients with bipolar disorder detected by phase-encoded 31P-MRS. Psychol. Med. 25, 557–566.
Kato, T., Stine, O. C., McMahon, F. J., and Crowe, R. R. (1997a). Increased levels of a mitochondrial DNA deletion in the brain of patients with bipolar disorder. Biol. Psychiatry 42, 871–875. doi: 10.1016/S0006-3223(97)00012-7
Kato, T., Winokur, G., McMahon, F. J., DePaulo, J. R., and Crowe, R. R. (1997b). Quantitative analysis of leukocyte mitochondrial DNA deletion in affective disorders. Biol. Psychiatry 42, 311–316. doi: 10.1016/S0006-3223(96)00377-0
Kato, T., Takahashi, S., Shioiri, T., and Inubushi, T. (1992). Brain phosphorous metabolism in depressive disorders detected by phosphorus-31 magnetic resonance spectroscopy. J. Affect. Disord. 26, 223–230. doi: 10.1016/0165-0327(92)90099-r
Kato, T., Takahashi, S., Shioiri, T., Murashita, J., Hamakawa, H., and Inubushi, T. (1994). Reduction of brain phosphocreatine in bipolar II disorder detected by phosphorus-31 magnetic resonance spectroscopy. J. Affect. Disord. 31, 125–133. doi: 10.1016/0165-0327(94)90116-3
Kato, T., and Takahashi, Y. (1996). Deletion of leukocyte mitochondrial DNA in bipolar disorder. J. Affect. Disord. 37, 67–73. doi: 10.1016/0165-0327(95)00050-x
Kazuno, A. A., Munakata, K., Kato, N., and Kato, T. (2008). Mitochondrial DNA-dependent effects of valproate on mitochondrial calcium levels in transmitochondrial cybrids. Int. J. Neuropsychopharmacol. 11, 71–78. doi: 10.1017/S1461145707007614
Kazuno, A. A., Munakata, K., Mori, K., Nanko, S., Kunugi, H., Nakamura, K., et al. (2009). Mitochondrial DNA haplogroup analysis in patients with bipolar disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B, 243–247. doi: 10.1002/ajmg.b.30804
Kazuno, A. A., Munakata, K., Nagai, T., Shimozono, S., Tanaka, M., Yoneda, M., et al. (2006). Identification of mitochondrial DNA polymorphisms that alter mitochondrial matrix pH and intracellular calcium dynamics. PLoS Genet. 2:e128. doi: 10.1371/journal.pgen.0020128
Keeney, P. M., Dunham, L. D., Quigley, C. K., Morton, S. L., Bergquist, K. E., and Bennett, J. P. Jr. (2009). Cybrid models of Parkinson’s disease show variable mitochondrial biogenesis and genotype-respiration relationships. Exp. Neurol. 220, 374–382. doi: 10.1016/j.expneurol.2009.09.025
Kenney, M. C., Chwa, M., Atilano, S. R., Falatoonzadeh, P., Ramirez, C., Malik, D., et al. (2014a). Inherited mitochondrial DNA variants can affect complement, inflammation and apoptosis pathways: insights into mitochondrial-nuclear interactions. Hum. Mol. Genet. 23, 3537–3551. doi: 10.1093/hmg/ddu065
Kenney, M. C., Chwa, M., Atilano, S. R., Falatoonzadeh, P., Ramirez, C., Malik, D., et al. (2014b). Molecular and bioenergetic differences between cells with African versus European inherited mitochondrial DNA haplogroups: implications for population susceptibility to diseases. Biochim. Biophys. Acta 1842, 208–219. doi: 10.1016/j.bbadis.2013.10.016
Kenney, M. C., Chwa, M., Atilano, S. R., Pavlis, J. M., Falatoonzadeh, P., Ramirez, C., et al. (2013). Mitochondrial DNA variants mediate energy production and expression levels for CFH, C3 and EFEMP1 genes: implications for age-related macular degeneration. PLoS One 8:e54339. doi: 10.1371/journal.pone.0054339
Khalifeh, S., Oryan, S., Khodagholi, F., Digaleh, H., Shaerzadeh, F., Maghsoudi, N., et al. (2016). Complexity of compensatory effects in Nrf1 knockdown: linking undeveloped anxiety-like behavior to prevented mitochondrial dysfunction and oxidative stress. Cell Mol. Neurobiol. 36, 553–563. doi: 10.1007/s10571-015-0236-0
Kim, H. K., Chen, W., and Andreazza, A. C. (2015). The potential role of the NLRP3 inflammasome as a link between mitochondrial complex I dysfunction and inflammation in bipolar disorder. Neural Plast. 2015:408136. doi: 10.1155/2015/408136
Konradi, C., Eaton, M., MacDonald, M. L., Walsh, J., Benes, F. M., and Heckers, S. (2004). Molecular evidence for mitochondrial dysfunction in bipolar disorder. Arch. Gen. Psychiatry 61, 300–308. doi: 10.1001/archpsyc.61.3.300
Konradi, C., Sillivan, S. E., and Clay, H. B. (2012). Mitochondria, oligodendrocytes and inflammation in bipolar disorder: evidence from transcriptome studies points to intriguing parallels with multiple sclerosis. Neurobiol. Dis. 45, 37–47. doi: 10.1016/j.nbd.2011.01.025
Korkiamaki, P., Kervinen, M., Karjalainen, K., Majamaa, K., Uusimaa, J., and Remes, A. M. (2013). Prevalence of the primary LHON mutations in Northern Finland associated with bilateral optic atrophy and tobacco-alcohol amblyopia. Acta Ophthalmol. 91, 630–634. doi: 10.1111/j.1755-3768.2012.02506.x
Kubota, M., Kasahara, T., Nakamura, T., Ishiwata, M., Miyauchi, T., and Kato, T. (2006). Abnormal Ca2+ dynamics in transgenic mice with neuron-specific mitochondrial DNA defects. J. Neurosci. 26, 12314–12324. doi: 10.1523/JNEUROSCI.3933-06.2006
Kumarasamy, S., Gopalakrishnan, K., Abdul-Majeed, S., Partow-Navid, R., Farms, P., and Joe, B. (2013). Construction of two novel reciprocal conplastic rat strains and characterization of cardiac mitochondria. Am. J. Physiol. Heart Circ. Physiol. 304, H22–H32. doi: 10.1152/ajpheart.00534.2012
Kurbalija Novicic, Z., Immonen, E., Jelic, M., AnEthelkovic, M., Stamenkovic-Radak, M., and Arnqvist, G. (2015). Within-population genetic effects of mtDNA on metabolic rate in Drosophila subobscura. J. Evol. Biol. 28, 338–346. doi: 10.1111/jeb.12565
Lane, N. (2011). Mitonuclear match: optimizing fitness and fertility over generations drives ageing within generations. Bioessays 33, 860–869. doi: 10.1002/bies.201100051
Latorre-Pellicer, A., Moreno-Loshuertos, R., Lechuga-Vieco, A. V., Sanchez-Cabo, F., Torroja, C., Acin-Perez, R., et al. (2016). Mitochondrial and nuclear DNA matching shapes metabolism and healthy ageing. Nature 535, 561–565. doi: 10.1038/nature18618
Lin, T. K., Lin, H. Y., Chen, S. D., Chuang, Y. C., Chuang, J. H., Wang, P. W., et al. (2012). The creation of cybrids harboring mitochondrial haplogroups in the Taiwanese population of ethnic Chinese background: an extensive in vitro tool for the study of mitochondrial genomic variations. Oxid. Med. Cell Longev. 2012:824275. doi: 10.1155/2012/824275
Loewen, C. A., and Ganetzky, B. (2018). Mito-nuclear interactions affecting lifespan and neurodegeneration in a Drosophila model of leigh syndrome. Genetics 208, 1535–1552. doi: 10.1534/genetics.118.300818
Ma, C., He, S., Li, P., Zhang, H., Li, W., and Li, Y. (2020). Negative association between caloric intake and estimated glomerular filtration rate in a chinese population: mediation models involving mitochondrial function. Gerontology 66, 439–446. doi: 10.1159/000508497
Machado, A. K., Pan, A. Y., da Silva, T. M., Duong, A., and Andreazza, A. C. (2016). Upstream pathways controlling mitochondrial function in major psychosis: a focus on bipolar disorder. Can. J. Psychiatry 61, 446–456. doi: 10.1177/0706743716648297
Magri, C., Gardella, R., Barlati, S. D., Valsecchi, P., Sacchetti, E., and Barlati, S. (2007). Mitochondrial DNA haplogroups and age at onset of schizophrenia. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 496–501. doi: 10.1002/ajmg.b.30496
Manji, H., Kato, T., Di Prospero, N. A., Ness, S., Beal, M. F., Krams, M., et al. (2012). Impaired mitochondrial function in psychiatric disorders. Nat. Rev. Neurosci. 13, 293–307. doi: 10.1038/nrn3229
Mattson, M. P., Gleichmann, M., and Cheng, A. (2008). Mitochondria in neuroplasticity and neurological disorders. Neuron 60, 748–766. doi: 10.1016/j.neuron.2008.10.010
McMahon, F. J., Chen, Y. S., Patel, S., Kokoszka, J., Brown, M. D., Torroni, A., et al. (2000). Mitochondrial DNA sequence diversity in bipolar affective disorder. Am. J. Psychiatry 157, 1058–1064. doi: 10.1176/appi.ajp.157.7.1058
Meiklejohn, C. D., Holmbeck, M. A., Siddiq, M. A., Abt, D. N., Rand, D. M., and Montooth, K. L. (2013). An incompatibility between a mitochondrial tRNA and its nuclear-encoded tRNA synthetase compromises development and fitness in Drosophila. PLoS Genet 9:e1003238. doi: 10.1371/journal.pgen.1003238
Mertens, J., Wang, Q. W., Kim, Y., Yu, D. X., Pham, S., Yang, B., et al. (2015). Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 527, 95–99. doi: 10.1038/nature15526
Mitchell, S. L., Neininger, A. C., Bruce, C. N., Chocron, I. M., Bregman, J. A., Estopinal, C. B., et al. (2017). Mitochondrial haplogroups modify the effect of diabetes duration and hba1c on proliferative diabetic retinopathy risk in patients with type 2 diabetes. Invest. Ophthalmol. Vis. Sci. 58, 6481–6488. doi: 10.1167/iovs.17-22804
Monpays, C., Deslauriers, J., Sarret, P., and Grignon, S. (2016). Mitochondrial dysfunction in schizophrenia: determination of mitochondrial respiratory activity in a two-hit mouse model. J. Mol. Neurosci. 59, 440–451. doi: 10.1007/s12031-016-0746-3
Montooth, K. L., Dhawanjewar, A. S., and Meiklejohn, C. D. (2019). Temperature-sensitive reproduction and the physiological and evolutionary potential for mother’s curse. Integr. Comp. Biol. 59, 890–899. doi: 10.1093/icb/icz091
Montooth, K. L., Meiklejohn, C. D., Abt, D. N., and Rand, D. M. (2010). Mitochondrial-nuclear epistasis affects fitness within species but does not contribute to fixed incompatibilities between species of Drosophila. Evolution 64, 3364–3379. doi: 10.1111/j.1558-5646.2010.01077.x
Morris, G., Walder, K., McGee, S. L., Dean, O. M., Tye, S. J., Maes, M., et al. (2017). A model of the mitochondrial basis of bipolar disorder. Neurosci. Biobehav. Rev. 74(Pt A), 1–20. doi: 10.1016/j.neubiorev.2017.01.014
Mossman, J. A., Biancani, L. M., Zhu, C. T., and Rand, D. M. (2016a). Mitonuclear epistasis for development time and its modification by diet in Drosophila. Genetics 203, 463–484. doi: 10.1534/genetics.116.187286
Mossman, J. A., Tross, J. G., Li, N., Wu, Z., and Rand, D. M. (2016b). Mitochondrial-nuclear interactions mediate sex-specific transcriptional profiles in Drosophila. Genetics 204, 613–630. doi: 10.1534/genetics.116.192328
Mossman, J. A., Tross, J. G., Jourjine, N. A., Li, N., Wu, Z., and Rand, D. M. (2017). Mitonuclear interactions mediate transcriptional responses to hypoxia in Drosophila. Mol. Biol. Evol. 34, 447–466. doi: 10.1093/molbev/msw246
Munakata, K., Tanaka, M., Mori, K., Washizuka, S., Yoneda, M., Tajima, O., et al. (2004). Mitochondrial DNA 3644T–>C mutation associated with bipolar disorder. Genomics 84, 1041–1050. doi: 10.1016/j.ygeno.2004.08.015
Murashita, J., Kato, T., Shioiri, T., Inubushi, T., and Kato, N. (2000). Altered brain energy metabolism in lithium-resistant bipolar disorder detected by photic stimulated 31P-MR spectroscopy. Psychol. Med. 30, 107–115. doi: 10.1017/s0033291799001439
Nashine, S., Chwa, M., Kazemian, M., Thaker, K., Lu, S., Nesburn, A., et al. (2016). Differential expression of complement markers in normal and AMD transmitochondrial cybrids. PLoS One 11:e0159828. doi: 10.1371/journal.pone.0159828
Ni, P., and Chung, S. (2020). Mitochondrial dysfunction in schizophrenia. Bioessays 42:e1900202. doi: 10.1002/bies.201900202
Nierenberg, A. A., Kansky, C., Brennan, B. P., Shelton, R. C., Perlis, R., and Iosifescu, D. V. (2013). Mitochondrial modulators for bipolar disorder: a pathophysiologically informed paradigm for new drug development. Aust. N. Z. J. Psychiatry 47, 26–42. doi: 10.1177/0004867412449303
Onyango, I., Khan, S., Miller, B., Swerdlow, R., Trimmer, P., and Bennett, P. Jr. (2006). Mitochondrial genomic contribution to mitochondrial dysfunction in Alzheimer’s disease. J. Alzheimers. Dis. 9, 183–193. doi: 10.3233/jad-2006-9210
Otsuka, I., Izumi, T., Boku, S., Kimura, A., Zhang, Y., Mouri, K., et al. (2017). Aberrant telomere length and mitochondrial DNA copy number in suicide completers. Sci. Rep. 7:3176. doi: 10.1038/s41598-017-03599-8
Park, C., and Park, S. K. (2012). Molecular links between mitochondrial dysfunctions and schizophrenia. Mol. Cells 33, 105–110. doi: 10.1007/s10059-012-2284-3
Peng, T. I., and Jou, M. J. (2010). Oxidative stress caused by mitochondrial calcium overload. Ann. N. Y. Acad. Sci. 1201, 183–188. doi: 10.1111/j.1749-6632.2010.05634.x
Pereira, C., Chavarria, V., Vian, J., Ashton, M. M., Berk, M., Marx, W., et al. (2018). mitochondrial agents for bipolar disorder. Int. J. Neuropsychopharmacol. 21, 550–569. doi: 10.1093/ijnp/pyy018
Picard, M., and McEwen, B. S. (2014). Mitochondria impact brain function and cognition. Proc. Natl. Acad. Sci. U.S.A. 111, 7–8. doi: 10.1073/pnas.1321881111
Picard, M., McEwen, B. S., Epel, E. S., and Sandi, C. (2018). An energetic view of stress: focus on mitochondria. Front. Neuroendocrinol. 49:72–85. doi: 10.1016/j.yfrne.2018.01.001
Pichaud, N., Ballard, J. W., Tanguay, R. M., and Blier, P. U. (2013). Mitochondrial haplotype divergences affect specific temperature sensitivity of mitochondrial respiration. J. Bioenerg. Biomembr. 45, 25–35. doi: 10.1007/s10863-012-9473-9
Pignataro, D., Francia, S., Zanetta, F., Brenna, G., Brandini, S., Olivieri, A., et al. (2017). A missense MT-ND5 mutation in differentiated Parkinson Disease cytoplasmic hybrid induces ROS-dependent DNA Damage Response amplified by DROSHA. Sci. Rep. 7:9528. doi: 10.1038/s41598-017-09910-x
Poyton, R. O., and McEwen, J. E. (1996). Crosstalk between nuclear and mitochondrial genomes. Annu. Rev. Biochem. 65, 563–607. doi: 10.1146/annurev.bi.65.070196.003023
Prata, D. P., Costa-Neves, B., Cosme, G., and Vassos, E. (2019). Unravelling the genetic basis of schizophrenia and bipolar disorder with GWAS: a systematic review. J. Psychiatr. Res. 114, 178–207. doi: 10.1016/j.jpsychires.2019.04.007
Quiros, P. M., Mottis, A., and Auwerx, J. (2016). Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 17, 213–226. doi: 10.1038/nrm.2016.23
Quiroz, J. A., Gray, N. A., Kato, T., and Manji, H. K. (2008). Mitochondrially mediated plasticity in the pathophysiology and treatment of bipolar disorder. Neuropsychopharmacology 33, 2551–2565. doi: 10.1038/sj.npp.1301671
Rand, D. M., and Mossman, J. A. (2020). Mitonuclear conflict and cooperation govern the integration of genotypes, phenotypes and environments. Philos. Trans. R. Soc. Lond. B Biol. Sci. 375:20190188. doi: 10.1098/rstb.2019.0188
Rand, D. M., Mossman, J. A., Zhu, L., Biancani, L. M., and Ge, J. Y. (2018). Mitonuclear epistasis, genotype-by-environment interactions, and personalized genomics of complex traits in Drosophila. IUBMB Life 70, 1275–1288. doi: 10.1002/iub.1954
Reeve, A. K., Ludtmann, M. H., Angelova, P. R., Simcox, E. M., Horrocks, M. H., Klenerman, D., et al. (2015). Aggregated alpha-synuclein and complex I deficiency: exploration of their relationship in differentiated neurons. Cell Death Dis. 6:e1820. doi: 10.1038/cddis.2015.166
Revesz, D., Verhoeven, J. E., Picard, M., Lin, J., Sidney, S., Epel, E. S., et al. (2018). Associations between cellular aging markers and metabolic syndrome: findings from the CARDIA study. J. Clin. Endocrinol. Metab. 103, 148–157. doi: 10.1210/jc.2017-01625
Ridout, K. K., Coe, J. L., Parade, S. H., Marsit, C. J., Kao, H. T., Porton, B., et al. (2020). Molecular markers of neuroendocrine function and mitochondrial biogenesis associated with early life stress. Psychoneuroendocrinology 116:104632. doi: 10.1016/j.psyneuen.2020.104632
Ridout, K. K., Parade, S. H., Kao, H. T., Magnan, S., Seifer, R., Porton, B., et al. (2019). Childhood maltreatment, behavioral adjustment, and molecular markers of cellular aging in preschool-aged children: a cohort study. Psychoneuroendocrinology 107, 261–269. doi: 10.1016/j.psyneuen.2019.05.015
Roberts, R. C. (2020). Mitochondrial dysfunction in schizophrenia: with a focus on postmortem studies. Mitochondrion 56, 91–101. doi: 10.1016/j.mito.2020.11.009
Robicsek, O., Ene, H. M., Karry, R., Ytzhaki, O., Asor, E., McPhie, D., et al. (2018). Isolated mitochondria transfer improves neuronal differentiation of schizophrenia-derived induced pluripotent stem cells and rescues deficits in a rat model of the disorder. Schizophr. Bull. 44, 432–442. doi: 10.1093/schbul/sbx077
Robin, E. D., and Wong, R. (1988). Mitochondrial DNA molecules and virtual number of mitochondria per cell in mammalian cells. J. Cell. Physiol. 136, 507–513. doi: 10.1002/jcp.1041360316
Rollins, B., Martin, M. V., Sequeira, P. A., Moon, E. A., Morgan, L. Z., Watson, S. J., et al. (2009). Mitochondrial variants in schizophrenia, bipolar disorder, and major depressive disorder. PLoS One 4:e4913. doi: 10.1371/journal.pone.0004913
Rossignol, D. A., and Frye, R. E. (2012). Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol. Psychiatry 17, 290–314. doi: 10.1038/mp.2010.136
Ryu, E., Nassan, M., Jenkins, G. D., Armasu, S. M., Andreazza, A., McElroy, S. L., et al. (2018). A genome-wide search for bipolar disorder risk loci modified by mitochondrial genome variation. Mol. Neuropsychiatry 3, 125–134. doi: 10.1159/000464444
Sawai, H., Takai-Igarashi, T., and Tanaka, H. (2015). Identification of collaborative activities with oxidative phosphorylation in bipolar disorder. Bioinformation 11, 207–216. doi: 10.6026/97320630011207
Scaini, G., Rezin, G. T., Carvalho, A. F., Streck, E. L., Berk, M., and Quevedo, J. (2016). Mitochondrial dysfunction in bipolar disorder: evidence, pathophysiology and translational implications. Neurosci. Biobehav. Rev. 68, 694–713. doi: 10.1016/j.neubiorev.2016.06.040
Schulmann, A., Ryu, E., Goncalves, V., Rollins, B., Christiansen, M., Frye, M. A., et al. (2019). Novel complex interactions between mitochondrial and nuclear DNA in schizophrenia and bipolar disorder. Mol. Neuropsychiatry 5, 13–27. doi: 10.1159/000495658
Sena, L. A., and Chandel, N. S. (2012). Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 48, 158–167. doi: 10.1016/j.molcel.2012.09.025
Shao, L., Martin, M. V., Watson, S. J., Schatzberg, A., Akil, H., Myers, R. M., et al. (2008). Mitochondrial involvement in psychiatric disorders. Ann. Med. 40, 281–295. doi: 10.1080/07853890801923753
Shrivastava, M., and Subbiah, V. (2016). Elevated caspase 3 activity and cytosolic cytochrome c in NT2 cybrids containing amyotrophic lateral sclerosis subject mtDNA. Int. J. Neurosci. 126, 839–849. doi: 10.3109/00207454.2015.1074902
Silva, D. F., Santana, I., Esteves, A. R., Baldeiras, I., Arduino, D. M., Oliveira, C. R., et al. (2013). Prodromal metabolic phenotype in MCI cybrids: implications for Alzheimer’s disease. Curr. Alzheimer Res. 10, 180–190. doi: 10.2174/1567205011310020008
Soh, J. W., Hotic, S., and Arking, R. (2007). Dietary restriction in Drosophila is dependent on mitochondrial efficiency and constrained by pre-existing extended longevity. Mech. Ageing Dev. 128, 581–593. doi: 10.1016/j.mad.2007.08.004
Srivastava, R., Faust, T., Ramos, A., Ishizuka, K., and Sawa, A. (2018). Dynamic changes of the mitochondria in psychiatric illnesses: new mechanistic insights from human neuronal models. Biol. Psychiatry 83, 751–760. doi: 10.1016/j.biopsych.2018.01.007
Stahl, E. A., Breen, G., Forstner, A. J., McQuillin, A., Ripke, S., Trubetskoy, V., et al. (2019). Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet. 51, 793–803. doi: 10.1038/s41588-019-0397-8
Stork, C., and Renshaw, P. F. (2005). Mitochondrial dysfunction in bipolar disorder: evidence from magnetic resonance spectroscopy research. Mol. Psychiatry 10, 900–919. doi: 10.1038/sj.mp.4001711
Substance Abuse and Mental Health Services Administration (2020). Key Substance use and Mental Health Indicators in the United States: Results from the 2019 National Survey on Drug Use and Health (HHS Publication No. PEP20-07-01-001, NSDUH Series H-55). Rockville, MD: Substance Abuse and Mental Health Services Administration.
Sun, X., Wang, J. F., Tseng, M., and Young, L. T. (2006). Downregulation in components of the mitochondrial electron transport chain in the postmortem frontal cortex of subjects with bipolar disorder. J. Psychiatry Neurosci. 31, 189–196.
Tavira, B., Gomez, J., Diaz-Corte, C., Llobet, L., Ruiz-Pesini, E., Ortega, F., et al. (2014). Mitochondrial DNA haplogroups and risk of new-onset diabetes among tacrolimus-treated renal transplanted patients. Gene 538, 195–198. doi: 10.1016/j.gene.2014.01.036
Taylor, R. W., and Turnbull, D. M. (2005). Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 6, 389–402. doi: 10.1038/nrg1606
Trevelyan, A. J., Kirby, D. M., Smulders-Srinivasan, T. K., Nooteboom, M., Acin-Perez, R., Enriquez, J. A., et al. (2010). Mitochondrial DNA mutations affect calcium handling in differentiated neurons. Brain 133(Pt 3), 787–796. doi: 10.1093/brain/awq023
Tsujii, N., Otsuka, I., Okazaki, S., Yanagi, M., Numata, S., Yamaki, N., et al. (2019). Mitochondrial DNA copy number raises the potential of left frontopolar hemodynamic response as a diagnostic marker for distinguishing bipolar disorder from major depressive disorder. Front. Psychiatry 10:312. doi: 10.3389/fpsyt.2019.00312
Tuppen, H. A., Blakely, E. L., Turnbull, D. M., and Taylor, R. W. (2010). Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta 1797, 113–128. doi: 10.1016/j.bbabio.2009.09.005
Tyrka, A. R., Parade, S. H., Price, L. H., Kao, H. T., Porton, B., Philip, N. S., et al. (2016). Alterations of mitochondrial DNA copy number and telomere length with early adversity and psychopathology. Biol. Psychiatry 79, 78–86. doi: 10.1016/j.biopsych.2014.12.025
Varga, N. A., Pentelenyi, K., Balicza, P., Gezsi, A., Remenyi, V., Harsfalvi, V., et al. (2018). Mitochondrial dysfunction and autism: comprehensive genetic analyses of children with autism and mtDNA deletion. Behav. Brain Funct. 14:4. doi: 10.1186/s12993-018-0135-x
Verma, P., Singh, A., Nthenge-Ngumbau, D. N., Rajamma, U., Sinha, S., Mukhopadhyay, K., et al. (2016). Attention deficit-hyperactivity disorder suffers from mitochondrial dysfunction. BBA Clin. 6, 153–158. doi: 10.1016/j.bbacli.2016.10.003
Veronese, N., Stubbs, B., Koyanagi, A., Vaona, A., Demurtas, J., Schofield, P., et al. (2019). Mitochondrial genetic haplogroups and cardiovascular diseases: data from the osteoarthritis initiative. PLoS One 14:e0213656. doi: 10.1371/journal.pone.0213656
Vyas, C. M., Ogata, S., Reynolds, C. F. III, Mischoulon, D., Chang, G., Cook, N. R., et al. (2020). Lifestyle and behavioral factors and mitochondrial DNA copy number in a diverse cohort of mid-life and older adults. PLoS One 15:e0237235. doi: 10.1371/journal.pone.0237235
Wallace, D. C. (2005). A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu. Rev. Genet. 39, 359–407. doi: 10.1146/annurev.genet.39.110304.095751
Wallace, D. C., Brown, M. D., and Lott, M. T. (1999). Mitochondrial DNA variation in human evolution and disease. Gene 238, 211–230. doi: 10.1016/s0378-1119(99)00295-4
Wang, D., Li, H., Du, X., Zhou, J., Yuan, L., Ren, H., et al. (2020). Circulating brain-derived neurotrophic factor, antioxidant enzymes activities, and mitochondrial DNA in bipolar disorder: an exploratory report. Front. Psychiatry 11:514658. doi: 10.3389/fpsyt.2020.514658
Wang, D., Li, Z., Liu, W., Zhou, J., Ma, X., Tang, J., et al. (2018). Differential mitochondrial DNA copy number in three mood states of bipolar disorder. BMC Psychiatry 18:149. doi: 10.1186/s12888-018-1717-8
Wang, J. F. (2007). Defects of mitochondrial electron transport chain in bipolar disorder: implications for mood-stabilizing treatment. Can. J. Psychiatry 52, 753–762. doi: 10.1177/070674370705201202
Washizuka, S., Ikeda, A., Kato, N., and Kato, T. (2003). Possible relationship between mitochondrial DNA polymorphisms and lithium response in bipolar disorder. Int. J. Neuropsychopharmacol. 6, 421–424. doi: 10.1017/S1461145703003778
Weidling, I. W., Wilkins, H. M., Koppel, S. J., Hutfles, L., Wang, X., Kalani, A., et al. (2020). Mitochondrial DNA manipulations affect tau oligomerization. J. Alzheimers. Dis. 77, 149–163. doi: 10.3233/JAD-200286
Weiss, H., Wester-Rosenloef, L., Koch, C., Koch, F., Baltrusch, S., Tiedge, M., et al. (2012). The mitochondrial Atp8 mutation induces mitochondrial ROS generation, secretory dysfunction, and beta-cell mass adaptation in conplastic B6-mtFVB mice. Endocrinology 153, 4666–4676. doi: 10.1210/en.2012-1296
Wong, J. Y. Y., Hu, W., Downward, G. S., Seow, W. J., Bassig, B. A., Ji, B. T., et al. (2017). Personal exposure to fine particulate matter and benzo[a]pyrene from indoor air pollution and leukocyte mitochondrial DNA copy number in rural China. Carcinogenesis 38, 893–899. doi: 10.1093/carcin/bgx068
Wu, S., Li, X., Meng, S., Fung, T., Chan, A. T., Liang, G., et al. (2019). Fruit and vegetable consumption, cigarette smoke, and leukocyte mitochondrial DNA copy number. Am. J. Clin. Nutr. 109, 424–432. doi: 10.1093/ajcn/nqy286
Yamaki, N., Otsuka, I., Numata, S., Yanagi, M., Mouri, K., Okazaki, S., et al. (2018). Mitochondrial DNA copy number of peripheral blood in bipolar disorder: the present study and a meta-analysis. Psychiatry Res. 269, 115–117. doi: 10.1016/j.psychres.2018.08.014
Yu, Q., Du, F., Douglas, J. T., Yu, H., Yan, S. S., and Yan, S. F. (2017). Mitochondrial dysfunction triggers synaptic deficits via activation of p38 MAP kinase signaling in differentiated alzheimer’s disease trans-mitochondrial cybrid cells. J. Alzheimers. Dis. 59, 223–239. doi: 10.3233/JAD-170283
Yu, Q., Fang, D., Swerdlow, R. H., Yu, H., Chen, J. X., and Yan, S. S. (2016). Antioxidants rescue mitochondrial transport in differentiated Alzheimer’s Disease trans-mitochondrial cybrid cells. J. Alzheimers. Dis. 54, 679–690. doi: 10.3233/JAD-160532
Yu, X., Gimsa, U., Wester-Rosenlof, L., Kanitz, E., Otten, W., Kunz, M., et al. (2009). Dissecting the effects of mtDNA variations on complex traits using mouse conplastic strains. Genome Res. 19, 159–165. doi: 10.1101/gr.078865.108
Yu, X., Koczan, D., Sulonen, A. M., Akkad, D. A., Kroner, A., Comabella, M., et al. (2008). mtDNA nt13708A variant increases the risk of multiple sclerosis. PLoS One 3:e1530. doi: 10.1371/journal.pone.0001530
Zaidi, A. A., and Makova, K. D. (2019). Investigating mitonuclear interactions in human admixed populations. Nat. Ecol. Evol. 3, 213–222. doi: 10.1038/s41559-018-0766-1
Zhao, D., Ding, Y., Lin, H., Chen, X., Shen, W., Gao, M., et al. (2019). Mitochondrial haplogroups N9 and G are associated with metabolic syndrome among human immunodeficiency virus-infected patients in China. AIDS Res. Hum. Retroviruses 35, 536–543. doi: 10.1089/AID.2018.0151
Zhou, H., Nie, K., Qiu, R., Xiong, J., Shao, X., Wang, B., et al. (2017). Generation and bioenergetic profiles of cybrids with East Asian mtDNA haplogroups. Oxid. Med. Cell Longev. 2017:1062314. doi: 10.1155/2017/1062314
Keywords: bipolar disorder, genetics, mitonuclear coevolution, mitonuclear coadaptation, mitonuclear incompatibility, mitonuclear interaction, epistasis
Citation: Gonzalez S (2021) The Role of Mitonuclear Incompatibility in Bipolar Disorder Susceptibility and Resilience Against Environmental Stressors. Front. Genet. 12:636294. doi: 10.3389/fgene.2021.636294
Received: 01 December 2020; Accepted: 22 February 2021;
Published: 16 March 2021.
Edited by:
Adriane R. Rosa, Federal University of Rio Grande do Sul, BrazilReviewed by:
Gabriel R. Fries, University of Texas Health Science Center at Houston, United StatesRichard S. Lee, Johns Hopkins University, United States
Copyright © 2021 Gonzalez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Suzanne Gonzalez, c2dvbnphbGV6MkBwZW5uc3RhdGVoZWFsdGgucHN1LmVkdQ==