 Parul Sharma1†
Parul Sharma1† Abhinav Jain
Abhinav Jain Vinod Scaria
Vinod Scaria
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Genet. , 13 May 2021
Sec. Applied Genetic Epidemiology
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.631340
Rare monogenic autoinflammatory diseases are a group of recurrent inflammatory genetic disorders caused due to genetic variants in over 37 genes. While a number of these disorders have been identified and reported in Middle Eastern populations, the carrier frequency of these genetic variants in the Middle Eastern population is not known. The availability of whole-genome and exome datasets of over 1,000 individuals from Qatar persuaded us to explore the genetic epidemiology of rare autoinflammatory genetic variants. We have systematically analyzed genetic variants in genome-scale datasets from Qatar with a compendium of variants associated with autoinflammatory diseases. The variants were systematically reclassified according to the American College of Medical Genetics and Genomics guidelines for interpretation of variant pathogenicity. Our analysis identified seven pathogenic and likely pathogenic variants with significant differences in their allele frequencies compared to the global population. The cumulative carrier frequency of these variants was found to be 2.58%. Furthermore, our analysis revealed that five genes, implicated in rare autoinflammatory diseases, were under natural selection. To the best of our knowledge, this is the first and most comprehensive study on the population-scale analysis and genetic epidemiology of genetic variants that cause rare autoinflammatory disease in Middle Eastern populations.
Autoinflammatory disorders are genetically heterogeneous groups of disorders caused due to the presence of disease-causing variants in the genes responsible for regulating the inflammatory response. However, monogenic autoinflammatory disorders are a group of Mendelian genetic disorders characterized by recurrent inflammatory episodes due to an abnormal innate immune system. Until recently, these disorders have only been defined by phenotypic traits including recurrent attacks of fever, abdominal pain, arthritis, skin rashes, or cutaneous signs and were known to exhibit overlapping symptoms with other diseases which has led to misdiagnoses in a large number of cases (Ben-Chetrit et al., 2018; Hausmann et al., 2018). Recent advancements in the understanding of the molecular basis of these disorders have resulted in an accurate classification that presently encompasses 20 disorders involving over 30 genes (Touitou et al., 2004; Ciccarelli et al., 2014; Martinez-Quiles and Goldbach-Mansky, 2018). The most common among all monogenic autoinflammatory diseases is Familial Mediterranean Fever (FMF). It is caused by the pathogenic variants in the MEFV gene and is highly prevalent in Middle Eastern countries (The International FMF Consortium, 1997; Salehzadeh, 2015). A recent study from our group has estimated the population-specific frequencies of MEFV variants associated with FMF among 2,000 Mediterranean individuals (Koshy et al., 2018).
There are multiple reports of autoinflammatory disorders from Middle Eastern countries, including reports from a single hospital in Riyadh where 34 patients were admitted due to autoinflammatory disorders over a span of 10 years (Alenazi et al., 2012). There are also multiple individuals from the Arab countries that are affected by Majeed syndrome (Majeed et al., 1989; Al-Mosawi et al., 2007; Alenazi et al., 2012). Over a span of 5 years, Aladbe’s group performed a retrospective study in Qatar on the clinical and genetic profiling of ~70 symptomatic and carrier individuals affected by the autoinflammatory disorder and reported the expansion of the autoinflammatory disorder in Qatar (Aladbe et al., 2013). In a web-based international registry of the autoinflammatory disorders i.e., Eurofever, composed of the baseline and clinical information of 1,880 patients affected with autoinflammatory disease from 31 countries, it was also found that the second largest number of patients belonged to the eastern and southern Mediterranean region (Toplak et al., 2012). Incidence reports from Sweden on autoinflammatory disorders and hereditary amyloidosis were found to be 98% of Eastern Mediterranean origin, mainly in young Syrian descendants (Hemminki et al., 2013). High consanguineous marriages in Arab countries provides an opportunity to find novel risk loci like LACC1 and LRBA that are associated with the monogenic juvenile idiopathic arthritis (JIA) and inflammatory bowel disease with combined immunodeficiency, respectively, in the Arab population (Alangari et al., 2012; Patel et al., 2014; Martorana et al., 2017).
The recent availability of sequencing data from Middle Eastern populations, which were not previously covered in global sequencing projects, motivates us to understand the genetic epidemiology of monogenic autoinflammatory diseases (Fakhro et al., 2016; Scott et al., 2016). In the present analysis, we performed an integrative analysis with extensive literature screening for classifying variants in autoinflammatory disorders. We further attempted to understand the prevalence of these disorders by studying the allele and genotype frequencies of the variants in the Qatar population. Our analysis points to significant differences in the allele frequencies even for the small population studied.
The analysis was performed on population-scale datasets which involves 1,005 Qatar Genomes and Exomes (Fakhro et al., 2016) and 1,111 Greater Middle East exomes (Scott et al., 2016).
The Qatar dataset comprises a total of 88 whole genome and 917 whole exome sequences and a total of 20,937,965 single nucleotide variants and indels that were identified in alignment to the hg19/GRCh37 version of the reference human genome. The sequenced individuals belong to seven different sub-populations within Qatar namely: European (EUR), South Asian (SA), Bedouin (BED), African Pygmy (AP), Arab (AB), Persian (PER), and Sub Saharan African (SAF) (Fakhro et al., 2016).
The GME Variome Project comprised of high-quality whole exome data of 1,111 unrelated individuals from six different regions of GME which includes Northwest Africa (NWA, n = 85), Northeast Africa (NEA, n = 423), Turkish Peninsula (TP, n = 140), Syrian Desert (SD, n = 81), Arabian Peninsula (AP, n = 214), and Persia and Pakistan (PP, n = 168). The dataset consists of a total of 689,297 SNVs and indels which were detected in alignment to the hg19/GRCh37 human reference genome (Scott et al., 2016).
Three independent datasets of genetic variants were compiled for the analysis. This includes ClinVar, Infevers, and the Human Gene Mutation Database (HGMD).
The ClinVar database [ClinVar version: 2018-02-25] variants, which were annotated as pathogenic or likely pathogenic, were retrieved (Landrum et al., 2018). These variants were further filtered based on a list of 37 rare autoinflammatory genes and then matched with the Qatar variants. The autoinflammatory genes and its associated disorders are tabulated in Supplementary Table 1.
Infevers (accessed on 2018-28-02) is a publicly available database of genetic variants implicated in autoinflammatory disorders. The database lists a total of 1,586 genetic variants for 30 genes associated with 31 autoinflammatory disorders (Milhavet et al., 2008) (Infevers). The rare autoinflammatory variants for 29 genes, excluding MEFV as it has already been mentioned (Milhavet et al., 2008; Koshy et al., 2018), were downloaded and compiled. These variants were further filtered for the variants identified in the Qatar dataset.
The Human Gene Mutation Database (HGMD public version 2018) is a comprehensive resource and a collection of genetic variants in human genes (Milhavet et al., 2008; Stenson et al., 2014; Koshy et al., 2018). The data for all 37 autoinflammatory genes was manually compiled from the public version of the HGMD and filtered for the variants identified in the Qatar dataset.
All the autoinflammatory variants from ClinVar, HGMD, and Infevers that were detected in the Qatar datasets were further annotated using ANNOVAR software (v. date 2017-07-17) for the functional annotation, which is composed of annotations from various databases (Wang et al., 2010). These databases include RefGene, dbnsfp33a, dbscsnv11, avsnp147, intervar_20170202, kaviar_20150923, and the GWAS catalog for the prediction of variant pathogenicity and other characteristics. There are databases for allele frequency across the global population which consist of esp6500si_all, exac03, and 1000g2015aug_all. We also annotated our variants using ClinVar (v. date 2018-02-25) which comprises human variations associated with a phenotype (Landrum et al., 2018).
The expert panel from the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) has put forward 28 criteria for variant classification into five broad categories i.e., pathogenic, likely pathogenic, benign, likely benign, and VUS. Each criterion, depending on the pathogenicity of the variant, was weighted as very strong (PVS1), strong (PS1-4), moderate (PM1-6), or supporting (PP1-5). A similar case for benign characteristics was weighted as stand-alone (BA1), strong (BS1-4), or supporting (BP1-7). These criteria in combination classify the variant into the five broad categories (Richards et al., 2015). The detailed protocol for annotation of variants and assigning weighted criteria is described in Supplementary Data Sheet 1.
Variants are assigned different criteria depending on various evidence from literature and other databases. These criteria were put together in the Genetic Variant Interpretation tool (Genetic Variant Interpretation Tool) which classifies them into five categories viz pathogenic, likely pathogenic, benign, likely benign, and VUS.
We employed four large datasets of global populations to compare allele frequencies that include:
The 1000 Genomes project (1000g2015aug_all) dataset which encompasses the genomes of 2,504 individuals from five major populations i.e., Europe (EUR), South Asia (SAS), Africa (AFR), East Asia (EAS), and America (AMR) (The 1000 Genomes Project Consortium, 2015).
The National Heart Lung and Blood Institute (NHLBI) Exome Sequencing Project (ESP) which comprises unrelated 2,203 African-American and 4,300 European-American, totaling 6,503 individuals’ exomes that have been sequenced (Fu et al., 2013).
The Genome Aggregation Database (gnomAD V2) which comprises data from 123,136 exomes and 15,496 whole genomes of unrelated individuals. These individuals belong to different ancestries i.e., African/African American, Latino, Ashkenazi Jewish, East Asian, Finnish, Non-Finish European, South Asian, and other (Karczewski et al., 2019). We used a combined allele frequency of the gnomAD V2 exomes and genomes dataset. There might be a possibility that the variants are genotyped in the genomes but are absent in the exomes, or vice-versa.
The updated version of the gnomAD V3.1 comprises genetic data from 76,156 whole genome sequences of unrelated individuals with >759 million short nuclear variants. These individuals belong to nine ancestral groups namely African/African-American, Amish, Latino/Admixed American, Ashkenazi Jewish, East Asian, Finnish, Non-Finnish European, Middle Eastern, South Asian, and other (Karczewski et al., 2019).
The differences in allele frequencies of variants, annotated as pathogenic or likely pathogenic by ACMG guidelines, were tested for statistically significant differences with the global population. We compared the allele frequencies of the Qatar and GME populations and their subpopulation to the gnomAD all V2. We used gnomAD v2, since gnomAD V2 encompasses all the variants that are annotated as pathogenic or likely pathogenic in the Qatar dataset. For statistical significance, we implemented Fisher’s exact test and data with a p-value less than 0.05 was considered significant.
Natural Selection of genes was predicted based on differences in the derived allele from the ancestral allele which was predicted using Integrated Haplotype Score (iHS), or by differences in the allele frequency between populations to deduce selection pressure i.e. Wright’s fixation index (Fst) (Vitti et al., 2013; Karczewski et al., 2019). iHS for variants in the gene should be >2 or <-2 to be predicted as naturally selected (MerrimanLab / selectionTools). So, we calculated iHS for all variants of the Qatar population using the selection tools pipeline (Cadzow et al., 2014), which is publicly available on GitHub, out of which the top 1% of variants were filtered. Those autoinflammatory genes which fall in the top 1% criteria of iHS have been taken up for further analysis and their Fst scores were calculated using the PLINK algorithm to infer selection pressure among populations (Purcell et al., 2007; Cadzow et al., 2014). We calculated the Fst score among populations i.e., Qatar population and its subpopulations with 1000 genome subpopulations (African, European and South Asian). The methodology adopted in this study is represented in Supplementary Figure 1.
We retrieved pathogenic and likely pathogenic genetic variants that encompass a total of 72,331 variants from the ClinVar database. These variants were further filtered based on the list of 37 genes associated with monogenic autoinflammatory diseases resulting in 270 variants. These variants were further overlapped with the Qatar dataset and five variants were retrieved. In a similar way, the Infevers database had 1,254 genetic variants from 29 rare autoinflammatory genes excluding MEFV [Accessed: 15/11/2017]. These Infevers genetic variants were identified in the Qatar dataset that resulted in an overlap of 95 variants. Similarly, for HGMD, a total of 1,209 genetic variants exist in 37 rare autoinflammatory genes. These variants were overlapped with the Qatar dataset and a total of 72 genetic variants were retrieved.
ClinVar, Infevers, and HGMD had overlapping variants, while three variants were common to ClinVar and HGMD, 22 variants were common to HGMD and Infevers, whereas there was no common variant to ClinVar and Infevers. There were also variants that were unique to the database i.e., one variant in ClinVar, 46 in HGMD, and 72 in Infevers. So, we had a total of 145 unique variants in Qatar which overlapped with the databases. Venn diagrams for variant numbers and databases are represented in Figure 1. The information for the total number of variants from each database and genes is tabulated in Supplementary Table 2.
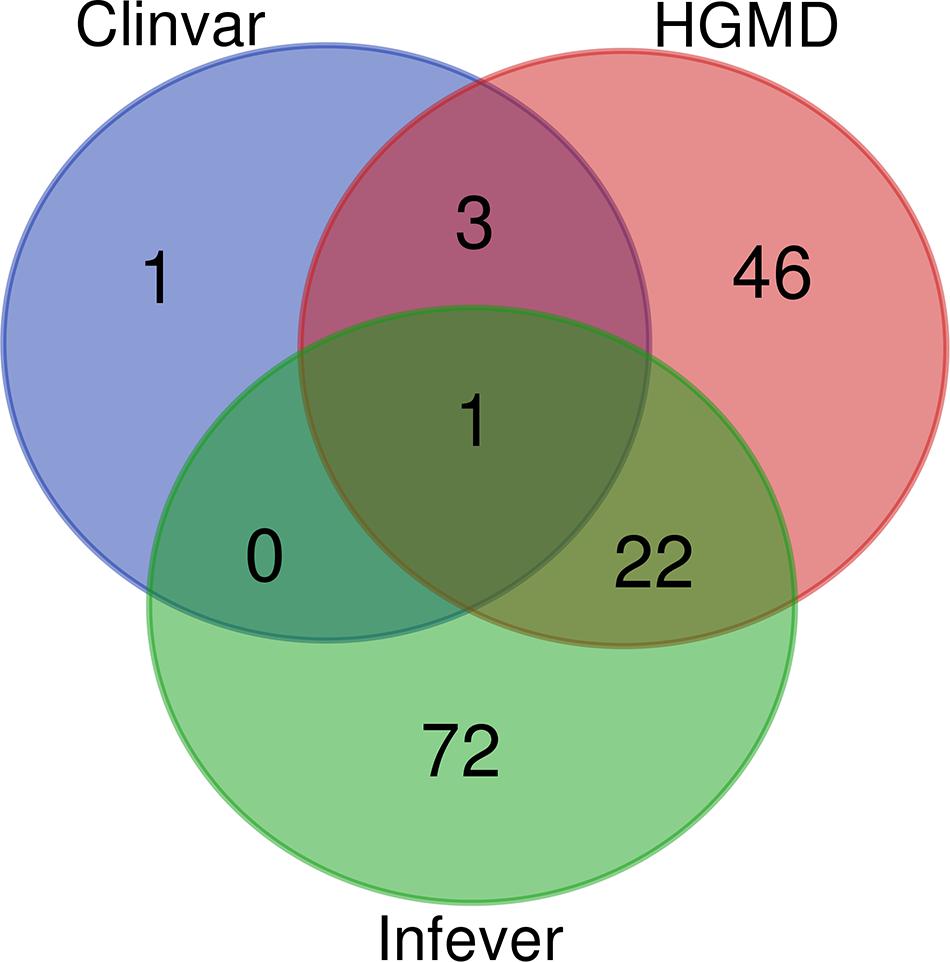
Figure 1. Venn Diagram for the variants present in HGMD, ClinVar, and Infevers.
We retrieved 145 unique Qatar genetic variants which overlapped with three databases. These were classified as one pathogenic and six likely pathogenic variants, while 92 variants were benign and likely benign, and 42 were VUS and VUS with conflicting evidence. The pathogenic and likely pathogenic variants are tabulated in Table 1. The detailed variant information is provided in Supplementary Table 3.
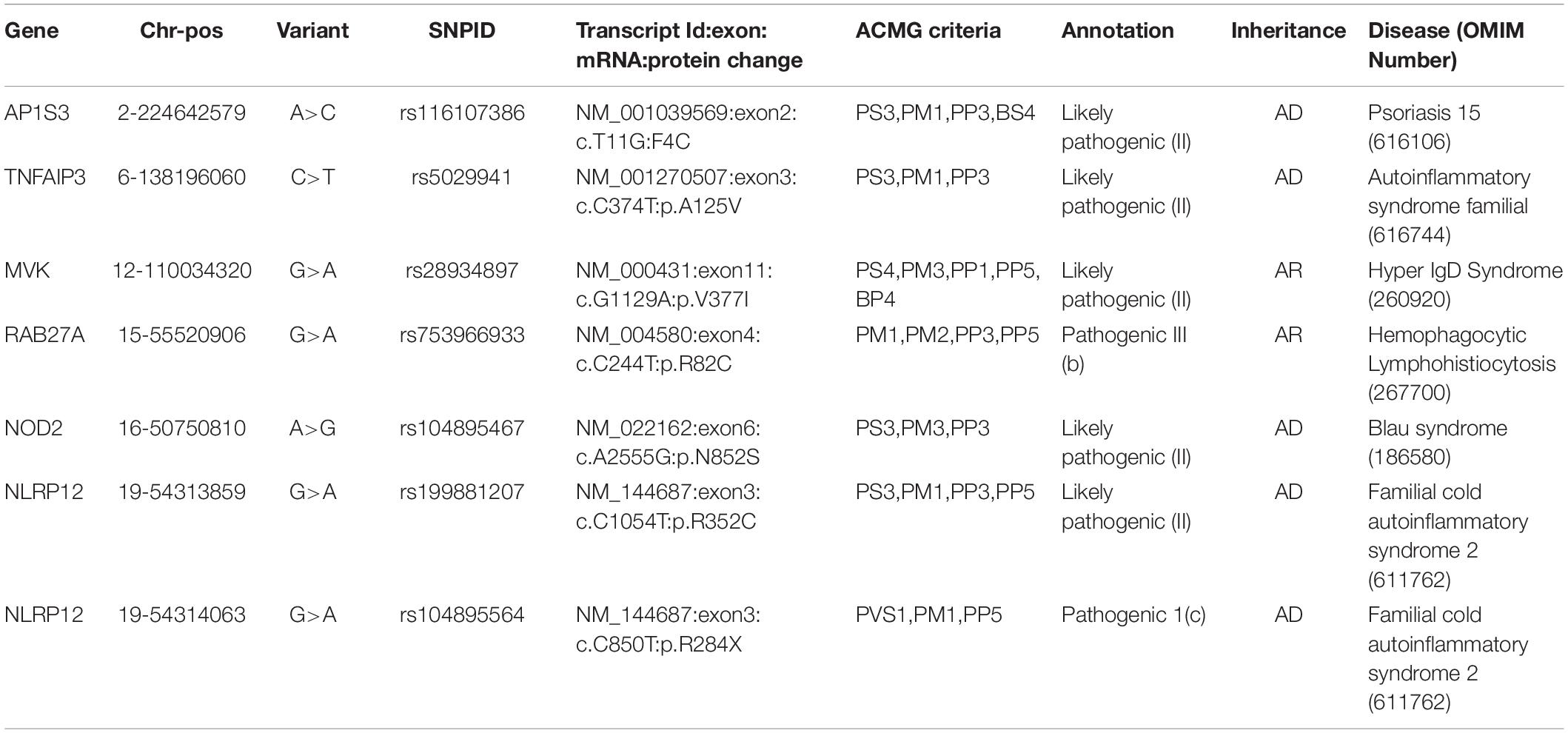
Table 1. Pathogenic and Likely Pathogenic variants annotated as per ACMG guidelines for interpretation of variants.
We implemented Fisher’s exact test to compare the allele frequencies of the Qatar and GME population and their subpopulations with the global dataset of gnomAD V2. We identified that five out of seven variants were significantly different (p-value < 0.05) in the Qatar population and its subpopulations, and two variants in GME population and its subpopulations. A likely pathogenic variant p.A125V rs5029941 in TNFAIP3 associated with the familial autoinflammatory, Behcet-like syndrome (OMIM: 616744) was found to be significantly high in the Sub-Saharan African subpopulation of Qatar compared to the gnomAD V2. In the Qatar Sub-Saharan African population, AF was found to be 1.6%, while in gnomAD V2 its AF was 0.16%. Another likely pathogenic variant p.V377I rs28934897 in the MVK gene associated with hyper-IgD syndrome (OMIM: 260920) was significantly enriched in the Qatar Arab subpopulation with AF 0.8% compared to the global AF i.e., absent in 1000 genomes, and ~0.15% in Esp6500, gnomAD V2, and gnomAD V3. Interestingly, this variant was also significantly enriched in the GME Arabian Peninsula (AP) subpopulation with an AF of 0.87% in comparison to the global frequency. An interesting pathogenic variant p.R82C rs753966933 in the RAB27A gene, associated with hemophagocytic lymphohistiocytosis (HLH) (OMIM number 267700), was found to be significantly enriched in the Qatar population and its Arab and Bedouin subpopulation in comparison to the global population. The AF in the Qatar population was found to be 0.5% and in Arab and Bedouin subpopulation its frequency is 2.7% and 0.1%, respectively. However, in the global population databases, this variant is absent in the 1000 Genome project, Esp6500, and gnomAD V3, while having a very low AF in gnomAD V2 i.e., 1.6 × 10–3%. This variant is also absent in the GME population and its subpopulation. A likely pathogenic variant p.R352C in NLRP12 rs199881207 was significantly enriched in two Qatar subpopulations i.e., European and South Asian, whose AF was 12.5% and 0.8%, respectively. However, it was absent in the 1000 genomes and had very low frequency in GME, Esp6500, gnomAD V2, and gnomAD V3 i.e., 0.05, 0.07, 0.04, and 0.02%, respectively. Another NLRP12 pathogenic variant p.R284X was found to be significantly enriched in Qatar and its Sub-Saharan African subpopulation with an AF of 0.5% and 2.9%, respectively, in comparison to the global population. The global population had low AF i.e., 0.2, 0.007, 0.015, and 0.04% in the 1000 Genome project, Esp6500, gnomAD V2, and gnomAD V3, respectively. Another likely pathogenic variant p.N852S in the NOD2 gene associated with Blau syndrome (OMIM number 186580), Crohn’s disease (OMIM number 266600), and Yao syndrome (OMIM number 617321) was significantly enriched in the GME population and its Northeast Africa (NEA) subpopulation in comparison to the global control population. Its allele frequency in GME and its NEA subpopulation was 0.3% and 0.5%, respectively, whereas in 1000 genome, Esp6500, gnomAD V2, and gnomAD V3 it was 0.04, 0.07, 0.1, and 0.07%, respectively. Comparison of the AF of the Qatar population and its subpopulation with GME and the global population dataset is represented in Figure 2. The comparison of the allele frequency of the Qatar population among the global population (1000 genome, Esp6500, gnomAD V2, and gnomAD V3) and GME is detailed in Supplementary Table 4.
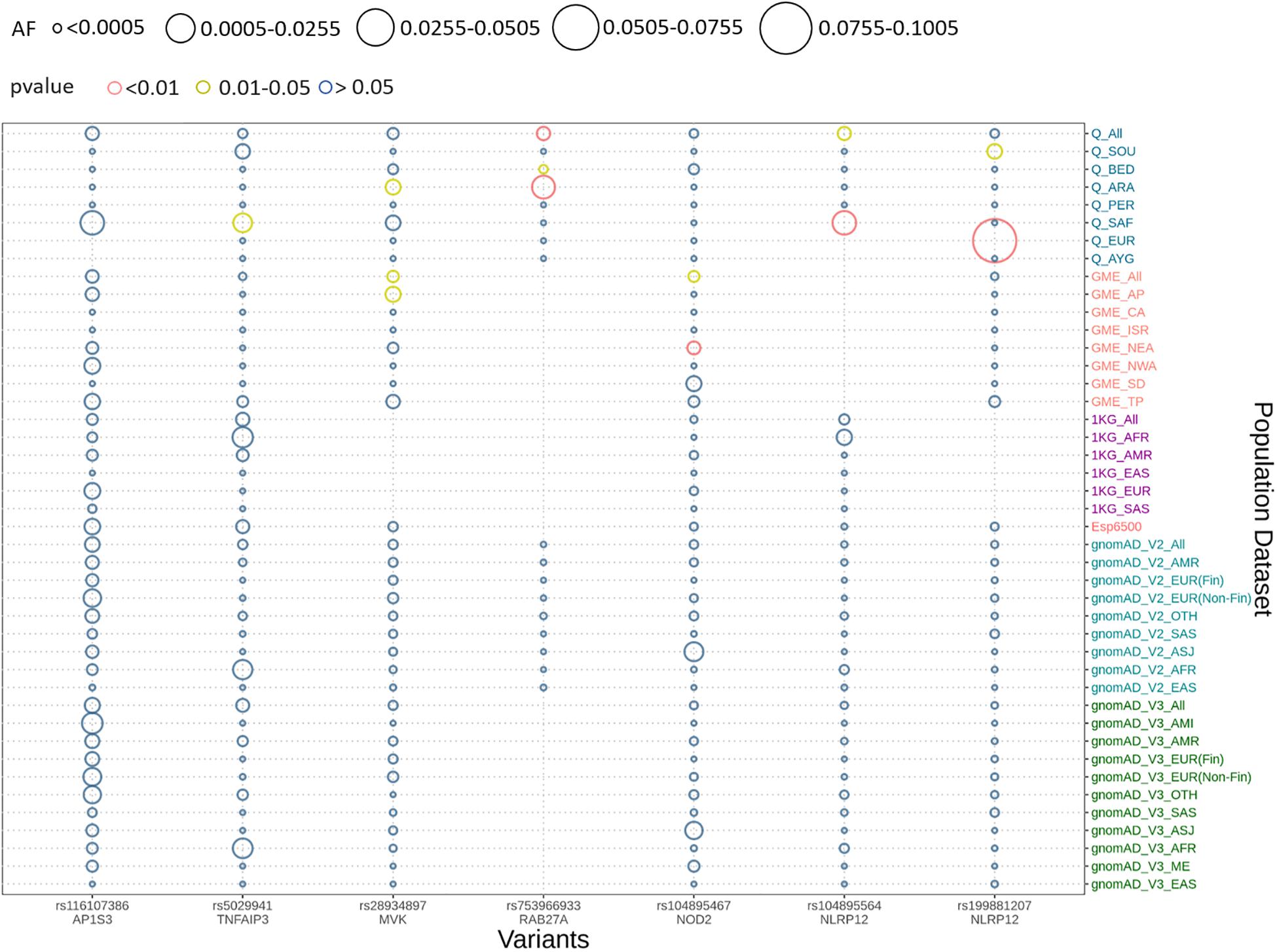
Figure 2. Comparison of allele frequency of pathogenic and likely pathogenic variants in the Qatar population with its subpopulation and GME and its subpopulation with 1000 genome, Esp6500, gnomAD V2, and gnomAD V3 and their subpopulations. Allele frequency highlighted with red for the p-values <0.01, yellow for p-value between 0.01 and 0.05 and blue for p-value >0.05.
We explored signals for natural selection in rare autoinflammatory genes. We took 1% of the top iHS scored variants and found that five rare autoinflammatory genes fall into the top 1% naturally selected variants. These five rare autoinflammatory genes were IL1RN, IL36RN, NLRP3, PSMB9, and RAB27A. Figure 3 represents the iHS and Fst plot for a natural selection of the RAB27A gene; we chose the RAB27A gene as one variant in RAB27A that had been annotated as likely pathogenic and had very high carrier allele frequency in comparison to the global control population.
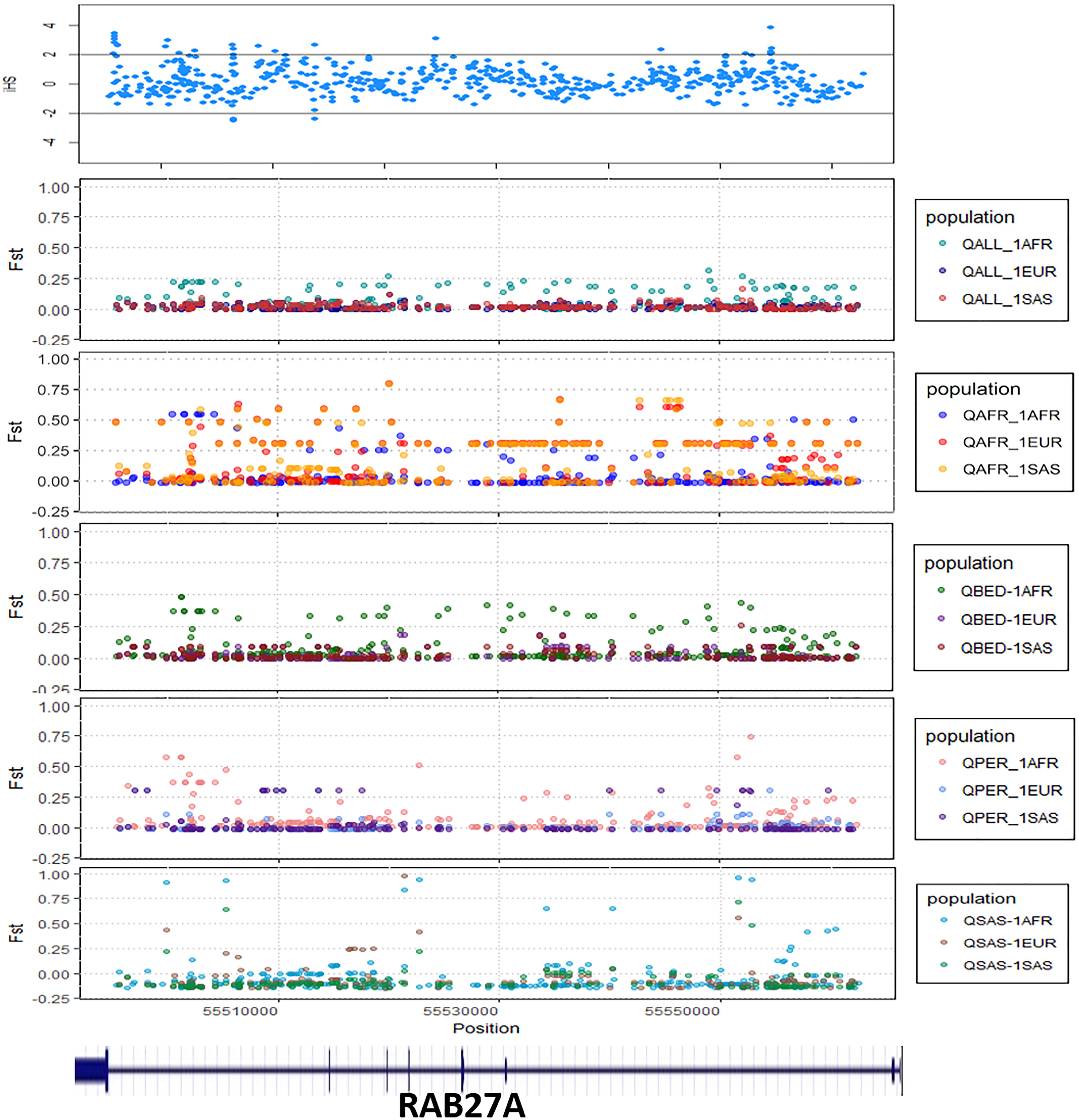
Figure 3. The Fst and iHS score depicted for RAB27A gene from the Qatari population (QALL, Qatar 1005 dataset; 1AFR, 1000 Genome AFRICAN; 1EUR, 1000 Genome EUROPEAN; 1SAS, 1000 Genome South Asian; QPER, Qatar-Persian subset; QBED, Qatar-Bedouin subset; QARB, Qatar-Arab-subset; QAFR, Qatar-African-subset; QSAS, Qatar-South-Asian-subset).
Monogenic autoinflammatory disorders are a group of Mendelian disorders that are caused due to the abnormal development and maturation of innate immune cells which results in unprovoked recurrent fever with rashes, systemic inflammation, pain in the chest, arthritis, and serositis (McDermott et al., 1999; Cush, 2013). The spectrum of diseases encompass over 30 distinct monogenic autoinflammatory diseases caused by pathogenic variants in 44 genes, as characterized by an expert panel of the Inborn Errors of Immunity Committee and Infevers (Touitou et al., 2004; Fakhro et al., 2016). A large number of cases of clinically and molecularly characterized autoinflammatory disorders have been reported in the literature, with the positivity of genetic confirmation of less than 10% including variants of uncertain significance (VUS) (Ter Haar et al., 2019). A significant number of autoinflammatory conditions have also been reported in the Middle Eastern population but the genetic epidemiology of autoinflammatory disorders is not yet known (Tunca and Ozdogan, 2005; de Jesus et al., 2015; Manthiram et al., 2017; Ter Haar et al., 2019). The available whole genome and whole exome sequence datasets of Middle Eastern populations were used to understand the genetic epidemiology of autoinflammatory disorders in the population (Fakhro et al., 2016; Scott et al., 2016).
In the present study, we compiled a comprehensive list of genes and genetic variants from a number of resources. A total of 37 genes associated with 35 autoinflammatory conditions formed the basis of the analysis (Touitou et al., 2004). We did not consider Familial Mediterranean Fever and the MEFV gene for the analysis, since an earlier work from our group comprehensively analyzed the genetic epidemiology of the genetic variants for this gene (Koshy et al., 2018). A total of 145 unique variants in the Qatar autoinflammatory variant, encompassing 917 exomes and 88 genomes were classified as per the ACMG/AMP guidelines for the interpretation of sequence variants. Systematic filtering and annotation by the ACMG/AMP guidelines resulted in seven pathogenic and likely pathogenic variants. The final list was composed of six nonsynonymous variants and one stop gain variant in five genes (one in MVK, one in RAB27A, one in NOD2, one in AP1S3, one in TNFAIP3, and two in NLRP12).
The nonsynonymous variant in AP1S3 i.e. Phe4Cys (p.F4C) rs116107386 associated with Psoriasis 15 (OMIM number 616106), annotated as likely pathogenic by the ACMG guidelines, had been functionally validated using transfection studies in HEK293 cells and HaCaT keratinocytes that showed the decreased activity of the mutant protein (Setta-Kaffetzi et al., 2014). The F4C variant is located in the functionally important domain of the AP1S3 protein, and it has been predicted as deleterious computationally. Another nonsynonymous likely pathogenic variant in TNFAIP3 i.e., Ala125Val (p.A125V), leads to familial autoinflammatory, Behcet-like syndrome (OMIM number 616744). Lodolce’s group transfected HEK293 cell lines with wild type and mutant alleles and found a decrease in enzymatic activity of TNFAIP3 to deubiquitinase the target protein TRAF2 in mutant allele (Lodolce et al., 2010). This variant also falls in a functionally important domain, which affects the functionality of the protein. The third nonsynonymous likely pathogenic variant in the MVK i.e., V377I, had been reported in various studies as a cause of Hyperimmunoglobulinemia D with Periodic Fever Syndrome (HIDS) (OMIM number 260920). Both of these diseases are rare and follow an autosomal recessive mode of inheritance. This variant was popularly known as the Dutch variant with a carrier frequency of 1:65 individuals (Houten et al., 2003). A number of studies have shown that this variant was present in homozygous or compound heterozygous states in multiple patients affected by HIDS with different ethnic backgrounds. Various in-vitro and in-vivo functional studies have proved that p.V377I is disease-causing with a very low allele frequency in control databases (Drenth et al., 1999; Houten et al., 1999, 2001; Cuisset et al., 2001; Levy et al., 2013; Govindaraj et al., 2020). The fourth nonsynonymous likely pathogenic variant has been found in RAB27A i.e., Arg82Cys (R82C), and had been predicted as a causal variant for hemophagocytic lymphohistiocytosis (HLH) (OMIM number 267700). This variant has very low allele frequency in the global population dataset and is located in a functional domain of the RAB27A. In 2016, Netter et al. (2016) also reported that a family has been severely affected due to homozygous p.R28C variant and less affected by heterozygous p.R82C. The functional assay also showed the significant effect of p.R82C on RAB27A binding activity to melanophilin and Munc13-4 (Cetica et al., 2015). A fifth nonsynonymous likely pathogenic variant was found in NOD2 i.e., Asn852Ser (p.N852S), which is associated with Blau syndrome (OMIM number 186580), Crohn’s disease (OMIM number 266600), and Yao syndrome (OMIM number 617321). These syndromes were associated with abnormal inflammation of the body. Horowitz et al. (2021) identified compound heterozygous variants in two affected individuals. The first individual was compound heterozygous for p.G908R and p.N852S and the second was for novel variant p.S506Vfs∗73 and p.G908R (Horowitz et al., 2021). The functional assessment had shown that p.N852S led to impaired NOD2 ligand muramyl dipeptide (MDP)-induced NF-κB activation which could lead to Crohn’s disease (Rivas et al., 2011). p.N852S had been co-segregated within affected Ashkenazi Jewish families with Crohn’s disease. It has been reported that 15% of Crohn’s disease patients of Ashkenazi Jewish ancestry had the p.N852S variant (Tukel et al., 2004). The sixth nonsynonymous likely pathogenic variant from NLRP12 i.e., Arg352Cys (p.R352C), is found to be associated with Familial cold autoinflammatory syndrome 2 (OMIM number 611762). In 2011 Jeru’s group performed an in-vivo experiment in which they transfected the HEK293 cell line with a wild-type plasmid and mutant R352C plasmid. They showed that the variant enhanced the signaling activity of procaspase 1 which leads to abnormal inflammation and causes NLRP12-Associated Periodic Fever Syndrome (Jéru et al., 2011). The R352C variant also falls in the functionally important domain of NLRP12. The seventh stop-gain pathogenic variant which falls in NLRP12 i.e., p.R248X causes Familial cold autoinflammatory syndrome 2 (OMIM number 611762). This stop gain variant leads to aberrant termination of protein synthesis which disrupts the integrity of protein structure and ultimately its function. NLRP12 was responsible for inhibiting the inflammatory response, but due to the stop-gain variant it was not regulated, which caused abnormal inflammation and periodic fever.
The allele frequencies of five out of seven variants classified as pathogenic or likely pathogenic were significantly different in the Qatar and GME population datasets in comparison with global populations. The allele frequencies of pathogenic or likely pathogenic variants showed concordance with another study previously performed in this region. In 2005, Mohammed Hammoudeh reported, for the first time, of an Arab child being affected by HIDS, with the homozygous variant p.V377I in the MVK gene (Hammoudeh, 2005). Moussa et al. (2015) also reported p.V377I segregation with HIDS in an Arabic family where two siblings were homozygous p.V377I and affected (Moussa et al., 2015). There were also various reports for a patient suffering from HIDS from the Middle East and nearby regions which include Turkey, Armenia, Kuwait, Israel, and Palestine (van der Hilst et al., 2008; Harel-Meir et al., 2009; Megged et al., 2011; Tas et al., 2012). In the Qatar population dataset, overall carrier allele frequency was 0.32% while the Arab subpopulation had a very high carrier allele frequency of 0.82%. Similarly, in the GME population dataset, carrier allele frequency was 0.35% while in the African Pygmy subpopulation, a high carrier allele frequency of 0.87% was observed. Another pathogenic variant p.R82C in RAB27A had a very high carrier allele frequency in the Qatar Arab subpopulation i.e., 2.7% while the allele frequency was 0.5% in the Qatar population dataset. A large study was conducted by Tukel et al. (2004) to find the frequency of Crohn’s disease-associated Ashkenazi, Sephardi, and Oriental Jewish Families in middle eastern countries which include Ashkenazi, Sephardi and Oriental Jews from Israel, Morocco, Turkey, Tunisia, Iraq, Kurds, Iran, Yemen, and Syria. They found that 15% of Ashkenazi Jews suffering from Crohn’s disease had the p.N852S variant (Tukel et al., 2004). Another study that involves p.N852S frequency in the Turkish population, but the N852S association with Crohn’s disease (Budak Diler and Yaraş, 2018) could not be found. In our study we found that p.N852S had high carrier allele frequency in GME subpopulations as 0.8% in the Syrian Desert, 0.5% in North-East Africa, and 0.3% in the Turkish Peninsula, and Qatar allele frequency was 0.1% and its subpopulation Bedouin was 0.2%.
The distinct frequencies of many variants prompted us to explore whether any of these genes showed signals of natural selection. We have adopted two parameters i.e., Wright Fixation score (Fst) and integrated Haplotype (iHS) score to understand the signal of the selection in the rare autoinflammatory genes. Our analysis revealed the RAB27A gene has high values of the Fst score in the Qatar African subpopulation in comparison to the global population. This marks the genetic differences of the Qatari subpopulation Africa with the global population as well as other Qatari subpopulations. Also, the positive iHS score for the multiple variants in the RAB27A gene represents the signal of selection of the RAB27A gene in the Qatar population. The RAB27A protein is mainly involved in the transportation of melanin for skin pigmentation. A previously reported study has shown a high correlation between RAB27A gene selection to the dark-pigmented melanocytes in comparison to the lightly-pigmented melanocytes. This makes the RAB27A gene a possible candidate to be positively selected for individuals of African descent in comparison to Caucasians (Yoshida-Amano et al., 2012). It also plays an important role in cytotoxic T lymphocytes for killing or neutralization of foreign invaders. RAB27A had been evolutionarily selected among different species, as editing levels of RAB27A showed the highest difference among humans and rhesus monkeys (Paz-Yaacov et al., 2010). Similarly, IL1RN was identified to be under selection in the European-American population (Akey et al., 2004; Jaffe et al., 2013) whereas IL36RN was found to be evolutionary conserved among different species (Lv et al., 2016). Other genes like NLRP3 and PSMB9 did not show any strong signals of natural selection or conservation.
In summary, our analysis integrating population-scale genomic datasets provide the first comprehensive insights into the genetic epidemiology of monogenic autoinflammatory diseases in Middle Eastern populations. Our analysis suggests distinct allele frequencies in subpopulations and also suggests that at least two genes show strong signals of natural selection in the middle eastern population. As more population-scale genomic datasets from the region become available, including from national initiatives like the Bahrain genome program and the Emirati genome initiative apart from the ongoing Qatar Genome project, would provide a higher resolution toward understanding frequencies of rarer genetic variants (Al-Ali et al., 2018).
The datasets for this study can be found in the NCBI Sequence Read Archive (SRA accessions: SRP060765, SRP061943, and SRP061463, accessible online at http://www.ncbi.nlm.nih.gov/Traces/study/?acc=SRP060765%2CSRP061943%2CSRP061463&go=go, SRA accession SRP061943).
PS collected the data. AJ performed the ACMG annotations, data analysis, and allele frequency comparison. VS oversaw the analysis. All authors contributed to writing the manuscript.
This study was funded by the Council of Scientific and Industrial Research (CSIR, India) through Grant MLP1809 (IndiGen). The authors acknowledge funding from the Council of Scientific and Industrial Research (CSIR, India) through Grant MLP1809 (IndiGen). AJ is a recipient of the Senior Research Fellowship from Council of Scientific and Industrial Research (CSIR, India). The funders had no role in the preparation of the manuscript or decision to submit.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Disha Sharma, Mukta Poojary, Bani Jolly, and Vishu Gupta for suggestions that enriched the manuscript. The authors acknowledge the researchers at Weill Cornell Medicine for sharing the genome and exome data sets without which this analysis was not possible.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.631340/full#supplementary-material
Supplementary Figure 1 | Schematic summarizing the data analysis pipeline utilized for this study.
Supplementary Table 1 | Genes and associated autoinflammatory diseases considered in the present analysis and their respective inheritance patterns.
Supplementary Table 2 | The total number of variants in the auto inflammatory genes from each database overlapped with the Qatar dataset.
Supplementary Table 3 | The detailed variant annotation and classification based on ACMG-AMP guidelines.
Supplementary Table 4 | Allele frequency of pathogenic and likely pathogenic variants compared with control global population (1000 genome, gnomAD V2, gnomAD V3, and Esp6500) using Fisher exact test (p < 0.05). BED, Bedouin; SAF, Sub-Saharan African; EUR, European; SOU, South Asian; APY, African Pygmy; ARA, ARAB; PER, Persian; NA, not applicable]. Significant values are marked with ∗.
Supplementary Data Sheet 1 | The ACMG-AMP guidelines for each attribute.
Akey, J. M., Eberle, M. A., Rieder, M. J., Carlson, C. S., Shriver, M. D., Nickerson, D. A., et al. (2004). Population history and natural selection shape patterns of genetic variation in 132 genes. PLoS Biol. 2:e286. doi: 10.1371/journal.pbio.0020286
Aladbe, B., Aly, A., Taha, R., Elshanti, H., Moussa, T., Al Amry, F., et al. (2013). P02-007 – Childhood autoinflammatory disorders in Qatar. Pediatr. Rheumatol. 11:A114. doi: 10.1186/1546-0096-11-s1-a114
Al-Ali, M., Osman, W., Tay, G. K., and AlSafar, H. S. (2018). A 1000 Arab genome project to study the Emirati population. J. Hum. Genet. 63, 533–536. doi: 10.1038/s10038-017-0402-y
Alangari, A., Alsultan, A., Adly, N., Massaad, M. J., Kiani, I. S., Aljebreen, A., et al. (2012). LPS-responsive beige-like anchor (LRBA) gene mutation in a family with inflammatory bowel disease and combined immunodeficiency. J. Allergy Clin. Immunol. 130, 481–488.e2.
Alenazi, A., Al Sonbul, A., Al Jumaah, S., Al Mehaidib, A., and Al-Mayouf, S. M. (2012). A retrospective review of autoinflammatory diseases in Saudi children at a rheumatology clinic. Ann. Saudi Med. 32, 43–48. doi: 10.5144/0256-4947.2012.43
Al-Mosawi, Z. S., Al-Saad, K. K., Ijadi-Maghsoodi, R., El-Shanti, H. I., and Ferguson, P. J. (2007). A splice site mutation confirms the role of LPIN2 in Majeed syndrome. Arthritis Rheum. 56, 960–964. doi: 10.1002/art.22431
Ben-Chetrit, E., Gattorno, M., Gul, A., Kastner, D. L., Lachmann, H. J., Touitou, I., et al. (2018). Consensus proposal for taxonomy and definition of the autoinflammatory diseases (AIDs): a Delphi study. Ann. Rheum. Dis. 77, 1558–1565. doi: 10.1136/annrheumdis-2017-212515
Budak Diler, S., and Yaraş, S. (2018). CTLA-4 (+49A/G) and NOD2/CARD15 (N852S) polymorphisms with inflammatory bowel disease in Turkish patients. Cell. Mol. Biol. 64, 97–101. doi: 10.14715/cmb/2018.64.11.18
Cadzow, M., Boocock, J., Nguyen, H. T., Wilcox, P., Merriman, T. R., and Black, M. A. (2014). A bioinformatics workflow for detecting signatures of selection in genomic data. Front. Genet. 5:293. doi: 10.3389/fgene.2014.00293
Cetica, V., Hackmann, Y., Grieve, S., Sieni, E., Ciambotti, B., Coniglio, M. L., et al. (2015). Patients with Griscelli syndrome and normal pigmentation identify RAB27A mutations that selectively disrupt MUNC13-4 binding. J. Allergy Clin. Immunol. 135, 1310–1318.e1.
Ciccarelli, F., De Martinis, M., and Ginaldi, L. (2014). An update on autoinflammatory diseases. Curr. Med. Chem. 21, 261–269. doi: 10.2174/09298673113206660303
Cuisset, L., Drenth, J. P., Simon, A., Vincent, M. F., van der Velde Visser, S., van der Meer, J. W., et al. (2001). Molecular analysis of MVK mutations and enzymatic activity in hyper-IgD and periodic fever syndrome. Eur. J. Hum. Genet. 9, 260–266. doi: 10.1038/sj.ejhg.5200614
de Jesus, A. A., Canna, S. W., Liu, Y., and Goldbach-Mansky, R. (2015). Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu. Rev. Immunol. 33, 823–874. doi: 10.1146/annurev-immunol-032414-112227
Drenth, J. P., Cuisset, L., Grateau, G., Vasseur, C., van de Velde-Visser, S. D., de Jong, J. G., et al. (1999). Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD study group. Nat. Genet. 22, 178–181. doi: 10.1038/9696
Fakhro, K. A., Staudt, M. R., Ramstetter, M. D., Robay, A., Malek, J. A., Badii, R., et al. (2016). The Qatar genome: a population-specific tool for precision medicine in the Middle East. Hum. Genome Var. 3:16016.
Fu, W., O’Connor, T. D., Jun, G., Kang, H. M., Abecasis, G., Leal, S. M., et al. (2013). Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 493, 216–220. doi: 10.1038/nature11690
Govindaraj, G. M., Jain, A., Peethambaran, G., Bhoyar, R. C., Vellarikkal, S. K., Ganapati, A., et al. (2020). Spectrum of clinical features and genetic variants in mevalonate kinase (MVK) gene of South Indian families suffering from Hyperimmunoglobulin D Syndrome. PLoS One 15:e0237999. doi: 10.1371/journal.pone.0237999
Hammoudeh, M. (2005). Hyperimmunoglobulinemia D syndrome in an Arab child. Clin. Rheumatol. 24, 92–94. doi: 10.1007/s10067-004-0953-0
Harel-Meir, M., Bujanover, Y., Berkun, Y., Anikster, Y., and Anikster, Y. (2009). Mevalonic aciduria in a child featuring hepatic fibrosis and novel mevalonate kinase mutations. Open Pediatr. Med. J. 3, 45–47. doi: 10.2174/1874309900903010045
Hausmann, J. S., Lomax, K. G., Shapiro, A., and Durrant, K. (2018). The patient journey to diagnosis and treatment of autoinflammatory diseases. Orphanet J. Rare Dis. 13:156.
Hemminki, K., Li, X., Försti, A., Sundquist, J., and Sundquist, K. (2013). Incidence of hereditary amyloidosis and autoinflammatory diseases in Sweden: endemic and imported diseases. BMC Med. Genet. 14:88. doi: 10.1186/1471-2350-14-88
Horowitz, J. E., Warner, N., Staples, J., Crowley, E., Murchie, R., Van Hout, C., et al. (2021). Mutation spectrum of NOD2 reveals recessive inheritance as a main driver of Early Onset Crohn’s Disease. Sci. Rep. 11:5595. doi: 10.1101/098574
Houten, S. M., Koster, J., Romeijn, G. J., Frenkel, J., Di Rocco, M., Caruso, U., et al. (2001). Organization of the mevalonate kinase (MVK) gene and identification of novel mutations causing mevalonic aciduria and hyperimmunoglobulinaemia D and periodic fever syndrome. Eur. J. Hum. Genet. 9, 253–259. doi: 10.1038/sj.ejhg.5200595
Houten, S. M., Kuis, W., Duran, M., de Koning, T. J., van Royen-Kerkhof, A., Romeijn, G. J., et al. (1999). Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nat. Genet. 22, 175–177. doi: 10.1038/9691
Houten, S. M., van Woerden, C. S., Wijburg, F. A., Wanders, R. J. A., and Waterham, H. R. (2003). Carrier frequency of the V377I (1129G>A) MVK mutation, associated with Hyper-IgD and periodic fever syndrome, in the Netherlands. Eur. J. Hum. Genet. 11, 196–200. doi: 10.1038/sj.ejhg.5200933
Jaffe, S., Normand, N., Jayaram, A., Orfanelli, T., Doulaveris, G., Passos, M., et al. (2013). Unique variation in genetic selection among Black North American women and its potential influence on pregnancy outcome. Med. Hypotheses 81, 919–922. doi: 10.1016/j.mehy.2013.08.015
Jéru, I., Le Borgne, G., Cochet, E., Hayrapetyan, H., Duquesnoy, P., Grateau, G., et al. (2011). Identification and functional consequences of a recurrent NLRP12 missense mutation in periodic fever syndromes. Arthritis Rheum. 63, 1459–1464. doi: 10.1002/art.30241
Karczewski, K. J., Francioli, L. C., Tiao, G., Cummings, B. B., Alföldi, J., Wang, Q., et al. (2019). Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv [Preprint] doi: 10.1101/531210
Koshy, R., Sivadas, A., and Scaria, V. (2018). Genetic epidemiology of familial Mediterranean fever through integrative analysis of whole genome and exome sequences from Middle East and North Africa. Clin. Genet. 93, 92–102. doi: 10.1111/cge.13070
Landrum, M. J., Lee, J. M., Benson, M., Brown, G. R., Chao, C., Chitipiralla, S., et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067.
Levy, M., Arion, A., Berrebi, D., Cuisset, L., Jeanne-Pasquier, C., Bader-Meunier, B., et al. (2013). Severe early-onset colitis revealing mevalonate kinase deficiency. Pediatrics 132, e779–e783.
Lodolce, J. P., Kolodziej, L. E., Rhee, L., Kariuki, S. N., Franek, B. S., McGreal, N. M., et al. (2010). African-derived genetic polymorphisms in TNFAIP3 mediate risk for autoimmunity. J. Immunol. 184, 7001–7009. doi: 10.4049/jimmunol.1000324
Lv, Z., Fan, J., Zhang, X., Huang, Q., Han, J., Wu, F., et al. (2016). Integrative genomic analysis of interleukin-36RN and its prognostic value in cancer. Mol. Med. Rep. 13, 1404–1412. doi: 10.3892/mmr.2015.4667
Majeed, H. A., Kalaawi, M., Mohanty, D., Teebi, A. S., Tunjekar, M. F., Al-Gharbawy, F., et al. (1989). Congenital dyserythropoietic anemia and chronic recurrent multifocal osteomyelitis in three related children and the association with Sweet syndrome in two siblings. J. Pediatr. 115, 730–734. doi: 10.1016/s0022-3476(89)80650-x
Manthiram, K., Zhou, Q., Aksentijevich, I., and Kastner, D. L. (2017). Erratum: Corrigendum: The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nature Immunol. 18, 1271–1271. doi: 10.1038/ni1117-1271a
Martinez-Quiles, N., and Goldbach-Mansky, R. (2018). Updates on autoinflammatory diseases. Curr. Opin. Immunol. 55, 97–105. doi: 10.1016/j.coi.2018.09.014
Martorana, D., Bonatti, F., Mozzoni, P., Vaglio, A., and Percesepe, A. (2017). Monogenic autoinflammatory diseases with mendelian inheritance: genes, mutations, and genotype/phenotype correlations. Front. Immunol. 8:344. doi: 10.3389/fimmu.2017.00344
McDermott, M. F., Aksentijevich, I., Galon, J., McDermott, E. M., Ogunkolade, B. W., Centola, M., et al. (1999). Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 97, 133–144. doi: 10.1016/s0092-8674(00)80721-7
Megged, O., Navon-Elkan, P., Anikster, Y., and Kleid, D. (2011). Recurrent Fever, arthritis, lymphadenopathy, and hepatosplenomegaly. Pediatr. Infect. Dis. J. 30, 182–185. doi: 10.1097/inf.0b013e3181f228b0
Milhavet, F., Cuisset, L., Hoffman, H. M., Slim, R., El-Shanti, H., Aksentijevich, I., et al. (2008). The infevers autoinflammatory mutation online registry: update with new genes and functions. Hum. Mutat. 29, 803–808. doi: 10.1002/humu.20720
Moussa, T., Aladbe, B., Taha, R. Z., Remmers, E. F., El-Shanti, H., and Fathalla, B. M. (2015). Overlap of familial Mediterranean fever and hyper-IgD syndrome in an Arabic kindred. J. Clin. Immunol. 35, 249–253. doi: 10.1007/s10875-015-0140-x
Netter, P., Chan, S. K., Banerjee, P. P., Monaco-Shawver, L., Noroski, L. M., Hanson, I. C., et al. (2016). A novel Rab27a mutation binds melanophilin, but not Munc13-4, causing immunodeficiency without albinism. J. Allergy Clin. Immunol. 138, 599–601.e3.
Patel, N., El Mouzan, M. I., Al-Mayouf, S. M., Adly, N., Mohamed, J. Y., Al Mofarreh, M. A., et al. (2014). Study of Mendelian forms of Crohn’s disease in Saudi Arabia reveals novel risk loci and alleles. Gut 63, 1831–1832. doi: 10.1136/gutjnl-2014-307859
Paz-Yaacov, N., Levanon, E. Y., Nevo, E., Kinar, Y., Harmelin, A., Jacob-Hirsch, J., et al. (2010). Adenosine-to-inosine RNA editing shapes transcriptome diversity in primates. Proc. Natl. Acad. Sci. U.S.A. 107, 12174–12179. doi: 10.1073/pnas.1006183107
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Ferreira, M. A. R., Bender, D., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Rivas, M. A., Beaudoin, M., Gardet, A., Stevens, C., Sharma, Y., Zhang, C. K., et al. (2011). Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 43, 1066–1073. doi: 10.1038/ng.952
Salehzadeh, F. (2015). Familial mediterranean fever in Iran: a report from FMF registration center. Int. J. Rheumatol. 2015:912137.
Scott, E. M., Halees, A., Itan, Y., Spencer, E. G., He, Y., Azab, M. A., et al. (2016). Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat. Genet. 48, 1071–1076. doi: 10.1038/ng.3592
Setta-Kaffetzi, N., Simpson, M. A., Navarini, A. A., Patel, V. M., Lu, H.-C., Allen, M. H., et al. (2014). AP1S3 mutations are associated with pustular psoriasis and impaired Toll-like receptor 3 trafficking. Am. J. Hum. Genet. 94, 790–797. doi: 10.1016/j.ajhg.2014.04.005
Stenson, P. D., Mort, M., Ball, E. V., Shaw, K., Phillips, A., and Cooper, D. N. (2014). The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet. 133, 1–9. doi: 10.1007/s00439-013-1358-4
Tas, D. A., Dınkcı, S., and Erken, E. (2012). Different clinical presentation of the hyperimmunoglobulin D syndrome (HIDS) (four cases from Turkey). Clin. Rheumatol. 31, 889–893. doi: 10.1007/s10067-011-1932-x
Ter Haar, N. M., Eijkelboom, C., Cantarini, L., Papa, R., Brogan, P. A., Kone-Paut, I., et al. (2019). Clinical characteristics and genetic analyses of 187 patients with undefined autoinflammatory diseases. Ann. Rheum. Dis. 78, 1405–1411. doi: 10.1136/annrheumdis-2018-214472
The 1000 Genomes Project Consortium (2015). A global reference for human genetic variation. Nature 526, 68–74. doi: 10.1038/nature15393
The International FMF Consortium (1997). Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial mediterranean fever. Cell 90, 797–807. doi: 10.1016/s0092-8674(00)80539-5
Toplak, N., Frenkel, J., Ozen, S., Lachmann, H. J., Woo, P., Koné-Paut, I., et al. (2012). An international registry on autoinflammatory diseases: the Eurofever experience. Ann. Rheum. Dis. 71, 1177–1182. doi: 10.1136/annrheumdis-2011-200549
Touitou, I., Lesage, S., McDermott, M., Cuisset, L., Hoffman, H., Dode, C., et al. (2004). Infevers: an evolving mutation database for auto-inflammatory syndromes. Hum. Mutat. 24, 194–198. doi: 10.1002/humu.20080
Tukel, T., Shalata, A., Present, D., Rachmilewitz, D., Mayer, L., Grant, D., et al. (2004). Crohn disease: frequency and nature of CARD15 mutations in Ashkenazi and Sephardi/Oriental Jewish families. Am. J. Hum. Genet. 74, 623–636. doi: 10.1086/382226
Tunca, M., and Ozdogan, H. (2005). Molecular and genetic characteristics of hereditary autoinflammatory diseases. Curr. Drug Targets Inflamm. Allergy 4, 77–80. doi: 10.2174/1568010053622957
van der Hilst, J. C. H., Bodar, E. J., Barron, K. S., Frenkel, J., Drenth, J. P. H., van der Meer, J. W. M., et al. (2008). Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine 87, 301–310. doi: 10.1097/md.0b013e318190cfb7
Vitti, J. J., Grossman, S. R., and Sabeti, P. C. (2013). Detecting natural selection in genomic data. Annu. Rev. Genet. 47, 97–120. doi: 10.1146/annurev-genet-111212-133526
Wang, K., Li, M., and Hakonarson, H. (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38:e164. doi: 10.1093/nar/gkq603
Keywords: autoinflammatory disease, Qatar, Middle East, genome, epidemiology
Citation: Sharma P, Jain A and Scaria V (2021) Genetic Landscape of Rare Autoinflammatory Disease Variants in Qatar and Middle Eastern Populations Through the Integration of Whole-Genome and Exome Datasets. Front. Genet. 12:631340. doi: 10.3389/fgene.2021.631340
Received: 19 November 2020; Accepted: 24 March 2021;
Published: 13 May 2021.
Edited by:
Dana C. Crawford, Case Western Reserve University, United StatesReviewed by:
Eda Tahir Turanli, National Institutes of Health Clinical Center, National Institutes of Health (NIH), United StatesCopyright © 2021 Sharma, Jain and Scaria. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vinod Scaria, dmlub2RzQGlnaWIuaW4=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.