 Binyi Yang
Binyi Yang Cheng Lei
Cheng Lei Danhui Yang
Danhui Yang Zhiping Tan
Zhiping Tan Ting Guo
Ting Guo Hong Luo
Hong Luo- 1Department of Pulmonary and Critical Care Medicine, The Second Xiangya Hospital, Central South University, Changsha, China
- 2Research Unit of Respiratory Disease, Central South University, Changsha, China
- 3Hunan Diagnosis and Treatment Center of Respiratory Disease, Changsha, China
- 4Department of Cardiovascular Surgery, Clinical Center for Gene Diagnosis and Therapy, The Second Xiangya Hospital of Central South University, Changsha, China
Background: Cystic fibrosis (CF) is an autosomal recessive disease caused by genetic variants of the cystic fibrosis transmembrane conductance regulator (CFTR) gene. It is a common hereditary disease in Caucasians while rare in the Chinese. Until now, only 87 Chinese patients have been reported with molecular confirmations. The variant spectrum and clinical features of Chinese CF patients are obviously different from those of Caucasians.
Materials and Methods: Whole-exome sequencing was applied to analyze the exome of three individuals who have only the typical CF phenotype in the respiratory system from two consanguineous families. The protein domain and structure analysis were applied to predict the impact of the variants. Sanger sequencing was applied to validate the candidate variants.
Results: A previously reported homozygous variant in CFTR (NM_000492.4: c.1000C > T, p.R334W) was identified in proband I. A novel homozygous variant in a polymorphic position (NM_000492.4: c.1409T > A, p.V470E) was identified in two individuals in the family II. The novel CFTR variant predicted to be disease-causing is the first, to the best of our knowledge, to be reported in CFTR. However, in vitro validation is still needed.
Conclusion: Our finding expands the variant spectrum of CFTR, reveals clearer clinical phenotype distinction and variant spectrum distinction between Chinese and Caucasian CF patients, and contributes to a more rapid genetic diagnosis and future genetic counseling.
Introduction
Cystic fibrosis (CF), first reported in 1938, is an inherited autosomal recessive disease involving multiple organs, including the respiratory, digestive, and reproductive systems (Andersen, 1938). Respiratory failure after bronchiectasis and infection is now the main cause of death in CF patients. CF is a common hereditary disease in Caucasians but is rare among Asians, especially the Chinese. The estimated incidence of the disease in Caucasian population is about 1/3,000, but no more than 100 cases that have been reported in China (Liu et al., 2020). The disease is mainly detected through abnormally high chloride concentrations in sweat and gene sequencing.
Cystic fibrosis is caused by variants of the cystic fibrosis trans-membrane conductance regulator (CFTR) gene. CFTR is a chloride and bicarbonate transport protein, encoded by the CFTR gene located at the long arm of chromosome 7, which has 27 exons. As CFTR is mainly expressed in various glandular tissues, the variants can cause abnormal ion transport and secretory function, subsequently leading to bronchiectasis, pancreatic insufficiency, and fertility dysfunction in males (Ratjen et al., 2015). More than 2,000 different variants have been identified in CFTR, and the most recent update from the CFTR2 database1 recorded a total of 442 variants including 360 CF-causing variants. All variants are divided into six classes according to their mechanisms and pathogenicity. The most prevalent variant in Caucasians, p.F508del, was classified in Class II(Ratjen et al., 2015).
Studies have shown that the clinical phenotypes and variant spectrum of CF patients in China and those of the Caucasian population are very different (Guo et al., 2018). There are approximately 90 Chinese CF patients who have been reported up to now. In recent years, however, as the understanding of CF has deepened and the methods of diagnosis have improved, a growing number of CF patients have been diagnosed and reported in China. Here, we used whole-exome sequencing to analyze the exome of three individuals who have only a typical respiratory system CF phenotype, from two consanguineous families, and found CFTR variants including a novel variant in a polymorphic position.
Materials and Methods
Ethical Compliance
This study was approved by the Review Board of the Second Xiangya Hospital of Central South University in China. Three patients diagnosed with bronchiectasis from two Chinese consanguineous families participated in the study. Written informed consent was obtained from all participants.
Whole-Exome Sequencing and Variants Filtering
Whole-exome sequencing was performed on three individuals from two consanguineous families. Peripheral blood samples (3–5 ml) from proband I in family I; proband IIa and his brother IIb in family II were obtained with informed consent, respectively. The genomic DNA was extracted from peripheral blood cells by DNeasy blood and tissue kit (QIAGEN # 69506). The exomes of the probands were captured by Agilent Sureselect Human All Exons V6 kit (Agilent, California, United States) and sequencing on Illumina Hiseq X-10 platform (Illumina Inc, San Diego, United States). The single-nucleotide variants (SNVs) and short insertions and deletions (INDELs) were filtered as follow as we describe (Supplementary Figure 1): (1) Variants in the 1,000 Genomes Project (1,000G2), NHLBI-ESP project3, and Exome Aggregation Consortium (ExAC4) with minor allele frequency (MAF) ≤ 0.01 were included. (2) Intergenic, intronic, and untranslated regions and synonymous variants were excluded from subsequent analyses. (3) Bioinformatics analyses (Sift, Polyphen-2, Mutationtaster, MutationAssessor) were used for the remaining variants. (4) The remaining data were flittered by bronchiectasis-related diseases, which include primary ciliary dyskinesia (PCD), cystic fibrosis, and primary immunodeficiency. (5) Runs of homozygosity (ROH) analysis was also performed due to the fact that all probands came from consanguineous families. The ROH analysis was based on the data from whole-exome sequencing data using the Automap algorithm (Quinodoz et al., 2021).
Variant Validation With Sanger Sequencing
Sanger sequencing was used to validate the candidate variants found in whole-exome sequencing. Primers for PCR amplification of fragments with individual variants were designed by Primer Premier 5 software (Premier Biosoft, Palo Alto, CA, United States). The sequences of the primers are listed in Supplementary Table 1.
Results
Clinical Summary
Family 1
Proband I was a 14-year-old girl whose parents are consanguineous (Figure 1). She had suffered from recurrent coughing and expectoration for 2 years and came to our clinic for 1 day due to acute hemoptysis. Physical examination showed moist rales in both lower lungs. CT (computed tomography) showed inflammation in bilateral maxillary sinus, ethmoid sinus, and sphenoid sinus, bronchiectasis in both lungs, and calcification of the liver (Figure 2A). Her nasal nitric oxide concentration (nNO) was 74 parts per billion (ppb). The pulmonary function test indicated that she had severe mixed pulmonary ventilation dysfunction (FEV1% predicted: 48.0%, FEV1/FVC: 80.5%), and the bronchial dilation test was negative. The sputum culture showed infection with Pseudomonas aeruginosa. After hemostasis, antibiotic, and expectorant treatment, her symptoms were relieved, and the patient was discharged.
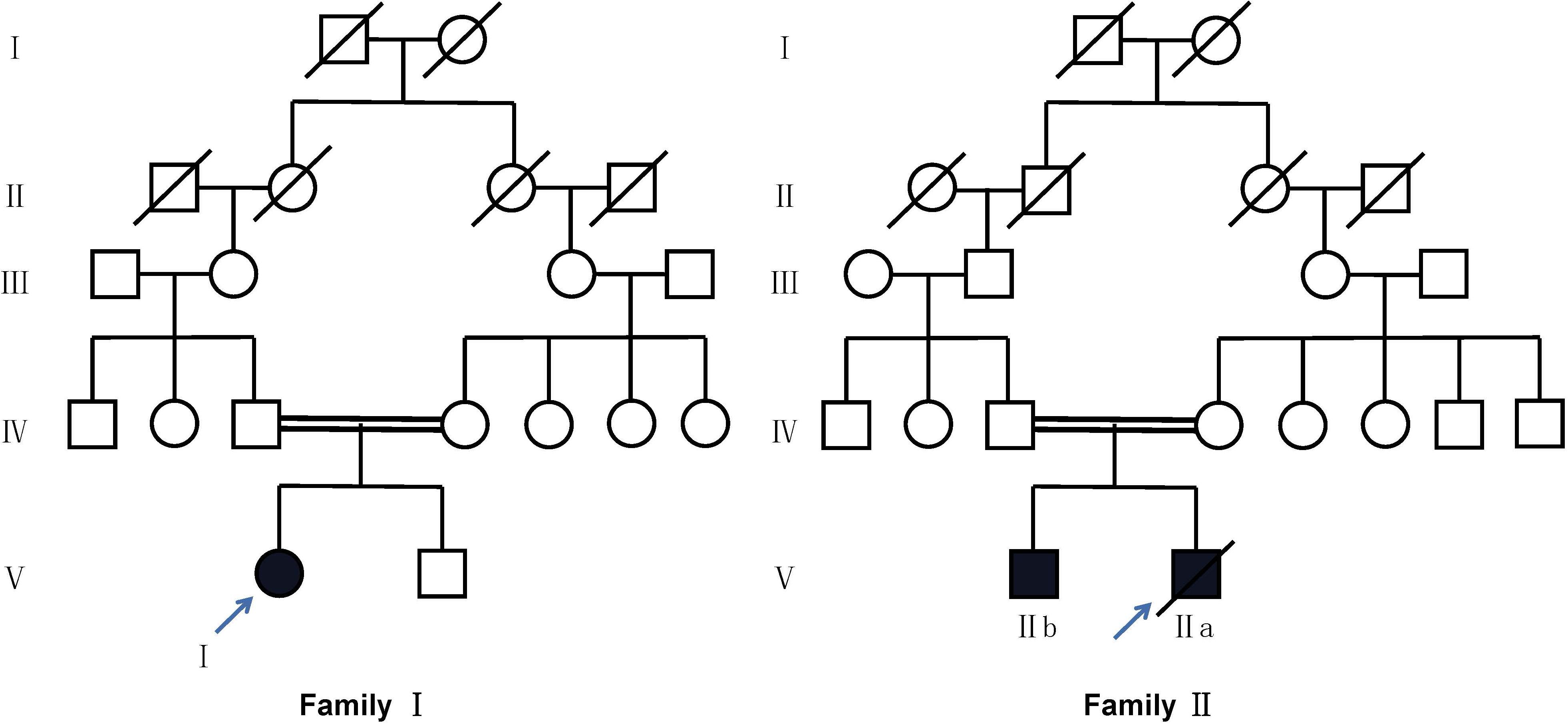
Figure 1. Pedigree of two families with CFTR variants. Circles refer to females. Squares refer to male subjects. Solid symbols refer to affected subjects. Crossed-out symbols refer to subjects who have passed away. The arrows indicate the probands.

Figure 2. High resolution computed tomography reveals signs of bronchiectasis in all probands [(A), proband I; (B), proband IIa; (C), patient IIb].
Family 2
Proband IIa was a 17-year-old teenager whose parents are also consanguineous (Figure 1). He had been coughing and expectoration repeatedly for 6 years and experiencing shortness of breath for 1 year, and he was admitted to the Respiratory Intensive Care Unit due to suddenly worsening symptoms. Physical examination revealed a barrel chest, some moist rales, and finger clubbing. Arterial blood gas analysis suggested hypercapnic acidosis. Chest radiography suggested bronchiectasis and infection. Color Doppler echocardiography showed mild mitral regurgitation. CT showed chronic inflammation of bilateral maxillary sinuses, an undeveloped sphenoid sinus, and left frontal sinus, along with diffuse bronchiectasis with patchy infection foci (Figure 2B). His nasal nitric oxide concentration (nNO) was 159 ppb. The patient was given ventilator and phlegm treatment. The sputum culture was positive for Pseudomonas aeruginosa. The cough and expectoration have not improved significantly after changing antibiotics several times. After sputum aspiration under fiberoptic bronchoscopy, the patient’s coughing and expectoration decreased, and his condition improved significantly. Soon after, the patient was discharged from the hospital.
IIb is the 5-year-old brother of IIa. His nitric oxide concentration (nNO) was 113 ppb. He had no symptoms except for digital clubbing and the CT suggested sinusitis, bronchiectasis, and calcification of the liver (Figure 2C).
Whole-Exome Sequencing Identified Two Variants in CFTR
The whole-exome sequencing was performed on three individuals, and the results were listed in Supplementary Table 2. Notably, 14.24, 7.09, and 7.25 Gb sequencing data were generated, and more than 99% of the targeted regions were covered, with a depth of more than 10×. Non-synonymous variants occurring in exons or splice sites (splicing junction 10 bp) were selected and analyzed after alignment. After the selection of rare variations (minor allele frequency <1%) related to bronchiectasis, one homozygous variant of cystic fibrosis transmembrane conductance regulator (CFTR) was identified in each family. The results of our ROH analysis confirmed the homozygous region in chromosome 7 contains the variants of CFTR. The detailed results of ROH analysis are shown in Supplementary Figure 2.
A homozygous variant of CFTR, c.1000C > T, p.R334W, which had been reported to be disease-causing, was found in proband I(Sheppard et al., 1993).
A novel variant of CFTR, c.1409T > A, p.V470E was found in proband IIa and IIb. It hadn’t been reported before and hadn’t been recorded on the Cystic Fibrosis Mutation Database5. The novel variant was predicted to be deleterious by SIFT and the CADD (CADD PHRED score = 24.8). Located at the exon 11, the variant may affect the function of the nucleotide binding domains 1 (NBD1) domain after translation (Figure 3). Swiss model was also used to predict the protein tertiary structure, the amino acid change from valine to glutamic acid might affect the stability of the helix (Figure 4).

Figure 3. Exon structure of the CFTR gene. Domain structure of the CFTR protein. The positions of variant c.1000C > T, p.R334W, and c.1409T > A, p.V470E. TMD, transmembrane domain; NBD, nucleotide binding domain; RD, regulatory (R) domain.
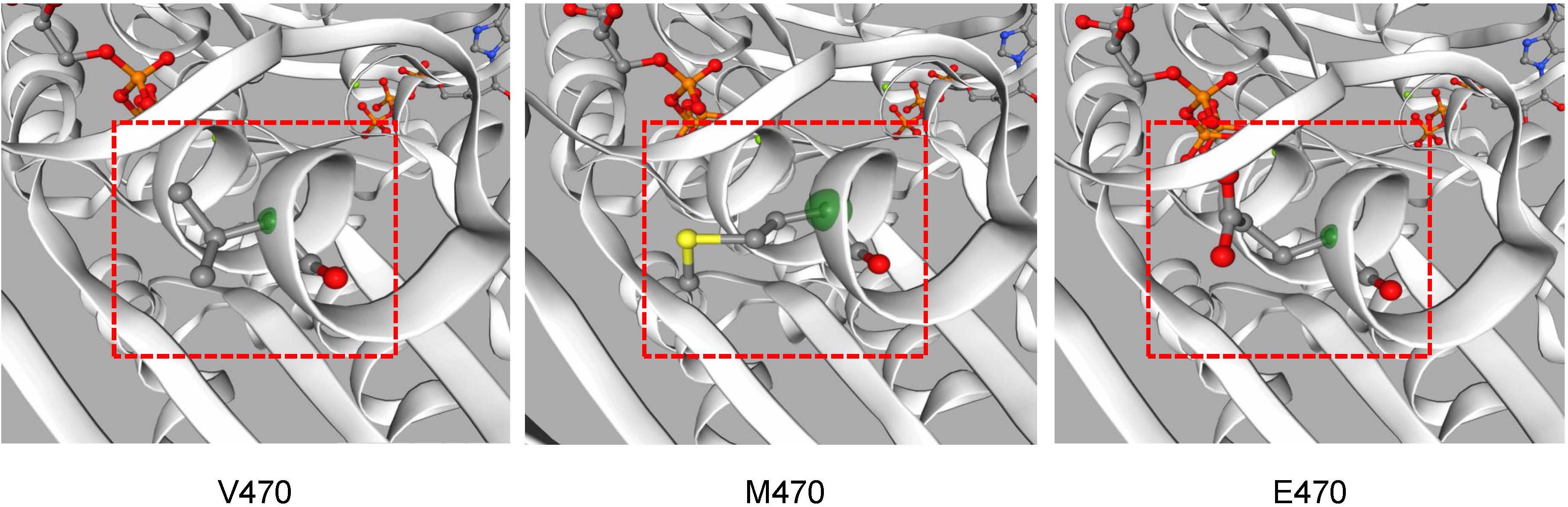
Figure 4. The CFTR protein tertiary structure was predicted by Swiss-model software. Different structures caused by amino acid changes are shown in position 470 of CFTR.
Sanger Sequencing for Variant Validation of CFTR
Sanger sequencing was used to validate the candidate variants identified by whole-exome sequencing, and segregation analyses were performed in both family members. Homozygous variant c.1000C > T, p.R334W in proband I and homozygous variant c.1409T > A, p.V470E in proband IIa and IIb were validated by Sanger sequencing. Sanger sequencing also confirmed that the healthy parents of the probands were all heterozygotes in corresponding positions (Figure 5).
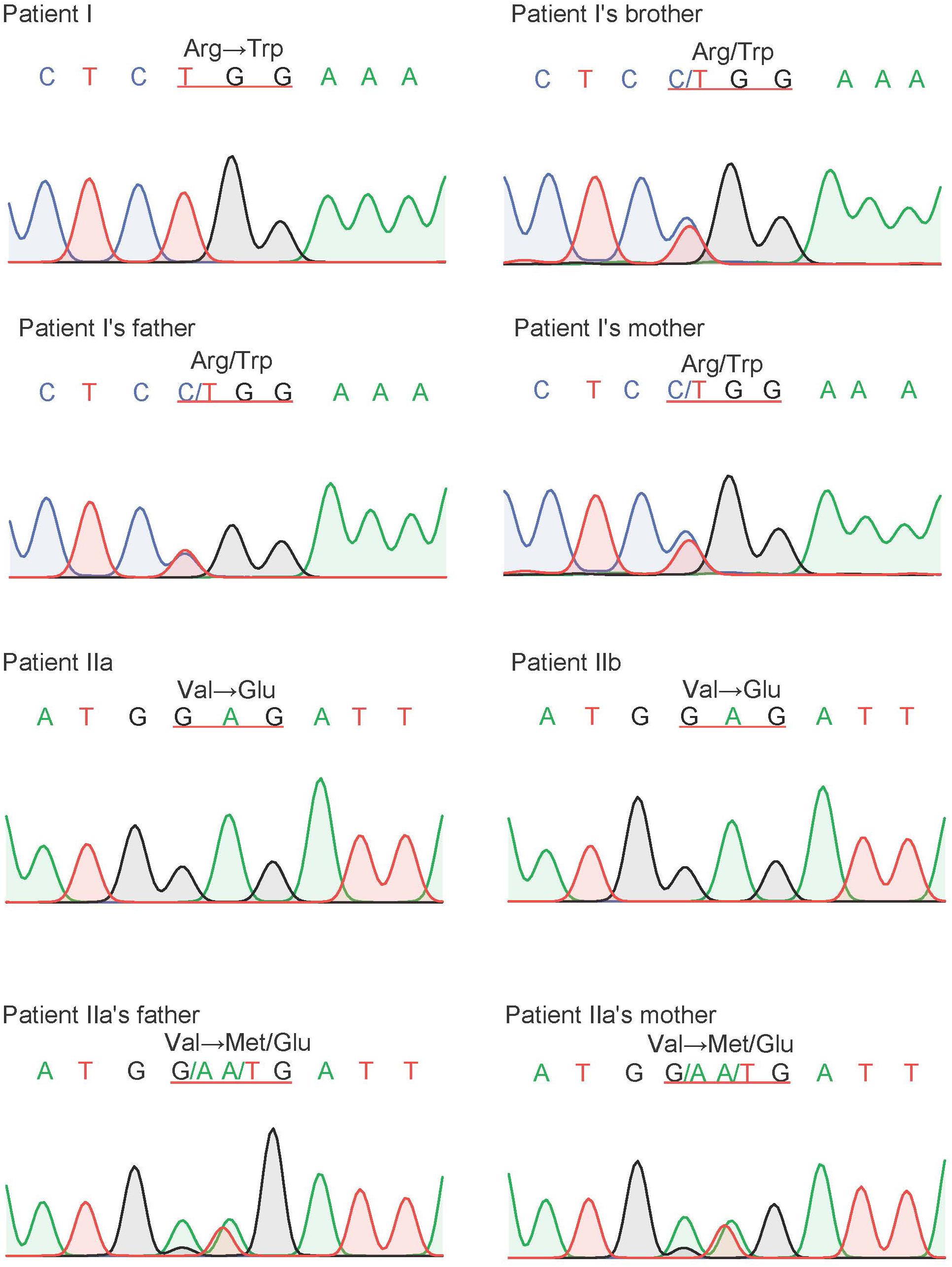
Figure 5. The result of Sanger sequencing of our probands and their family members. The variant of CFTR, c.1000C > T, p.R334W, were found in proband I and c.1409T > A, p.V470E were found in proband IIa and IIb. All of our probands are homozygous, and their parents are heterozygotes.
Discussion
In this study, whole-exome sequencing was performed to identify 2 variants in CFTR in 2 families. The parents of patients in both families are heterozygously married and are close relatives. A novel homozygous variant (c.1409T > A, p.V470E) was found in both patients in family II. It was located in the NBD1 domain of CFTR and may disturb the structure and function of ATP binding. Another reported homozygous variant (c.1000C > T, p.R334W) found in patient I was considered to be the cause of her respiratory disease.
The novel variant c.1409T > A, p.V470E in exon 11, which may affect the function of the NBD1 domain after translation, was not present in the current Cystic Fibrosis Mutation Database or the Human Gene Mutation Database6. Notably, this position is a polymorphic position of M470V (c.1408G > A) variants and is related to bronchiectasis and chronic pancreatitis by affecting structural stability of NBD1 and intrinsic chloride channel activity when it is combined with other variants (Lee et al., 2003; Yang et al., 2018). The V470 allele can reduce the activity of the CFTR protein chloride channel, which might help to reduce the disease damage and decrease the rate of diarrhea, primary sclerosing cholangitis, and prostate cancer (Henckaerts et al., 2009; Qiao et al., 2008). M470 allele is significantly associated with an increasing risk of chronic pancreatitis in both Asian and Caucasian populations (Zhou et al., 2020), which also contributes to a low birth rate through the congenital bilateral absence of the vas deferens (CBAVD) (Kosova et al., 2010). The variants in sequence among different races and species well reflect the natural selection during human evolution (Maria Ciminelli et al., 2011; Figure 6). The functional difference and species evolution caused by the polymorphic variants at this site also indicate that the variant V470E at this site has a high possibility of affecting the function of CFTR and causing disease. Both patients in family II have the typical cystic fibrosis clinical phenotype, and brother IIb, with no infection or symptoms, still had significant CT imaging findings of bronchiectasis and digital clubbing. This suggested a possible relationship between the variant and cystic fibrosis. According to ACMG guidelines, the novel variant was classified as the variant of uncertain significance (VUS), meeting the following criteria from the ACMG guidelines: PM2, PP1, PP3, and PP4 (Richards et al., 2015). Nevertheless, the phenotype-genotype association is hard to define completely because of the small sample size. In vitro validation is still in need for confirmation of this variant’s role in cystic fibrosis.

Figure 6. Sequence alignment of mammalian CFTR proteins. The R334 is located in a highly conserved amino acid, and the V470 is located at a variant position according to the MutationTaster.
The variant found in proband I was a reported variant c.1000C > T, p.R334W, present in the common Caucasian CFTR mutation-screening panels (Gasparini et al., 1991). According to the database of CFTR2, there are 429 recorded data of alleles in this position, and only 40% of patients suffered from pancreatic insufficiency. Like most patients with this site variant, proband I showed no symptoms other than respiratory symptoms such as bronchiectasis and sinusitis. This variant is located in the eighth exon and the sixth transmembrane region, which is close to the gate situated between amino acid residues 337 and 344 (Beck et al., 2008; Gao and Hwang, 2015; Figure 4). Because it only affects the conductivity of the channel that decreases the flow of ions, the phenotype of patients is often comparably less severe. The variant was defined as a mild variant (Class IV) based on the pathogenic mechanism and severity of bronchus and pancreas (Ratjen et al., 2015).
The survival rate of CF patients has been greatly improved with the rapid development of treatments. The CFTR potentiator, ivacaftor, the first drug approved to be used in CF patients, is now approved to be used in many class III and class IV variants. Ivacaftor is also effective for the most common pathogenic variant, p.F508del, when combined with corrector lumacaftor. However, patient I in our study has the class IV variant R334W and is not a target of ivacaftor. New research showed that phosphodiesterase (PDE) inhibitors RPL554 (Verona Pharma) can elevate intracellular cAMP in R334W Fisher rat thyroid (FRT) cells. This finding supports the therapeutic potential of RPL554 for CF patients with class III/IV variants including R334W (Turner et al., 2020). In addition, gene and molecular therapy for some mutations is also a research hot spot. At present, the main difficulty is to find a suitable vector (Ratjen et al., 2015).
Cystic fibrosis is the most common and potentially fatal genetic disease in Caucasians, though it is rare in China. Here, we reviewed the 90 cases of Chinese CF patients with DNA sequencing (Supplementary Table 3). Like the previous review, only a few patients had pancreas insufficiency and CBVAD, and most patients have been diagnosed with bronchiectasis and lung infections (Shen et al., 2016). According to the results of a sputum culture, chronic infection of Pseudomonas aeruginosa was found in 62% of patients. Compared with the low prevalence of ABPA in CF patients in Europe, more than 25% of Chinese CF patients met the criteria for ABPA. Sweat testing has always been the gold standard for CF diagnosis; however, it has only been carried out in hospitals in some large cities. Only 66 of 90 cases had the data of sweat conductivity test. Different from the most common pathogenic variants p.F508del in Europe and America, c.2909G > A (p.G970D) was the most common pathogenic variant in China among all the variants. Research has shown that the CFTR variant spectrum of CF patients in China is different from that in Caucasians in Europe and America since some variants, such as c.1766 + 5G > T, only occur in Chinese people (Xu et al., 2017). It is necessary for us to establish a variant database of Chinese CF patients since most variants in Chinese CF patients have not been present in the common Caucasian CFTR mutation-screening panels. For patients with atypical clinical symptoms, it is difficult to make a clear diagnosis at an early stage without sweat tests or genetic testing, explaining the low diagnosis rate of CF in China. In the last decade, the number of reports of cystic fibrosis patients in China has increased significantly, and more cystic fibrosis patients and data will be discovered and reported in the future, after awareness and diagnosis techniques of the disease are improved.
Conclusion
Our study used whole-exome sequencing to identify two homozygous variants of CFTR, including a novel variant (c.1409T > A, p.V470E) and a reported variant (c.1000C > T, p.R334W), in three individuals from two consanguineous families. The existence of the novel variant (c.1409T > A, p.V470E), which has never been reported, implies the association between the polymorphism of the site and the disease and indicates its changes in human evolution. In addition, our findings in the patient with variant c.1000C > T, p.R334W are the same as that of previous studies that showed that the phenotype identified by this variant is less severe. Although the current functional test and drug experiment for these variants have not achieved decisive results, we do think that they are necessary. Our review of Chinese CF patients also suggests that doctors should pay more attention to the atypical symptoms of Chinese CF patients. Our result expands the variant spectrum of CFTR and contributes to a more rapid genetic diagnosis and future genetic counseling.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Ethics Statement
The studies involving human participants were reviewed and approved by Review Board of the Second Xiangya Hospital of Central South University in China. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author Contributions
HL and ZT conceived and designed the experiments. BY, CL, and DY performed the experiments and analyzed the data. TG collected samples and the clinical data. BY and CL wrote the manuscript. All authors critically reviewed, revised, and approved the final version of the manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (81770002 and 8207003 to HL, 81470445 to ZT, and 81900002 to TG), the Natural Science Foundation of Hunan Province, China (2020JJ5805 to TG), the Hunan Province Health Commission Scientific Research Project (B2019159 to HL), the Science and Technology Program of Changsha, China (kq1901120 to HL), the Xiangya Clinical Big Data System Construction Project in Pulmonary Inflammatory Disease of Central South University, and the National Key Clinical Specialty Construction Projects of China.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the patients for participating in the present study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.631221/full#supplementary-material
Footnotes
- ^ https://cftr2.org
- ^ http://www.1000genomes.org
- ^ http://evs.gs.washington.edu/EVS/
- ^ https://gnomad.broadinstitute.org/
- ^ http://www.genet.sickkids.on.ca
- ^ http://www.hgmd.cf.ac.uk/ac/index.php
References
Andersen, D. H. (1938). Cystic Fibrosis of the pancrease and its relation to celiac dsease. Am. J. Dis. Child. 56, 344–399. doi: 10.1001/archpedi.1938.01980140114013
Beck, E. J., Yang, Y., Yaemsiri, S., and Raghuram, V. (2008). Conformational changes in a pore-lining helix coupled to cystic fibrosis transmembrane conductance regulator channel gating. J. Biol. Chem. 283, 4957–4966. doi: 10.1074/jbc.m702235200
Gao, X., and Hwang, T. C. (2015). Localizing a gate in CFTR. Proc. Natl. Acad. Sci. U. S. A. 112, 2461–2466. doi: 10.1073/pnas.1420676112
Gasparini, P., Nunes, V., Savoia, A., Dognini, M., Morral, N., Gaona, A., et al. (1991). The search for south European cystic fibrosis mutations: identification of two new mutations, four variants, and intronic sequences. Genomics 10, 193–200. doi: 10.1016/0888-7543(91)90500-e
Guo, X., Liu, K., Liu, Y., Situ, Y., Tian, X., Xu, K. F., et al. (2018). Clinical and genetic characteristics of cystic fibrosis in CHINESE patients: a systemic review of reported cases. Orphanet J. Rare Dis. 13:224.
Henckaerts, L., Jaspers, M., Van Steenbergen, W., Vliegen, L., Fevery, J., Nuytten, H., et al. (2009). Cystic fibrosis transmembrane conductance regulator gene polymorphisms in patients with primary sclerosing cholangitis. J. Hepatol. 50, 150–157. doi: 10.1016/j.jhep.2008.07.033
Kosova, G., Pickrell, J. K., Kelley, J. L., McArdle, P. F., Shuldiner, A. R., Abney, M., et al. (2010). The CFTR Met 470 allele is associated with lower birth rates in fertile men from a population isolate. PLoS Genet. 6:e1000974. doi: 10.1371/journal.pgen.1000974
Lee, J. H., Choi, J. H., Namkung, W., Hanrahan, J. W., Chang, J., Song, S. Y., et al. (2003). A haplotype-based molecular analysis of CFTR mutations associated with respiratory and pancreatic diseases. Hum. Mol. Genet. 12, 2321–2332. doi: 10.1093/hmg/ddg243
Liu, K., Xu, W., Xiao, M., Zhao, X., Bian, C., Zhang, Q., et al. (2020). Characterization of clinical and genetic spectrum of Chinese patients with cystic fibrosis. Orphanet J. Rare Dis. 15:150.
Maria Ciminelli, B., Bombieri, C., Ciccacci, C., Belpinati, F., Pompei, F., Maselli, R., et al. (2011). Anthropological features of the CFTR gene: its variability in an African population. Ann. Hum. Biol. 38, 203–209. doi: 10.3109/03014460.2010.507554
Qiao, D., Yi, L., Hua, L., Xu, Z., Ding, Y., Shi, D., et al. (2008). Cystic fibrosis transmembrane conductance regulator (CFTR) gene 5T allele may protect against prostate cancer: a case-control study in Chinese Han population. J. Cyst. Fibros. 7, 210–214. doi: 10.1016/j.jcf.2007.07.011
Quinodoz, M., Peter, V. G., Bedoni, N., Royer Bertrand, B., Cisarova, K., Salmaninejad, A., et al. (2021). AutoMap is a high performance homozygosity mapping tool using next-generation sequencing data. Nat. Commun. 12:518.
Ratjen, F., Bell, S. C., Rowe, S. M., Goss, C. H., Quittner, A. L., and Bush, A. (2015). Cystic fibrosis. Nat. Rev. Dis. Primers 1:15010.
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Shen, Y., Liu, J., Zhong, L., Mogayzel, P. J. Jr., Zeitlin, P. L., Sosnay, P. R., et al. (2016). Clinical Phenotypes and Genotypic Spectrum of Cystic Fibrosis in Chinese Children. J. Pediatr. 171, 269–276.e1.
Sheppard, D. N., Rich, D. P., Ostedgaard, L. S., Gregory, R. J., Smith, A. E., and Welsh, M. J. (1993). Mutations in CFTR associated with mild-disease-form Cl- channels with altered pore properties. Nature 362, 160–164. doi: 10.1038/362160a0
Turner, M. J., Luo, Y., Thomas, D. Y., and Hanrahan, J. W. (2020). The dual phosphodiesterase 3/4 inhibitor RPL554 stimulates rare class III and IV CFTR mutants. Am. J. Physiol. Lung Cell. Mol. Physiol. 318, L908–L920.
Xu, J., Yin, Y., Zhang, L., Zhang, J., Yuan, S., and Zhang, H. (2017). Four case reports of Chinese cystic fibrosis patients and literature review. Pediatr. Pulmonol. 52, 1020–1028. doi: 10.1002/ppul.23744
Yang, Z., Hildebrandt, E., Jiang, F., Aleksandrov, A. A., Khazanov, N., Zhou, Q., et al. (2018). Structural stability of purified human CFTR is systematically improved by mutations in nucleotide binding domain 1. Biochim. Biophys. Acta Biomembr. 1860, 1193–1204. doi: 10.1016/j.bbamem.2018.02.006
Keywords: cystic fibrosis, CFTR, bronchiectasis, Chinese, genetic variants, phenotype
Citation: Yang B, Lei C, Yang D, Tan Z, Guo T and Luo H (2021) Whole-Exome Sequencing Identified CFTR Variants in Two Consanguineous Families in China. Front. Genet. 12:631221. doi: 10.3389/fgene.2021.631221
Received: 19 November 2020; Accepted: 12 May 2021;
Published: 02 July 2021.
Edited by:
María L. Couce, Complejo Hospitalario Universitario de Santiago, SpainReviewed by:
Jeffrey Dennis Calhoun, Northwestern University, United StatesSaumel Ahmadi, Washington University in St. Louis, United States
Copyright © 2021 Yang, Lei, Yang, Tan, Guo and Luo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Luo, ZHJsdW9ob25nQGNzdS5lZHUuY24=; Ting Guo, Z3VvdGluZ3h5QGNzdS5lZHUuY24=
†These authors have contributed equally to this work