
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet., 11 February 2021
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.625959
This article is part of the Research TopicGenetics of Familial Hypercholesterolemia: New InsightView all 17 articles
 Arman Moradi1
Arman Moradi1 Majid Maleki2
Majid Maleki2 Zahra Ghaemmaghami2
Zahra Ghaemmaghami2 Zahra Khajali2
Zahra Khajali2 Feridoun Noohi2
Feridoun Noohi2 Maryam Hosseini Moghadam2
Maryam Hosseini Moghadam2 Samira Kalyinia2
Samira Kalyinia2 Seyed Javad Mowla1
Seyed Javad Mowla1 Nabil G. Seidah3
Nabil G. Seidah3 Mahshid Malakootian2*
Mahshid Malakootian2*Familial hypercholesterolemia (FH) is a common, yet underdiagnosed, genetic disorder characterized by lifelong elevated low-density lipoprotein cholesterol levels, which can increase the risk of early-onset coronary artery disease (CAD). In the present study, we screened the nucleotide variations of the LDLR and PCSK9 genes, as well as a part of the APOB gene, in Iranian patients with FH and premature CAD to find the genetic cause of the disorder. Fifteen unrelated individuals with a clinical diagnosis of FH and premature CAD were recruited. Direct DNA sequencing was applied to screen the whole coding exons and exon–intron boundaries of the LDLR and PCSK9 genes and the main parts of their introns, together with exon 26 of the APOB gene. The pathogenicity of the identified mutations was investigated via either segregation analyses in the family or in silico predictive software. Six different point mutations (p.Cys148Tyr, p.Cys216Tyr, p.Cys302Trp, p.Cys338Trp, p.Leu479Gln, and p.G593Afs∗72) in LDLR and a double mutation (p.Asp172His and p.Ala53Val) in both LDLR and PCSK9 genes were identified in seven families with clinically diagnosed FH (43%), whereas no pathogenic mutations were found in eight families with clinically diagnosed FH. This study is the first to identify 1 pathogenic mutation in the LDLR gene (c.1014C > G [p.Cys338Trp]) and to cosegregate it from the affected individual in the family. No mutations were found in the APOB gene, whereas several silent mutations/polymorphisms were identified in the LDLR and PCSK9 genes. Genetic testing and reports on nucleotide alterations in the Iranian population are still limited. Our findings not only further confirm the significant role of FH in the incidence of premature CAD but also enlarge the spectrum of LDLR and PCSK9 variations and exhibit the heterogeneity of FH in Iranians. In patients with no mutation in the examined genes, the disease could be begotten either by a polygenic cause or by gene defects occurring in other related genes and regions not targeted in this study.
Familial hypercholesterolemia (FH) is a monogenic disorder of the metabolism of low-density lipoprotein cholesterol (LDL-C) and is characterized by lifelong elevated levels of LDL particles and LDL-C arterial deposits (Khachadurian, 1964; Brown and Goldstein, 1974). Untreated patients carry the risk of early onset coronary artery disease (premature CAD) and the augmented risk of cardiovascular events (Nordestgaard et al., 2013; Cuchel et al., 2014b; Gidding et al., 2015; Krogh et al., 2016). The current prevalence of FH is estimated to be 1:311 individuals in the general population (Beheshti et al., 2020; Hu et al., 2020).
This monogenic disorder is inherited in two forms of autosomal dominant and autosomal recessive (De Castro-Orós et al., 2010; Cuchel et al., 2014a). The autosomal dominant form of FH is mostly due to the heterozygous and homozygous states of pathogenic variants in the low-density lipoprotein receptor (LDLR), apolipoprotein B (APOB), and proprotein convertase subtilisin/kexin type 9 (PCSK9) genes. Between 85 and 90% of the mutations harbored in LDLR that result in defective LDL receptors attenuate LDL-C clearance from the blood and, consequently, raise LDL-C plasma levels (Varret and Rabès, 2012).
Likewise, mutations in the APOB gene weaken the binding of LDL to LDL receptors, and gain-of-function mutations in the PCSK9 gene account for 5–15% of cases with FH (El Khoury et al., 2017; Sharifi et al., 2017). PCSK9, a serine protease, mediates the internalization and degradation of LDLR in the lysosome via binding to LDL receptors, leading to diminished LDLR recycling and impaired clearance of LDL-C particles from the plasma (Seidah, 2017). Recently, next-generation sequencing has demonstrated the FH phenotype due to occasional dominant mutations in apolipoprotein E (APOE) or signal-transducing adaptor family member 1 (STAP1), as well (Nordestgaard et al., 2013; Cuchel et al., 2014b; Fouchier et al., 2014; Berberich and Hegele, 2019).
The autosomal recessive form of FH is frequently in consequence of pathogenic mutations in the low-density lipoprotein receptor adaptor protein (LDLRAP) gene (Cuchel et al., 2014a). Nevertheless, whole-exome sequencing has revealed that mutations in the lysosomal acid lipase A (LIPA), ATP-binding cassette sub-family G member 5 and 8 (ABCG5/8), and cholesterol 7 alpha-hydroxylase (CYP7A1) genes phenotypically cause hypercholesterolemia similar to FH in the recessive status (Brautbar et al., 2015; Berberich and Hegele, 2019).
At present, the diagnosis of FH is frequently based on clinical phenotypes, with the most extensively utilized FH clinical criteria being the Simon Broome register (SBR) (Simon Broome Register Group., 1991) and the Dutch Lipid Clinic Network System (DLCNS) (Nordestgaard et al., 2013). In this study, we analyzed the possible mutations of the LDLR, APOB, and PCSK9 genes in Iranian patients suffering from both FH and premature CAD.
The present study enrolled 15 unrelated patients with clinically diagnosed FH and premature CAD who met the SBR and/or DLCNS criteria. The demographic characteristics, clinical features, and cholesterol levels of the study population are presented in Table 1. The SB and DLCNS criteria encompass the levels of total cholesterol (TC) and LDL-C, the presence of tendon xanthomas, and a family history of hypercholesterolemia, premature CAD, or cardiovascular events in a first- and/or second-degree relative.
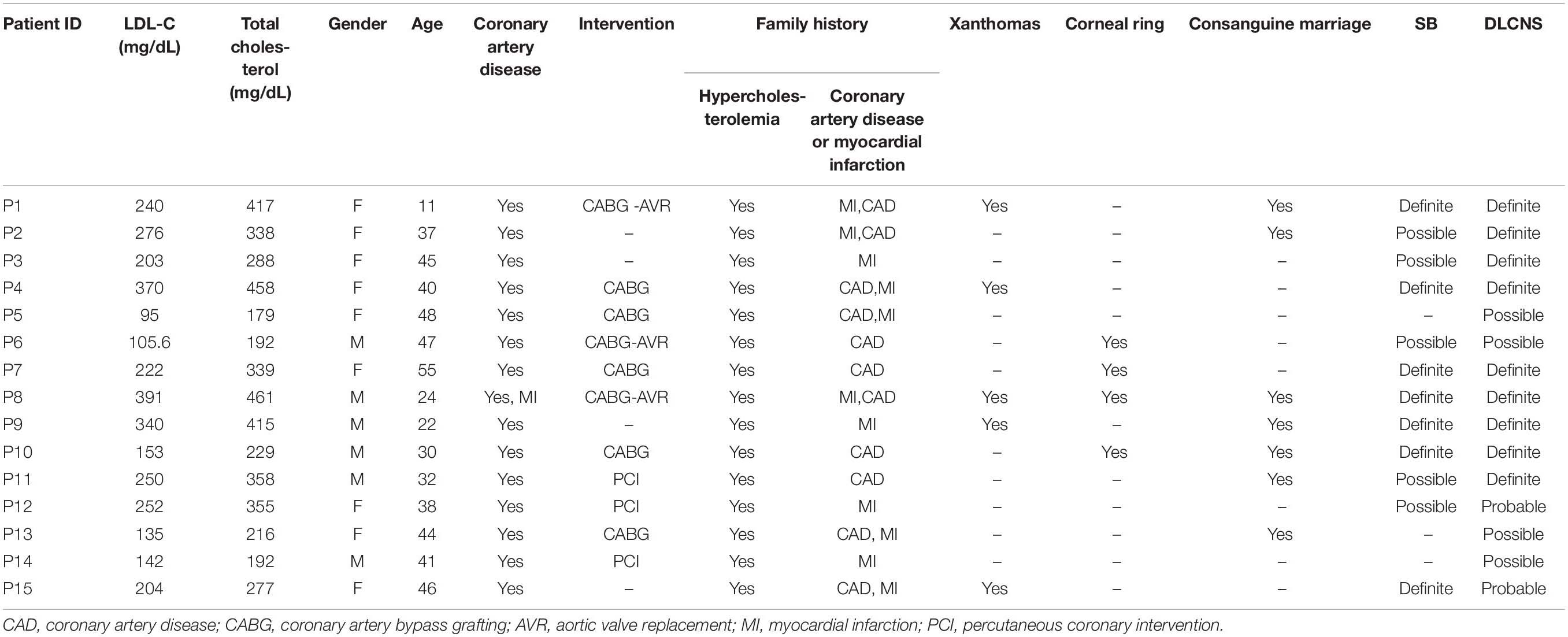
Table 1. Clinical characteristics of the enrolled patients.
For the purposes of the segregation analysis, the family members of some patients (if available) were involved in the study. The study protocol was approved by the Ethics Committee of Rajaie Cardiovascular Medical and Research Center (RHC.AC.IR.REC.1396.62), and it was conducted in accordance with the Helsinki Declaration.
Peripheral blood samples (3–5 mL) were collected from the patients and the available family members in EDTA tubes for DNA extraction. Genomic DNA was isolated from peripheral blood using the Exgene Blood SV Mini Kit (GeneAll Seoul, South Korea). The quantity of the extracted DNA was measured with the NanoDrop Spectrophotometer (Thermo Fisher Scientific, United States).
For the amplification of the whole coding exons, exon–intron junctions, and the main parts of the introns of the LDLR (NG_009060) and PCSK9 (NG_009061) genes, specific oligonucleotides were designed (Supplementary Table 1).
The forward primer 5′-AGCCTCACCTCTTACTTTTCC ATT-3′ and the reverse primer 5′-CTTTGCTTGTATGTTCT CCGTTGGT-3′ were used to amplify exon 26 of the APOB gene (307 base pairs in length), which creates the common FH-causing mutations (p.Arg3500Gln, p.Arg3500Trp, p.Arg3531Cys, and p.His3543Tyr).
The primers were designed using Gene Runner (Gene Runner 6.5.50), PerlPrimer (PerlPrimer 1.1.21), Primer3 (Primer3, 4.1.0), and OligoAnalyzer (OligoAnalyzer 3.1) software tools.
The polymerase chain reaction (PCR) test was performed according to an individual setup for each set of oligos, covering the aforementioned regions of the LDLR, PCSK9, and APOB genes. Sanger sequencing was done for all the PCR products using the ABI 3500 DNA Sequencer (Applied Biosystems, CA, United States).
BioEdit software (BioEdit 7.2.1) was applied to analyze the sequencing outcomes. The identified variants of the nucleotides were analyzed through the UCSC Genome Browser1, ClinVar2, UCL3, and LOVD4 databases.
Additionally, SIFT5, PROVEAN6, PolyPhen-27, and MutationTaster8 in silico predictive software tools were utilized to scrutinize the pathogenesis of the detected mutations.
Fifteen unrelated individuals with clinically diagnosed FH and premature CAD who met the SB and/or DLCNS criteria were recruited in the current study. According to the SB criteria, of the 15 patients, seven had definite FH and five possible FH. Based on the DLCNS criteria, of the 15 patients, nine had definite, two probable, and four possible FH (Table 1). The study population’s demographic characteristics, clinical features, and cholesterol levels are demonstrated in Table 1. Almost all the recruited patients had premature CAD unrelated to congenital heart diseases, hypertension, arrhythmias, or any other cardiovascular diseases.
Direct DNA sequencing of the whole coding exons, with their flanking intron sequences, of the LDLR and PCSK9 genes was performed. Also examined was the presence of the mutations (p.Arg3500Gln, p.Arg3500Trp, p.Arg3531Cys, and p.His3543Tyr) in the gene coding for APOB. The FH-causing variants were identified in seven (46.6%) individuals of the 15 screened patients, whereas no mutation was detected in eight patients clinically diagnosed with FH and premature CAD. Among the identified mutations, six occurred in the LDLR gene, whereas both LDLR and PCSK9 genes were mutated in one patient (P1). The seven detected mutations were harbored in five different exons (4, 6, 7, 10, and 12) of the LDLR gene (Table 2 and Figure 1), and a p.Ala53Val mutation in the PCSK9 gene was located in exon 1. The bioinformatics analysis of all the identified mutations in the LDLR gene categorized them as deleterious, probably damaging, and disease causing (Table 2).
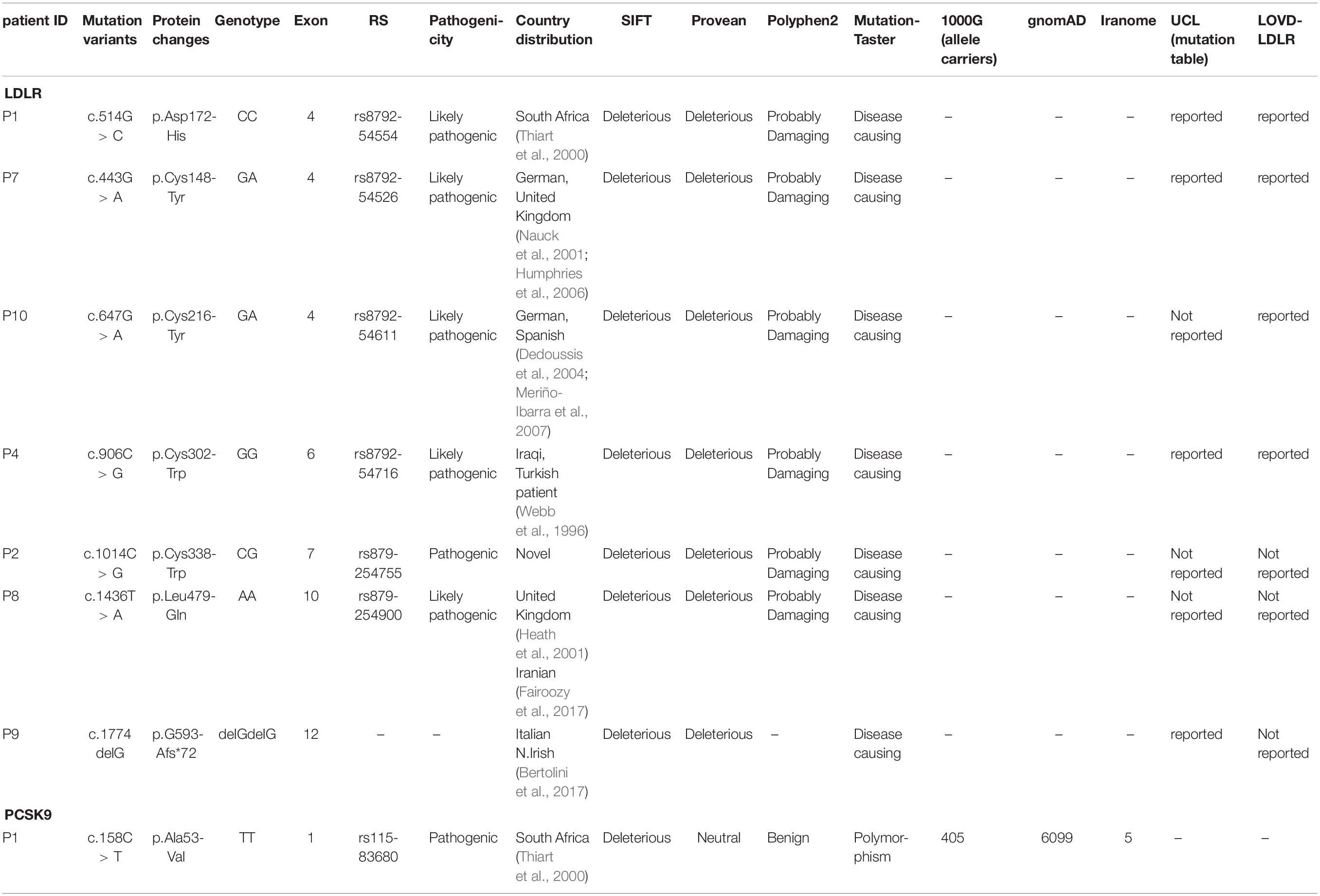
Table 2. The pathogenic mutations of LDLR and PCSK9 genes detected in this study.
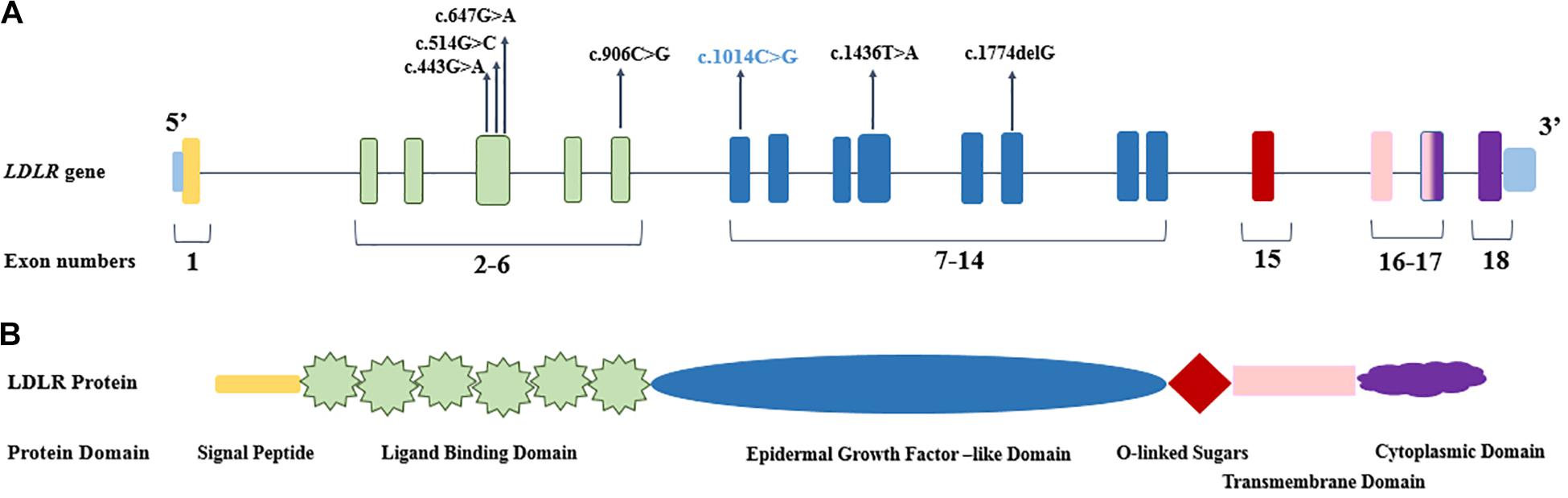
Figure 1. The genomic and protein organization of LDLR. (A) The location of identified pathogenic mutations demonstrated in the different exons of LDLR gene. The novel mutation presented in blue color. (B) The domain organization of LDLR protein, which corresponded to LDLR coding exons (with the common colors).
Among these seven probands, four (P1, P4, P8, and P9) carried the homozygous FH-causing variants and three probands (P2, P7, and P10) carried a mutation in the heterozygous status. Our data showed that all the mutations in the LDLR gene (p.Asp172His, p.Cys338Trp, p.Cys302Trp, p.Cys148Tyr, p.Leu479Gln, and p.Cys216Tyr) and the mutation in the PCSK9 gene (p.Ala53Val) were of the missense type, except for 1 mutation in the LDLR gene (p.G593Afs∗72), which was nonsense. The p.Asp172His, p.Cys302Trp, p.Cys148Tyr, p.Cys216Tyr, and p.G593Afs∗72 mutations in the LDLR gene and the p.Ala53Val mutation in the PCSK9 gene have been reported in populations other than Iranians, whereas the p.Leu479Gln change has been reported previously in an Iranian patient. Segregation analyses were done on the available family members of the study population.
The homozygous nucleotide variants (c.514G > C [p.Asp172His]), harbored in exon 4 of the LDLR gene, and c.158C > T [p.Ala53Val], located in exon 1 of the PCSK9 gene) were identified in an 11-year-old girl, who had undergone heart valve replacement and had high levels of TC (417 mg/dL), LDL-C (240 mg/dL), and xanthomas, with the latter seen in her knees and elbows and between her fingers. The segregation analysis confirmed that these mutations were disease-causing variants and presented in the proband’s parents and two brothers. Both of her parents and both of her siblings were heterozygous for the LDLR mutation, although they had different statuses vis-à-vis the PCSK9 mutation. The c.158C > T PCSK9 variant was in a heterozygous form in both parents but was inherited in the homozygous status in the elder brother. The younger brother, 2 years of age, was a wild type for this nucleotide change (Figure 2). The parents and the siblings had elevated cholesterol levels as well (father: TC = 306 mg/dL and LDL-C = 228 mg/dL; mother: TC = 245 mg/dL and LDL-C = 187 mg/dL; elder brother: TC = 223 mg/dL and LDL-C = 191 mg/dL; and younger brother: TC = 233 mg/dL and LDL-C = 177.8 mg/dL). The mother and father of the proband were first cousins, and there was a positive family history of myocardial infarction, cardiovascular diseases, and hypercholesterolemia in this pedigree (Figure 2). The recognized PCSK9 mutation in this family was reported as a loss-of-function mutation.
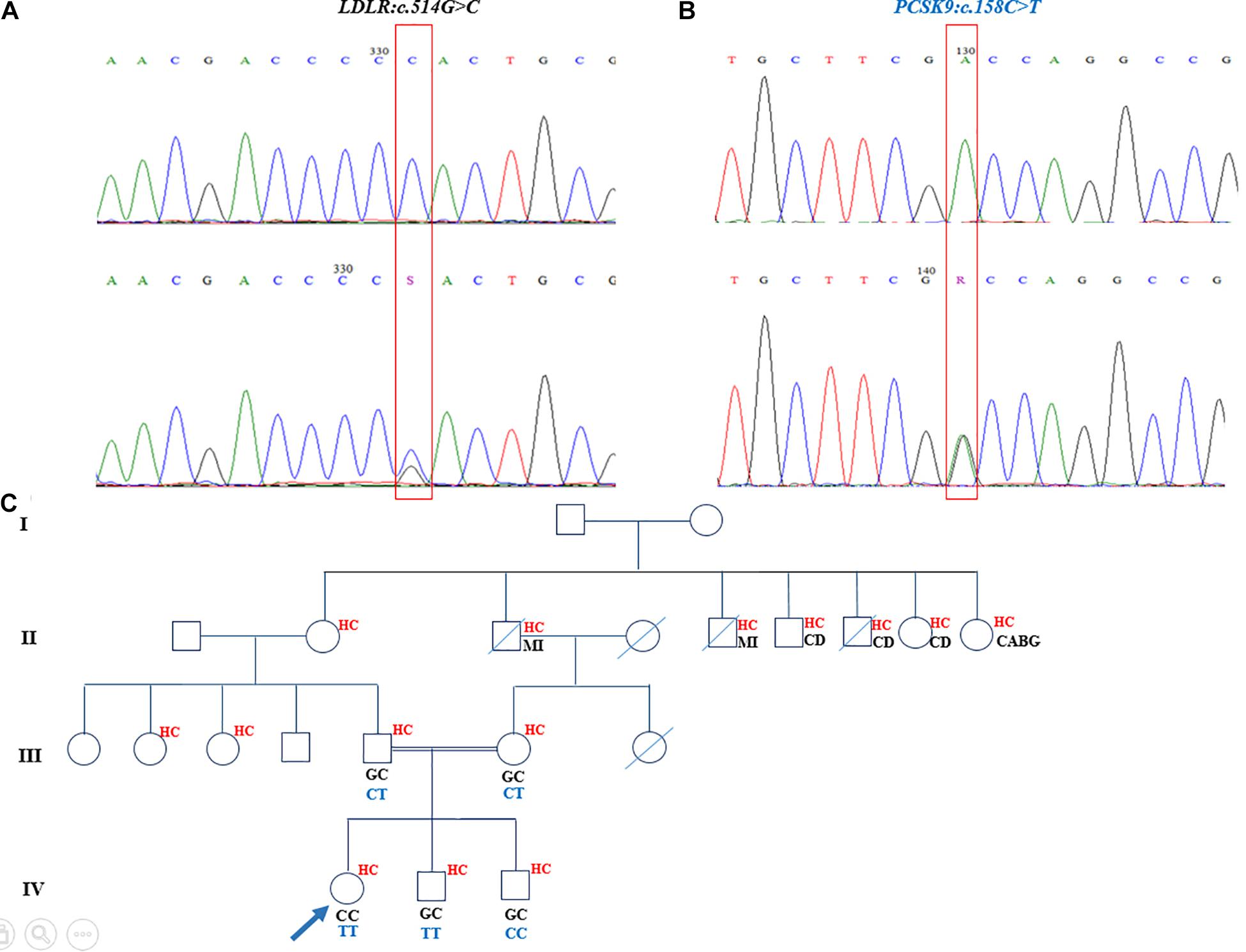
Figure 2. Sequencing analysis and pedigree of proband who had double mutations in LDLR and PCSK9 genes. (A) The homozygote and heterozygote nucleotide changes at c.514G > C in LDLR demonstrated in upper chromaogram and lower chromatogram, respectively. (B) The loss-of-function mutation of PCSK9 in homozygote and heterozygote states depicted correspondingly in upper panel and lower panel. (C) The genotype of proband and her family members for LDLR and PCSK9 nucleotide changes demonstrated, respectively, in black and dark blue in the pedigree. The family history of proband exhibited positive history of hypercholesterolemia and cardiovascular diseases and myocardial infarction. The mother and father of the proband are first cousins. MI, myocardial infarction; HC, hypercholesterolemia; CD, cardiovascular diseases; CABG, coronary artery bypass grafting.
Another homozygous mutation (c.1436T > A) in exon 10 of the LDLR gene was found in a 24-year-old man (P8). The patient had undergone coronary artery bypass graft (CABG) surgery and valve replacement and had a high, uncontrollable cholesterol level (TC = 461 mg/dL and LDL-C = 391 mg/dL) with detectable xanthomas. This nucleotide variant changes leucine in position 479 to glutamine, which is deleterious, probably damaging, and disease causing according to different in silico programs. The patient’s parents had a consanguineous marriage and a positive history of myocardial infarction, CABG, and hypercholesterolemia. The cosegregation analysis confirmed that this variant was an FH-causing mutation in the family. The proband’s father, mother, and brother were heterozygous for the mutation, whereas his sister was a wild type (Supplementary Figure 1).
A nonsense homozygous nucleotide variant (c.1774delG [p.G593Afs∗72]) was identified in a 22-year-old man (Supplementary Figure 2). This variant, harbored in exon 12 of the LDLR gene, was a frameshift mutation, where the deletion of G led to a shift in the reading frame, created a stop codon 72 amino acids downstream, and produced a truncated protein. The DNA sequencing of the LDLR gene demonstrated five G nucleotides in positions 1774–1778, making two glycines at a protein level (592-593 amino acids). Nonetheless, the deletion of G in any of the aforementioned positions altered the second glycine to alanine and generated stop codon 72 amino acids downstream. The software tools of SIFT and MutationTaster predicted this variant to be deleterious and disease causing, respectively. The pedigree analysis of this proband revealed a consanguineous marriage in the family. His mother and father were first cousins with a positive paternal family history of hypercholesterolemia and myocardial infarction. The proband had two younger siblings, who were apparently not affected. The father, who was the only available individual from the family, was heterozygous for this deletion. As the proband’s parents were first cousins, and both he and his father were correspondingly homozygous and heterozygous for the variant, the mother of the family, who had hypercholesterolemia, was probably heterozygous for this variant as well.
Additionally, 34 nucleotide variants, dedicated to the exons, introns, and 3-prime untranslated region (3′-UTR) of the LDLR gene, were detected in the 15 examined patients (Supplementary Table 2). All the nucleotide variants previously described were categorized as benign, likely benign, and with other allele. Moreover, 31 nucleotide variants were identified in the PCSK9 gene; most of them were categorized as benign, likely benign, with other allele, and with uncertain significance allele (Supplementary Table 3). Among them, one exonic (c.1233G > A), one intronic (c.1863+20C > G), and two 3′-UTR nucleotide variants (c.∗863 A > G and c.∗980 A > G) have not been previously reported. None of the mutations of the APOB gene (p.Arg3500Gln, p.Arg3500Trp, p.Arg3531Cys, and p.His3543Tyr) was detected in our 15 screened patients.
One novel nucleotide variant (c.1014C > G) was detected and cosegregated in the available family members as well. The in silico mutation prediction tools of SIFT, PROVEAN, PolyPhen-2, and MutationTaster were applied to predict the functionality of the aforementioned variant. The c.1014C > G heterozygous missense mutation was found in a 39-year-old woman. This mutation corresponded to exon 7 of the LDLR gene and changed cysteine (C) amino acid in position 338 to tryptophan (W). SIFT, PROVEAN, PolyPhen-2, and MutationTaster predicted this variant to be deleterious, probably damaging, and disease causing, respectively. The family pedigree of the proband featured a positive history of hypercholesterolemia and myocardial infarction on the maternal side (Figure 3). The patient had three sisters and a deceased brother. Two younger sisters, aged 36 and 33 years, suffered from hypercholesterolemia as well; the youngest sister, however, was seemingly normal. The only available individual from the family was the proband’s 36-year-old sister, who was also heterozygous for this mutation. The mother, who also had hypercholesterolemia, passed away at the age of 60 years due to myocardial infarction. She probably carried the mutation. The proband and her sister, who carried the heterozygous mutation, had consanguineous marriages with their first cousins.
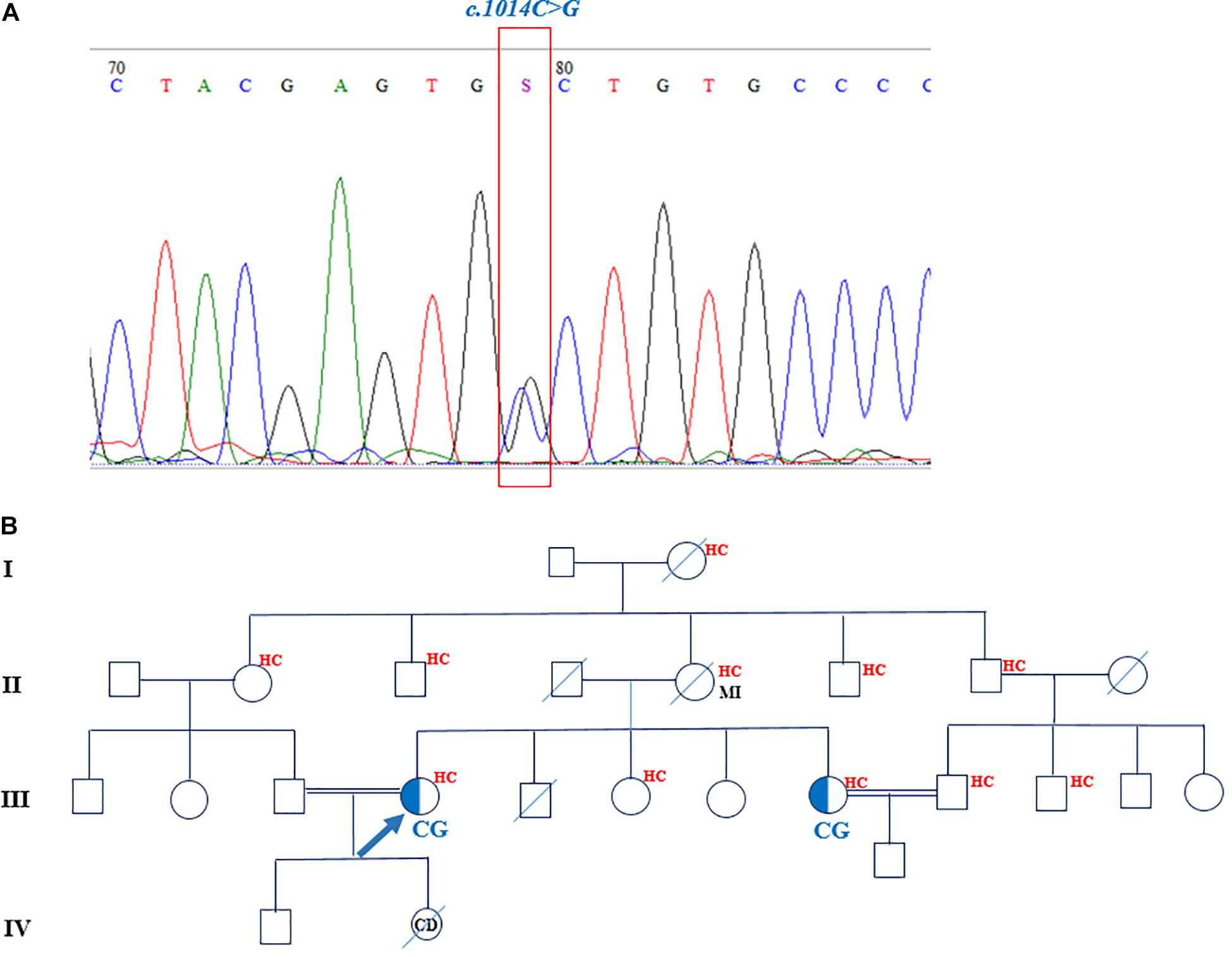
Figure 3. The chromatogram and pedigree of patient carried the novel mutation in LDLR gene. (A) The identified c.1014C > G nucleotide change shown in a chromatogram. (B) The pedigree of proband demonstrated the positive family history of hypercholesterolemia and myocardial infarction. The sister of proband is also heterozygote for the mutation. MI, myocardial infarction; HC, hypercholesterolemia.
Frequent LDLR, APOB, and PCSK9 gene mutations have been described in patients suffering from hypercholesterolemia; however, genetic data on the Iranian population are still limited (Jensen et al., 1996; Fardesfahani et al., 2005; Fardesfahani and Khatami, 2010; Farrokhi et al., 2011; Fairoozy et al., 2017; Ekrami et al., 2018; Nikkhooy et al., 2018; Tajamolian et al., 2018).
In this study, we found eight different mutations among 15 patients with clinically diagnosed FH and premature CAD. Among the study population, one patient had double mutations in both LDLR and PCSK9 genes. The present study is the first investigation to report the c.1014C > G mutation. With the exception of the c.1436T > A mutation, which has been previously reported in Iran (Fairoozy et al., 2017), all the other mutations have already been reported in populations other than Iranians. Among seven mutations in the LDLR gene, only four nucleotide variants are reported in the UCL-FH database (Table 2), and none of the pathogenic mutations can be found in 1000Genome, gnomAD, and Iranome databases (Table 2).
Previous research on the Iranian population (Jensen et al., 1996; Fardesfahani et al., 2005; Fardesfahani and Khatami, 2010; Khan et al., 2011; Fairoozy et al., 2017; Ekrami et al., 2018; Nikkhooy et al., 2018; Tajamolian et al., 2018) has identified 15 different mutations harbored in exons 3, 4, 9, 10, 11, 12, 14, and 17 of the LDLR gene (Supplementary Table 4). In 2017, an investigation drawing upon target next-generation sequencing identified seven different mutations (C.389C > G, c.1436T > A, c.1599G > A, c.1729T > C, c.2001_2002delinsGT, and c.2146dupG) that were not previously reported in the Iranian population (Fairoozy et al., 2017). Two different mutations (c.285C > G and c.415G > C) were found in another study conducted on a sample of the Iranian population (Ekrami et al., 2018). The c.445G > T, c. 1246C > T, c.1478-1479delCT, c.660-661InsCC, c.105A > T, and c.1474G > A mutations have also been reported in different studies (Fardesfahani et al., 2005; Fardesfahani and Khatami, 2010; Khan et al., 2011; Nikkhooy et al., 2018; Tajamolian et al., 2018).
The detected mutations in the LDLR gene in the present study were scattered over exons 4, 6, 7, 10, and 12, and they encoded the functional domain, the ligand-binding domain, and the epidermal growth factor (EGF)–like of the mature LDLR protein (Figure 1). The prevalence of the identified mutations was uppermost in exon 4 (3/7 = 43%) of the LDLR gene (Heath et al., 2001; Villéger et al., 2002). Exon 4, along with exons 5 to 7, of the LDLR gene encodes the ligand-binding domain of the LDLR protein; nevertheless, exon 4 encodes the critical ligand-binding section of the protein, and any changes in the sequence may affect the folding of the protein and change the functionality of the mature protein. The consequences of missense mutations in this exon may lessen the receptor affinity of LDLR toward both APOE and APOB (Esser et al., 1988). Our data revealed c.514C > G, c.443G > A, and c.647G > A mutations in exon 4; none of these mutations has been previously reported among Iranians. Based on the previously detected mutations in the Iranian population, 5 of the 15 (33.3%) identified mutations occurred in exon 4 of LDLR (Supplementary Table 4). Accumulating evidence has demonstrated that individuals carrying mutations in exon 4 have a more severe FH phenotype and congenital heart disease risks; mutations in this region are therefore predominantly deleterious to receptor function (Gudnason et al., 1994; Graham et al., 1999; Humphries et al., 2006). The c.443G > A and c.647G > A mutations have both been described in Germany, the United Kingdom, and Spain (Nauck et al., 2001; Dedoussis et al., 2004; Humphries et al., 2006; Meriño-Ibarra et al., 2007).
The genetic analysis on an 11-year-old girl from the southern Iranian city of Kerman demonstrated a homozygous loss-of-function mutation in the PCSK9 gene (c.158C > T [p.Ala53Val]) and a pathogenic homozygous mutation in the LDLR gene (c.514C > G [p.Asp172His]). The c.158C > T variant in PCSK9 has also been previously reported in South Africa (Thiart et al., 2000). Several studies have revealed that loss-of-function mutations in PCSK9 culminate in low LDL-C levels and lead to a diminished risk of CAD (Benjannet et al., 2012; Seidah, 2017; Bayona et al., 2020). Apropos the effect of loss-of-function PCSK9 variants, some clinical trials have also confirmed that PCSK9 inhibitors lower LDL-C concentrations in the serum (Cohen et al., 2006; Stein et al., 2014; Vuorio et al., 2016; Naeli et al., 2017). Although the index patient carried the homozygous loss-of-function mutation in PCSK9, she presented severe phenotypes such as an uncontrollable cholesterol level; several elbows, knees, and hip xanthomas; and aortic valve disease, which necessitated valve replacement surgery. The reason for the patient’s presentations may have been completely defective LDLR activity owing to homozygous mutations. In addition, she was the offspring of a consanguineous marriage between a couple who presented with moderate hypercholesterolemia but had no cardiovascular diseases. The parents carried both mutations in LDLR and PCSK9 in heterozygous forms. Thus, it seems that the loss-of-function mutation in PCSK9 has a protective effect against the development of the severe forms of phenotypes (Seidah, 2019) in such individuals, notwithstanding the heterozygous mutation in LDLR. The proband had two brothers, who also carried the heterozygous mutation in the LDLR gene. In regard to the PCSK9 gene, the elder brother, similar to the parents, featured a heterozygous mutation, whereas the younger one was a wild type.
To the best of our knowledge, this is the first report on the coincidental inheritance of a pathologic mutation in LDLR (p.Asp172His) and a loss-of-function mutation (p.Ala53Val) in PCSK9 in an Iranian family. The loss-of-function mutation (c.137G > T [p.Arg46Leu]) in PCSK9 and a pathologic mutation in LDLR (c.902A > G [p.Asp301Gly]), both in the heterozygous form, have been previously reported in a Caucasian family (Bayona et al., 2020).
The c.906C > G FH-causing variant (p.Cys302Trp), which was found in this study in exon 6 of LDLR, has also been previously identified in an Iraqi–Turkish patient (Webb et al., 1996). The 40-year-old woman carrying this mutation originated from the north of Iran. Given the close geographical and cultural links between Iraq, Turkey, and Iran, it is probable that the two patients shared some common founder mutations.
The novel mutation found in this study (c.1014C > G), which is located in exon 7 of the LDLR gene, changes cysteine to tryptophan. According to the LOVD database, approximately 3% of the total 3731 variants of LDLR happen on exon 7 (Fokkema et al., 2005). This variant has not been previously identified; still, the same location features another change that has been previously reported (c.1014C > A). This variation, which is a stop-gained mutation, alters cysteine in position 338 to the termination code and creates a truncated protein (Kotzer and Baudhuin, 2009). Exon 7, along with exons 8–12, of the LDLR gene participates in the production of the EGF precursor homology domain of LDLR. This domain of LDLR shares such conserved sequences as cysteine-rich sequences, YWTD repeats, and EGF repeats to EGF (Davis et al., 1987; Springer, 1998). Furthermore, the EGF precursor-like domain comprised two EGF homology domains (EGF-A and EGF-B), dispersed from the third EGF-like domain by a B-propeller domain. Exon 7 of LDLR encodes the EGF-A part of the EGF-like domain of LDLR, which encompasses 40-amino-acid residues and six cysteine amino acids, forming three disulfide bonds (Springer, 1998; Jeon et al., 2001). In the molecular mechanisms of LDLR recycling in hepatocytes, PCSK9 binds to the EGF-A domain of LDLR in a PH- and calcium-dependent manner and through lysosomal degradation decreases the total LDLR level in the liver (Zhang et al., 2007; Bottomley et al., 2009). A deficiency in this region interferes with the release of the binding ligand of the internalized LDLR and inhibits its recycling to the cell surface (Davis et al., 1987). On account of the fact that this missense mutation exists in a cysteine residue, it may result in misfolding and defective protein receptors. In addition, all in silico predictive programs describe this variant as deleterious, probably damaging, and disease causing. The segregation analysis of this variant in the index patient’s sister, who had hypercholesterolemia, confirmed the pathogenicity of this variant as well.
The point mutations (p.leu479Gln and p.G593Afs∗72) were respectively harbored in exons 10 and 12, which encode different parts of the LDLR EGF precursor-like domain. Exon 10 encodes part of a 5-repeat region of 40–60 amino acids that has a conserved motif (YWTD). This part is also known to play a role in the intracellular trafficking and recycling of LDLR, which explains why mutations in this section are acknowledged to be the cause of transport-defective or recycling-defective proteins (Brown and Goldstein, 1974; Davis et al., 1987). The p.leu479Gln mutation has been previously described in Iran (Fairoozy et al., 2017) and the United Kingdom (Heath et al., 2001). Our segregation analysis of this mutation in the first degree-relatives of the proband (P8) confirmed the pathogenicity of the mutation too. The translation product of the allele carrying the frameshift mutation (p.G593Afs∗72) leads to the truncation of the proteins of 663 amino acids and is deemed a receptor-negative mutation (Bertolini et al., 2017). With respect to the pathogenicity of this mutation, confirmed via the molecular diagnosis of the variation in the family of the proband (P9), the result of the screening of the family demonstrated that the father carried this mutation in a heterozygous state. It is also probable that the proband’s mother was also heterozygous for this variation. This mutation was originally found in Italian and Northern Ireland populations (Bertolini et al., 2017).
Furthermore, we succeeded in detecting 31 different nucleotide variants in the PCSK9 gene. While most of them have been previously reported, there are some unreported variants scattered in exons, introns, and 3′-UTRs (Supplementary Table 3). The UTR parts (5′ and 3′) include regulatory elements that interact with such different regulatory factors as transcription factors and microRNAs. Altering the nucleotides in the UTRs of genes may interfere with their interactions and causes the misexpression of their targeted gene (Chatterjee and Pal, 2009; Azad et al., 2020). The existing evidence notwithstanding, this hypothesis still requires more functional analyses to be confirmed.
In the present study, we detected none of the common APOB mutations, suggesting that the frequency of APOB mutations in Iran may differ from that in other countries. Further research on this point is warranted.
Among our study population, which comprised 15 subjects with clinically diagnosed FH and premature CAD, we detected eight patients who were negative for FH mutations. Accumulating evidence has shown that, in addition to mutations in the LDLR and PCSK9 genes, mutations in other single or multiple genes can contribute to the creation of the FH phenotype (Smilde et al., 2001; Nordestgaard et al., 2013; Cuchel et al., 2014b; Fouchier et al., 2014; Brautbar et al., 2015; Berberich and Hegele, 2019). The accumulation of common small-effect LDL-C–raising alleles can elevate LDL-C levels and, consequently, cause FH (Talmud et al., 2013). Accordingly, the absence of mutations in the FH-mutation–negative probands in our study may be attributable to nucleotide alterations in other genes, nucleotide variations in other parts of the APOB gene (not addressed in this study), or a combination of LDL-C-raising alleles. Be that as it may, previous studies on other diseases have demonstrated that the presence and combinations of some polymorphisms can render individuals more susceptible to disease (Khajali and Khajali, 2014; Mesic et al., 2019; Pinheiro et al., 2019).
We succeeded in finding several nucleotide variations/polymorphisms in both LDLR and PCSK9 scattered in exons, introns, and 5′–3′ UTRs. These nucleotide variations/polymorphisms could be the combination of different polymorphisms, predisposing FH-mutation–negative individuals to hypercholesterolemia.
The results of the present study should be interpreted in light of its limitations. First, our small sample size precluded an accurate estimation of the prevalence of the mutations in the Iranian population. Budget constraints also limited our investigation to the screening of only the LDLR and PCSK9 genes and a part of the APOB gene. Indubitably, many other hitherto reported and unreported genes may be the underlying causes of FH-mutation negativity. A large insertion or deletion in the LDLR gene causes FH in 5–10% of patients (Hobbs et al., 1985; Goldmann et al., 2010); we were, unfortunately, unable to detect such mutations. Finally, what has thus far hampered research on polygenic FH in Iran is, aside from financial restraints, the dearth of data on the frequency of polymorphisms in the Iranian population.
The existing literature contains a paucity of information regarding genetic testing on nucleotide alterations in the Iranian population. The finding of seven different mutations in this study, along with other studies on samples of the Iranian population, demonstrates the high degree of genetic heterogeneity and the wide spectrum of mutations in Iranians. This domain undoubtedly requires further in-depth research. In daily practice, patients presenting with premature CAD are rarely screened for the genetic analysis of FH; this is a missed opportunity for preventive therapy. Given that the first-degree relatives of patients with FH have a 50% chance of affliction, a targeted molecular genetic screening of individuals with premature CAD and FH is an effective strategy both to prevent cardiovascular diseases in their nascent stages and to confer more desirable management plans.
The accession number of the novel and reported variants are as follows:
LDLR
c.1014C>G variant: [ClinVar]: [SCV001446309]
c.443G>A variant: [ClinVar]: [SCV001467722]
c.647G>A variant: [ClinVar]: [SCV001467723.1]
c.906C>G variant: [ClinVar]: [SCV001467724.1]
c.1436T>A variant: [ClinVar]: [SCV001467725.1]
c.1774delG variant: [ClinVar]: [SUB8843875, processing]
c.514G>C variant: [ClinVar]: [SUB8843863, processing]
PCSK9
c.1233G>A variant: [ClinVar]: [SCV001450531]
c.1863+20C>G variant: [ClinVar]: [SCV001450532]
c.∗863A>G variant: [ClinVar]: [SCV001450537]
c.∗980A>G variant: [ClinVar]: [SCV001450538]
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The studies involving human participants were reviewed and approved by Rajaie Cardiovascular, Medical and Research Center Ethics Committees. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
AM: experiment design, lab work, data production, data interpretation, and MS edit. MMale, ZG, ZK, and FN: clinical evaluation of patients, data interpretation, and MS edit. MMo: lab work, data production, and MS edit. SK: data interpretation and MS edit. SM and NS: experiment design, data interpretation, and MS edit. MMala: experiment design, data interpretation, writing the first draft of MS, and manuscript final edit. All authors contributed to the article and approved the submitted version.
This work was supported in part by a research grant to Dr. Malakootian from Iran’s National Science Foundation (94017890), the Research Deputyship of Rajaie Cardiovascular Medical and Research Center (94112), and this work was also supported in part by a CIHR Foundation grant (NGS: # 148363) and a Canada Research Chair in Precursor Proteolysis (NGS: # 950-231335).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank the patients and families who contributed to this project. We are grateful to Mrs. Niloofar Naderi for her technical assistance.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.625959/full#supplementary-material
Supplementary Figure 1 | Sequencing analysis and pedigree of proband with c.1436T>A nucleotide change in LDLR gene. (A) The mutation found in proband in a homozygote form (upper chromatogram) and identified in a heterozygote statue in father of the patient (lower chromatogram). (B) The pedigree of the proband demonstrated the consanguineous marriage, positive family history of hypercholesterolemia, myocardial infarction and cardiovascular diseases. The genotypes of his sibling and his parent exhibited in dark blue color as well. MI, HC, and CD stand for myocardial infarction, hypercholesterolemia, and cardiovascular diseases, respectively.
Supplementary Figure 2 | The chromatogram and pedigree of patient who carried the c.1774delG mutation in LDLR gene. (A) The upper chromatogram demonstrated the sequence in wild type. The homozygote deletion mutation in nucleotide 1774 exhibited in middle chromatogram and the heterozygote form depicted in lower chromatogram. (B) The pedigree of the proband showed the consanguineous marriage and positive family history of hypercholesterolemia and MI. MI and HC stand for Myocardial infarction and Hypercholesterolemia, respectively.
Supplementary Table 1 | The sequences of all designed oligonucleotides utilized in this study to perform the PCRs and Sanger sequencing.
Supplementary Table 2 | All nucleotide variations of LDLR gene found in this study.
Supplementary Table 3 | All nucleotide variations of PCSK9 gene found in this study.
Supplementary Table 4 | The previously reported pathogenic mutations in Iranian population.
Azad, F. M., Arabian, M., Maleki, M., and Malakootian, M. (2020). Small Molecules with Big Impacts on Cardiovascular Diseases. Biochem. Genet. 58, 359–383. doi: 10.1007/s10528-020-09948-z
Bayona, A., Arrieta, F., Rodríguez-Jiménez, C., Cerrato, F., Rodríguez-Nóvoa, S., Fernández-Lucas, M., et al. (2020). Loss-of-function mutation of PCSK9 as a protective factor in the clinical expression of familial hypercholesterolemia: A case report. Medicine 99:e21754. doi: 10.1097/md.0000000000021754
Beheshti, S. O., Madsen, C. M., Varbo, A., and Nordestgaard, B. G. (2020). Worldwide Prevalence of Familial Hypercholesterolemia. Meta Analy. Million Sub. 75, 2553–2566. doi: 10.1016/j.jacc.2020.03.057
Benjannet, S., Hamelin, J., Chrétien, M., and Seidah, N. G. (2012). Loss-and gain-of-function PCSK9 variants cleavage specificity, dominant negative effects, and low density lipoprotein receptor (LDLR) degradation. J. Biol. Chem. 287, 33745–33755.
Berberich, A. J., and Hegele, R. A. (2019). The complex molecular genetics of familial hypercholesterolaemia. Nat. Rev. Cardiol. 16, 9–20. doi: 10.1038/s41569-018-0052-6
Bertolini, S., Pisciotta, L., Fasano, T., Rabacchi, C., and Calandra, S. (2017). The study of familial hypercholesterolemia in Italy: A narrative review. Atheroscl. Suppl. 29, 1–10. doi: 10.1016/j.atherosclerosissup.2017.07.003
Bottomley, M. J., Cirillo, A., Orsatti, L., Ruggeri, L., Fisher, T. S., Santoro, J. C., et al. (2009). Structural and biochemical characterization of the wild type PCSK9-EGF (AB) complex and natural familial hypercholesterolemia mutants. J. Biol. Chem. 284, 1313–1323. doi: 10.1074/jbc.m808363200
Brautbar, A., Leary, E., Rasmussen, K., Wilson, D. P., Steiner, R. D., and Virani, S. (2015). Genetics of familial hypercholesterolemia. Curr. Atheroscl. Rep. 17:20.
Brown, M. S., and Goldstein, J. L. (1974). Familial hypercholesterolemia: defective binding of lipoproteins to cultured fibroblasts associated with impaired regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity. Proc. Natl. Acad. Sci. U. S. A. 71, 788–792. doi: 10.1073/pnas.71.3.788
Chatterjee, S., and Pal, J. K. (2009). Role of 5′-and 3′-untranslated regions of mRNAs in human diseases. Biol. Cell 101, 251–262. doi: 10.1042/bc20080104
Cohen, J. C., Boerwinkle, E., Mosley, T. H. Jr., and Hobbs, H. H. (2006). Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 354, 1264–1272. doi: 10.1056/nejmoa054013
Cuchel, M., Bruckert, E., Ginsberg, H., Raal, F., Santos, R., Hegele, R., et al. (2014a). European Atherosclerosis Society Consensus Panel on Familial Hypercholesterolaemia. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur. Heart J. 35, 2146–2157.
Cuchel, M., Bruckert, E., Ginsberg, H. N., Raal, F. J., Santos, R. D., Hegele, R. A., et al. (2014b). Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur. Heart J. 35, 2146–2157.
Davis, C. G., Goldstein, J. L., Südhof, T. C., Anderson, R. G., Russell, D. W., and Brown, M. S. (1987). Acid-dependent ligand dissociation and recycling of LDL receptor mediated by growth factor homology region. Nature 326, 760–765. doi: 10.1038/326760a0
De Castro-Orós, I., Pocoví, M., and Civeira, F. (2010). The genetic basis of familial hypercholesterolemia: inheritance, linkage, and mutations. Appl. Clin. Genet. 3:53. doi: 10.2147/tacg.s8285
Dedoussis, G. V., Genschel, J., Bochow, B., Pitsavos, C., Skoumas, J., Prassa, M., et al. (2004). Molecular characterization of familial hypercholesterolemia in German and Greek patients. Hum. Mutat. 23, 285–286. doi: 10.1002/humu.9218
Ekrami, M., Torabi, M., Ghafouri-Fard, S., Mowla, J., Soltani, B. M., Hashemi-Gorji, F., et al. (2018). Genetic analysis of iranian patients with familial hypercholesterolemia. Iran. Biomed. J. 22:117.
El Khoury, P., Elbitar, S., Ghaleb, Y., Abou Khalil, Y., Varret, M., Boileau, C., et al. (2017). PCSK9 mutations in familial hypercholesterolemia: from a groundbreaking discovery to anti-PCSK9 therapies. Curr. Atheroscl. Rep. 19:49.
Esser, V., Limbird, L., Brown, M. S., Goldstein, J. L., and Russell, D. W. (1988). Mutational analysis of the ligand binding domain of the low density lipoprotein receptor. J. Biol. Chem. 263, 13282–13290. doi: 10.1016/s0021-9258(18)37702-0
Fairoozy, R., Futema, M., Vakili, R., Abbaszadegan, M., Hosseini, S., Aminzadeh, M., et al. (2017). The genetic spectrum of familial hypercholesterolemia (FH) in the Iranian population. Scient. Rep. 7, 1–9.
Fardesfahani, P., and Khatami, S. (2010). Familial hypercholesterolemia in Iran: A novel frameshift mutation in low density lipoprotein receptor (LDLR) gene.
Fardesfahani, P., Rouhi, D. S., Khatami, S., Zeinali, C., and Taghikhani, M. (2005). A Novel Mutation in exon 4 of the low density lipoprotein (LDL) receptor gene in an Iranian Familial Hypercholesterolemia patient. Iran. Biomed. J. 3:9.
Farrokhi, E., Shayesteh, F., Mobarakeh, S. A., Dehkordi, F. R., Samani, K. G., and Chaleshtori, M. H. (2011). Molecular characterization of Iranian patients with possible familial hypercholesterolemia. Ind. J. Clin. Biochem. 26, 244–248. doi: 10.1007/s12291-011-0113-7
Fokkema, I. F., den Dunnen, J. T., and Taschner, P. E. (2005). LOVD: easy creation of a locus-specific sequence variation database using an “LSDB-in-a-box” approach. Hum. Mutat. 26, 63–68. doi: 10.1002/humu.20201
Fouchier, S. W., Dallinga-Thie, G. M., Meijers, J. C., Zelcer, N., Kastelein, J. J., Defesche, J. C., et al. (2014). Mutations in STAP1 are associated with autosomal dominant hypercholesterolemia. Circul. Res. 115, 552–555. doi: 10.1161/circresaha.115.304660
Gidding, S. S., Ann Champagne, M., de Ferranti, S. D., Defesche, J., Ito, M. K., Knowles, J. W., et al. (2015). The agenda for familial hypercholesterolemia: a scientific statement from the American Heart Association. Circulation 132, 2167–2192. doi: 10.1161/cir.0000000000000297
Goldmann, R., Tichý, L., Freiberger, T., Zapletalová, P., Letocha, O., Soška, V., et al. (2010). Genomic characterization of large rearrangements of the LDLR gene in Czech patients with familial hypercholesterolemia. BMC Med. Genet. 11, 1–8. doi: 10.1186/1471-2350-11-115
Graham, C. A., McClean, E., Ward, A. J., Beattie, E. D., Martin, S., O’Kane, M., et al. (1999). Mutation screening and genotype: phenotype correlation in familial hypercholesterolaemia. Atherosclerosis 147, 309–316.
Gudnason, V., Day, I., and Humphries, S. (1994). Effect on plasma lipid levels of different classes of mutations in the low-density lipoprotein receptor gene in patients with familial hypercholesterolemia. Arterioscl. Thromb. J. Vascul. Biol. 14, 1717–1722. doi: 10.1161/01.atv.14.11.1717
Heath, K. E., Humphries, S. E., Middleton-Price, H., and Boxer, M. (2001). A molecular genetic service for diagnosing individuals with familial hypercholesterolaemia (FH) in the United Kingdom. Eur. J. Hum. Genet. 9, 244–252. doi: 10.1038/sj.ejhg.5200633
Hobbs, H. H., Lehrman, M. A., Yamamoto, T., and Russell, D. W. (1985). Polymorphism and evolution of Alu sequences in the human low density lipoprotein receptor gene. Proc. Natl. Acad. Sci. 82, 7651–7655. doi: 10.1073/pnas.82.22.7651
Hu, P., Dharmayat, K. I., Stevens, C. A., Sharabiani, M. T., Jones, R. S., Watts, G. F., et al. (2020). Prevalence of familial hypercholesterolemia among the general population and patients with atherosclerotic cardiovascular disease: a systematic review and meta-analysis. Circulation 141, 1742–1759. doi: 10.1161/circulationaha.119.044795
Humphries, S. E., Cranston, T., Allen, M., Middleton-Price, H., Fernandez, M. C., Senior, V., et al. (2006). Mutational analysis in UK patients with a clinical diagnosis of familial hypercholesterolaemia: relationship with plasma lipid traits, heart disease risk and utility in relative tracing. J. Mole. Med. 84, 203–214. doi: 10.1007/s00109-005-0019-z
Jensen, H., Jensen, L., Hansen, P., Faergeman, O., and Gregersen, N. (1996). An Iranian-Armenian LDLR frameshift mutation causing familial hypercholesterolemia. Clin. Genet. 49, 88–90. doi: 10.1111/j.1399-0004.1996.tb04334.x
Jeon, H., Meng, W., Takagi, J., Eck, M. J., Springer, T. A., and Blacklow, S. C. (2001). Implications for familial hypercholesterolemia from the structure of the LDL receptor YWTD-EGF domain pair. Nat. Struct. Biol. 8, 499–504.
Khachadurian, A. K. (1964). The inheritance of essential familial hypercholesterolemia. Am. J. Med. 37, 402–407. doi: 10.1016/0002-9343(64)90196-2
Khajali, F., and Khajali, Z. (2014). L-carnitine supplementation decreases lipid peroxidation and improves cardiopulmonary function in broiler chickens reared at high altitude. Acta Veter. Hungar. 62, 489–499. doi: 10.1556/avet.2014.022
Khan, S. P., Ghani, R., Ahmed, K. Z., and Yaqub, Z. (2011). Genetic basis for diagnosis of novel mutation of LDL receptor gene. Int. J. Collabor. Res. Int. Med. Publ. Health 3, 0–0.
Kotzer, M., and Baudhuin, L. (2009). Novel human pathological mutations. Gene symbol: LDLR. Disease: hypercholesterolemia. Hum. Genet. 126:352.
Krogh, H. W., Mundal, L., Holven, K. B., and Retterstøl, K. (2016). Patients with familial hypercholesterolaemia are characterized by presence of cardiovascular disease at the time of death. Eur. Heart J. 37, 1398–1405. doi: 10.1093/eurheartj/ehv602
Meriño-Ibarra, E., Puzo, J., Jarauta, E., Cenarro, A., Recalde, D., García-Otín, A., et al. (2007). Hyperlipoproteinaemia (a) is a common cause of autosomal dominant hypercholesterolaemia. J. Inher. Metab. Dis. 30:970. doi: 10.1007/s10545-007-0585-z
Mesic, A., Rogar, M., Hudler, P., Bilalovic, N., Eminovic, I., and Komel, R. (2019). Characterization and risk association of polymorphisms in Aurora kinases A, B and C with genetic susceptibility to gastric cancer development. BMC Cancer 19:919.
Naeli, P., Mirzadeh Azad, F., Malakootian, M., Seidah, N. G., and Mowla, S. J. (2017). Post-transcriptional regulation of PCSK9 by miR-191, miR-222, and miR-224. Front. Genet. 8:189. doi: 10.3389/fgene.2017.00189
Nauck, M., Köster, W., Dörfer, K., Eckes, J., Scharnagl, H., Gierens, H., et al. (2001). Identification of recurrent and novel mutations in the LDL receptor gene in German patients with familial hypercholesterolemia. Hum. Mutat. 18, 165–166. doi: 10.1002/humu.1171
Nikkhooy, M., Foroughmand, A. M., Galehdari, H., and Yazdankhah, S. (2018). The First Report of GAC< inline-formula id=. Zahedan J. Res. Med. Sci. 8:20.
Nordestgaard, B. G., Chapman, M. J., Humphries, S. E., Ginsberg, H. N., Masana, L., Descamps, O. S., et al. (2013). Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur. Heart J. 34, 3478–3490. doi: 10.1093/eurheartj/eht273
Pinheiro, D. S., Santos, R. S., Jardim, P. C. V., Silva, E. G., Reis, A. A., Pedrino, G. R., et al. (2019). The combination of ACE I/D and ACE2 G8790A polymorphisms revels susceptibility to hypertension: A genetic association study in Brazilian patients. PLoS One 14:e0221248. doi: 10.1371/journal.pone.0221248
Seidah, N. G. (2017). The PCSK9 revolution and the potential of PCSK9-based therapies to reduce LDL-cholesterol. Glob. Cardiol. Sci. Pract. 2017:e201702.
Seidah, N. G. (2019). The elusive inhibitory function of the acidic N-terminal segment of the prodomain of PCSK9: the plot thickens. J. Mol. Biol. 431, 904–907. doi: 10.1016/j.jmb.2019.01.015
Sharifi, M., Futema, M., Nair, D., and Humphries, S. E. (2017). Genetic architecture of familial hypercholesterolaemia. Curr. Cardiol. Rep. 19:44.
Simon Broome Register Group. (1991). Risk of fatal coronary heart disease in familial hypercholesterolaemia. BMJ 303, 893–896. doi: 10.1136/bmj.303.6807.893
Smilde, T. J., van Wissen, S., Awollersheim, H., Trip, M. D., Kastelein, J., and Stalenhoef, A. F. (2001). Effect of aggressive versus conventional lipid lowering on atherosclerosis progression in familial hypercholesterolemia (ASAP): a prospective, randomised, double-blind trial. Lancet 357, 577–581. doi: 10.1016/s0140-6736(00)04053-8
Springer, T. A. (1998). An extracellular β-propeller module predicted in lipoprotein and scavenger receptors, tyrosine kinases, epidermal growth factor precursor, and extracellular matrix components. J. Mole. Biol. 283, 837–862. doi: 10.1006/jmbi.1998.2115
Stein, E. A., Giugliano, R. P., Koren, M. J., Raal, F. J., Roth, E. M., Weiss, R., et al. (2014). Efficacy and safety of evolocumab (AMG 145), a fully human monoclonal antibody to PCSK9, in hyperlipidaemic patients on various background lipid therapies: pooled analysis of 1359 patients in four phase 2 trials. Eur. Heart J. 35, 2249–2259. doi: 10.1093/eurheartj/ehu085
Tajamolian, M., Kolahdouz, P., Nikpour, P., Forouzannia, S. K., Sheikhha, M. H., and Yazd, E. F. (2018). A rare missense mutation and a polymorphism with high frequency in LDLR gene among iranian patients with familial hypercholesterolemia. Adv. Biomed. Res. 7:37.
Talmud, P. J., Shah, S., Whittall, R., Futema, M., Howard, P., Cooper, J. A., et al. (2013). Use of low-density lipoprotein cholesterol gene score to distinguish patients with polygenic and monogenic familial hypercholesterolaemia: a case-control study. Lancet 381, 1293–1301. doi: 10.1016/s0140-6736(12)62127-8
Thiart, R., Scholtz, C. L., Vergotine, J., Hoogendijk, C. F., de Villiers, J. N. P., Nissen, H., et al. (2000). Predominance of a 6 bp deletion in exon 2 of the LDL receptor gene in Africans with familial hypercholesterolaemia. J. Med. Genet. 37, 514–519. doi: 10.1136/jmg.37.7.514
Varret, M., and Rabès, J.-P. (2012). Missense mutation in the LDLR gene: a wide spectrum in the severity of familial hypercholesterolemia. Mutat. Hum. Genet. Dis. 73, 55–74.
Villéger, L., Abifadel, M., Allard, D., Rabès, J. P., Thiart, R., Kotze, M. J., et al. (2002). The UMD-LDLR database: additions to the software and 490 new entries to the database. Hum. Mutat. 20, 81–87. doi: 10.1002/humu.10102
Vuorio, A., Watts, G. F., and Kovanen, P. T. (2016). Initiation of PCSK9 inhibition in patients with heterozygous familial hypercholesterolaemia entering adulthood: a new design for living with a high-risk condition? Eur. Heart J. 37, 1353–1356. doi: 10.1093/eurheartj/ehw010
Webb, J. C., Sun, X.-M., McCarthy, S., Neuwirth, C., Thompson, G., Knight, B., et al. (1996). Characterization of mutations in the low density lipoprotein (LDL)-receptor gene in patients with homozygous familial hypercholesterolemia, and frequency of these mutations in FH patients in the United Kingdom. J. Lip. Res. 37, 368–381. doi: 10.1016/s0022-2275(20)37623-9
Zhang, D.-W., Lagace, T. A., Garuti, R., Zhao, Z., McDonald, M., Horton, J. D., et al. (2007). Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J. Biol. Chem. 282, 18602–18612. doi: 10.1074/jbc.m702027200
Keywords: pre-mature CAD, Familial Hypercholesterolemia, loss-of-function, LDLR, PCSK9 (proprotein convertase subtilisin kexin type 9)
Citation: Moradi A, Maleki M, Ghaemmaghami Z, Khajali Z, Noohi F, Moghadam MH, Kalyinia S, Mowla SJ, Seidah NG and Malakootian M (2021) Mutational Spectrum of LDLR and PCSK9 Genes Identified in Iranian Patients With Premature Coronary Artery Disease and Familial Hypercholesterolemia. Front. Genet. 12:625959. doi: 10.3389/fgene.2021.625959
Received: 04 November 2020; Accepted: 11 January 2021;
Published: 11 February 2021.
Edited by:
Alpo Juhani Vuorio, University of Helsinki, FinlandReviewed by:
Maria Paola Lombardi, University of Amsterdam, NetherlandsCopyright © 2021 Moradi, Maleki, Ghaemmaghami, Khajali, Noohi, Moghadam, Kalyinia, Mowla, Seidah and Malakootian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mahshid Malakootian, TWFsYWtvb3RpYW5AcmhjLmFjLmly; bWFoc2hpZC5tYWxha29vdGlhbkBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.