 Janelle Jiminez
Janelle Jiminez Edouard Timsit
Edouard Timsit Karin Orsel
Karin Orsel Frank van der Meer
Frank van der Meer Le Luo Guan
Le Luo Guan Graham Plastow
Graham Plastow- 1Department of Agricultural, Food and Nutritional Science, Livestock Gentec, University of Alberta, Edmonton, AB, Canada
- 2Department of Production Animal Health, Faculty of Veterinary Medicine, University of Calgary, Calgary, AB, Canada
- 3Simpson Ranch Chair in Beef Cattle Health and Wellness, University of Calgary, Calgary, AB, Canada
- 4Ceva Santé Animale, Libourne, France
- 5Department of Ecosystem and Public Health, Faculty of Veterinary Medicine, University of Calgary, Calgary, AB, Canada
Bovine respiratory disease (BRD) is one of the main factors leading to morbidity and mortality in feedlot operations in North America. A complex of viral and bacterial pathogens can individually or collectively establish BRD in cattle, and to date, most disease characterization studies using transcriptomic techniques examine bronchoalveolar and transtracheal fluids, lymph node, and lung tissue as well as nasopharyngeal swabs, with limited studies investigating the whole-blood transcriptome. Here, we aimed to identify differentially expressed (DE) genes involved in the host immune response to BRD using whole blood and RNA sequencing. Samples were collected from heifers (average arrival weight = 215.0 ± 5.3 kg) with (n = 25) and without (n = 18) BRD at a commercial feedlot in Western Canada. RNAseq analysis showed a distinct whole-blood transcriptome profile between BRD and non-BRD heifers. Further examination of the DE genes revealed that those involved in the host inflammatory response and infectious disease pathways were enriched in the BRD animals, while gene networks associated with metabolism and cell growth and maintenance were downregulated. Overall, the transcriptome profile derived from whole blood provided evidence that a distinct antimicrobial peptide-driven host immune response was occurring in the animals with BRD. The blood transcriptome of the BRD animals shows similarities to the transcriptome profiles obtained from lung and bronchial lymph nodes in other studies. This suggests that the blood transcriptome is a potential diagnostic tool for the identification of biomarkers of BRD infection and can be measured in live animals and used to further understand infection and disease in cattle. It may also provide a useful tool to increase the understanding of the genes involved in establishing BRD in beef cattle and be used to investigate potential therapeutic applications.
Introduction
Bovine respiratory disease (BRD) is one of the main causes of morbidity and mortality in beef cattle in North America (USDA, 2011). Beef cattle of all ages can be affected with BRD; however, they are most affected on or soon after entry into the feedlot (Babcock et al., 2010). This timing of infection is most likely due to the animal’s exposure to a wide range of pathogens that takes place at a time when various stressors (weaning, transportation, and commingling) negatively affect their immune system (Caswell, 2014; Timsit et al., 2016).
Although respiratory pathogens (mainly viruses and bacteria) and factors predisposing cattle to BRD are relatively well understood (Taylor et al., 2010), the host response and its relationship with disease outcomes to BRD, such as the host’s ability to maintain performance regardless of pathogen burden, needs to be further investigated (Van Eenennaam et al., 2014; Mulder and Rashidi, 2017). For instance, in cattle infected with respiratory pathogens, it is currently difficult to determine which cattle will exhibit visual and clinical signs of BRD or even require an antimicrobial treatment (Timsit et al., 2011b; Wolfger et al., 2015). Transcriptome analysis can lead to insights into disease processes, and biomarkers to assess disease states, progression, and prognosis. Thus far, transcriptomic techniques have examined bronchoalveolar fluids, lung tissue, and sputum samples of cattle with or without BRD (Aich et al., 2009; Rai et al., 2015; Behura et al., 2017; Johnston et al., 2019), but there is much less information on the whole-blood transcriptome (Lindholm-Perry et al., 2018; Sun et al., 2020). In comparison with lung tissue biopsies, blood is easier to obtain and can be collected repeatedly throughout the production period and can give real-time results, instead of postmortem conclusions. Furthermore the host immune response detected in the blood can reflect those responses occurring at the site of infection (Kawayama et al., 2016; Vinther et al., 2016).
Therefore, the objective of this study was to use RNA sequencing to analyze the whole-blood transcriptome of feedlot cattle with or without BRD. We hypothesized that animals exhibiting BRD would show a specific pattern of response in their blood transcriptome and that such patterns will provide further insight into the host immune response. Furthermore, variation in the blood transcriptome of animals with and without BRD could potentially provide markers of resistance or resilience markers for future application in breeding or management.
Materials and Methods
Ethics Statement
This study was conducted in accordance to the Canadian Council of Animal Care (2009) guidelines and recommendations (CCAC, 2009). All experimental procedures were reviewed and approved by the University of Calgary Veterinary Sciences Animal Care Committee (AC15-0109).
Animals
Mixed-breed beef heifers at high risk of developing BRD (i.e., recently weaned, commingled, and auction-market derived) were enrolled between November 2015 and January 2016 at a commercial feedlot in Southern Alberta, Canada. At on-arrival processing, heifers received a subcutaneous injection of a long-acting macrolide (tulathromycin, Draxxin, 2.5 mg/kg, Zoetis, Kirkland, QC, Canada) and were weighed and vaccinated against infectious bovine herpes virus-1 (BoHV-1), bovine viral diarrhea virus (BVDV) (types I and II), bovine parainfluenza-3 (PI3V), bovine respiratory syncytial virus (BRSV), Mannheimia haemolytica, Histophilus somni, and clostridial pathogens. They were also dewormed with a pour-on ivermectin solution. In addition, they received a prostaglandin F2α analog to induce abortion, as per standard feedlot procedure. Heifers were fed in large outdoor dirt-floor pens with approximately 250–300 animals per pen. They were fed twice daily, a concentrate barley-based receiving/growing diet formulated to meet or exceed nutrient requirements. This diet contained 25 ppm of monensin (Rumensin 200, Elanco, Guelph, ON, Canada) and 35 ppm of chlortetracycline (Aureomycin 220, Zoetis). Each morning before feeding, bunks were visually inspected, and feed deliveries were adjusted to ensure that sufficient feed was available for ad libitum consumption. At approximately 30 days after arrival, cattle received another vaccination against infectious BoHV-1, BVDV types I and II, PI3V, BRSV, and a growth implant. Finally, cattle were individually weighed at approximately 120 days on feed (DOF). Average daily gain (ADG) was calculated using the difference between arrival weight and weight at blood sampling, divided by the DOF.
Case Definition
Animals were retrospectively identified as BRD positive based on clinical examination and serum haptoglobin concentration. Heifers with at least one visual BRD sign, a rectal temperature ≥40°C, abnormal lung sounds detected at auscultation, a serum haptoglobin concentration ≥0.25 g/L, and no prior treatment against BRD or other diseases during the feeding period (i.e., first BRD occurrence) were defined as BRD cases. Heifers that had no visual signs of BRD, a rectal temperature <40°C, no abnormal lung sounds detected at auscultation, a serum haptoglobin concentration <0.25 g/L, and no history of treatment against BRD or other disease during the feeding period were treated as healthy controls, which were classified as non-BRD (NB) animals for transcriptome analysis.
Study Design
Heifers were observed daily by experienced pen checkers for detection of clinical illness during the first 60 days from entry. Cattle with one or more visual signs of BRD (e.g., depression, nasal or ocular discharge, cough, tachypnea, or dyspnea) were removed from the pens by pen checkers and, if not previously treated for BRD or another disease during the feeding period, were clinically examined by an experienced veterinarian (ET) and a blood sample collected. For every heifer suspected of having BRD, one or two visually healthy cattle (no visual signs of BRD or other disease) were selected as pen-matched contemporary controls (for convenience, these animals were close to the gate or to the apparently sick animal, etc.) examined as for the BRD animals (if not previously treated for BRD or another disease during the feeding period).
Clinical examinations included assessment of visual signs of respiratory disease (cf. above), determination of respiratory rate and rectal temperature, and a complete lung auscultation using a conventional stethoscope to detect abnormal lung sounds (e.g., increased bronchial sounds, crackles, and wheezes). Two blood samples from each animal were collected at the same time by jugular vein puncture to determine (i) serum haptoglobin concentration [plastic serum tubes; Becton Dickinson, ON (Timsit et al., 2011a)] and (ii) the whole-blood transcriptome (Tempus tubes; Thermo Fisher Scientific, ON). Heifers with at least one visual BRD sign and a rectal temperature ≥40°C received an antibiotic treatment intramuscularly in combination with non-steroidal anti-inflammatory drugs (e.g., 40 mg/kg of florfenicol and 2.2 mg/kg of flunixin, 2 ml/15 kg, Resflor, Merck Animal Health) after sample collection, in accordance with feedlot treatment protocols.
Determination of Serum Haptoglobin Concentration
Serum haptoglobin concentrations were determined in duplicate using a commercially available kit (Tridelta Phase Range Haptoglobin assay, Tridelta Development) as described (Timsit et al., 2011a). The working range was 0.0–2.5 g/L.
Total RNA Isolation and mRNA Library Preparation
Total RNA was isolated from bovine blood using a Preserved Blood RNA Purification Kit (Norgen Biotek Corp, Thorold, ON, Canada), and the quality of RNA was measured using the 2200 RNA ScreenTape TapeStation System (Agilent Technologies Inc., Cedar Creek, TX, United States) producing RNA integrity (RIN) values ranging from 8.0 to 9.8. To prepare the mRNA cDNA libraries, 1.0 μg of total RNA was used from each sample using the TruSeq RNA Library Preparation kit v2 (Illumina, San Diego, CA, United States). Poly A-containing mRNA was enriched from the total RNA using poly-T oligo attached beads and fragmented for first-strand cDNA synthesis, followed by second-strand synthesis. The ends were repaired, and 3′ end adenylation and adapter ligation were performed for each library. Following these steps, libraries were polymerase chain reaction (PCR) amplified, validated using the Bioanalyzer (Agilent Technologies Inc., Cedar Creek, TX, United States), and finally normalized and pooled. Unique indices were used for all samples, and libraries were pooled and sequenced paired end (2 × 100 bp) on four separate lanes on a HiSeq 4000 platform, and sequencing was performed at McGill University and Genome Quebec Innovation Center (Montreal, QC, Canada). In total, 43 samples were used to generate paired-end sequences, and their raw reads were used for downstream analyses.
Transcriptome Data Analysis
Raw reads were analyzed for quality and adapter sequence presence using FastQC (v0.11.8), and adapter sequences were removed using Trimmomatic (v0.39). These cleaned-up sequences were mapped and aligned to the Bos taurus reference genome (ARS-UCD1.2.98) using STAR (v2.7.1a) with default settings (Dobin et al., 2013), and read counts were generated using FeatureCounts (SubRead v1.6.4). The counts were then analyzed using the Bioconductor packages EdgeR and DESeq in the R (v3.5.2) software environment. Counts per million (CPM) was used to evaluate expression, and transcripts with CPM > 2 were considered as expressed.
Differential Gene Expression Analysis
Differential gene expression results were obtained using EdgeR to compare animals with BRD (n = 25) with NB (n = 18) using the following parameters: P-value < 0.05 were adjusted to a 0.01 cutoff (P-adj), with a log fold change (Log2FC) > 2, with log CPM > 2. The data were also filtered with the “keep” command to keep samples with CPM ≥ 2 in at least 18 samples, as the number of samples in the NB group was 18 (Robinson et al., 2010). This value represents genes that are expressed in all the samples measured, and the dataset was normalized with the trimmed mean of M-values (TMM) normalization. To test for differential expression between the BRD and NB animals, the factors of “brd” and “pen” were used to test the difference in expression between the animals. The NB animals were set as the reference in this design model, and the read count data were fitted to a negative binomial general linear model (GLM) representing the design. Prior to fitting the model, the “Common,” “Trended,” and “Tagwise” negative binomial dispersion were estimated, and the biological coefficient of variation was calculated at 78% with a dispersion ratio of 0.61. Statistical tests were then performed for the coefficient relating to the BRD animals, and the top differentially expressed (DE) genes (DEGs) between the BRD and NB samples were ranked by P-value and absolute log2FC. In total, three different DEG analyses were performed: the total DEGs with read counts from both the BRD and NB animals (total DEGs, n = 43, coef = pen); BRD DEGs (n = 25, coef = cluster); and NB DEGs (n = 18, coef = pen). A “cluster” coefficient was also added for the BRD animals representing the three subgroups of the BRD samples differentiated by principal component analysis (PCA) determination of clustered samples (Cluster; n = 3).
Ingenuity Pathway Analysis
Network and pathway analyses were analyzed using Ingenuity Pathway Analysis (IPA)1 (Qiagen, 2000–2019) software. This core analysis tool was used to identify gene pathways, disease, and networks using the gene expression data calculated by EdgeR. Input files of expression data included DEGs from all animals (n = 43) and the BRD-only animals (n = 25).
Statistical Analyses
Statistical analysis used the R software package. P-values ≤ 0.05 were used to indicate significance, while false discovery rate (FDR) values were set at 0.05 for the adjusted P-values, unless otherwise stated. Both EdgeR and IPA incorporate statistical analyses into their analysis packages, and those values were reported. For ADG, rectal temperatures, and DOF, a Wilcoxon rank sum test with continuity correction was used to compare the BRD and NB animal values in the Dplyr package.
Results
Confirmation of Disease Status
Forty-four heifers (average arrival weight = 215.0 ± 5.3 kg) were enrolled to the study and were clinically examined and sampled between November 11 and December 11, 2015. Of these, 25 were classified as BRD positive and 18 were classified as NB based on clinical examination and serum haptoglobin concentration. One control heifer was removed from the study, as it had a serum haptoglobin concentration of 3.6 g/L (i.e., ≥0.25 g/L). Heifers with BRD had higher (P < 0.001) average rectal temperatures of 40.6 ± 0.03°C, than NB heifers averaging 39.3 ± 0.14°C (Supplementary Table 1). Furthermore, the ADG in the NB heifers was considerably higher (P < 0.001) than in the BRD heifers, which on average gained less weight (P < 0.001) from the time they arrived to the feedlot to the time they were enrolled in the study (Supplementary Table 1).
Total Gene Expression Data Summary of All Bovine Respiratory Disease Animals Compared With All Non-bovine Respiratory Disease Animals
A total of 1.51 billion raw reads were generated for the mRNA libraries, and after trimming, an average of 31 M reads per sample was used for alignment (Supplementary Table 2). The read-mapping rates ranged from 75.27 to 92.09%, and on average approximately 25 M reads were uniquely mapped per sample (Supplementary Table 3). In total, EdgeR analysis identified 11,966 genes, with 3,075 downregulated and 3,236 upregulated when comparing the BRD with NB samples (n = 43) using BRD as the coefficient to determine DEGs; 6,311 total DEG, log2FC > 2, P-adj < 0.05. To explore the difference between the expression profiles of the NB and BRD samples, PCA was used to analyze the differences and similarities between the samples. The PCA showed that whole-blood transcriptome profiles of BRD cattle were separated from the NB profiles with 54% of the variation attributed to PC1 (Figure 1). Four samples appear as outliers in the PCA plot: two BRD samples and two NB samples (Figure 1). As might be expected from this result, the number of DEGs in the NB group was relatively small (n = 33 DEGs; total transcripts = 11, 787), whereas thousands of DEGs were identified within the BRD samples, which had a total of 13,404 transcripts identified.
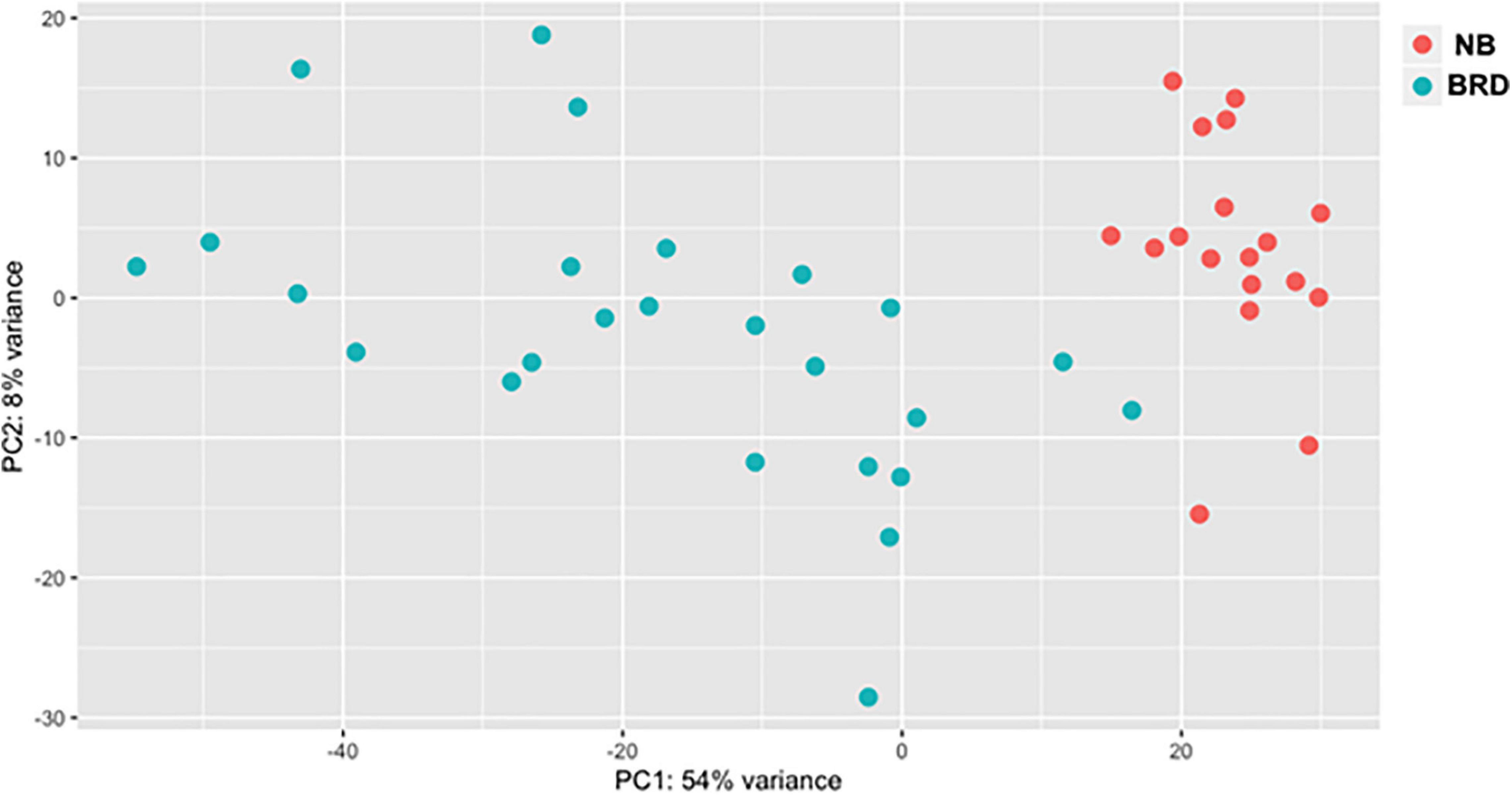
Figure 1. Principal component analysis (PCA) plot comparing differences in total differentially expressed (DE) gene populations between bovine respiratory disease (BRD) and non-BRD (NB) animals. PCA plot displaying differing clustering patterns between heifers displaying clinical signs of BRD (blue) and non-BRD animals (red). Plot was designed using normalized counts (n = 43), using the variable stabilization transformation for the PlotPCA tool in DEGSeq.
Identification of the Differentially Expressed Genes Between Bovine Respiratory Disease and Non-bovine Respiratory Disease Animals
To investigate the host response due to BRD infection, the top ranked DEGs were identified by comparing the DEGs between the NB and BRD samples. Table 1 shows the genes with the highest logFC values using the NB animal expression as the reference. Major immune genes such as interleukin (IL)1 receptor 2 (IL1R2), complement factor B (CFB), and IL3 receptor subunit alpha (IL3RA) were identified in the top 10 upregulated DEGs, with TNF alpha induced protein 6 (TNFAIP6) and IL12B evident in the top 30 upregulated DEGs. Furthermore, haptoglobin (HP), lipocalin (LCN2), serpin peptidase inhibitor (SERPINB4), and S100 calcium-binding proteins (S100A9 and S100A8) were also among the top expressed genes in the BRD animals (Table 1). The top downregulated DEGs when comparing the BRD with NB animals (Table 2) belonged to hemoglobin synthesis pathways, including alpha globin (HBA), beta globin (HBB), mu globin (HBM), and aminolevulinic acid synthase (ALAS2). The enriched genes (upregulated in the BRD animals) belong to immune response pathways, as well as gastrointestinal, inflammatory, infectious, and respiratory disease pathways (not shown).

Table 1. Top enriched total differentially expressed (DE) genes identified when comparing all bovine respiratory disease (BRD) with all non-BRD (NB) animals.
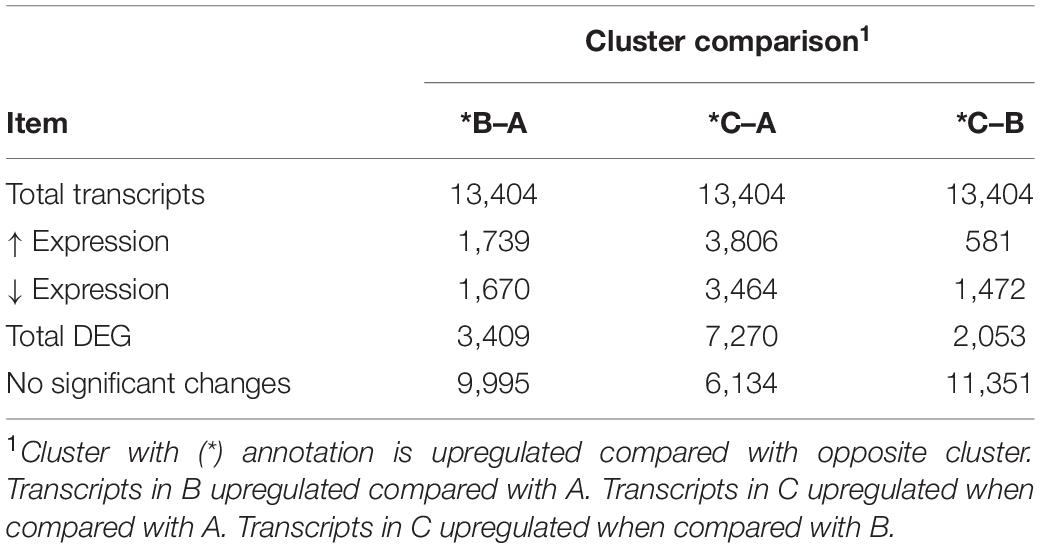
Table 2. Summary of differentially expressed (DE) genes between bovine respiratory disease (BRD) clusters.
Analysis of Bovine Respiratory Disease Clusters and Differentially Expressed Genes
As the BRD samples were more dispersed in the PCA than those from NB (Figure 1), gene expression in the 25 BRD animals was investigated further. Three distinct subsets or clusters were identified within the BRD samples (Figure 2). These clusters were not associated with serum haptoglobin level or rectal temperature at clinical examination (Supplementary Table 1).
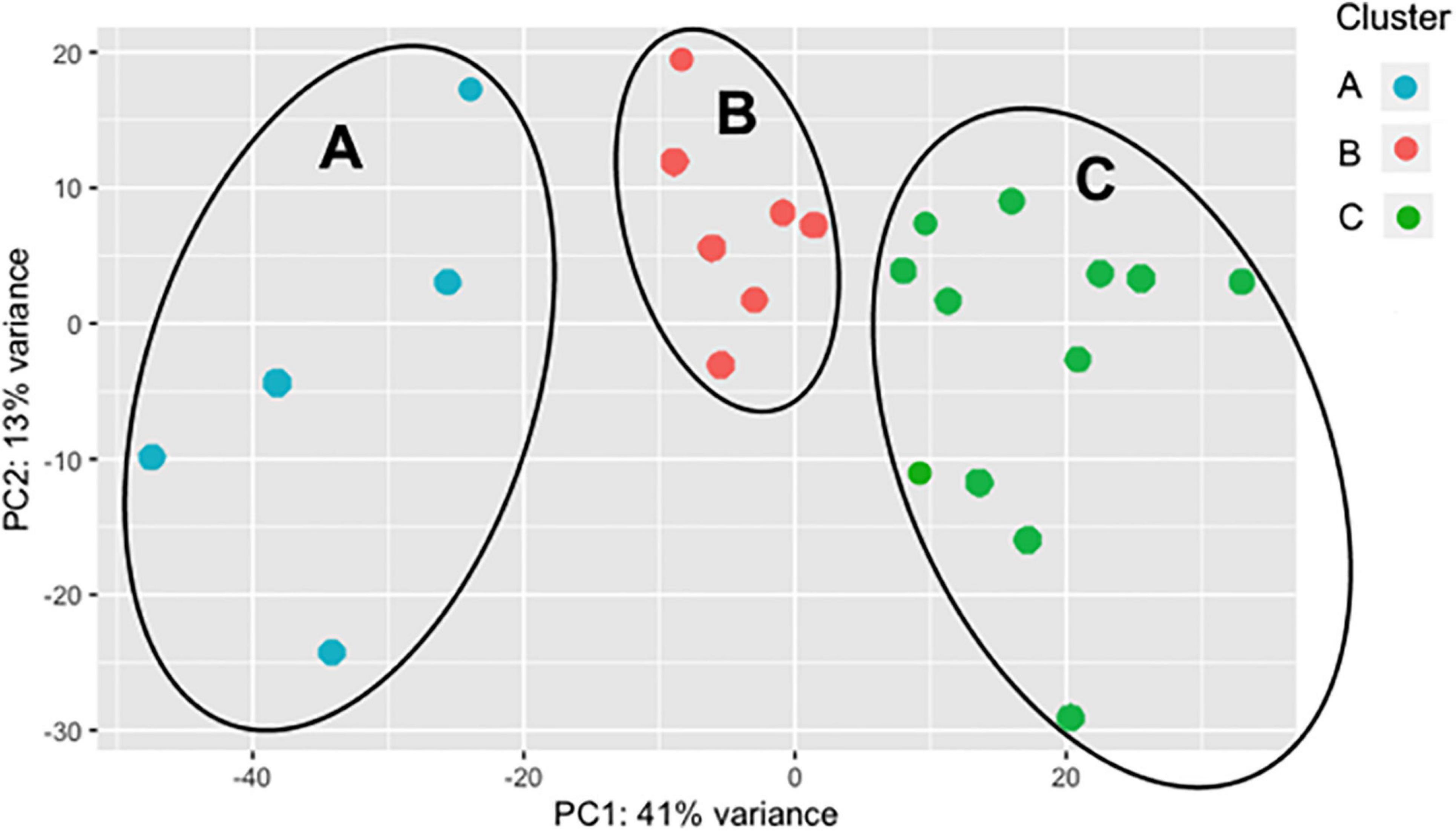
Figure 2. Principal component analysis (PCA) displaying clustering of bovine respiratory disease (BRD) samples. Cluster dendrogram identifying groups in the BRD population (n = 25) that are similar to one another based on gene expression. The BRD data is further subdivided into three distinct clusters.
Differentially expressed gene values were calculated within the BRD samples (n = 25) and compared with one another for DEG profile, with cluster used as the coefficient to determine DEGs; log2FC > 2, P-adj < 0.05. A total of 13,404 DEGs were identified in these samples (Table 2). As Cluster A appeared to be the most distinct, Cluster A read counts were compared with those in Clusters B and C. With the use of logFC > 2, P-value < 0.05, P-adj < 0.01, when compared with A, 109 DEGs common to Clusters B and C were identified (34 upregulated and 74 downregulated). There were 273 DEGs unique to Cluster C and 18 to Cluster B when compared with Cluster A. The top upregulated genes unique to Cluster B included multidrug resistance protein 4, duodenase-1, and trefoil factor 2, while the top downregulated genes were all from the keratin family (Table 3). For Cluster C, upregulated genes included cornifin B-like, solute carrier family 6, and serine protease 50, while thy-1 cell surface antigen and leucine-rich alpha-2-glycoproteins were downregulated (Table 3). When compared with animals in Cluster B and C, animals in Cluster A showed increased expression of genes encoding bovine antimicrobial peptides. Specifically, cathelicidin-2 (CATH2), CATH3, CATH5, and CATH6 were upregulated in Cluster A (Table 4). These genes had high logFC values (>log2), and genes for other antimicrobial peptides such as enteric beta defensin (EBD) and beta-defensin 4A (DEFB4) were also upregulated in Cluster A, when compared with those in B and C. Genes downregulated in Cluster A when compared with Cluster B and C are shown in Table 5. Further analysis using the core analysis function in IPA shows the pathway involved in viral infection as one of the top disease pathways according to z-score in the comparison between Cluster A animals with Clusters B and C (Figure 3). The highly activated genes in this comparison include LCN2, S1008A, and CFB, with bovine cathelicidin antimicrobial peptide (CAMP) having the highest experimental log ratio value as identified through IPA (Table 6).
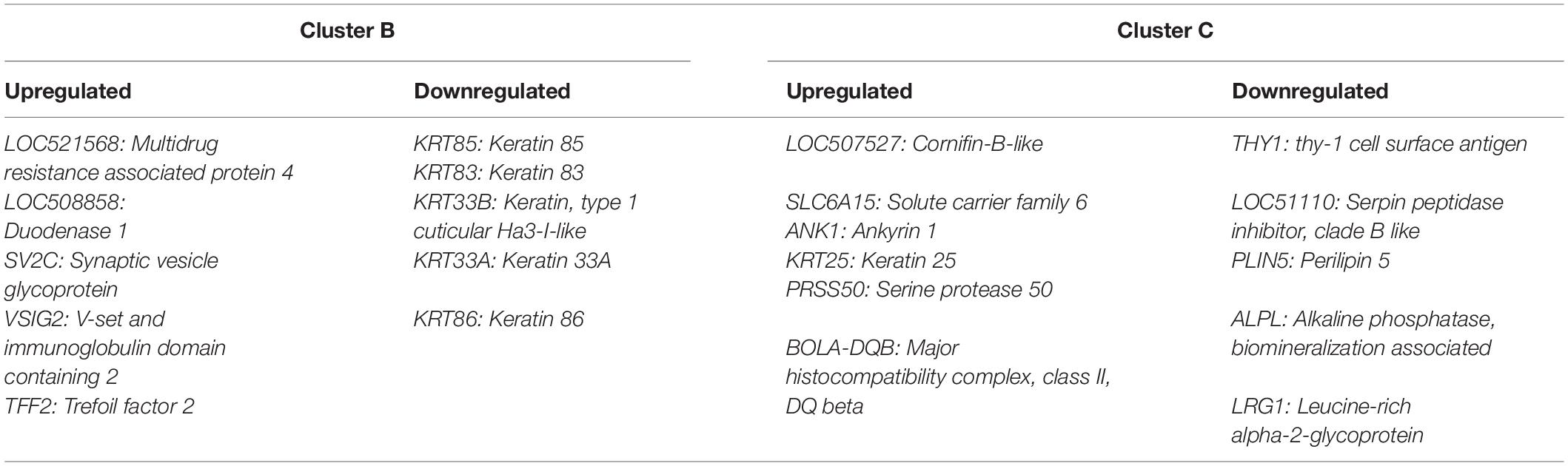
Table 3. Unique genes of interest in Clusters B and C.

Table 4. Top enriched bovine respiratory disease (BRD) differentially expressed (DE) genes in Cluster A compared with Clusters B and C.
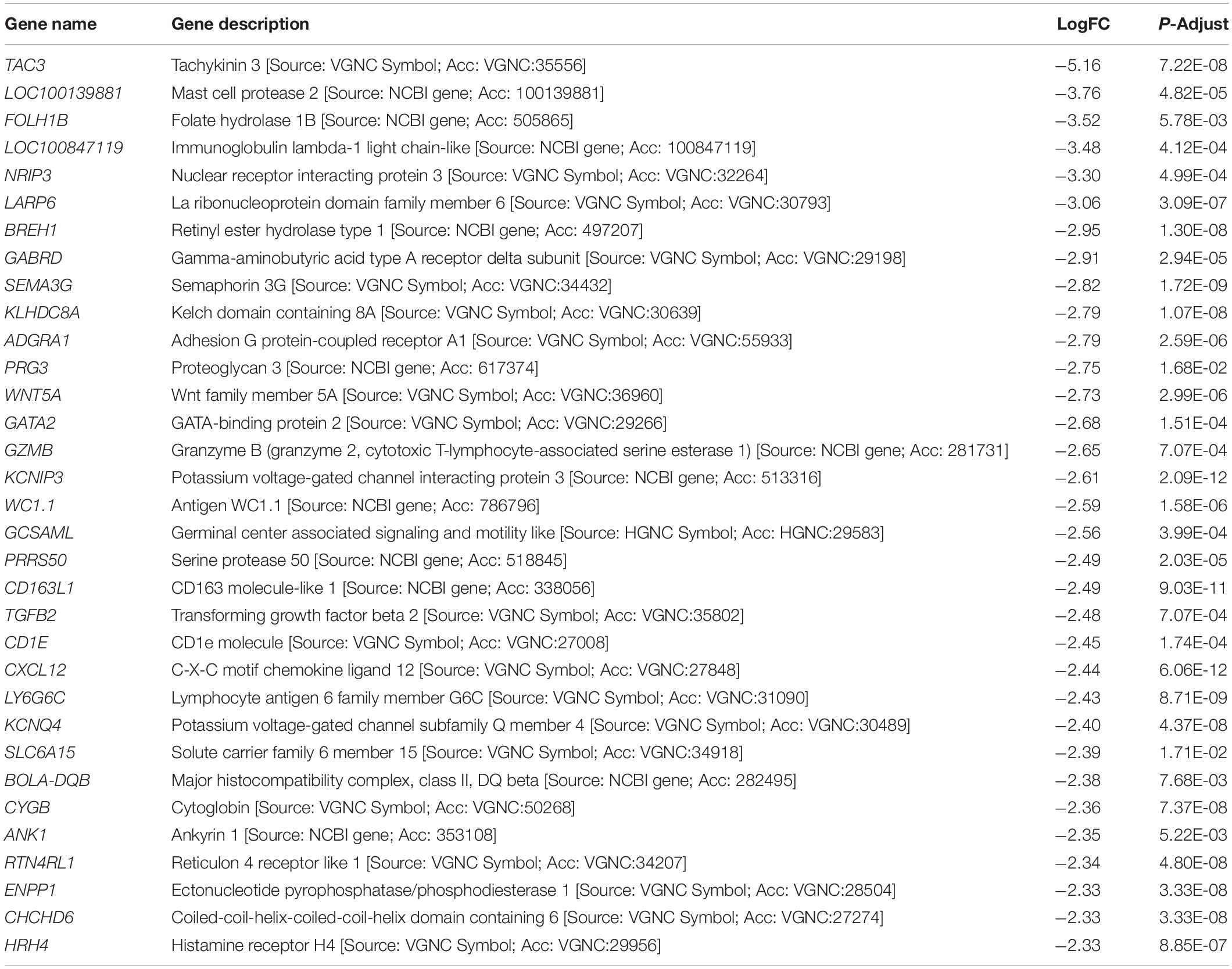
Table 5. Genes downregulated in Cluster A when compared with Clusters B and C.
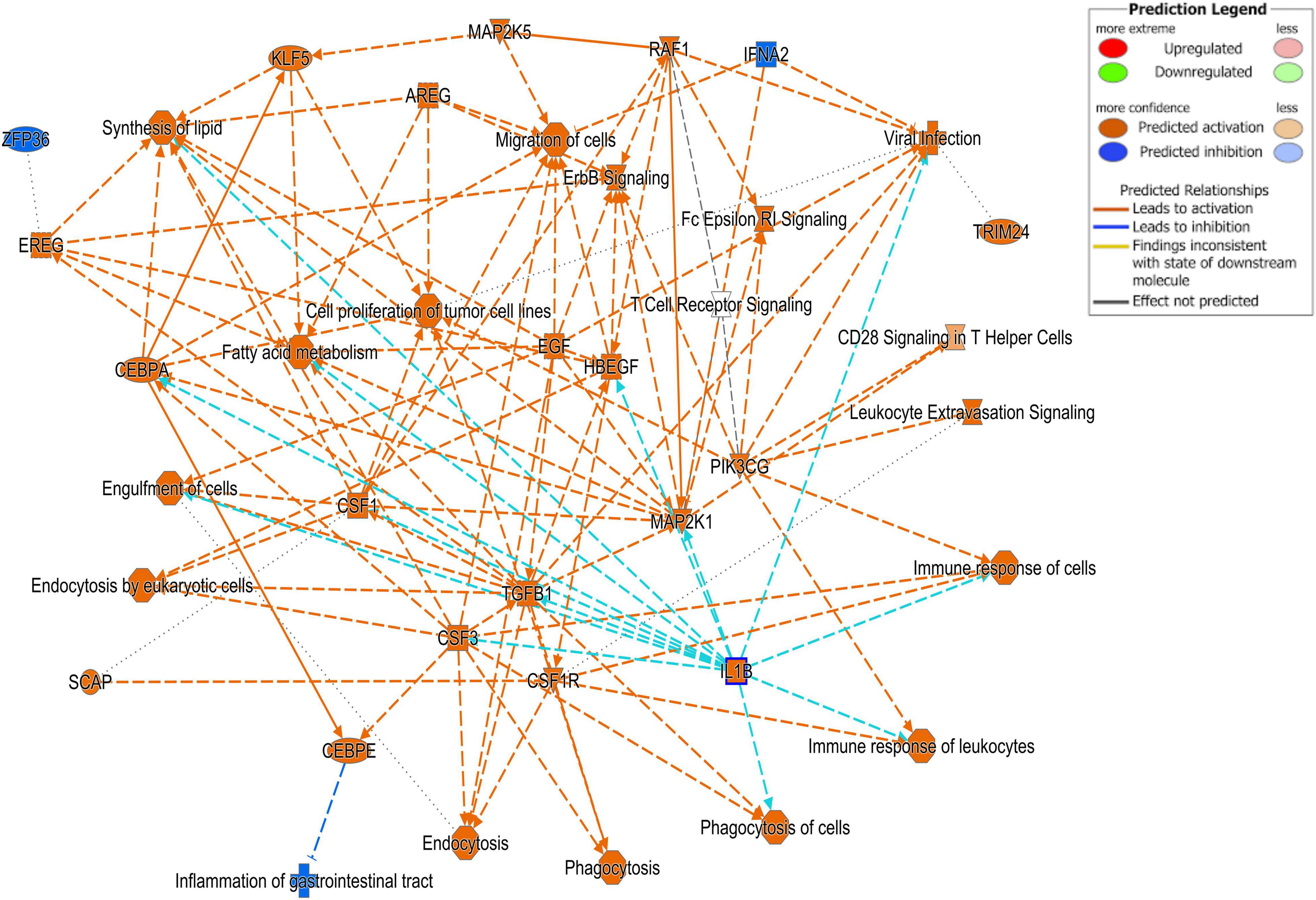
Figure 3. Ingenuity Pathway Analysis (IPA). Cell signaling pathways involved in the viral response pathway identified as one of the top disease pathways according to z-score in the comparison between Cluster A animals and Clusters B and C.
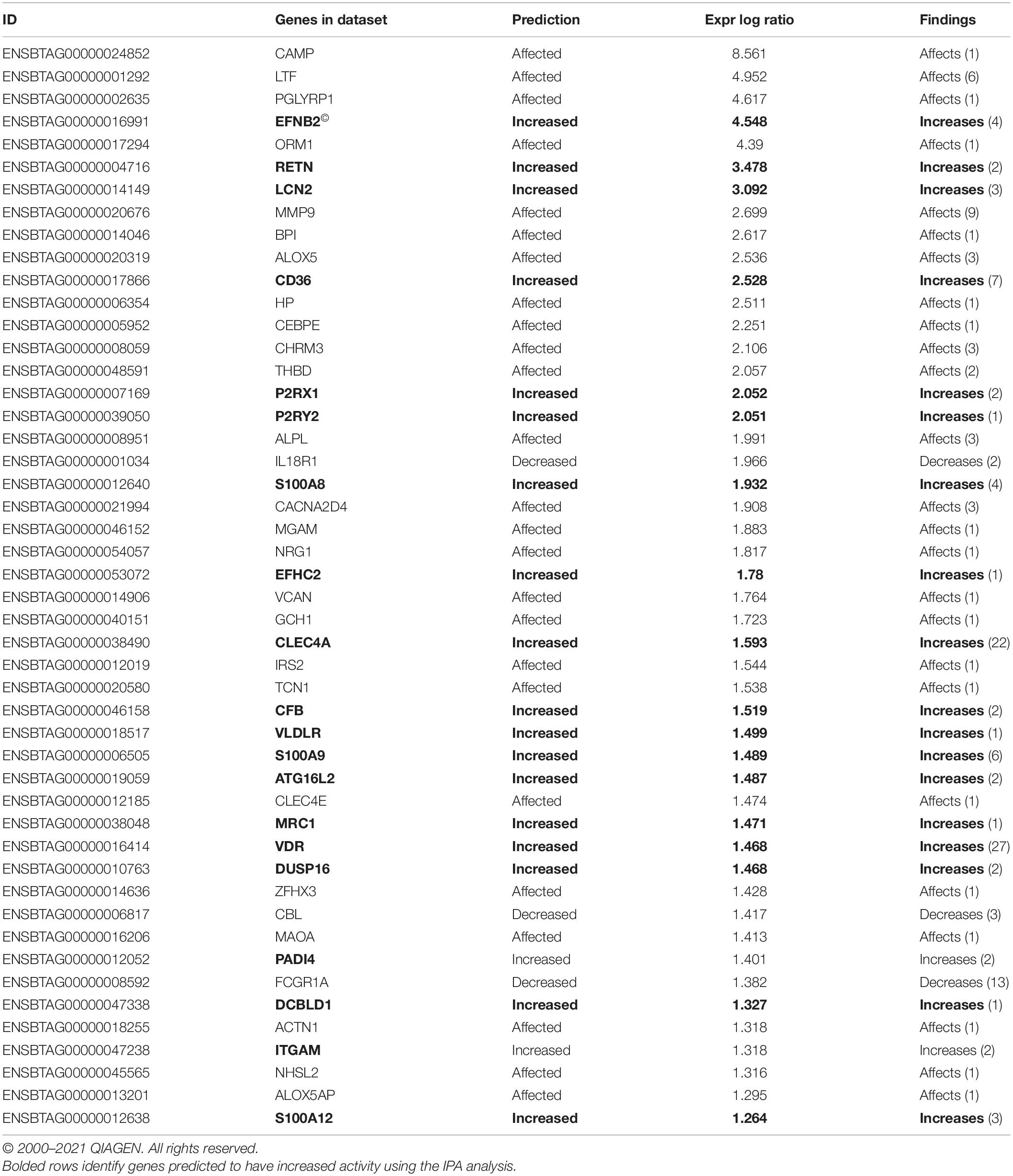
Table 6. Ingenuity Pathway Analysis (IPA) list of genes predicted to affect viral infection in Cluster A compared with B and C.
Comparison With Related Studies
In order to determine the validity of our results, finding similarities in gene expression to related studies was also a goal of our analysis. Three studies in particular also investigated gene expression in response to cattle with BRD using the blood and bronchial lymph node transcriptome. The work done by Johnston et al. (2019) showed similarities to our work in the clear separation observed when plotting the gene expression pattern between control and infected animals, and also in the identification of genes related to acute phase protein expression (Supplementary Table 4). Additionally, Sun et al. (2020) identified enriched expression of genes belonging to heme biosynthesis, acute phase response signaling, and granzyme B signaling, which was also observed in our results (Supplementary Table 4). Finally, Scott et al. (2020), who also investigated the blood transcriptome, found similarities with the highly upregulated genes found here including CATH2, LRG1, and CFB, as well as decreased expression of ALOX15 and GZMB.
Discussion
Most previous studies investigating BRD have used fluids and tissues located at the main sites of infection for BRD pathogens, such as bronchial lymph nodes (Tizioto et al., 2015; Johnston et al., 2019), lung tissue (Rai et al., 2015; Chen et al., 2016; Behura et al., 2017), and lymph fluid (Gershwin et al., 2015), and have reported various immune-related genes enriched at each site of infection. In addition, these studies have collected these fluids and tissues at postmortem examination. Only a few studies (Lindholm-Perry et al., 2018; Scott et al., 2020) use RNA extracted from blood for gene expression analysis despite the relative ease of its sampling from live animals. We therefore applied a functional genomics approach to investigate changes in the whole-blood transcriptome, making two different comparisons; the first examined the difference in gene expression between all the BRD and NB animals, while the second explored the larger variation observed among the BRD animals.
As anticipated, we found that gene expression profiles in whole blood varied between animals diagnosed with BRD and those not exhibiting clinical signs of BRD. Analysis of the differential gene expression between phenotypically healthy cattle (NB) and those with BRD showed that, as with the tissues at infection sites, the major pathways activated in cattle with BRD were also associated with the host immune response.
The BRD animals also had lower expression of genes involved in hemoglobin synthesis. For example, HBA1, HBA2, HBB, and ALAS2 were all downregulated in the BRD animals. These genes are involved in erythropoiesis and are regulated by iron availability (Chiabrando et al., 2014). Iron homeostasis is involved in oxygen transport, cellular respiration, and metabolic processes (Ali et al., 2017). The regulation of iron concentration in blood also plays an important role in modulating bacterial infection and contributes to the progression of lung disease (Roehrig et al., 2006; Ali et al., 2017). During bacterial infection, neutrophils maintain iron homeostasis by releasing LCN2 and lactoferrin (LTF) to sequester free iron (Ali et al., 2017) and protect the lung from oxidative stress induced by iron and HBA and HBB molecules (Tubsuwan et al., 2011). Furthermore, LCN2 decreases iron availability to limit the growth of pathogenic bacteria (Xiao et al., 2017; Pokorska et al., 2019). Pasteurella multocida express outer membrane protein receptors for iron-binding proteins, and the expression of these proteins increases during conditions of iron restriction (Prado et al., 2005). Animals with BRD show decreased expression of genes for hemoglobin and iron-binding proteins and regulators and an increase in genes for iron maintenance proteins (i.e., LCN2 and LTF) that are released from neutrophils as a response to infection. In both comparisons of gene expression (BRD vs. NB and within the BRD animals), LCN2 expression was increased while in the BRD vs. NB comparison, expression of genes encoding iron-binding proteins was lowered.
Bovine respiratory disease is multifactorial (Taylor et al., 2010), and etiological diagnosis of BRD is difficult if not impossible to reach in a field setting (Pardon and Buczinski, 2020). Major BRD pathogens such as Mannheimia haemolytica, P. multocida, Haemophilus somnus, or Mycoplasma bovis can be isolated from both healthy and sick animals (Angen et al., 2009; Timsit et al., 2017, 2018). Furthermore, multiple BRD pathogens (i.e., viruses and bacteria) are often detected at the same time in the same animal (Angen et al., 2009; Fulton et al., 2009), and it is impossible to determine which ones are causing lung lesions and associated clinical signs without performing a postmortem examination (Fulton and Confer, 2012). This explains why identification of the individual microbial and viral species was not performed in this study.
Although identification of the individual microbial and viral species was not performed in this study, we may be able to infer what agents were present by comparing the gene expression results with those from specific challenge studies. For example, Tizioto et al. (2015) performed single pathogen challenges with the common pathogens in the BRD complex and examined gene expression in bronchial lymph nodes of these animals (Tizioto et al., 2015). The patterns of enriched genes in the blood transcriptome in this study share similar gene characteristics with previous investigations. For example, S100A8, S100A9, and matrix metallopeptidase 9 were highly expressed in all of the specific challenges independent of pathogen (Rai et al., 2015; Tizioto et al., 2015). An increase in expression of S100A8 and S100A9 is also associated with toll-like receptor 4 (TLR4) binding (Wang et al., 2016). TLR4 forms complexes that lead to recruitment of members of IL1 receptor signaling to sites of infection (Bhattacharyya et al., 2013). Interestingly we also found upregulation of IL1R2 and IL1RAP in the blood of the BRD animals. Expression of IL1 and IL1RAP become elevated in the host when intracellular pathogens are present (Peters et al., 2013), and both viral and bacterial pathogens can often increase the expression of this cytokine to promote a cytotoxic T cell-mediated response. We also found increased expression of SERPINB4, which encodes a protein located in the skin, mucous membranes, and respiratory system to prevent pathogens from crossing epithelial barriers (Geiger et al., 2015).
A second comparison analyzed the differences within the BRD samples and compared the differences between the identified clusters. Expression of several genes encoding antimicrobial peptides was increased in Cluster A compared with Clusters B and C. These included the genes such as LTF, and several encoding cathelicidins (CATH2, CATH3, CATH5, and CATH6). LTF functions as an antimicrobial molecule but also has immunomodulatory qualities (Drago-Serrano et al., 2017), suggesting a potential therapeutic role for this protein. Cathelicidins are defined as host defense peptides that are highly expressed in bovine granulocytes and located at mucosal surfaces in the lungs, lymphoid tissues, and intestines of the host (Baumann et al., 2017). Expression of four of the seven known bovine cathelicidin genes, CATH2, 3, 5, and 6, was increased in the BRD animals. These peptides have been detected and isolated from sick animals and are generally not present in healthy tissues (Tomasinsig et al., 2002). Therefore, their identification as the top genes with the greatest fold-change increases in the BRD Cluster A suggests a strong host immune response in this group of affected animals. It has also been reported that M. haemolytica causes the induction of bovine beta-defensins, especially in animals with subacute and chronic infection (Fales-Williams et al., 2002), and we observed enteric beta-defensin as well as beta-defensin 4A among the top expressed genes in the BRD animals. It can be concluded that the expression of these defensin genes is indicative of chronic infection (Bhattacharyya et al., 2013) or simply the result of the host defense response stimulating helper T cell type 1 (TH1) and helper T cell type 2 (TH2) responses to help clear infection (Gurao et al., 2017).
The overall abundance of gamma delta T cells in ruminants is higher than in other species, and in non-ruminants, this cell subset has been associated with increasing production of TH2 cytokines (Plattner and Hostetter, 2011). Although this association has not been observed in ruminants, it has been reported that a CD163 relative, Workshop Cluster 1 (WC1), plays an important role in gamma delta T cell regulation in cattle (Herzig et al., 2010; Plattner and Hostetter, 2011), especially in young calves. This T cell subset also facilitates protective immunity following vaccinations (Davis et al., 1996; Guzman et al., 2012) and has been described to be involved in increased expression of major histocompatibility complex (MHC) class II on WC1+ cells through interaction with dendritic cells during Mycobacterium bovis infection (Price and Hope, 2009). When comparing Cluster A with Clusters B and C, expression of WC1, WC1.1, WC1.3, and WC1-12 was significantly decreased in Cluster A. Animals in Cluster A showed lowered expression of WC1 genes that directly promote antigen presentation and regulation of alpha beta T cells and CD4/CD8 antigens on WC1+ T cells (Ackermann et al., 2010). This suggests that the BRD animals in Cluster A were displaying lower antigen presentation and T cell regulation, suggesting that they may have been infected with a greater pathogen load that hinders the host immune response in comparison with that in the animals in Clusters B and C. Furthermore, as there was also an increase in the host antimicrobial response in Cluster A, these animals may also have had a unique pathogen subset leading to BRD than the animals in Clusters B and C.
Animals in Cluster A also exhibited a decrease in the expression of GZMB, which has many established roles in stimulating the cytotoxic T cell response and limiting viral replication in the host (Johnston et al., 2019). Granzyme B, in addition to leukotriene C4, IL4, and IL13, are involved in mediating allergic and asthmatic reactions in humans (Plattner and Hostetter, 2011). Basophil granulocytes are the major effector molecules in a TH2 immune response and are the source for leukotriene C4, IL4, and IL13. IL3 specifically leads to the synthesis of GZMB and contributes to the basophil granule population in the TH2 immune response (Tschopp et al., 2006), and it is one of the most potent cytokines with the longest duration of action (Tschopp et al., 2006). Therefore, the decreased expression of GZMB suggests that the animals in Cluster A had a lowered host immune response to infection than the animals in Clusters B and C.
Conclusion
In conclusion, the results suggest that the blood transcriptome provides a useful resource to investigate the biology of BRD in feedlot cattle. The whole-blood transcriptome may only give a general overview of the health status, e.g., severe infection from a systemic immune response compared with that from the response reported in tissues at the site of infection. However, results from the BRD subsets (Clusters A, B, and C) do show some similarities with gene expression results using tissue and fluids isolated directly from the sites of infection, as well as other studies that also used RNA sequencing to identify BRD in tissues and blood. Analysis of the pathogens present in the sampled animals may allow this commonality to be explored further. For example, it may be that specific pathways and genes expressed in whole blood are associated with individual pathogens, which could assist in directing targeted therapeutic treatments. Such transcriptome data may also provide information on potential therapeutic targets for BRD infection. Investigation of the WC1+ cell subset and cathelicidin antimicrobial peptides could be useful in this respect. Gene expression analysis of whole blood from BRD and NB cases provides new insights for understanding host response to infection and suggests that there is significant value in using blood for BRD studies. This approach is supported by recent results obtained by Scott et al. (2020) as well as Sun et al. (2020); however, in the future, we could increase the validity of our findings by screening more animals for the genes identified in this study using qPCR. Furthermore, genes upregulated in healthy animals may also be related to protective mechanisms that reduce an individual’s susceptibility to BRD, and this warrants further investigation, as our findings put genes related to leukotriene biosynthesis and granzyme expression into this class of protective genes.
Data Availability Statement
The RNAseq data are available at NCBI Gene Expression Omnibus (GEO) database under accession number GSE162156.
Ethics Statement
The animal study was reviewed and approved by University of Calgary Veterinary Sciences Animal Care Committee, AC15-0109. Written informed consent was obtained from the owners for the participation of their animals in this study.
Author Contributions
GP, ET, and KO designed the project and obtained the funding. ET designed the field trial and collected the samples. JJ prepared and analyzed the sequencing data. LG provided advice on RNAseq analysis. GP, JJ, and ET interpreted the results and drafted the manuscript. All authors contributed to the writing and revisions of the manuscript and approved the final manuscript.
Funding
This publication is the result of research supported by the “Genomic Approaches to the Control of Bovine Respiratory Disease Complex (BRDC)” Grant, provided by Genome Alberta, Calgary, AB, Canada, through the Applied Livestock Genomics Program 2014, project code A222. This research is also part of the AMR–One Health Consortium, funded by the Major Innovation Fund Program of the Alberta Ministry of Economic Development, Trade and Tourism.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
JJ thanks Eoin O’Hara and Arun Kommadath for help with transcriptomic analyses. GP acknowledges the support of Alberta Innovates.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.627623/full#supplementary-material
Abbreviations
ADG, average daily gain; ALAS, aminolevulinic acid synthase; BoHV-1, bovine herpes virus-1; BRD, bovine respiratory disease; BRSV, bovine respiratory syncytial virus; BVDV, bovine viral diarrhea virus; CATH, cathelicidin; CFB, complement factor B; CPM, counts per million; DE, differentially expressed; DEFB, beta-defensin; DOF, days on feed; EBD, enteric beta defensin; FDR, false discovery rate; GLM, general linear model; GZM, granzyme; HB, globin; HP, haptoglobin; IL, interleukin; LCN, lipocalin; LTF, lactoferrin; MHC, major histocompatibility complex; MMP, matrix metallopeptidase; NB, non-BRD; PCA, principal component analysis; PCR, polymerase chain reaction; PI3V, bovine parainfluenza-3; RIN, RNA integrity value; SERPINB, serpin peptidase inhibitor; S100A, S100 calcium-binding protein; TLR, toll-like receptor; TMM, trimmed mean of M-values; TNFAIP, tumor necrosis factor alpha induced protein; WC, workshop cluster.
Footnotes
References
Ackermann, M. R., Derscheid, R., and Roth, J. A. (2010). Innate immunology of bovine respiratory disease. Vet. Clin. North Am. Food Anim. Pract. 26, 215–228. doi: 10.1016/j.cvfa.2010.03.001
Aich, P., Babiuk, L. A., Potter, A. A., and Griebel, P. (2009). Biomarkers for prediction of bovine respiratory disease outcome. Omi. A J. Integr. Biol. 13, 199–209. doi: 10.1089/omi.2009.0012
Ali, M. K., Kim, R. Y., Karim, R., Mayall, J. R., Martin, K. L., Shahandeh, A., et al. (2017). Role of iron in the pathogenesis of respiratory disease. Int. J. Biochem. Cell Biol. 88, 181–195. doi: 10.1016/j.biocel.2017.05.003
Angen, O., Thomsen, J., Larsen, L. E., Larsen, J., Kokotovic, B., Heegaard, P. M. H., et al. (2009). Respiratory disease in calves: microbiological investigations on trans-tracheally aspirated bronchoalveolar fluid and acute phase protein response. Vet. Microbiol. 137, 165–171. doi: 10.1016/j.vetmic.2008.12.024
Babcock, A. H., Renter, D. G., White, B. J., Dubnicka, S. R., and Scott, H. M. (2010). Temporal distributions of respiratory disease events within cohorts of feedlot cattle and associations with cattle health and performance indices. Prev. Vet. Med. 97, 198–219. doi: 10.1016/J.PREVETMED.2010.09.003
Baumann, A., Kiener, M. S., Haigh, B., Perreten, V., and Summerfield, A. (2017). Differential ability of bovine antimicrobial cathelicidins to mediate nucleic acid sensing by epithelial cells. Front. Immunol. 8:59. doi: 10.3389/fimmu.2017.00059
Behura, S. K., Tizioto, P. C., Kim, J., Grupioni, N. V., Seabury, C. M., Schnabel, R. D., et al. (2017). Tissue tropism in host transcriptional response to members of the bovine respiratory disease complex. Sci. Rep. 7:17938. doi: 10.1038/s41598-017-18205-0
Bhattacharyya, S., Kelley, K., Melichian, D. S., Tamaki, Z., Fang, F., Su, Y., et al. (2013). Toll-like receptor 4 signaling augments transforming growth factor-β responses: a novel mechanism for maintaining and amplifying fibrosis in scleroderma. Am. J. Pathol. 182, 192–205. doi: 10.1016/j.ajpath.2012.09.007
Caswell, J. L. (2014). Failure of respiratory defenses in the pathogenesis of bacterial pneumonia of cattle. Vet. Pathol. 51, 393–409. doi: 10.1177/0300985813502821
CCAC (2009). CCAC Guidelines on: The Care and Use of Farm Animals in Research, Teaching, and Testing. Available online at: https://www.ccac.ca/Documents/Standards/Guidelines/Farm_Animals.pdf (accessed November 1, 2020).
Chen, J., Yang, C., Tizioto, P. C., Huang, H., Lee, M. O. K., Payne, H. R., et al. (2016). Expression of the bovine NK-Lysin gene family and activity against respiratory pathogens. PLoS One 11:e0158882. doi: 10.1371/journal.pone.0158882
Chiabrando, D., Mercurio, S., and Tolosano, E. (2014). Heme and erythropoieis: more than a structural role. Haematologica 99, 973–983. doi: 10.3324/haematol.2013.091991
Davis, W. C., Brown, W. C., Hamilton, M. J., Wyatt, C. R., Orden, J. A., Khalid, A. M., et al. (1996). Analysis of monoclonal antibodies specific for the γδ TcR. Vet. Immunol. Immunopathol. 52, 275–283. doi: 10.1016/0165-2427(96)05578-X
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., et al. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. doi: 10.1093/bioinformatics/bts635
Drago-Serrano, M. E., Campos-Rodríguez, R., Carrero, J. C., and de la Garza, M. (2017). Lactoferrin: balancing ups and downs of inflammation due to microbial infections. Int. J. Mol. Sci. 18:501. doi: 10.3390/ijms18030501
Fales-Williams, A. J., Gallup, J. M., Ramírez-Romero, R., Brogden, K. A., and Ackermann, M. R. (2002). Increased anionic peptide distribution and intensity during progression and resolution of bacterial pneumonia. Clin. Diagn Lab. Immunol. 9, 28L–32L. doi: 10.1128/CDLI.9.1.28-32.2002
Fulton, R. W., and Confer, A. W. (2012). Laboratory test descriptions for bovine respiratory disease diagnosis and their strengths and weaknesses: gold standards for diagnosis, do they exist? Can. Vet. J. 53, 754–761.
Fulton, R. W., Blood, K. S., Panciera, R. J., Payton, M. E., Ridpath, J. F., Confer, A. W., et al. (2009). Lung pathology and infectious agents in fatal feedlot pneumonias and relationship with mortality, disease onset, and treatments. J. Vet. Diagn. Invest. 21, 464–477. doi: 10.1177/104063870902100407
Geiger, M., Wahlmüller, F., and Furtmüller, M. (eds). (2015). The Serpin Family: Proteins with Multiple Functions in Health and Disease. Cham: Springer. doi: 10.1007/978-3-319-22711-5
Gershwin, L. J., Van Eenennaam, A. L., Anderson, M. L., McEligot, H. A., Shao, M. X., Toaff-Rosenstein, R., et al. (2015). Single pathogen challenge with agents of the bovine respiratory disease complex. PLoS One 10:e0142479. doi: 10.1371/journal.pone.0142479
Gurao, A., Kashyap, S. K., and Singh, R. (2017). β-defensins: an innate defense for bovine mastitis. Vet. World 10, 990–998. doi: 10.14202/vetworld.2017.990-998
Guzman, E., Price, S., Poulsom, H., and Hope, J. (2012). Bovine γδ T cells: cells with multiple functions and important roles in immunity. Vet. Immunol. Immunopathol. 148, 161–167. doi: 10.1016/J.VETIMM.2011.03.013
Herzig, C. T., Waters, R. W., Baldwin, C. L., and Telfer, J. C. (2010). Evolution of the CD163 family and its relationship to the bovine gamma delta T cell co-receptor WC1. BMC Evol. Biol. 10:181. doi: 10.1186/1471-2148-10-181
Johnston, D., Earley, B., McCabe, M. S., Lemon, K., Duffy, C., McMenamy, M., et al. (2019). Experimental challenge with bovine respiratory syncytial virus in dairy calves: bronchial lymph node transcriptome response. Sci. Rep. 9, 1–13. doi: 10.1038/s41598-019-51094-z
Kawayama, T., Kinoshita, T., Matsunaga, K., Kobayashi, A., Hayamizu, T., Johnson, M., et al. (2016). Responsiveness of blood and sputum inflammatory cells in Japanese COPD patients, non-COPD smoking controls, and non-COPD nonsmoking controls. Int. J. Chron. Obstruct. Pulmon. Dis. 11, 295–303. doi: 10.2147/COPD.S95686
Lindholm-Perry, A. K., Kuehn, L. A., McDaneld, T. G., Miles, J. R., Workman, A. M., Chitko-Mckown, C. G., et al. (2018). Complete blood count data and leukocyte expression of cytokine genes and cytokine receptor genes associated with bovine respiratory disease in calves. BMC Res. Notes 11:786. doi: 10.1186/s13104-018-3900-x
Mulder, H. A., and Rashidi, H. (2017). Selection on resilience improves disease resistance and tolerance to infections1. J. Anim. Sci. 95, 3346–3358. doi: 10.2527/jas.2017.1479
Pardon, B., and Buczinski, S. (2020). Bovine respiratory disease diagnosis: what progress has been made in infectious diagnosis? Vet. Clin. North Am. Food Anim. Pract. 36, 425–444. doi: 10.1016/j.cvfa.2020.03.005
Peters, V. A., Joesting, J. J., and Freund, G. G. (2013). IL-1 receptor 2 (IL-1R2) and its role in immune regulation. Brain Behav. Immun. 32, 1–8. doi: 10.1016/j.bbi.2012.11.006
Plattner, B. L., and Hostetter, J. M. (2011). Comparative gamma delta T cell immunology: a focus on mycobacterial disease in cattle. Vet. Med. Int. 2011:214384.
Pokorska, J., Piestrzyńska-Kajtoch, A., Kułaj, D., Ochrem, A., and Radko, A. (2019). Polymorphism of bovine lipocalin-2 gene and its impact on milk production traits and mastitis in Holstein Friesian cattle. Electron. J. Biotechnol. 40, 17–21. doi: 10.1016/J.EJBT.2019.04.004
Prado, M. E., Dabo, S. M., and Confer, A. W. (2005). Immunogenicity of iron-regulated outer membrane proteins of Pasteurella multocida A:3 in cattle: molecular characterization of the immunodominant heme acquisition system receptor (HasR) protein. Vet. Microbiol. 105, 269–280. doi: 10.1016/j.vetmic.2004.11.009
Price, S. J., and Hope, J. C. (2009). Enhanced secretion of interferon-gamma by bovine gammadelta T cells induced by coculture with Mycobacterium bovis-infected dendritic cells: evidence for reciprocal activating signals. Immunology 126, 201–208. doi: 10.1111/j.1365-2567.2008.02889.x
Rai, A. N., Epperson, W. B., and Nanduri, B. (2015). Application of functional genomics for bovine respiratory disease diagnostics. Bioinform. Biol. Insights 9, 13–23. doi: 10.4137/BBi.s30525
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Roehrig, S. C., Gunkel, N., Tran, H. Q., Selzer, P. M., Spehr, V., and Ullrich, H. J. (2006). The response of Mannheimia haemolytica to iron limitation: implications for the acquisition of iron in the bovine lung. Vet. Microbiol. 121, 316–329. doi: 10.1016/j.vetmic.2006.12.013
Scott, M. A., Woolums, A. R., Swiderski, C. E., Perkins, A. D., Nanduri, B., Smith, D. R., et al. (2020). Whole blood transcriptomic analysis of beef cattle at arrival identifies potential predictive molecules and mechanisms that indicate animals that naturally resist bovine respiratory disease. PLoS One 15:e0227507. doi: 10.1371/journal.pone.0227507
Sun, H.-Z., Srithayakumar, V., Jiminez, J., Jin, W., Hosseini, A., Raszek, M., et al. (2020). Longitudinal blood transcriptomic analysis to identify molecular regulatory patterns of bovine respiratory disease in beef cattle. Genomics 112, 3968–3977. doi: 10.1016/j.ygeno.2020.07.014
Taylor, J. D., Fulton, R. W., Lehenbauer, T. W., Step, D. L., and Confer, A. W. (2010). The epidemiology of BRD: what is the evidence for preventive measures? Can. Vet. J. 51, 1351. doi: 10.1128/CMR.16.1.79
Timsit, E., Assié, S., Quiniou, R., Seegers, H., and Bareille, N. (2011a). Early detection of bovine respiratory disease in young bulls using reticulo-rumen temperature boluses. Vet. J. 190, 136–142. doi: 10.1016/j.tvjl.2010.09.012
Timsit, E., Bareille, N., Seegers, H., Lehebel, A., and Assié, S. (2011b). Visually undetected fever episodes in newly received beef bulls at a fattening operation: occurrence, duration, and impact on performance. J. Anim. Sci. 89, 4272–4280. doi: 10.2527/jas.2011-3892
Timsit, E., Hallewell, J., Booker, C., Tison, N., Amat, S., and Alexander, T. W. (2017). Prevalence and antimicrobial susceptibility of Mannheimia haemolytica, Pasteurella multocida, and Histophilus somni isolated from the lower respiratory tract of healthy feedlot cattle and those diagnosed with bovine respiratory disease. Vet. Microbiol. 208, 118–125. doi: 10.1016/j.vetmic.2017.07.013
Timsit, E., Holman, D. B., Hallewell, J., and Alexander, T. W. (2016). The nasopharyngeal microbiota in feedlot cattle and its role in respiratory health. Anim. Front. 6, 44–50. doi: 10.2527/af.2016-0022
Timsit, E., Workentine, M., van der Meer, F., and Alexander, T. (2018). Distinct bacterial metacommunities inhabit the upper and lower respiratory tracts of healthy feedlot cattle and those diagnosed with bronchopneumonia. Vet. Microbiol. 221, 105–113. doi: 10.1016/j.vetmic.2018.06.007
Tizioto, P. C., Kim, J. W., Seabury, C. M., Schnabel, R. D., Gershwin, L. J., Van Eenennaam, A. L., et al. (2015). Immunological response to single pathogen challenge with agents of the bovine respiratory disease complex: an RNA-sequence analysis of the bronchial lymph node transcriptome. PLoS One 10:e0131459. doi: 10.1371/journal.pone.0131459
Tomasinsig, L., Scocchi, M., Di Loreto, C., Artico, D., and Zanetti, M. (2002). Inducible expression of an antimicrobial peptide of the innate immunity in polymorphonuclear leukocytes. J. Leukoc. Biol. 72, 1003–1010.
Tschopp, C. M., Spiegl, N., Didichenko, S., Lutmann, W., Julius, P., Virchow, J. C., et al. (2006). Granzyme B, a novel mediator of allergic inflammation: its induction and release in blood basophils and human asthma. Blood 108, 2290–2299. doi: 10.1182/blood-2006-03-010348
Tubsuwan, A., Munkongdee, T., Jearawiriyapaisarn, N., Boonchoy, C., Winichagoon, P., Fucharoen, S., et al. (2011). Molecular analysis of globin gene expression in different thalassaemia disorders: individual variation of βE pre-mRNA splicing determine disease severity. Br. J. Haematol. 154, 635–643. doi: 10.1111/j.1365-2141.2011.08770.x
USDA (2011). USDA Feedlot 2011 “Part IV: Health and Health Management on U.S. Feedlots with a Capacity of 1,000 or More Head” USDA–APHIS–VS–CEAH–NAHMS. Fort Collins, CO #638.0913. Available online at: https://www.aphis.usda.gov/aphis/ourfocus/animalhealth/monitoring-and-surveillance/nahms/nahms_feedlot_studies (accessed November 1, 2020).
Van Eenennaam, A., Neibergs, H., Seabury, C., Taylor, J., Wang, Z., Scraggs, E., et al. (2014). Results of the BRD CAP project: progress toward identifying genetic markers associated with BRD susceptibility. Anim. Heal Res. Rev. 15, 157–160. doi: 10.1017/S1466252314000231
Vinther, A. M. L., Heegaard, P. M. H., Skovgaard, K., Buhl, R., Andreassen, S. M., and Andersen, P. H. (2016). Characterization and differentiation of equine experimental local and early systemic inflammation by expression responses of inflammation-related genes in peripheral blood leukocytes. BMC Vet. Res. 12:83. doi: 10.1186/s12917-016-0706-8
Wang, Y., Zhang, Z., Zhang, L., Li, X., Lu, R., Xu, P., et al. (2016). S100A8 promotes migration and infiltration of inflammatory cells in acute anterior uveitis. Sci. Rep. 6:36140. doi: 10.1038/srep36140
Wolfger, B., Schwartzkopf-Genswein, K. S., Barkema, H. W., Pajor, E. A., Levy, M., and Orsel, K. (2015). Feeding behavior as an early predictor of bovine respiratory disease in North American feedlot systems. J. Anim. Sci. 93, 377–385. doi: 10.2527/jas.2013-8030
Keywords: bovine respiratory disease, differentially expressed genes (DEGs), host immune response, innate immunity, RNA sequencing
Citation: Jiminez J, Timsit E, Orsel K, van der Meer F, Guan LL and Plastow G (2021) Whole-Blood Transcriptome Analysis of Feedlot Cattle With and Without Bovine Respiratory Disease. Front. Genet. 12:627623. doi: 10.3389/fgene.2021.627623
Received: 09 November 2020; Accepted: 08 February 2021;
Published: 08 March 2021.
Edited by:
Fabiana Quoos Mayer, Institute of Veterinary Research Desidério Finamor (IPVDF), BrazilReviewed by:
Ricardo Zanella, The University of Passo Fundo, BrazilJose Vargas, The University of Passo Fundo, Brazil
Copyright © 2021 Jiminez, Timsit, Orsel, van der Meer, Guan and Plastow. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Graham Plastow, cGxhc3Rvd0B1YWxiZXJ0YS5jYQ==