 Katrina V. Good
Katrina V. Good John B. Vincent
John B. Vincent Juan Ausió
Juan Ausió- 1Department of Biochemistry and Microbiology, University of Victoria, Victoria, BC, Canada
- 2Molecular Neuropsychiatry & Development (MiND) Lab, Centre for Addiction and Mental Health, Campbell Family Mental Health Research Institute, Toronto, ON, Canada
- 3Institute of Medical Science, University of Toronto, Toronto, ON, Canada
- 4Department of Psychiatry, University of Toronto, Toronto, ON, Canada
Mutations in methyl CpG binding protein 2 (MeCP2) are the major cause of Rett syndrome (RTT), a rare neurodevelopmental disorder with a notable period of developmental regression following apparently normal initial development. Such MeCP2 alterations often result in changes to DNA binding and chromatin clustering ability, and in the stability of this protein. Among other functions, MeCP2 binds to methylated genomic DNA, which represents an important epigenetic mark with broad physiological implications, including neuronal development. In this review, we will summarize the genetic foundations behind RTT, and the variable degrees of protein stability exhibited by MeCP2 and its mutated versions. Also, past and emerging relationships that MeCP2 has with mRNA splicing, miRNA processing, and other non-coding RNAs (ncRNA) will be explored, and we suggest that these molecules could be missing links in understanding the epigenetic consequences incurred from genetic ablation of this important chromatin modifier. Importantly, although MeCP2 is highly expressed in the brain, where it has been most extensively studied, the role of this protein and its alterations in other tissues cannot be ignored and will also be discussed. Finally, the additional complexity to RTT pathology introduced by structural and functional implications of the two MeCP2 isoforms (MeCP2-E1 and MeCP2-E2) will be described. Epigenetic therapeutics are gaining clinical popularity, yet treatment for Rett syndrome is more complicated than would be anticipated for a purely epigenetic disorder, which should be taken into account in future clinical contexts.
Introduction
The term epigenetics has gained much popularity and has gathered the attention of many researchers in recent years. Yet, the term has, at times, been loosely used and quite often in an ambiguous way (Greally, 2018).
In the right context, epi (beyond)-genetics is defined as gene expression alterations resulting from a change in the DNA/chromatin structure which does not involve a change in the underlying DNA nucleotide sequence (i.e., mutations). At the molecular level, this can be elicited by chemical post-replication/post-translational “tags” that mark DNA (Bird, 1993; Greenberg and Bourc’his, 2019), histones (Bannister and Kouzarides, 2011) (the main protein component of chromatin) or other chromosomal and non-chromosomal proteins. These “tags” (for instance DNA methylation (Greenberg and Bourc’his, 2019) or protein modifications) are written and erased through the action of different enzymes (writers and erasers) and read by transcriptional regulatory cofactors (readers) (Marmorstein and Zhou, 2014; Seto and Yoshida, 2014). Such is the case for the methyl CpG binding protein 2 (MeCP2), a DNA methylation reader protein. This protein, initially thought to have repressor activity (Nan et al., 1997), is now recognized to have both transcriptionally repressive and activating functions through its interaction with different cofactors (Yasui et al., 2007; Chahrour et al., 2008). The protein is able to recognize (or read) DNA and histone methylation marks (Lewis et al., 1992; Thambirajah et al., 2012; Lee et al., 2020) and, hence, it acts as a methylation-dependent transcriptional modulator within the context of chromatin (Li et al., 2020). DNA methylation dysregulation is one of the hallmarks of diseases such as cancer (Baylin and Jones, 2016), and MeCP2 mutations can alter the reading of this mark, as in RTT (Kriaucionis and Bird, 2003), where it can impact the normal activity of cells. Interestingly, MeCP2 has been recognized as a bona fide oncogene and has been involved in many cancers (Neupane et al., 2015). In any such instance, these diseases often have a genetic origin with downstream epigenetic effects (Yoon et al., 2020).
From structural and functional perspectives, MeCP2 is a good example of an intrinsically disordered protein (IDP) (Dunker et al., 2001; Hite et al., 2012), given its relatively low contents of secondary and tertiary structure organization in solution. Despite this, the protein can be divided into several well-defined structural/functional domains (Ghosh et al., 2010): NTD; N-terminal; MBD, methyl binding; ID, intervening; TRD, transcription repression; NID, NCoR interaction; and CTD, C-terminal; domains (see Figure 1). The TRD includes the nuclear localization sequence (NLS) [amino acids 253–271 in MeCP2-E2 nomenclature (Figure 1B)]. Genetic mutations in the coding region of the X-chromosome-linked MECP2 gene alter the ability with which its encoded protein MeCP2 binds to DNA within the context of chromatin. In particular, those mutations affecting the MBD of the protein (Figure 1) which affect the stability (Kucukkal et al., 2015) and affinity (Yang et al., 2016) of its DNA binding and which represents an important aspect of this review.
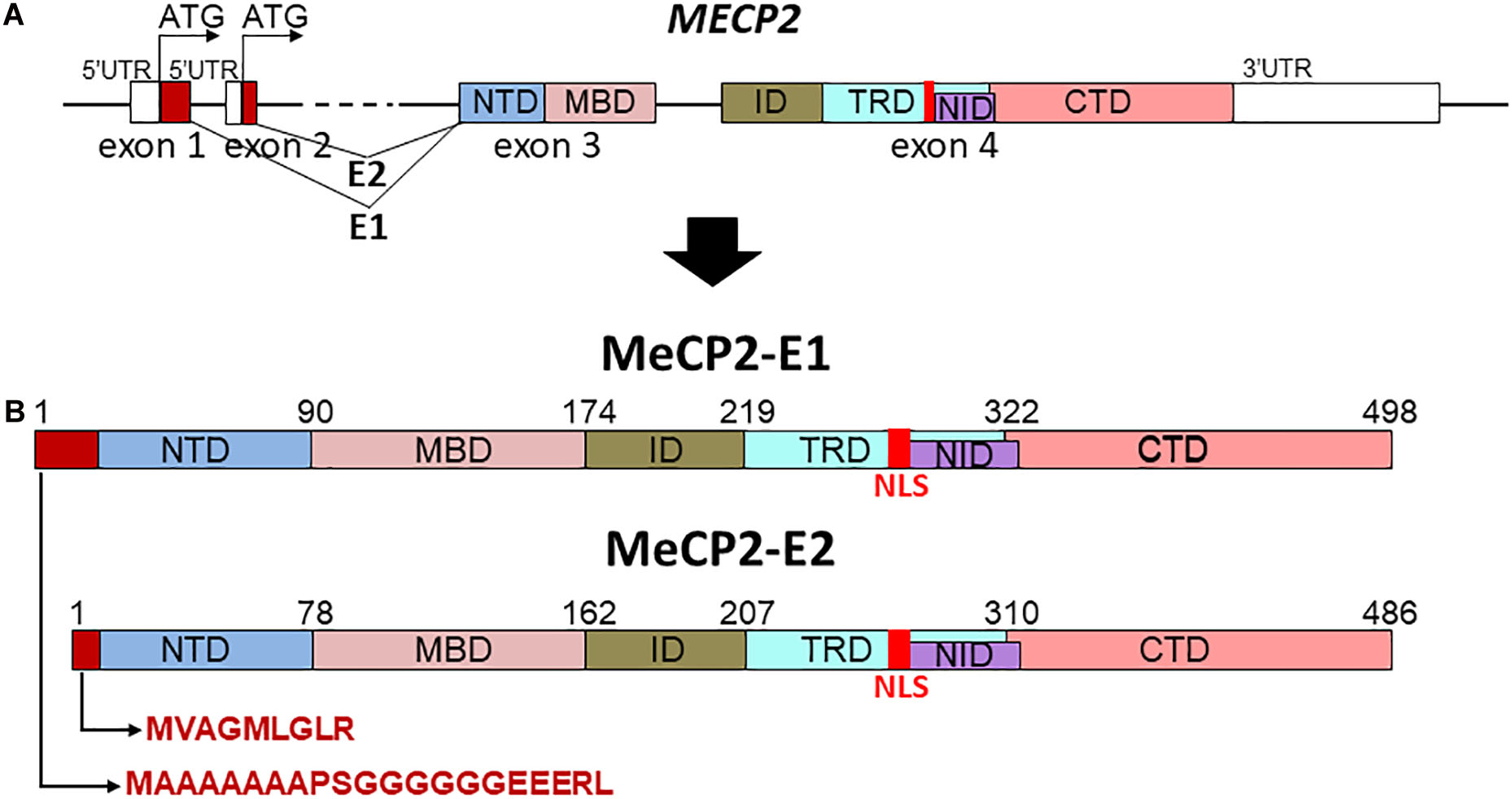
Figure 1. Schematic representation of MECP2 gene organization (A) and different protein domains of the MeCP2-E1 and MeCP2-E2 isoforms (B). NTD, N-terminal; MBD, methyl binding; ID, intervening; TRD, transcription repression; NID, NCoR interaction; CTD, C-terminal domains; NLS, nuclear localization signal.
Besides its ability to bind methylated DNA and histones, MeCP2 was earlier recognized (Jeffery and Nakielny, 2004) and more recently confirmed to be (Castello et al., 2016) an RNA binding protein. This less studied facet of MeCP2 will be described next. Following this, we will focus on an equally less understood role of this protein, namely its function in tissues other than those within the brain, and will finally conclude this review with the controversial potential physiological relevance of the two isoforms, MeCP2-E1 and MeCP2-E2 (Figure 1), in RTT (Liyanage and Rastegar, 2014; Martínez de Paz et al., 2019). Because the MeCP2-E2 isoform was the first to be discovered (Lewis et al., 1992), the mutations observed in RTT originally referred to this isoform. Therefore, the amino acid numbers (mutations) referring to the protein sequence of MeCP2 used in the following sections will be those of this isoform (unless otherwise indicated).
Subsections
Genetic Origin of Rett Syndrome
Most of the nucleotide transition mutations in Rett syndrome are the C > T type that take place at CpG hotspots (Wan et al., 1999), and likely reflect variable site methylation in the male germline (Cheadle et al., 2000). Hence, all the sporadic mutations, which represent more than 99% of the individuals affected by this syndrome (Chahrour and Zoghbi, 2007), and which involve de novo mutations (Comings, 1986) of the MECP2 gene, are of paternal origin (Trappe et al., 2001). This paternal origin may be explained by a combination of the elevated levels of methylation and mitotic divisions in the male germline (Driscoll and Migeon, 1990; Shahbazian and Zoghbi, 2002). As in the case of other C > T transition mutations, this is likely to involve methyl cytosine oxidative deamination of abnormally methylated cytosines (Tomatsu et al., 2004).
Rett syndrome is almost exclusively a disease that affects girls (XX), yet is not a disease with epigenetic inheritance, such as Prader-Willi syndrome and Angelman syndrome, where the clinical outcome depends on whether a mutation is transmitted from a paternal or maternal chromosome, and RTT mutations are not epigenetic mutations (epimutations) per se. Rather, RTT mutations have epigenetic consequences, as MeCP2 is considered to be a reader of epigenetic signals. Although considered a disease affecting girls, this is not completely exclusive. The vast majority of mutations that lead to RTT occur de novo in paternal germline cells (Cheadle et al., 2000), and these can only be transmitted to female offspring and never to males. De novo MECP2 mutations can occasionally be transmitted from mothers, or inherited from mothers who either have mild cognitive impairment or are asymptomatic, due to skewed X-inactivation favoring expression from their wild-type allele.
Rett syndrome clinical features include regression of motor and communicative skills after 6–18 months of apparently normal development [the reader is referred to Einspieler and Marchik (2019) and Banerjee et al. (2019)] for comprehensive descriptions and review of typical and atypical RTT clinical features. While females born with a de novo (from mother’s or father’s germ cells) or inherited (from a carrier mother) MECP2 RTT mutation may have a wide spectrum of severity, males with the same MECP2 mutations typically have much severer consequences, with a more rapid progression of symptoms and lower average age of death (Neul et al., 2019). However, these may be ameliorated in the presence of an additional X chromosome (Klinefelter’s syndrome), or where the mutation is a somatic mosaic rather than germline. Also, there are reports of males with MECP2 mutations that are not known pathogenic RTT mutations who are affected, but not with classical RTT (Neul et al., 2019), and some where the clinical presentation includes psychiatric disorders such as schizophrenia (Cohen et al., 2002; Villard, 2007; McCarthy et al., 2014; Curie et al., 2017; Sheikh et al., 2018), bipolar disorder (Sheikh et al., 2016), and Asperger’s (Curie et al., 2017).
MeCP2 Mutations and the High Complexity of MeCP2 Stability
Rett syndrome can arise from a number of missense, nonsense, frame shift, splice site, and start codon mutations as well as larger deletions that can lead to a range of phenotypes with varying degrees of severity (Chahrour and Zoghbi, 2007). From the protein structural point of view (Figure 1B), MeCP2 mutations can be grouped into three main broad categories. The first corresponds to mutations that affect the NTD, a second, and corresponding to a very significant group of RTT phenotypes, are those that affect the MBD, and a third, those affecting the rest of the molecule. This classification is not arbitrary as the NTD has been shown to modulate the ability of MeCP2 (through the MBD) to interact with DNA (Martínez de Paz et al., 2019) as well as to influence the turn-over rate of the protein (Sheikh et al., 2017; Martínez de Paz et al., 2019), and hence mutations within this region can affect these parameters. With the MBD being the only structurally ordered portion of MeCP2, mutations within the MBD can affect the tertiary structure (folding) (Figure 2B; Kucukkal et al., 2015) of this region and hence its binding affinity (Yang et al., 2016). Many of the remaining mutations are located in the C-terminal domain (Moncla et al., 2002; Bebbington et al., 2010) and can affect the interactions of MeCP2 with many of its diverse interaction partners (Lyst et al., 2013), including the chromatin (Nikitina et al., 2007a) itself and RNA (see following section).
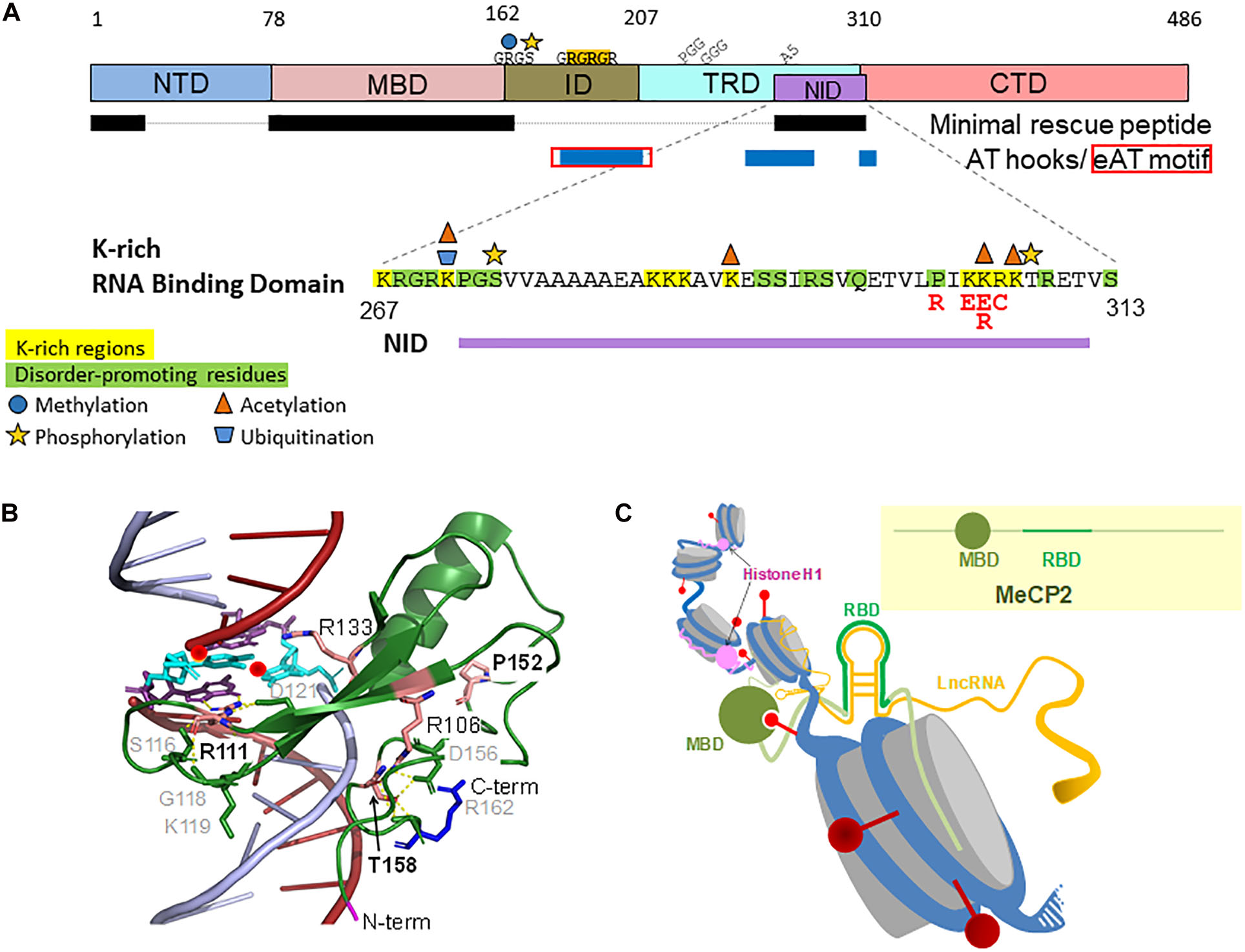
Figure 2. Placement and amino acid sequence and PTMs of the RNA Binding Domain (RBD) within the MeCP2-E2 domains. The main missense RTT mutations are in red (A). Detail of the DNA-bound MBD three dimensional structure (Ho et al., 2008) (Protein Data Bank PDB ID 5BT2) (Berman et al., 2000) (B). Cartoon representation of RNA-bound MeCP2 within a chromatin context (C). Red spheres, DNA methylation; Gray cylinders, nucleosomes.
The first attempts to study the functional correlation between RTT MeCP2 mutations and the impairment to DNA methyl binding and transcriptional regulation activities were carried out in the late Alan Wolffe’s lab (Yusufzai and Wolffe, 2000) soon after the discovery of their involvement in this disease (Amir et al., 1999). These initial results were subsequently followed by a detailed characterization of the binding affinity alterations caused by several missense mutations within MBD (Ballestar et al., 2000) and set the framework for the type of work which will be described in the following sections.
MeCP2 N-Terminal Mutations
Although not as frequent as the mutations affecting the MBD or TRD at their C-termini (Shah and Bird, 2017; Spiga et al., 2019), several NTD mutations have been described to date and are more often than not associated with a typical RTT phenotype (Saunders et al., 2009). However, as with mutations elsewhere in MeCP2, other factors, skewing of X-inactivation, for example, play a role. Without the ability to compare clinical severity across a larger number of RTT girls with the same N-terminal mutation, and without identification of males with these mutations, it is difficult to draw any firm conclusions. However, a study of clinical severity, albeit with only five cases with N-terminal mutations, of which four displayed typical RTT, suggests on average lower clinical severity in comparison to the common nonsense mutations and missense mutations such as T158M (Cuddapah et al., 2014). It is important to note that the two isoforms differ at the N-terminal sequences, with the MeCP2-E2 N-terminus encoded by exon 2, and the slightly longer MeCP2-E1 N-terminus coming from exon 1 (Figure 1). It is also worth emphasizing that, until recently, no unequivocal RTT mutations have been reported within exon 2 (or indeed for any clinical entity). Very recently, however, there is a report of a NM_004992:c.7G > C; p.Ala3Pro Rett mutation in exon 2 (Wen et al., 2020). Given its rarity, it will be important to assess the molecular effects of this variant to confirm its true pathogenicity.
The MeCP2-E1 N-terminus contains polyGGC and polyGGA stretches that encode stretches of alanine and glycine residues, respectively. In-frame insertions and deletions within the polyalanine and polyglycine regions of exon 1 (Figure 1B) have been identified. Although initially they were suggested to be a relatively frequent cause of intellectual disability or developmental delay (Harvey et al., 2007), with the current availability of exome sequence data from large control populations1 it is likely that these in-frame indels are unrelated to disease.
Although genuine disease-causing mutations within exon 1 would appear to affect the MeCP2-E1 isoform exclusively, this is not necessarily the case. The first mutation reported in exon 1, an 11 bp frameshifting deletion, was described when the MeCP2-E1 isoform itself was first reported (Mnatzakanian et al., 2004). Since then, the same mutations have been reported in multiple studies (Saunders et al., 2009), and in one study it was shown that, while there was no disruption of transcription of the MeCP2-E2 mRNA, there was interference with, and reduction of translation of MeCP2-E2 protein (Saxena et al., 2006), leading to the possibility that mutations within the MeCP2-E1 N-terminus affect both major isoforms.
However, subsequently, several classic Rett patients were identified with mutations affecting the start codon in exon 1 (Gauthier et al., 2005; Saunders et al., 2009). Levels of mRNA for MeCP2-E1 and E2 were unaffected, and peripheral blood lymphocytes were still positive for MeCP2 antibodies, and thus presumably only able to generate the MeCP2-E2 protein (Gianakopoulos et al., 2012). Additionally, a genuine RTT missense mutation (A2V) within the same N-terminal region of MeCP2-E1 was reported in two patients (Fichou et al., 2009; Saunders et al., 2009). The mutation resulted in an RTT phenotype characterized by severe epilepsy, cognitive impairment and developmental delay, in addition to microcephaly and no language in one patient (Fichou et al., 2009), and is described as classic Rett syndrome in the other (Saunders et al., 2009). Importantly, neither the transcriptional nor the translational properties of MeCP2-E2 were affected as observed in fibroblasts or lymphocytes obtained from the patients (Fichou et al., 2009; Gianakopoulos et al., 2012). An apparent synonymous or silent mutation in exon 1, p.Gly16Gly, that was shown to trigger usage of a cryptic splice donor resulting in a frameshift and premature truncation for the MeCP2-E1 isoform but with no predicted effect on the MeCP2-E2 isoform has also been documented (Sheikh et al., 2013). A study on the cellular and molecular effects caused by the A2V mutation showed that, while it neither impacted the localization of the MeCP2-E1 isoform nor its co-localization with chromatin, it affected the N-terminal co- and post-translational modifications that regulate the physiological turnover of the protein. Complete N-methionine excision (NME) and evidence of excision of multiple alanine residues from the N-terminal poly-alanine stretch of wild type (WT) MeCP2-E1 was observed, whereas the A2V mutant exhibited only partial NME of either methionine or valine and reduced N-acetylation (NA). This resulted in different in vitro protein degradation rates between the WT and the mutant. Indeed, a higher proteasomal degradation activity was observed for MeCP2-E1-A2V compared with that of WT MeCP2-E1 (Sheikh et al., 2017). Hence, the etiopathology of this mutation is likely due to a reduced bio-availability of MeCP2 resulting from the defective co-post-translational N-terminal modifications that lead to a faster degradation of the A2V mutant (Sheikh et al., 2017).
Apart from A2V, there are no other published reports of exon 1 missense mutations. In fact, there are remarkably few examples of missense mutations within the N-terminal region leading up to the MBD. One of these few rarities is A59P. The A59P mutation was described in three Tunisian RTT patients with variable scores of clinical severity (Kharrat et al., 2015). Despite the intrinsically disordered organization of the MeCP2 NTD, such an amino acid change was predicted to have an important structural effect on the overall conformation of the protein backbone. However, the structural role and protein stability properties affected remain to be determined. This also applies to all the other NTD mutations described above, with the exception of A2V, and hints to the complexity of the molecular mechanisms probably involved.
MeCP2 Missense Mutations
Missense mutations represent the most abundant mutations in RTT [over 70% (Spiga et al., 2019)] and mainly affect the MBD (residues 78 to 162, Figure 1B) where they make up to approximately 45% of the cases (Ghosh et al., 2008), underscoring the primary role of the MBD in the function of this protein. This domain corresponds to the main structured part within this intrinsically disordered protein (Dunker et al., 2001; Ausió et al., 2014), and is the only region that has been amenable to crystallization (Ho et al., 2008). The MBD crystal structure has provided an excellent resource for the analysis of the structural alterations caused by mutations within this region.
Because of their high occurrence, these mutations have been studied extensively using in vitro structural, in situ cell culture and in vivo mouse model approaches (Tillotson and Bird, 2019). Within the first category, we have already referred to the early pioneering work on R106W, R133C, F155S, and T158M by Ballestar et al. (2000). This structural work was followed by a study of R106W, R111G, R133C, F155S, and T158M, which, in the absence of crystallographic MBD information (Ho et al., 2008), used NMR and provided a more detailed molecular characterization of the structural changes resulting from these mutants (Free et al., 2001). It was noticed that the R133C mutation affected DNA binding without changing the MBD structure, thus highlighting, for the first time, the relevance for proper DNA binding of the basic amino acids at the MBD-DNA interface (Spiga et al., 2019). These studies were ensued by a later characterization of the same mutants using a combination of biophysical techniques that included fluorescence spectroscopy and circular dichroism. They allowed the authors to correlate the magnitude of the structural changes elicited by each mutant to the severity of the associated RTT phenotypes (Ghosh et al., 2008). Due to the complexity of the structural work, initial studies focused on some of the most prevalent RTT mutations and only more recently have been extended to other mutants such as the Y120D and to their binding to mCH (where H = A, T, or C) (Sperlazza et al., 2017), and in particular to mCA (Gabel et al., 2015). Although no structural differences were observed between the binding of the MeCP2 mutants to mCG versus mCA (Sperlazza et al., 2017), this study was very relevant as, immediately after birth, during neuron differentiation, mCH (Lister et al., 2013) and MeCP2 (Kaufmann et al., 2005b; Olson et al., 2014) concomitantly increase during a changing methylation landscape that may account for the onset of RTT (Lavery and Zoghbi, 2019). From the structural point of view, the different missense MBD-RTT associated mutations can change either the stability of the MBD, its DNA binding affinity, or both to a different extent. In this regard, a couple of recent exhaustive structural studies summarize this in a way that clusters these mutations into three different groups (Kucukkal et al., 2015; Yang et al., 2016) (see Table 1 and Figure 3).

Table 1. Classification of MeCP2 MBD missense mutations according to their structural characteristics (Yang et al., 2016).
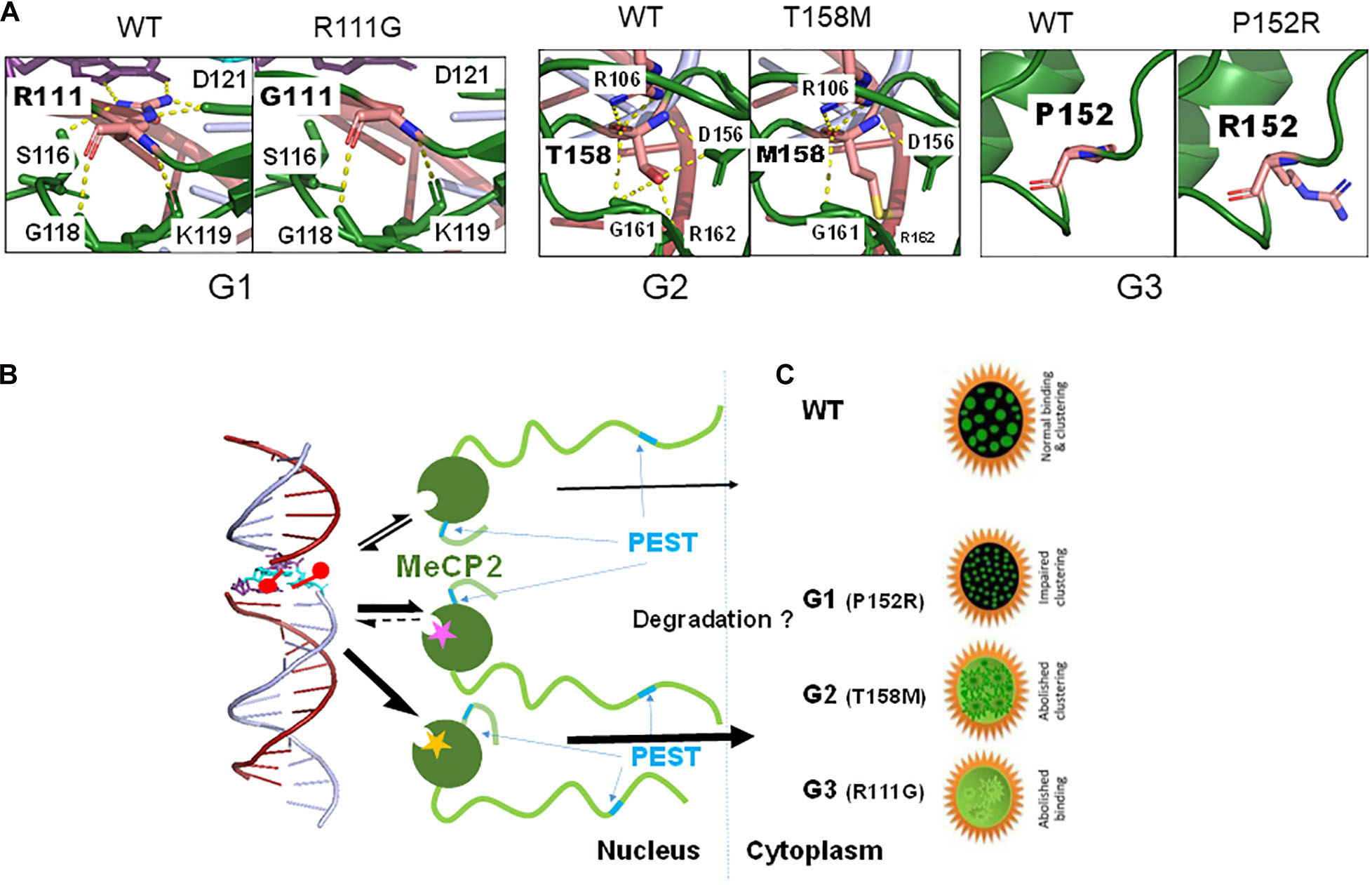
Figure 3. Effect of a few MBD missense mutations [groups G1 to G3 (Yang et al., 2016)] on MeCP2 structure (stability) (A), binding affinity (magenta star: G1; yellow star: G3) (B) and nuclear distribution (C).
The information from the structural studies has been complemented by in situ experiments in different cell culture settings. A few representative papers covering a wide spectrum of mutations have been published by Kudo et al. (2003), and by Agarwal et al. (2007). In addition to stability and affinity of binding, these studies have also focused on the clustering ability of MeCP2 around the pericentromeric heterochromatin. Importantly, the work has also provided insight to the altered distribution within the nuclear/cytoplasmic compartments caused by the MeCP2 mutations (Figure 3C). As with the in vitro structural work, an attempt has been made to correlate the observations made with clinical severity (Sheikh et al., 2016; Figure 3C). Interestingly, while some of the MeCP2 protein mutants resulting from MBD mutations (i.e., R133C and A140V) are still able to bind to chromatin, their interaction with ATRX is fully compromised (Nan et al., 2007). ATRX is a protein member of the ATP-dependent SWI/SNF family of chromatin remodeling complexes (Pazin and Kadonaga, 1997). This additional disruption of a functionally relevant protein-protein interaction underscores the molecular mechanistic complexity of some of these mutations.
In more recent years, a few knock-in mice models have been produced for the Y120D, R133C and T158M/A mutations as well as two transgenic models for the R111G and R306C mutations (Heckman et al., 2014). These models have provided useful information from an in vivo perspective. This work has revealed that, in instances such as T158M, where the mutation significantly decreases the amount of MeCP2 in the nucleus, the RTT phenotype can be rescued by increasing the expression of the T158M mutant (Lamonica et al., 2017). Moreover, the decrease in the mutated MeCP2 was shown to be due to proteasomal degradation (Lamonica et al., 2017). This decrease is reminiscent of that observed for the truncated form of MeCP2 expressed in the Jaenisch (Mecp2 TM 1.1Jae/Mmcd) mouse model (Stuss et al., 2013), in which exon 3 of MeCP2 is deleted (Figure 1A) such that most the MBD is lacking (Chen et al., 2001).
MeCP2 Nonsense and C-Terminal Mutations
In this section, we include all the mutants affecting MeCP2 beyond its MBD. These include mutations affecting the ID, TRD, and CTD domains that encompass the NID and RNA binding domain (RBD) (Figure 3A). The C-terminal region defined in this way, is also where MeCP2 mutations pertaining to other brain disorders such as schizophrenia, which involves the ID, (Sheikh et al., 2018; Chen et al., 2020) take place. Many of the RTT nonsense mutations occur within this region and its most significant missense mutations take place in the TRD (Figure 3A). From a structural perspective, mutations within this region have been less extensively studied, in contrast to those of the MBD. This is particularly true as it pertains to the nonsense and frameshift mutations leading to early termination. In this regard, a hint into some of their potential molecular effects can be envisaged from the early structural work carried out in the late Alan Wolffe’s lab (Chandler et al., 1999; Yusufzai and Wolffe, 2000) which used C-terminally truncated versions of MeCP2 to show that the CTD facilitates its binding to nucleosomes (Chandler et al., 1999) and hence to chromatin. A more detailed follow up, carried out by Nikitina et al., highlighted the role of residues 295–486 for chromatin interaction (Nikitina et al., 2007b), and showed that R294X failed to produce the nucleosome–nucleosome interactions (Nikitina et al., 2007a) that are observed with the native form of the protein. Disruption of the inter-nucleosome interactions may play an important role in the intrinsic ability of MeCP2 to organize chromatin into chromocenters (Brero et al., 2005; Agarwal et al., 2011; Ausio, 2016; Wang et al., 2020). The RTT nonsense mutations, R168X, R255X, R270X, and R294X, which together account for about 90% of all nonsense mutations (Krishnaraj et al., 2017) were recently shown to disrupt the ability of MeCP2 to cluster heterochromatin (Li et al., 2020) in a way that progressively decreased with the proximity of the MeCP2 truncation to the C-terminus (Wang et al., 2020). These results agree with the clinical observations which indicate that severity of the C-terminal truncations decreases with its proximity to the carboxy-terminus of MeCP2, with individuals with the R168X mutation being more severely affected than those with R294X (a mild RTT mutation) or other more C-terminal mutations (Neul et al., 2008; Bebbington et al., 2010; Cuddapah et al., 2014). They also agree well with the recently described critical role of the unstructured MeCP2 CTD in conjunction with the MBD to form heterochromatin condensates (Li et al., 2020). Of note, the R255X and R270X mutations fall within the NLS region, however, the molecular relevance of this is not clear, especially since NLS inactivation does not affect the progression of the disease in a RTT mouse model (Lyst et al., 2018).
Given the confounding effects resulting from the mosaic expression of MeCP2 in females (XX), boys (XY) with RTT allow for a better correlation between mutation and phenotype severity. In this regard, boys with truncation or frameshift mutations before or including residue R270 exhibit neonatal encephalopathy and death, whereas males with the same type of mutations beyond G273 survive. Using R270X and G273X mouse models, the breaking point was shown to be due to the disruption of an AT-hook 2 HMGA1 (high mobility group)-like domain in MeCP2 that was found to be critical for chromatin maintenance and α-thalassemia mental retardation X-linked protein (ATRX) localization in the nervous system (Baker et al., 2013), underscoring again the multifaceted role of MeCP2 in chromatin organization. As with the chromatin architectural HMGA1 non-histone protein (Reeves, 2001), MeCP2 contains three homologous AT hook domains (Ausió et al., 2014) that provide the molecule with DNA binding properties involved in chromatin clustering and heterochromatin organization. Interestingly, HMGAs play an important role in the regulation of the neurogenic potential of neuron precursor cells, and their expression is lost during neuron differentiation (Tyssowski et al., 2014).
In addition to all the above, the possibility exists that the deleterious consequences of the nonsense mutations giving rise to the truncated forms of MeCP2 may also be partially indirect in nature. Indeed, analyses of the histone PTMs in lymphocytes from RTT patients showed a decrease in the levels of acetylation of lysines 9 and 14 of histone H3 (Kaufmann et al., 2005a). These analyses are interesting, and add to the promise of biomarker discovery in RTT patient lymphocytes, but unfortunately they do not provide insight into the potential molecular mechanisms involved. In experiments carried out on clonal cell cultures from an RTT female with the R168X mutant and cells from a male hemizygote for the frameshift mutation 803delG (V288X), both sets of mutant cells exhibited histone H4 hyperacetylation specifically associated with increased acetylation of lysine 16 (H4K16ac) (Wan et al., 2001). In a different study using a mouse model of Rett syndrome expressing MeCP2 truncated by introducing a stop codon after codon 308 (Mecp2308/y), a 2–3 fold increase in histone H3 acetylation was observed in cortex (Shahbazian M. et al., 2002). The changes in global acetylation observed in these studies might have important alterations in gene expression and in both instances had been attributed to the inability of these truncated versions of the expressed protein to recruit the MeCP2-associated histone deacetylase (HDAC) complexes (Nan et al., 1998; Jones et al., 2001). However, the relation of this acetylation to the MeCP2-dependent HDAC recruitment is surprising, as the null mouse model lacking the expression of MeCP2 does not exhibit any differences in histone H3 or H4 acetylation (Urdinguio et al., 2007). A more plausible explanation would be that MeCP2 might have a developmental-dependent downstream effect on gene expression (Thatcher and Lasalle, 2006) which is altered in different ways in the presence of different truncated MeCP2 forms and in different tissues.
The most important C-terminal missense mutations (in terms of frequency): P302R, K304E, K305R, and R306C occur in the TRD within the NID (Figure 3A). In this regard, R306C represents one of the most frequent mutations observed in RTT with 245 (5.1%) RTT cases reported (Krishnaraj et al., 2017). R306C suppresses MeCP2 binding to the nuclear receptor co-repressor (NCoR)-mediated recruitment of HDACs, which is also severely compromised by any of these four RTT mutations (Kruusvee et al., 2017). Yet, despite the functional relevance to RTT attributed to the MeCP2 NID (Tillotson et al., 2017), and despite the main functional role of MeCP2 in the brain being to recruit the NCoR1/2 co-repressor complex to methylated DNA sites in this tissue (Tillotson and Bird, 2019), mutations within this region correspond to some of the clinically milder RTT forms reported (Schanen et al., 2004; Cuddapah et al., 2014; Neul et al., 2014). Also, the genomic sites to which the NCoR1/2 complex is recruited (Connolly and Zhou, 2019) and their relative abundance are still unknown.
Other molecular implications for the mutations within the MeCP2 C-terminus could arise from the fact that the CTD had been shown early on to contain a WW domain binding region (WDR) encompassing amino acids 325–498. This region is responsible for the interaction of MeCP2 with group II WW-containing domains in splicing factors FBP11 and HYPC (Buschdorf and Stratling, 2004). Several missense mutations and small C-terminal INDELs causing truncations occur within this region (Kyle et al., 2018; Lavery and Zoghbi, 2019; Spiga et al., 2019), the most prominent being E395K (Krishnaraj et al., 2017). However, structural information for any of these is still lacking.
Proteasomal Degradation and the PEST Sequences
As has already been mentioned above for various MeCP2 mutations, several of the anomalous cellular levels and pathological aspects involved arise from alterations in the proteasomal degradation processing of these mutants (Lamonica et al., 2017; Sheikh et al., 2017). Whether the proteasome activity takes place in the nucleus or in the cytoplasm is not yet clear and it may be dependent on the type of mutation. While both the N terminus-dependent pathway and the lysine-dependent (PEST) degradation pathways can occur in the nucleus, the latter occurs more actively in the cytoplasm (Lingbeck et al., 2003). Hence, the cellular localization of the MeCP2 protein molecules to be degraded might be mutation dependent. Indeed, the MBD by itself is important for nuclear localization (Lyst et al., 2018) and hence MeCP2 partitioning within the cell might be affected by impairment of its ability to bind to its methylated DNA target.
Regardless of the cellular compartment where the degradation of MeCP2 mutants takes place, the ubiquitin proteasome system (UPS) plays a very important role in neurological diseases, including cognitive disorders like RTT (Lehman, 2009). Mesenchymal stromal cells from a heterozygous RTT female mouse model null for MeCP2 (Mecp2tm1.1Bird), which mimics partial MeCP2 loss of function, have been shown to exhibit increased proteasome activity (Squillaro et al., 2019). Similar observations associated with changes in cellular ubiquitination have been described for peripheral blood lymphomonocytes from RTT patients (Pecorelli et al., 2013).
In view of all of this, we propose here a mechanism for MeCP2 degradation of missense mutations that relies on the presence of two PEST domains in MeCP2 (Thambirajah et al., 2009; Figure 3B). These domains consist of consensus sequences enriched in proline, glutamate, serine, and threonine (PEST) residues, which act as a recognition signal for rapid degradation by the 26S UPS (Rogers et al., 1986; Rechsteiner and Rogers, 1996). The two PEST domains of MeCP2 are N-terminally and C-terminally located at amino acid residues 73–94 and 389–426, respectively (Thambirajah et al., 2009). However, whether this mechanism would apply to all the RTT mutations it is not clear and, as in the case of the N-terminal mutations, additional mechanisms may also apply (Sheikh et al., 2017). While limited detailed information is available for some of the N-terminal (Sheikh et al., 2017) and MBD missense (Lamonica et al., 2017) mutations, information in this regard on mutations occurring at other MeCP2 domain locations is significantly lacking, and information on stability and binding affinity it is only known in a few instances. Such is the case of the reduced DNA binding affinity of the R306C mutation described in the previous section, which affects the TRD/NID (Figure 3A). Using a mouse model for this mutation, Heckman et al. have conclusively shown that, beyond the impairment of binding the repressive NCoR complex (Kruusvee et al., 2017), alteration of the basic cluster of basic amino acids (304–309) within the RNA binding domain (Figure 3A) by R306C lowers the binding affinity of the mutant protein by MeCP2 binding sequences in vivo (Heckman et al., 2014). Importantly, it might also disrupt the interaction of the protein with RNA as will be described next.
MeCP2 as an RNA Interacting Protein
MeCP2 RNA Binding Domain(s)
Direct interaction between MeCP2 and RNA was first shown by in vitro electrophoretic mobility assays (Jeffery and Nakielny, 2004). MeCP2 shifted mouse immunoglobulin mRNA and Xenopus U1 spliceosomal small nuclear (sn)RNA, but not human tRNA or Xenopus 5 S rRNA, suggesting that RNA binding is not promiscuous. Removal of an RG repeat motif C-terminal to the MBD abolished RNA binding (Figure 2A). Of note, double stranded (ds), but not single stranded (ss) RNA was shown to compete with methylated DNA, implying mutually exclusive binding to methylated DNA or dsRNA. Despite these promising but preliminary results, very little further research has been performed to characterize MeCP2-RNA interactions. It is becoming increasingly clear that protein interactions with both DNA and RNA are key to almost all nuclear processes, particularly because of the emerging regulatory roles played by lncRNAs (Hudson and Ortlund, 2014). RG repeat motifs are well established RNA binding modules (Thandapani et al., 2013). The binding preference for these motifs is debated, but evidence tends toward affinity for G-quadruplexes or GC-rich dsRNA, where arginine residues form hydrogen bonds with guanines (Jarvelin et al., 2016). This allows for almost transcriptome-wide binding possibilities, where specificity might be modulated by structural context and/or residues commonly occurring near RG repeats, such as the PGG, GGG, and polyalanine residues found in MeCP2 (Figure 2A; Chong et al., 2018). MeCP2 has a relatively short RG repeat, which is generally associated with low RNA binding affinity. An AT-hook domain overlaps the RG repeat, which was later characterized as a non-canonical extended AT-hook (eAT-hook) (Figure 2A; Filarsky et al., 2015). eAT-hook proteins have an order of magnitude greater affinity for dsRNA than DNA, and long stem-loop RNA structures are preferred over short hairpins (Figure 2C).
Advanced proteome-wide screens of RNA binding proteins (RBPs) have identified MeCP2 as an important RBP (He et al., 2016; Trendel et al., 2019). Recent comprehensive in vivo capture of RBPs identified a lysine-rich non-canonical RNA binding motif within the TRD of MeCP2 (Castello et al., 2016). RNA binding domains (RBDs) that lack sequence homology to known RBDs are being found with increasing frequency, and are presently characterized by basic (R and K) and disorder-promoting (R, G, P, S, and Q in MeCP2) residues. K-rich regions in DNA binding domains are thought to allow “hopping” or “sliding” to specific sequences; however, K-rich RNA binding domains characterized to date imply RNA structure over sequence-specific binding (Wilson et al., 1998; Takeuchi et al., 2009; Castello et al., 2016; Figure 2C). Most non-canonical RBDs are also enriched in having DNA and protein interaction surfaces, suggesting competition between RNA binding and other molecular interactions at these regions. This agrees with the K-rich RBD of MeCP2 overlapping its NID, a region within which RTT-causing missense mutations are also enriched (Figure 2A). RTT-like phenotype rescue in knock-in Mecp2-null mice expressing a minimal MeCP2 protein lacking the N- and C-terminal regions as well as the intervening region suggest that just the MBD and NID are necessary and sufficient for MeCP2 function (Figure 2A; Tillotson et al., 2017). The CTD probably plays a minor functional role, as its absence caused mild intellectual phenotypes. The discrepancy between a minimal peptide and diverse functionality of the protein has been reconciled by concluding that the dominant role of MeCP2 is to mediate transcriptional repression via NCoR complex recruitment to methylated DNA. An alternative explanation could be that the regions containing the MBD and/or NID mediate some degree of protein multifunctionality (Ghosh et al., 2010). Surface plasmon resonance assays show that MeCP2-TBLR1 (the direct binding subunit of the NCoR complex) interaction is relatively weak (KD 9.5 ± 0.5 μM), which could be due to a lack of physiological context in vitro, or it could indicate transient binding (Kruusvee et al., 2017). Evidence for NCoR mutations specifically causing Rett syndrome is lacking (Sakaguchi et al., 2018; Zaghlula et al., 2018). Instead, they can cause intellectual disabilities, rather than RTT, per se, similar to misregulation of other MeCP2-associated processes such as mRNA splicing or miRNA biogenesis, suggesting comparable importance of MeCP2’s differing roles (Young et al., 2005; Ha and Kim, 2014). Given a growing catalog of diverse regulatory RNAs, the presence of a flexible RBD within the indispensable NID region of MeCP2 is a good candidate to explain its functional multiplicity. Moreover, combinatorial action between the K-rich RBD and the eAT-hook/RG repeat region of MeCP2 could add to the complexity.
Post-translational modifications (PTMs) are another regulatory mechanism of RBPs, some of which MeCP2 may share (Xu et al., 2019). Conservation analysis of PTMs found within RBDs showed that phosphoserine is often immediately preceded by a conserved glycine, and phosphothreonine can often be found 5 amino acids upstream from a conserved serine such as that seen on MeCP2 residues S274 and T308 (Figure 2A; Castello et al., 2016). Both PTMs have been experimentally determined (Bellini et al., 2014). Differential activity-dependent mechanisms determine phosphorylation of S274 (protein kinase A) and T308 (membrane depolarization), where phosphorylated T308 abrogates NCoR binding, potentially also representing differential activity-dependent mechanisms of RNA binding regulation (Ebert et al., 2013). MeCP2 is also ubiquitinated (K271) and acetylated (K271, K289, K305 or 307) at sites within the RBD (Gonzales et al., 2012; Pandey et al., 2015). The roles of these PTMs are unclear, but ubiquitination can influence protein conformation, and acetylated K305 could be important for protein function, as indicated by the acetyl-defective RTT mutations, K305E/R (Ausió et al., 2014). Methylation at R162 within the RG repeat motif could affect affinity for RNA, as is common for other RGG/RG proteins (Blackwell and Ceman, 2012; Guo et al., 2014). PTMs outside of RBDs can also modulate RBP function, allowing for further positive or negative regulation of RNA processing and fate (Lovci et al., 2016). This may also be true for MeCP2, similar to how activity-dependent phosphorylation outside the MBD bi-directionally regulates DNA binding (Tillotson and Bird, 2019). Altogether, the available data support MeCP2-RNA binding, but the role(s) RNA may play, and how the K-rich and/or RG repeat motifs are involved, can only be speculated as of now.
mRNA Splicing Regulation
Methyl CpG binding protein 2 has been shown to increase exon inclusion of a CD44 minigene reporter through RNA-dependent interaction with the YB-1 splice factor in HeLa and Neuro2A cells (Young et al., 2005). MeCP2 pulls-down YB-1 through TRD residues, but a C-terminal RTT-causing truncation, MeCP2-308X, binds less efficiently to YB-1, reducing exon inclusion. MeCP2 also immunoprecipitates CD44 precursor (pre-) mRNA, and MeCP2-308X abrogates this binding. Given that MeCP2’s putative RBDs do not overlap with the CTD (see above), the pre-mRNA interaction may be indirect, or direct pre-mRNA binding could require CTD-protein binding, PTMs, or structural context. Wild type and Mecp2308/Y mice have significantly altered genome-wide alternative splicing (AS), including that of Dlx5 and Cdk10 – direct targets of MeCP2-mediated repression and activation, respectively. Activity-dependent dephosphorylation at Serine pS80 enhances MeCP2-YB-1 interaction, suggesting MeCP2-dependent splicing regulation occurs in the brain (Gonzales et al., 2012).
In addition to YB-1, MeCP2 binds many splicing factors in different contexts, primarily through the CTD, and to a lesser extent the transcriptional repression domain (TRD) (Table 2; Buschdorf and Stratling, 2004; Long et al., 2011; Maxwell et al., 2013). The RNA-binding or RTT relevance of these interactions also vary, or are unknown. MeCP2 assembles with pre-mRNA processing factor 3 (Prpf3) and serologically defined colon cancer antigen gene 1 (Sdccag1) to pre- and mature mRNA of the MeCP2 gene targets Cdk10 and Frg1 (Long et al., 2011). The number of documented MeCP2 splice factor interactions continues to grow, with one article even reporting that the majority of MeCP2-bound proteins are involved in RNA splicing and processing (Cheng et al., 2017). The spliceosome is a massive macromolecular complex with many auxiliary proteins providing context-specific AS, so it is unsurprising that such variation exists and that concrete ties to RTT pathology have been difficult to make.
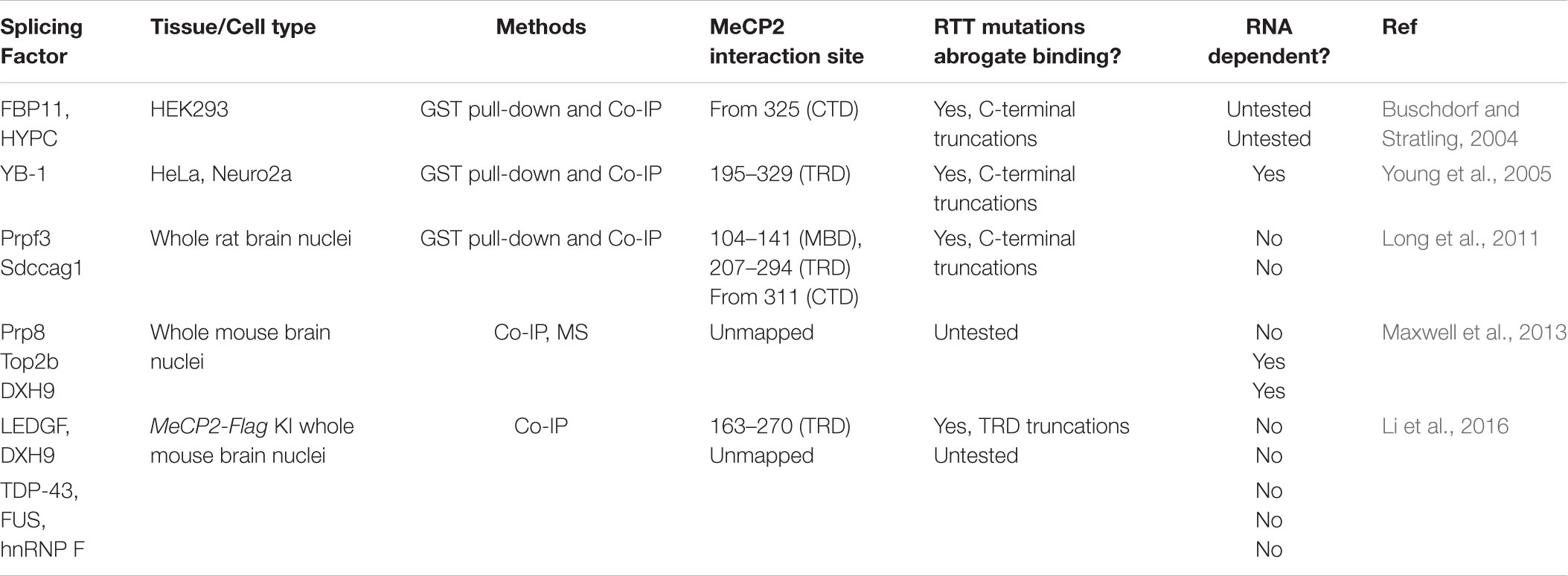
Table 2. MeCP2 interacting splicing factors.
C-terminal truncations account for ∼10% of RTT cases, yet significant functional relevance has yet to be attributed to this domain. Several truncations tested in the articles above coincide with known Rett syndrome genotypes, suggesting correlation with splice factor binding.
Most reported MeCP2-mediated AS events are cassette exon inclusion and intron exclusion (Young et al., 2005; Wong et al., 2017; Osenberg et al., 2018). Intron exclusion events were aberrant for MeCP2 target gene transcripts, Dlx5 and Cdk10, in Mecp2308/Y mice (Young et al., 2005). RNA-seq analysis in Mecp2 KO mouse cortex, however, suggests bidirectional roles in several types of AS (Li et al., 2016). In addition to protein-RNA-mediated regulation, AS is intimately tied with DNA methylation, and MeCP2 binds methylated exonic DNA, stalling RNA Polymerase II (RNAPII), thus reducing the chance of skipping alternative exons (Maunakea et al., 2013). These initial findings did not distinguish methylation (5mC) from hydroxymethylation (5hmC). However, more discriminatory experiments reveal enrichment of 5hmC at exon-intron boundaries in neurons, whereas 5mC exon-intron enrichment is prevalent in non-neuronal cells, supporting a role for 5hmC in MeCP2-mediated AS in the brain (Khare et al., 2012; Wen et al., 2014; Li et al., 2016). This agrees with MeCP2 enrichment on exon-intron gene boundaries and on 5hmC at active neuronal genes during postnatal development (Kinde et al., 2015; Li et al., 2016).
Alternative splicing is a highly conserved process allowing for greater protein and ncRNA diversity than provided by individual genes (Weyn-Vanhentenryck et al., 2018). A plethora of splice factors with different expression profiles is essential for correct AS during development. Cassette exon inclusion increases in mouse brain during development, which correlates with the increase in MeCP2 expression (Olson et al., 2014; Weyn-Vanhentenryck et al., 2018). MeCP2 interaction with the spliceosome and with pre-mRNA occur primarily through the CTD, suggesting some RTT pathologies from C-terminal truncations could, to some extent, derive from aberrant AS. Of note, during the writing of this manuscript, two research articles related to MeCP2’s role in AS were published. In the first, quantitative assessment of high-quality sequencing datasets found little variation in global AS as a result of differential MeCP2 and/or DNA methylation levels (Chhatbar et al., 2020). However, in the second article, MeCP2 was found to be required for maintaining mature hippocampal AS profiles, and to regulate splicing of specific neuronal genes in the hippocampus during memory consolidation (Brito et al., 2020). These two recent papers underscore the still long road ahead in understanding MeCP2’s role in AS, as well as the importance of careful context-specific interpretation of MeCP2 studies moving forward.
miRNA Biogenesis and Binding
MicroRNAs (miRNAs) represent an important class of ∼22 nucleotide molecules with key roles in regulating the translation of the proteome (Ha and Kim, 2014). Genome-wide miRNA expression levels are aberrant in the brains of Rett syndrome patients, and offer a potential tool to measure RTT disease progression and treatment response (Wu et al., 2010; Sheinerman et al., 2019). Processing primary (pri-) miRNA into precursor (pre-) miRNA by the nuclear microprocessor complex before export to the cytoplasm is the key regulatory step in determining mature miRNA levels in the cell (Conrad et al., 2014). The core microprocessor proteins are Drosha, which cleaves the pri-miRNA, and DiGeorge syndrome critical region 8 (DGCR8), which provides RNA binding affinity to Drosha (Ha and Kim, 2014). Additional development and cell-type specific co-factors regulate microprocessor activity. In addition to MeCP2, a major atypical RTT-pathogenic protein, FOXG1, is recruited to Drosha to influence miRNA biogenesis, implicating the importance of this process to RTT pathology (Weise et al., 2019).
At a resting state, phosphorylated MeCP2 pS80 inhibits miRNA biogenesis in cultured rat cortical neurons by binding and sequestering DGCR8; activity-dependent dephosphorylation reduces this interaction, allowing miRNA processing to proceed (Cheng et al., 2014). In Mecp2-KO mice, miR-134 increases, resulting in decreased levels of its targets involved in neuronal development and plasticity, in addition to reduced dendritic growth. Another group corroborated MeCP2 regulation of miRNA processing through microprocessor interaction, but with some key differences that require reconciliation (Tsujimura et al., 2015). Here, MeCP2 was found to positively regulate miRNA levels in neurons and neural stem cells (NSCs). The authors posited that the different state of MeCP2 phosphorylation, which varies between neuron types and brain regions, could explain the seemingly opposite observations. A global screen of significantly reduced miRNAs in Mecp2-KO neurons and NSCs identified miR-199a as important to RTT pathophysiology due to its positive regulation of mechanistic target of rapamycin (mTOR) signaling. MeCP2-mediated miR-199a biogenesis results in targeted inhibition of mTOR inhibitors SIRT1, HIF1a, and PDE4D. SIRT1 deacetylates MeCP2, adding the possibility of feedback regulation by acetylation level, in addition to phosphorylation (Zocchi and Sassone-Corsi, 2013). Furthermore, phosphorylation of DGCR8 by the mTOR-kinase ERK increases its stability (Herbert et al., 2013). RNA immunoprecipitation (RIP) shows either direct or indirect in vivo interaction of MeCP2 with pri-miR199a-1 and pri-miR199a-2. Similar to a proposed mechanism of alternative splicing (see above), MeCP2 binds to methylated miRNA gene boundaries, stalling RNAPII, and enhancing miRNA biogenesis by permitting access to processing machinery (Glaich et al., 2019). It could be that methylation level and MeCP2-DNA binding promote miRNA biogenesis, such as with miR199a, whereas unmethylated miRNA genes are subject to alternative MeCP2-mediated miRNA biogenesis suppression, like in Cheng et al. (2014). The above data offer tenuous support of direct MeCP2-RNA binding in miRNA processing regulation. It is intriguing to speculate a correlation between MeCP2 interaction with paraspeckle lncRNA, NEAT1, which can scaffold Drosha and DGCR8 to peripheral paraspeckle proteins, resulting in the regulation of miRNA biogenesis (Jiang et al., 2017). MeCP2 is known to bind the long isoform of NEAT1 in the brain (see below) (Cheng et al., 2018).
In addition to pri-miRNAs, RIP identifies 87 mature nuclear miRNAs associated with MeCP2 in primary mouse cortical cells (Khan et al., 2017). All MeCP2-interacting miRNA target gene sets are inhibited in Mecp2-null mouse cerebellum, implying an inhibitory role of MeCP2 on mature miRNAs, thus positively targeting gene expression. In addition to the canonical role of miRNAs in mRNA decay, nuclear miRNAs are associated with transcriptional repression and activation, as well as alternative splicing (Roberts, 2014).
miRNAs are essential to mammalian cell function, and their aberrant regulation as a result of MeCP2 mutations likely contributes to RTT phenotypes, but the exact interplay of molecular interactions, and whether RNA is directly involved, is complex and remains unclear.
lncRNA Interactions
Long non-coding RNAs (lncRNAs) are >200 base molecules with low coding potential, the varying species of which are involved at every processing step in the nucleus (Zhang et al., 2019). Tissue-specific expression patterns of lncRNAs during development are highly dynamic, allowing diverse outcomes, and are thus unsurprisingly aberrant in RTT (Petazzi et al., 2013; Hosseini et al., 2019). Protein-lncRNA interactions occur with all major classes of epigenetic modifying complexes (Betancur, 2016). Notably, all known RNA-binding subunits of lncRNA-interacting epigenetic complexes lack a canonical RNA binding region, and have at least some level of disorder, similar to MeCP2. Currently, there are four lncRNAs whose interaction with MeCP2 has been reported: Evf2, RNCR3, Neat1L, and HSATII, as discussed below.
During embryonic GABAergic neuron development, Evf2 recruits MeCP2 and the transcriptional activator distal-less homeobox 1 (DLX1) to the Dlx5/6 homeotic gene cluster, and inhibits DNA methylation there to facilitate antagonism between the two proteins (Berghoff et al., 2013). Evf2 deletion leads to impaired synaptic connectivity, and Mecp2-KO as well as common RTT mutations present GABAergic defects (Horike et al., 2005; Schule et al., 2007). Evf2 also recruits the SWI/SNF-like chromatin remodeling complex protein brahma-related gene 1 (BRG1) to Dlx5/6, which has overlapping protein and RNA binding motifs, similar to MeCP2 (Cajigas et al., 2015). These data led to an important speculation that global DNA binding proteins that paradoxically cause specific intellectual phenotypes when dysregulated, such as MeCP2 in RTT or BRG1 in Coffin-Siris syndrome, may be regulated by specific lncRNAs like Evf2. RNA immunoprecipitation of mouse cerebellum robustly pulled down the retinal non-coding RNA (Rncr3), followed by metastasis associated lung adenocarcinoma transcript 1 (Malat1) (Maxwell et al., 2013). Rncr3 is upregulated during retinal development, whereas its expression is reduced in Mecp2-null mice (Blackshaw et al., 2004). Rncr3–/– mice display hindlimb clasping, small brain size, and aberrant axonal sprouting, similar to RTT phenotypes, but these phenotypes are attributed to miR-124a, which is expressed from the Rncr3 gene (Sanuki et al., 2011).
Aberrant expression of pericentric HSATII RNA occurs in several cancers, and recruits polycomb group complex 1 (PRC1) as well as MeCP2 and its protein partner Sin3a into large nuclear condensates (Landers et al., 2020). These epigenetic factors are sequestered from regular function, facilitating genomic instability and cancer development. HSATII RNA is also aberrantly enriched in Parkinson’s disease patient blood samples, and MeCP2 regulates pericentric heterochromatin regions in neurons in an RNA-dependent manner, suggesting a role for HSATII RNA with MeCP2 in intellectual disorders (Billingsley et al., 2019; Marano et al., 2019). A paper published during the preparation of this manuscript showed that MeCP2 and major satellite RNA cooperate to organize pericentric heterochromatin, and that RNA interaction depends on the TRD (Fioriniello et al., 2020). This is consistent with the presence of an RBD overlapping MeCP2’s NID, as mentioned earlier.
Huntington’s disease (HD) studies found that MeCP2 binds and inhibits the long isoform of nuclear enriched abundant transcript 1 (Neat1L) lncRNA in various neuronal and brain tissue types (Cheng et al., 2018). MeCP2 inhibits NEAT1L through RNA rather than DNA interaction in wild type cells, whereas MeCP2 is reduced in HD, and increased NEAT1L levels protect against the mutant HTT gene. NEAT1L increases expression of anti-inflammatory and growth factors including peroxisome proliferator activated receptor-γ (PPARG), Nuclear Factor Kappa B Subunit 1 (NFκB1), and brain-derived neurotrophic factor (BDNF), which are also targets of MeCP2-mediated repression (Martinowich et al., 2003; Mann et al., 2010; Kishi et al., 2016). The short (3,735 nt, Neat1S) and long (22,741 nt, Neat1L) NEAT1 transcripts are ubiquitously expressed in mammalian cells, and the long isoform is required for the formation of massive ribonucleoprotein paraspeckles, which are heavily implicated in transcriptional regulation (Yamazaki et al., 2020). Neat1, Malat1, and Evf2 are upregulated upon neuronal differentiation, similar to MeCP2 (Olson et al., 2014; Roberts et al., 2014). Overall, the data point toward important corollary roles for lncRNAs and MeCP2 during brain development. Further research of their interactions with MeCP2 are integral to understanding how they may relate to RTT. Of note, RNA has been shown to promote the formation of spatial compartments in the nucleus (Quinodoz et al., 2020), and might assist MeCP2 in its formation of heterochromatin condensates (Sheikh et al., 2016; Li et al., 2020).
Histone PTMs and lncRNAs
Given that MeCP2 repression occurs primarily through HDAC recruitment, it would be expected that MeCP2 deletion invariably increases histone acetylation levels. However, as we discussed earlier (section “MeCP2 Non-sense and C-Terminal Mutations”), studies to date have been conflicting (Wan et al., 2001; Balmer et al., 2002; Kaufmann et al., 2005a; Thatcher and Lasalle, 2006; Lilja et al., 2013). As explored in the previous section, lncRNAs are a promising means to explain disparate functions of the same protein complexes. Proteome-wide analysis of RNA-dependent protein complex formation shows that the Sin3a complex, including HDACs 1 and 2, requires RNA, whereas the NCoR1 complex, including HDAC3, forms independent of RNA (Caudron-Herger et al., 2019). This is consistent with mutually exclusive MeCP2 binding to RNA or NCoR, as suggested by their overlapping binding domains (Figure 1). Moreover, Sin3a has been shown to bind lncRNAs in the brain (Dharap et al., 2013). Whether there is any specific binding and regulation of MeCP2 together with Sin3a and its associated HDACs by lncRNA, however, has yet to be determined.
In addition to methylated DNA, MeCP2 has been shown to interact with histone methylation marks associated with constitutive and facultative heterochromatin: di-methylated histone H3 lysine 9 (H3K9me2) and tri-methylated histone H3 lysine 27 (H3K27me3), respectively, in mouse brain nuclear extracts (Thambirajah et al., 2012). A recent report shows that MeCP2 preferentially binds nucleosomes with H3K27me3 via the MBD (Lee et al., 2020; Figure 2A). Also, the genomic distribution of the DNA and histone methylation marks overlap, and MeCP2 differentially regulates transcription depending on H3K27me3 and H3K9ac profiles. MeCP2 also associates with histone methyltransferases G9a, protein arginine methyltransferase 6 (PRMT6), and euchromatic histone-lysine N-methyltransferase 1 (EHMT-1) (Dhawan et al., 2011; Xue et al., 2013; Subbanna et al., 2014). MeCP2 increases H3K9 methylation in mouse fibroblasts (Fuks et al., 2003). The Neat1 lncRNA interacts with EHMT-1 at select genes in neuronal cells, and Neat1 knockdown decreases H3K9me2 and is associated with increased memory formation (Butler et al., 2019). Little is known about the relationship between MeCP2 and H3K27me3 in the context of RTT, except the previously mentioned findings in the brain, and contrasting findings by Zachariah et al. (2012) where MeCP2 overlaps more with constitutive than facultative heterochromatin marks in primary mouse cortical neurons. These contradictory findings may be explained by the different contexts, or the fact that H3K27me3 levels were compared to different normalizers. Both constitutive (H3K9me3 marked) and facultative (H3K27me3 marked) heterochromatin domains have been shown to be regulated or stabilized to some extent by lncRNA (Yang et al., 2015; Thakur et al., 2020). Despite convoluted results, MeCP2 has continued to be found associated with various histone PTMs and chromatin-modifying enzymes over the years. Whether they are relevant to altered gene expression profiles in RTT patients is still unclear, and will require scrupulous context-specific examination in the future to form conclusions. Potential regulation by previously unconsidered factors like lncRNAs adds to the complexity of the issue.
Functional Roles of MeCP2 Beyond the Brain
Because of MeCP2 multi-functionality as well as its high abundance in the brain, alterations of MeCP2 have been involved in almost every single neurodevelopmental and neurodegenerative disorder of this organ (Ausio, 2016). Nevertheless, besides the brain, the protein is quite abundant in several other tissues, for instance in the lungs (Shahbazian M.D. et al., 2002), and the implications of MeCP2 mutations for RTT within this context represent one of the less studied areas in RTT research. It is thus highly possible that several symptoms observed in RTT do not simply arise from neurological disorders, but are also caused in part by disfunctional cellular regulation (Kyle et al., 2018) in organs other than the brain. Indeed, RTT patients often develop breathing issues and one of the most abundant causes of death in RTT is related to respiratory failure (Ehrhart et al., 2016).
In what follows, we will provide a few examples where MeCP2 has been shown to have an involvement that transcends the neural system and which might be of relevance to RTT.
MeCP2 plays an important role in the modulation of the immune system by influencing the expression of the transcription factor FOXP3 (fork head box P3) – a master regulator of T-helper and T-reg cells (Li et al., 2014). Thus, MeCP2 mutations can contribute to the pathogenesis of inflammatory disease in RTT. Moreover, intestinal isolates from RTT subjects show the presence of an altered microbiota and altered production of short chain fatty acids (Strati et al., 2016), and the presence of proinflammatory strains of Candida parapsilosis (Strati et al., 2018). The alteration in the microbiota may also contribute to the gastrointestinal pathophysiology such as constipation status (Motil et al., 2012; Strati et al., 2016). MeCP2 can also contribute to the development of rheumatoid arthritis (Miao et al., 2013).
Cardiac arrhythmia is one of the factors contributing to the greater than expected occurrence of sudden death in RTT individuals (Acampa and Guideri, 2006). Although the molecular involvement of MeCP2 is not completely understood, it appears that dysregulation of Rho GTPase cytoskeletal and inflammation mediated chemokine and cytokine signaling pathway genes are involved (Wang et al., 2018).
Osteopenia is another early symptom that RTT patients are at a risk of developing, and which is dependent on the MeCP2 mutation type (Caffarelli et al., 2020). Although the use of RTT murine models suggest an epigenetic regulation of bone (O’Connor et al., 2009) which might involve RANKL (receptor activator of nuclear factor-κB ligand) (Kitazawa and Kitazawa, 2007), which negatively regulates osteoblast differentiation and bone formation in bone marrow mesenchymal stem cells (Cao, 2018). Yet, the detailed molecular mechanism(s) by which MeCP2 is involved are not understood. This example underscores how the studies on the effects of MeCP2 mutations in non-neural cell types are still in their relative infancy, as this area of MeCP2 research remains understudied.
Metabolic dysfunction also represents an important component of RTT (Kyle et al., 2018). In this regard, it was recently shown that MeCP2 plays an important role in the regulation of liver homeostasis through a molecular mechanism that involves the targeting the NCoR1/HDAC3 complex to lipogenic gene targets in hepatocytes (Kyle et al., 2016). This underscores the relevance of the MeCP2/HDAC complex outside the neuronal realm. Moreover, lipid metabolism is a more approachable therapeutic target, offering the potential to alleviate the symptoms associated with altered metabolism in RTT patients (Kyle et al., 2016).
Although some of the RTT peripheral organ-related symptoms described above might also have a neuronal component (Cronk et al., 2016), the transcriptional regulatory role of MeCP2 of specific genes within the context of the cell types of the particular organs affected indicates an important role of the MeCP2 mutations within each specific tissue. As in the case of the impaired response to stimuli and stressors observed in RTT (Pillion et al., 2003; Rose et al., 2019), such specificity might also be MeCP2 isoform-dependent, as will be discussed in the next section.
Do MeCP2-E1 and MeCP2-E2 Isoforms Play a Role in RTT?
Not only is the function of MeCP2 important in tissues other than the brain, but also within the brain its role transcends (Ausio, 2016, 2018) that of mere involvement in neurodevelopmental and neurodegenerative disorders (Tan and Zoghbi, 2019). For example, under healthy conditions, the levels of MeCP2 in mouse have been shown to change in a circadian cycle-dependent way (Martinez de Paz et al., 2015; Figure 4) – a mechanism which is likely regulated by miR-132/212 (Mendoza-Viveros et al., 2017) in response to the metabolism-dependent circadian cycle changes of the epigenome (Haws et al., 2020). This might have consequences for RTT (Tsuchiya et al., 2015). Indeed, a circadian rhythm disruption has been described in a mouse model of RTT, and disruption of the cycle was observed in fibroblasts from RTT patients (Li et al., 2015). RTT patients are known to frequently experience sleep disorders (McArthur and Budden, 1998). However, the implications of MeCP2 in the circadian cycle are undoubtedly much broader, and the system has allowed us to gain some insight into the different functionality of the E1 and E2 isoforms (Martínez de Paz et al., 2019).
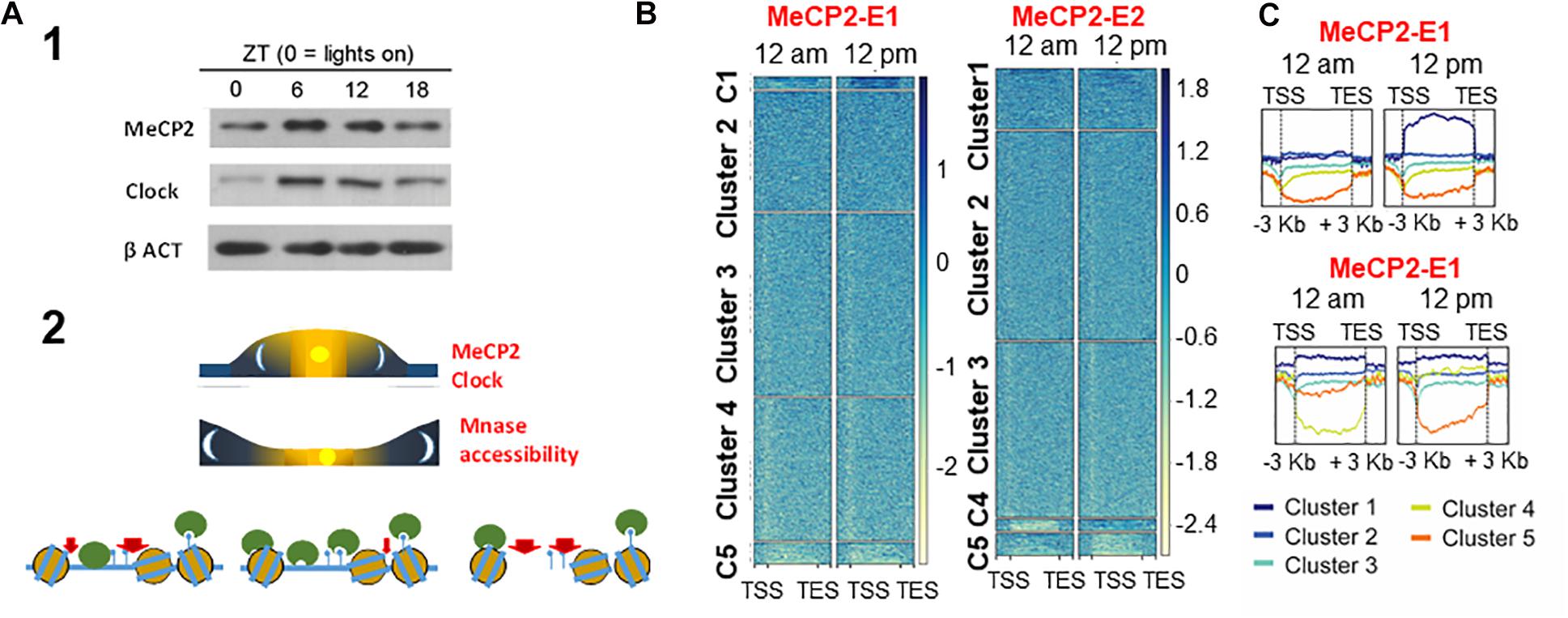
Figure 4. Circadian-dependent MeCP2 (A-1) and resulting chromatin changes (A-2) and diurnal dynamic change heatmaps for MeCP2-E1 and MecP2-E2 isoform gene occupancy (B), divided into 5 clusters, with isoform-specific enrichment on the different clustered genes over time shown in (C). MeCP2-E1 and MeCP2 enrichment differences in the different clusters in (A) (Martinez de Paz et al., 2015; Martínez de Paz et al., 2019).
Ironically, for several years most of the research on MeCP2 was carried out with the E2 isoform, which was the first to be identified (Lewis et al., 1992). However, it was not until almost 12 years later that a previously unknown MeCP2 isoform, which is much more highly expressed in human brain than its MeCP2-E2 counterpart, was discovered first in humans (Mnatzakanian et al., 2004), and then several months later in mice (Kriaucionis and Bird, 2004). The two isoforms are the product of alternative splicing. Despite an initially conflicting nomenclature, it was agreed that the longer E1 isoform corresponds to the encoded form starting at exon 1 (skipping exon 2) whereas the E2 isoform was the one encoded starting at exon 2 (Figure 1A). The physiological relevance of the two different isoforms of MeCP2 [MeCP2-E1 and MeCP2-E2 (Figure 1B)] and in particular, the relevance of the E2 form to RTT have been very controversial (Itoh et al., 2012). However, it is worth emphasizing that RTT mutations have never been identified within exon 2. In all these considerations, however, it is important to recognize that the ratio between the two isoforms and their overall abundance varies significantly from tissue to tissue (Mnatzakanian et al., 2004), and particularly in mature brain, MeCP2-E2 is present at a much lower ratio (approximately 15 fold less) than MeCP2-E1 (Martínez de Paz et al., 2019) as a result of their differential gene expression (Mnatzakanian et al., 2004).
In what follows, we will discuss the information available in support of a different specialized functional involvement of the two isoforms. As we mentioned in the previous sections, the occurrence so far of mutations in the amino acid distinctive MeCP2-E1 NTD suggests that only this isoform is relevant to RTT. However, as it was also mentioned earlier, mutations in exon 1 interfere with the translation of the MeCP2-E2 isoform (Saxena et al., 2006) and the possibility exists for other mutations along the MeCP2 protein to have a similar effect. At the gene level, the 5′ and 3′ UTRs of each isoform have been shown to be differently regulated in a cell type and development-dependent way (Liyanage et al., 2019; Rodrigues et al., 2020). At the 3′UTR, the transcripts undergo alternative polyadenylation affecting the length of these regions, ranging from 0.1 to 8.6 kb, with a preferential association of MeCP2-E1 with the longest 3′UTR (Rodrigues et al., 2020). Although how these differences regulate the expression of the isoforms is not clearly understood, they represent important targets for the binding of regulatory miRNAs and RBPs (Rodrigues et al., 2016). At the 5′ UTRs, it was reported that DNA methylation is significantly correlated with the differential expression of the two isoforms in neurons and astrocytes in a sex-dependent way, with higher levels of DNA methylation corresponding to lower levels of their expression (Liyanage et al., 2019).
As it was mentioned at the beginning of this section, recently, we took advantage of the circadian oscillation of MeCP2 to gain a functional insight on the role of the MeCP2-E1 and MeCP2-E2 isoforms. ChIP-seq analysis, taking advantage of the availability of isoform-specific antibodies, showed a differential binding site preference. MeCP2 isoform-specific enrichments were found to be mainly involved in ligand-receptor interaction in E1 and ribosomal proteins in E2 (Martínez de Paz et al., 2019). Of note, analysis of brains from RTT patients carrying MeCP2 mutations showed abnormal ribosome biogenesis (Olson et al., 2018).
At the protein level, a biophysical analysis using isothermic titration calorimetry and fluorescence spectroscopy analyses of the interaction of the E1 and E2 NTD-MBD protein region (Figure 1B) with methylated and unmethylated DNA showed a 10-fold higher affinity and higher structural stability of this region for the E2 isoform compared to the E1 counterpart. Half-life, MS, and FRAP (fluorescence recovery after photobleaching) analysis consistently reported a higher dynamic turnover of MeCP2-E1 compared to MeCP2-E2 (Sheikh et al., 2017; Martínez de Paz et al., 2019). Moreover, using isoform-specific antibodies, a proteomic analysis of the proteins interacting with the two isoforms revealed that, while both isoforms appear to be involved in similar processes, they act through different sets of protein partners. Of interest was the enriched association found between E1 and β-tubulin and microtubule-associated proteins (Martínez de Paz et al., 2019). The extent of overlap observed is, to a certain degree, unsurprising. It has been recently shown that MeCP2-E2 is able to partially compensate for the lack of the E1 isoform in a male case of RTT phenotype (Takeguchi et al., 2020). However, the association of E1 with tubulin remains intriguing. MeCP2 deficiency and mutations have been shown to affect microtubule stability (Delepine et al., 2013) and ciliogenesis (Frasca et al., 2020), respectively, through an indirect association between MeCP2 alteration and HDAC6 deacetylation of tubulin (Gold et al., 2015), though the molecular mechanisms are not yet clearly understood.
In conclusion, while the two MeCP2 isoforms may have a significant extent of generic overlapping functionality as a result of their extensive overlapping primary structure (Figure 1), they nevertheless exhibit important distinctive functional traits. Their effect(s) may depend on their different stoichiometry and overall abundance in different tissues as well as on the alteration of the mechanisms regulating their gene expression (Rodrigues et al., 2020), and their potential implications for RTT should not to be overlooked. Indeed, it has been recently shown that, in human brain, the MeCP2E1/E2-BDNF-miR132 homeostasis regulatory network is region-dependent and is altered in RTT patients (Pehjan et al., 2020).
Epigenetic Therapeutics
The area of therapeutics for RTT has become quite crowded over recent years. There are two main approaches, firstly that of addressing downstream effects of the MECP2 mutation, for instance attempting to upregulate genes under MeCP2’s control, such as BDNF or IGF1 [reviewed in Vashi and Justice (2019)], or neurotransmitter pathways such as NMDA receptors, or downstream target K+/Cl– co-transporter 2 (KCC2) (Tang et al., 2019). The second approach is to target MECP2 directly, either through gene therapy, delivering a functional version of the MECP2 gene exogenously, for example using adenoviral delivery systems [e.g., (Tillotson et al., 2017)], through correcting the mutation at the level of genomic DNA, for example using CRISPR/cas9 editing, or at the RNA level by programmable RNA editing (e.g., Sinnamon et al., 2020), or using compounds such as aminoglycosides to enable “read-through” of MECP2 nonsense mutations [e.g., (Merritt et al., 2020)].
Attempts at epigenetic therapies for RTT, also within this second category of direct targeting of MECP2, would aim at the upregulation of MECP2 expression. This in itself is a somewhat hazardous approach, as MECP2 over-expression may also have severe developmental repercussions, as witnessed in MECP2-duplication syndrome (MIM # 300260). Since RTT is almost exclusively in females, who carry one normal copy of the MECP2 gene along with a mutated copy, one approach that is being considered is reactivating the silent X [reviewed in Vashi and Justice (2019)]. Naturally occurring X-chromosomal inactivation (XCI) randomly silences one or other of the two X-chromosomes possessed by females epigenetically. This process allows dosage compensation of X-linked genes, which helps maintain the expression of most X-linked genes at a similar level to males.
Skewed XCI may favor the wild-type (WT) allele and hence expression of WT MECP2, in which cases RTT symptoms are milder, or, with extreme skewing, asymptomatic. If XCI skewing favors the mutant allele, expression of mutant MECP2 is favored, and RTT symptoms would be more severe. XCI is an epigenetic process that occurs through the expression of an X-linked non-coding RNA, Xist. One potential therapeutic strategy for RTT (and other X-linked disorders) involves reactivating the inactive X chromosome in order to increase expression of WT MECP2, which should compensate for the loss of function (and/or expression) of the mutant MECP2. The drawback here is that X reactivation could potentially increase dosage of other X-linked genes to pathogenic levels, and so the challenge is to reactivate only MECP2 or MECP2 and its immediate genomic neighbors. Studies are still at an early stage, and there have been a number of high-throughput screens to identify molecules that can reactivate MECP2 expression from the inactive X chromosome [e.g., (Minkovsky et al., 2015; Lessing et al., 2016; Sripathy et al., 2017)]. Subsequently, in one study, researchers used a small-molecule inhibitor of DNA methylation, 5-aza-2′-deoxycytidine, together with an antisense oligonucleotide knock-down of Xist RNA in vitro, that significantly upregulated MECP2 expression, and in vivo using Xist knockout mice together with the 5-aza-2′-deoxycytidine-induced inhibition of DNA methylation successfully reactivated the inactive (Carrette et al., 2018a). In heterozygous Mecp2 knockout mice with a mutation in Tsix, the antisense regulator of Xist, the phenotype observed resembled that of severely affected knockout null male mice, and demonstrated that small increases (5–10%) in WT MeCP2 protein expression can have dramatic improvements on the phenotype (Carrette et al., 2018b). The Tsix/Mecp2 mouse model generated in this study may prove to be an excellent preclinical model for evaluating the effects of XCI-based epigenetic therapeutic compounds.
Author Contributions
KG, JV, and JA contributed equally to the preparation, writing, and revision of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant of the Ontario Rett Syndrome Association (ORSA) to JV and JA, and by a grant from the Canadian Institutes of Health Research; (CIHR grant MOP-130417) to JA, CVG is the recipient of a Natural Sciences and Engineering Research Council of Canada (NSERC) CGS-M fellowship.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ATRX, α-thalassemia, mental retardation, X-linked protein; AS, alternative splicing; BDNF, brain-derived neurotrophic factor; BRG1, brahma-related gene 1; CD44, cluster of differentiation 44; Cdk10, cyclin-dependent kinase 10; ChIP, chromatin immune-precipitation; CTD, C-terminal domain; DGCR8, DiGeorge syndrome Critical Region 8; DNMT1, DNA methyltransferase 1; DLX1, distal-less homeobox 1; Dlx5, distal-less homeobox 5; eAT-hook, extended AT-hook; FBP11, formin-binding protein 11; FOXG1, fork head box G1; FOXP3, fork head box P3; FRG1, FSHD region gene 1; GABA, gamma aminobutyric acid; EHMT, euchromatic histone-lysine N-methyltransferase; ERK, extracellular signal-regulated kinase; EVF2, Rattus norvegicus non-coding RNA; HIF1a, hypoxia inducible factor 1 alpha; hmC, hydroxymethyl cytosine; HD, Huntington’s disease; HDAC, histone deacetylase; HMG, high mobility group; HSATII, human satellite II; HTT, huntingtin; HYPC, huntingtin yeast partner C; ID, intervening domain; IDP, intrinsically disordered protein; INDEL, insertions and deletion; lncRNA, long non-coding RNA; Malat1, metastasis-associated lung adenocarcinoma transcript 1; MBD, methyl biding domain; MeCP2, methyl CpG binding protein 2; mTOR, mechanistic target of rapamycin; NCoR, nuclear receptor co-repressor; ncRNA, non-coding RNA; NEAT1, nuclear enriched abundant transcript 1; NF κ B1, nuclear factor kappa B subunit 1; NID, NCoR interaction; NLS, nuclear localization sequence; NME, N-methionine excision; NTD, N-terminal domain; NSC, neural stem cell; PDE4D, cAMP-specific 3′-,5′-cyclic phosphodiesterase 4D; PEST, enriched in proline, glutamate, serine, threonine; PPARG, peroxisome proliferator activated receptor- γ; PRC1, polycomb group complex 1; PRMT6, protein arginine methyltransferase 6; PRPF3, pre-mRNA processing factor 3; PTM, post-translational modification; RANKL, receptor activator of nuclear factor-κB ligand; RBD, RNA binding domain; RBP, RNA binding protein; RIP, RNA immunoprecipitation; RNCR3, retinal non-coding RNA 3; RTT, Rett syndrome; Sin3A, switch independent 3 gene encoded protein a; SDCCAG1, serologically defined colon cancer antigen gene 1; SIRT1, Sirtuin 1; SMRT, silencing mediator of retinoic acid and thyroid hormone receptor; SWI/SNF, switch of the mating type/sucrose non-fermenting; TBLR1, transducin beta-Like 1X-Related protein 1; TRD, transcription repression; UPS, ubiquitin–proteasome system; UTR, untranslated region; WWDR, WW domain binding region; YB-1, Y box binding protein 1.
Footnotes
References
Acampa, M., and Guideri, F. (2006). Cardiac disease and Rett syndrome. Arch. Dis. Child. 91, 440–443. doi: 10.1136/adc.2005.090290
Agarwal, N., Becker, A., Jost, K. L., Haase, S., Thakur, B. K., Brero, A., et al. (2011). MeCP2 Rett mutations affect large scale chromatin organization. Hum. Mol. Genet. 20, 4187–4195. doi: 10.1093/hmg/ddr346
Agarwal, N., Hardt, T., Brero, A., Nowak, D., Rothbauer, U., Becker, A., et al. (2007). MeCP2 interacts with HP1 and modulates its heterochromatin association during myogenic differentiation. Nucleic Acids Res. 35, 5402–5408. doi: 10.1093/nar/gkm599
Amir, R. E., Van Den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23, 185–188. doi: 10.1038/13810
Ausio, J. (2016). MeCP2 and the enigmatic organization of brain chromatin. Implications for depression and cocaine addiction. Clin. Epigenetics 8:58.
Ausio, J. (2018). Role of MeCP2 in neurological disorders: current status and future perspectives. Epigenomics 10, 5–8. doi: 10.2217/epi-2017-0128
Ausió, J., De Paz, A., and Esteller, M. (2014). MeCP2: the long trip from a chromatin protein to neurological disorders. Trends Mol. Med. 20, 487–498. doi: 10.1016/j.molmed.2014.03.004
Baker, S. A., Chen, L., Wilkins, A. D., Yu, P., Lichtarge, O., and Zoghbi, H. Y. (2013). An AT-hook domain in MeCP2 determines the clinical course of Rett syndrome and related disorders. Cell 152, 984–996. doi: 10.1016/j.cell.2013.01.038
Ballestar, E., Yusufzai, T. M., and Wolffe, A. P. (2000). Effects of Rett syndrome mutations of the methyl-CpG binding domain of the transcriptional repressor MeCP2 on selectivity for association with methylated DNA. Biochemistry 39, 7100–7106. doi: 10.1021/bi0001271
Balmer, D., Arredondo, J., Samaco, R. C., and Lasalle, J. M. (2002). MECP2 mutations in Rett syndrome adversely affect lymphocyte growth, but do not affect imprinted gene expression in blood or brain. Hum. Genet. 110, 545–552. doi: 10.1007/s00439-002-0724-4
Banerjee, A., Miller, M. T., Li, K., Sur, M., and Kaufmann, W. (2019). Towards a better diagnosis and treatment of Rett syndrome: a model synaptic disorder. Brain 142, 239–248. doi: 10.1093/brain/awy323
Bannister, A. J., and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Nat. Publish. Group 21, 381–395. doi: 10.1038/cr.2011.22
Baylin, S. B., and Jones, P. A. (2016). Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 8:a019505. doi: 10.1101/cshperspect.a019505
Bebbington, A., Percy, A., Christodoulou, J., Ravine, D., Ho, G., Jacoby, P., et al. (2010). Updating the profile of C-terminal MECP2 deletions in Rett syndrome. J. Med. Genet. 47, 242–248. doi: 10.1136/jmg.2009.072553
Bellini, E., Pavesi, G., Barbiero, I., Bergo, A., Chandola, C., Nawaz, M. S., et al. (2014). MeCP2 post-translational modifications: a mechanism to control its involvement in synaptic plasticity and homeostasis? Front. Cell. Neurosci. 8:236. doi: 10.3389/fncel.2014.00236
Berghoff, E. G., Clark, M. F., Chen, S., Cajigas, I., Leib, D. E., and Kohtz, J. D. (2013). Evf2 (Dlx6as) lncRNA regulates ultraconserved enhancer methylation and the differential transcriptional control of adjacent genes. Development 140, 4407–4416. doi: 10.1242/dev.099390
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The protein data bank. Nucleic Acids Res. 28, 235–242.
Betancur, J. G. (2016). Pervasive lncRNA binding by epigenetic modifying complexes–The challenges ahead. Biochim. Biophys. Acta 1859, 93–101. doi: 10.1016/j.bbagrm.2015.10.009
Billingsley, K. J., Lattekivi, F., Planken, A., Reimann, E., Kurvits, L., Kadastik-Eerme, L., et al. (2019). Analysis of repetitive element expression in the blood and skin of patients with Parkinson’s disease identifies differential expression of satellite elements. Sci. Rep. 9:4369.
Bird, A. P. (1993). Functions for DNA methylation in vertebrates. Cold. Spring Harb. Symp. Quant. Biol. 58, 281–285. doi: 10.1101/sqb.1993.058.01.033
Blackshaw, S., Harpavat, S., Trimarchi, J., Cai, L., Huang, H., Kuo, W. P., et al. (2004). Genomic analysis of mouse retinal development. PLoS Biol. 2:E247. doi: 10.1371/journal.pbio.0020247
Blackwell, E., and Ceman, S. (2012). Arginine methylation of RNA-binding proteins regulates cell function and differentiation. Mol. Reprod. Dev. 79, 163–175.
Brero, A., Easwaran, H. P., Nowak, D., Grunewald, I., Cremer, T., Leonhardt, H., et al. (2005). Methyl CpG-binding proteins induce large-scale chromatin reorganization during terminal differentiation. J. Cell Biol. 169, 733–743. doi: 10.1083/jcb.200502062
Brito, D. V. C., Karaca, K. G., Kupke, J., Frank, L., and Oliveira, A. M. M. (2020). MeCP2 gates spatial learning-induced alternative splicing events in the mouse hippocampus. Mol. Brain 13:156.
Buschdorf, J. P., and Stratling, W. H. (2004). A WW domain binding region in methyl-CpG-binding protein MeCP2: impact on Rett syndrome. J. Mol. Med. 82, 135–143. doi: 10.1007/s00109-003-0497-9
Butler, A. A., Johnston, D. R., Kaur, S., and Lubin, F. D. (2019). Long noncoding RNA NEAT1 mediates neuronal histone methylation and age-related memory impairment. Sci. Signal. 12:eaaw9277. doi: 10.1126/scisignal.aaw9277
Caffarelli, C., Gonnelli, S., Pitinca, M. D. T., Camarri, S., Al Refaie, A., Hayek, J., et al. (2020). Methyl-CpG-binding protein 2 (MECP2) mutation type is associated with bone disease severity in Rett syndrome. BMC Med. Genet. 21:21. doi: 10.1186/s12881-020-0960-2
Cajigas, I., Leib, D. E., Cochrane, J., Luo, H., Swyter, K. R., Chen, S., et al. (2015). Evf2 lncRNA/BRG1/DLX1 interactions reveal RNA-dependent inhibition of chromatin remodeling. Development 142, 2641–2652. doi: 10.1242/dev.126318
Cao, X. (2018). RANKL-RANK signaling regulates osteoblast differentiation and bone formation. Bone Res. 6:35.
Carrette, L. L. G., Blum, R., Ma, W., Kelleher, R. J. III, and Lee, J. T. (2018a). Tsix-Mecp2 female mouse model for Rett syndrome reveals that low-level MECP2 expression extends life and improves neuromotor function. Proc. Natl. Acad. Sci. U.S.A. 115, 8185–8190. doi: 10.1073/pnas.1800931115
Carrette, L. L. G., Wang, C. Y., Wei, C., Press, W., Ma, W., and Kelleher, R. J. III et al. (2018b). A mixed modality approach towards Xi reactivation for Rett syndrome and other X-linked disorders. Proc. Natl. Acad. Sci. U.S.A. 115, E668–E675.
Castello, A., Fischer, B., Frese, C. K., Horos, R., Alleaume, A. M., Foehr, S., et al. (2016). Comprehensive identification of RNA-binding domains in human cells. Mol. Cell. 63, 696–710. doi: 10.1016/j.molcel.2016.06.029
Caudron-Herger, M., Rusin, S. F., Adamo, M. E., Seiler, J., Schmid, V. K., Barreau, E., et al. (2019). R-DeeP: proteome-wide and quantitative identification of RNA-dependent proteins by density gradient ultracentrifugation. Mol. Cell. 75, 184.e10–199.e10.
Chahrour, M., Jung, S. Y., Shaw, C., Zhou, X., Wong, S. T., Qin, J., et al. (2008). MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 320, 1224–1229. doi: 10.1126/science.1153252
Chahrour, M., and Zoghbi, H. Y. (2007). The story of Rett syndrome: from clinic to neurobiology. Neuron 56, 422–437. doi: 10.1016/j.neuron.2007.10.001
Chandler, S. P., Guschin, D., Landsberger, N., and Wolffe, A. P. (1999). The methyl-CpG binding transcriptional repressor MeCP2 stably associates with nucleosomal DNA. Biochemistry 38, 7008–7018. doi: 10.1021/bi990224y
Cheadle, J. P., Gill, H., Fleming, N., Maynard, J., Kerr, A., Leonard, H., et al. (2000). Long-read sequence analysis of the MECP2 gene in Rett syndrome patients: correlation of disease severity with mutation type and location. Hum. Mol. Genet. 9, 1119–1129. doi: 10.1093/hmg/9.7.1119
Chen, C. H., Cheng, M. C., Huang, A., Hu, T. M., Ping, L. Y., and Chang, Y. S. (2020). Detection of rare Methyl-CpG binding protein 2 gene missense mutations in patients with schizophrenia. Front. Genet. 11:476. doi: 10.3389/fgene.2020.00476
Chen, R. Z., Akbarian, S., Tudor, M., and Jaenisch, R. (2001). Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 27, 327–331. doi: 10.1038/85906
Cheng, C., Spengler, R. M., Keiser, M. S., Monteys, A. M., Rieders, J. M., Ramachandran, S., et al. (2018). The long non-coding RNA NEAT1 is elevated in polyglutamine repeat expansion diseases and protects from disease gene-dependent toxicities. Hum. Mol. Genet. 27, 4303–4314.
Cheng, T. L., Chen, J., Wan, H., Tang, B., Tian, W., Liao, L., et al. (2017). Regulation of mRNA splicing by MeCP2 via epigenetic modifications in the brain. Sci. Rep. 7:42790.
Cheng, T. L., Wang, Z., Liao, Q., Zhu, Y., Zhou, W. H., Xu, W., et al. (2014). MeCP2 suppresses nuclear microRNA processing and dendritic growth by regulating the DGCR8/Drosha complex. Dev. Cell 28, 547–560. doi: 10.1016/j.devcel.2014.01.032
Chhatbar, K., Cholewa-Waclaw, J., Shah, R., Bird, A., and Sanguinetti, G. (2020). Quantitative analysis questions the role of MeCP2 as a global regulator of alternative splicing. PLoS Genet. 16:e1009087. doi: 10.1371/journal.pgen.1009087
Chong, P. A., Vernon, R. M., and Forman-Kay, J. D. (2018). RGG/RG motif regions in RNA binding and phase separation. J. Mol. Biol. 430, 4650–4665. doi: 10.1016/j.jmb.2018.06.014
Cohen, D., Lazar, G., Couvert, P., Desportes, V., Lippe, D., Mazet, P., et al. (2002). MECP2 mutation in a boy with language disorder and schizophrenia. Am. J. Psychiatry 159, 148–149. doi: 10.1176/appi.ajp.159.1.148-a
Comings, D. E. (1986). The genetics of Rett syndrome: the consequences of a disorder where every case is a new mutation. Am. J. Med. Genet. Suppl. 1, 383–388. doi: 10.1002/ajmg.1320250540
Connolly, D. R., and Zhou, Z. (2019). Genomic insights into MeCP2 function: a role for the maintenance of chromatin architecture. Curr. Opin. Neurobiol. 59, 174–179. doi: 10.1016/j.conb.2019.07.002
Conrad, T., Marsico, A., Gehre, M., and Orom, U. A. (2014). Microprocessor activity controls differential miRNA biogenesis in vivo. Cell. Rep. 9, 542–554. doi: 10.1016/j.celrep.2014.09.007
Cronk, J. C., Derecki, N. C., Litvak, V., and Kipnis, J. (2016). Unexpected cellular players in Rett syndrome pathology. Neurobiol. Dis. 92, 64–71. doi: 10.1016/j.nbd.2015.05.005
Cuddapah, V. A., Pillai, R. B., Shekar, K. V., Lane, J. B., Motil, K. J., Skinner, S. A., et al. (2014). Methyl-CpG-binding protein 2 (MECP2) mutation type is associated with disease severity in Rett syndrome. J. Med. Genet. 51, 152–158. doi: 10.1136/jmedgenet-2013-102113
Curie, A., Lesca, G., Bussy, G., Manificat, S., Arnaud, V., Gonzalez, S., et al. (2017). Asperger syndrome and early-onset schizophrenia associated with a novel MECP2 deleterious missense variant. Psychiatr. Genet. 27, 105–109. doi: 10.1097/ypg.0000000000000165
Delepine, C., Nectoux, J., Bahi-Buisson, N., Chelly, J., and Bienvenu, T. (2013). MeCP2 deficiency is associated with impaired microtubule stability. FEBS Lett. 587, 245–253. doi: 10.1016/j.febslet.2012.11.033
Dharap, A., Pokrzywa, C., and Vemuganti, R. (2013). Increased binding of stroke-induced long non-coding RNAs to the transcriptional corepressors Sin3A and coREST. ASN Neuro 5, 283–289.
Dhawan, S., Georgia, S., Tschen, S. I., Fan, G., and Bhushan, A. (2011). Pancreatic beta cell identity is maintained by DNA methylation-mediated repression of Arx. Dev. Cell 20, 419–429. doi: 10.1016/j.devcel.2011.03.012
Driscoll, D. J., and Migeon, B. R. (1990). Sex difference in methylation of single-copy genes in human meiotic germ cells: implications for X chromosome inactivation, parental imprinting, and origin of CpG mutations. Somat. Cell. Mol. Genet. 16, 267–282. doi: 10.1007/bf01233363
Dunker, A. K., Lawson, J. D., Brown, C. J., Williams, R. M., Romero, P., Oh, J. S., et al. (2001). Intrinsically disordered protein. J. Mol. Graph. Modell. 19, 26–59.
Ebert, D. H., Gabel, H. W., Robinson, N. D., Kastan, N. R., Hu, L. S., Cohen, S., et al. (2013). Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature 499, 341–345. doi: 10.1038/nature12348
Ehrhart, F., Coort, S. L., Cirillo, E., Smeets, E., Evelo, C. T., and Curfs, L. M. (2016). Rett syndrome - biological pathways leading from MECP2 to disorder phenotypes. Orphanet. J. Rare Dis. 11:158.
Einspieler, C., and Marchik, P. B. (2019). Regression in Rett syndrome: developmental pathways to its onset. Neurosci. Biobehav. Rev. 98, 320–332. doi: 10.1016/j.neubiorev.2019.01.028
Fichou, Y., Nectoux, J., Bahi-Buisson, N., Rosas-Vargas, H., Girard, B., Chelly, J., et al. (2009). The first missense mutation causing Rett syndrome specifically affecting the MeCP2_e1 isoform. Neurogenetics 10, 127–133. doi: 10.1007/s10048-008-0161-1
Filarsky, M., Zillner, K., Araya, I., Villar-Garea, A., Merkl, R., Langst, G., et al. (2015). The extended AT-hook is a novel RNA binding motif. RNA Biol. 12, 864–876. doi: 10.1080/15476286.2015.1060394
Fioriniello, S., Csukonyi, E., Marano, D., Brancaccio, A., Madonna, M., Zarrillo, C., et al. (2020). MeCP2 and major satellite forward RNA cooperate for pericentric heterochromatin organization. Stem Cell Rep. 15, 1317–1332. doi: 10.1016/j.stemcr.2020.11.006
Frasca, A., Spiombi, E., Palmieri, M., Albizzati, E., Valente, M. M., Bergo, A., et al. (2020). MECP2 mutations affect ciliogenesis: a novel perspective for Rett syndrome and related disorders. EMBO Mol. Med. 12:e10270.
Free, A., Wakefield, R. I., Smith, B. O., Dryden, D. T., Barlow, P. N., and Bird, A. P. (2001). DNA recognition by the methyl-CpG binding domain of MeCP2. J. Biol. Chem. 276, 3353–3360.
Fuks, F., Hurd, P. J., Deplus, R., and Kouzarides, T. (2003). The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 31, 2305–2312. doi: 10.1093/nar/gkg332
Gabel, H. W., Kinde, B., Stroud, H., Gilbert, C. S., Harmin, D. A., Kastan, N. R., et al. (2015). Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 522, 89–93. doi: 10.1038/nature14319
Gauthier, J., De Amorim, G., Mnatzakanian, G. N., Saunders, C., Vincent, J. B., Toupin, S., et al. (2005). Clinical stringency greatly improves mutation detection in Rett syndrome. Can. J. Neurol. Sci. 32, 321–326. doi: 10.1017/s0317167100004200
Ghosh, R. P., Horowitz-Scherer, R. A., Nikitina, T., Gierasch, L. M., and Woodcock, C. L. (2008). Rett syndrome-causing mutations in human MeCP2 result in diverse structural changes that impact folding and DNA interactions. J. Biol. Chem. 283, 20523–20534. doi: 10.1074/jbc.m803021200
Ghosh, R. P., Nikitina, T., Horowitz-Scherer, R. A., Gierasch, L. M., Uversky, V. N., Hite, K., et al. (2010). Unique physical properties and interactions of the domains of methylated DNA binding protein 2. Biochemistry 49, 4395–4410. doi: 10.1021/bi9019753
Gianakopoulos, P. J., Zhang, Y., Pencea, N., Orlic-Milacic, M., Mittal, K., Windpassinger, C., et al. (2012). Mutations in MECP2 exon 1 in classical Rett patients disrupt MECP2_e1 transcription, but not transcription of MECP2_e2. Am. J. Med. Genet. B Neuropsychiatr. Genet. 159B, 210–216. doi: 10.1002/ajmg.b.32015
Glaich, O., Parikh, S., Bell, R. E., Mekahel, K., Donyo, M., Leader, Y., et al. (2019). DNA methylation directs microRNA biogenesis in mammalian cells. Nat Commun 10:5657.
Gold, W. A., Lacina, T. A., Cantrill, L. C., and Christodoulou, J. (2015). MeCP2 deficiency is associated with reduced levels of tubulin acetylation and can be restored using HDAC6 inhibitors. J. Mol. Med. 93, 63–72. doi: 10.1007/s00109-014-1202-x
Gonzales, M. L., Adams, S., Dunaway, K. W., and Lasalle, J. M. (2012). Phosphorylation of distinct sites in MeCP2 modifies cofactor associations and the dynamics of transcriptional regulation. Mol. Cell. Biol. 32, 2894–2903. doi: 10.1128/mcb.06728-11
Greally, J. M. (2018). A user’s guide to the ambiguous word ‘epigenetics’. Nat. Rev. Mol. Cell Biol. 19, 207–208. doi: 10.1038/nrm.2017.135
Greenberg, M. V. C., and Bourc’his, D. (2019). The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 20, 590–607. doi: 10.1038/s41580-019-0159-6
Guo, A., Gu, H., Zhou, J., Mulhern, D., Wang, Y., Lee, K. A., et al. (2014). Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol. Cell. Proteomics 13, 372–387. doi: 10.1074/mcp.o113.027870
Ha, M., and Kim, V. N. (2014). Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 15, 509–524.
Harvey, C. G., Menon, S. D., Stachowiak, B., Noor, A., Proctor, A., Mensah, A. K., et al. (2007). Sequence variants within exon 1 of MECP2 occur in females with mental retardation. Am. J. Med. Genet. B Neuropsychiatr. Genet. 144B, 355–360. doi: 10.1002/ajmg.b.30425
Haws, S. A., Leech, C. M., and Denu, J. M. (2020). Metabolism and the epigenome: a dynamic relationship. Trends Biochem. Sci. 45, 731–747. doi: 10.1016/j.tibs.2020.04.002
He, C., Sidoli, S., Warneford-Thomson, R., Tatomer, D. C., Wilusz, J. E., Garcia, B. A., et al. (2016). High-resolution mapping of RNA-binding regions in the nuclear proteome of embryonic stem cells. Mol. Cell. 64, 416–430. doi: 10.1016/j.molcel.2016.09.034
Heckman, L. D., Chahrour, M. H., and Zoghbi, H. Y. (2014). Rett-causing mutations reveal two domains critical for MeCP2 function and for toxicity in MECP2 duplication syndrome mice. eLife 3:e02676.
Herbert, K. M., Pimienta, G., Degregorio, S. J., Alexandrov, A., and Steitz, J. A. (2013). Phosphorylation of DGCR8 increases its intracellular stability and induces a progrowth miRNA profile. Cell Rep. 5, 1070–1081. doi: 10.1016/j.celrep.2013.10.017
Hite, K. C., Kalashnikova, A. A., and Hansen, J. C. (2012). Coil-to-helix transitions in intrinsically disordered methyl CpG binding protein 2 and its isolated domains. Protein Sci. 21, 531–538. doi: 10.1002/pro.2037
Ho, K. L., Mcnae, I. W., Schmiedeberg, L., Klose, R. J., Bird, A. P., and Walkinshaw, M. D. (2008). MeCP2 binding to DNA depends upon hydration at methyl-CpG. Mol. Cell. 29, 525–531. doi: 10.1016/j.molcel.2007.12.028
Horike, S., Cai, S., Miyano, M., Cheng, J. F., and Kohwi-Shigematsu, T. (2005). Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 37, 31–40. doi: 10.1038/ng1491
Hosseini, E., Bagheri-Hosseinabadi, Z., De Toma, I., Jafarisani, M., and Sadeghi, I. (2019). The importance of long non-coding RNAs in neuropsychiatric disorders. Mol. Aspects Med. 70, 127–140. doi: 10.1016/j.mam.2019.07.004
Hudson, W. H., and Ortlund, E. A. (2014). The structure, function and evolution of proteins that bind DNA and RNA. Nat. Rev. Mol. Cell Biol. 15, 749–760. doi: 10.1038/nrm3884
Itoh, M., Tahimic, C. G., Ide, S., Otsuki, A., Sasaoka, T., Noguchi, S., et al. (2012). Methyl CpG-binding protein isoform MeCP2_e2 is dispensable for Rett syndrome phenotypes but essential for embryo viability and placenta development. J. Biol. Chem. 287, 13859–13867. doi: 10.1074/jbc.m111.309864
Jarvelin, A. I., Noerenberg, M., Davis, I., and Castello, A. (2016). The new (dis)order in RNA regulation. Cell Commun. Signal. 14:9.
Jeffery, L., and Nakielny, S. (2004). Components of the DNA methylation system of chromatin control are RNA-binding proteins. J. Biol. Chem. 279, 49479–49487. doi: 10.1074/jbc.m409070200
Jiang, L., Shao, C., Wu, Q. J., Chen, G., Zhou, J., Yang, B., et al. (2017). NEAT1 scaffolds RNA-binding proteins and the Microprocessor to globally enhance pri-miRNA processing. Nat. Struct. Mol. Biol. 24, 816–824. doi: 10.1038/nsmb.3455
Jones, P. L., Wade, P. A., and Wolffe, A. P. (2001). Purification of the MeCP2/histone deacetylase complex from Xenopus laevis. Methods Mol. Biol. 181, 297–307. doi: 10.1385/1-59259-211-2:297
Kaufmann, W. E., Jarrar, M. H., Wang, J. S., Lee, Y.-J. M., Reddy, S., Bibat, G., et al. (2005a). Histone modifications in Rett syndrome lymphocytes: a preliminary evaluation. Brain Dev. 27, 331–339. doi: 10.1016/j.braindev.2004.09.005
Kaufmann, W. E., Johnston, M. V., and Blue, M. E. (2005b). MeCP2 expression and function during brain development: implications for Rett syndrome’s pathogenesis and clinical evolution. Brain Dev. 27, (Suppl. 1), S77–S87.
Khan, A. W., Ziemann, M., Rafehi, H., Maxwell, S., Ciccotosto, G. D., and El-Osta, A. (2017). MeCP2 interacts with chromosomal microRNAs in brain. Epigenetics 12, 1028–1037. doi: 10.1080/15592294.2017.1391429
Khare, T., Pai, S., Koncevicius, K., Pal, M., Kriukiene, E., Liutkeviciute, Z., et al. (2012). 5-hmC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nat. Struct. Mol. Biol. 19, 1037–1043. doi: 10.1038/nsmb.2372
Kharrat, M., Hsairi, I., Fendri-Kriaa, N., Kenoun, H., Othmen, H. B., Ben Mahmoud, A., et al. (2015). A novel mutation p.A59P in N-terminal domain of methyl-CpG-binding protein 2 confers phenotypic variability in 3 cases of tunisian rett patients: clinical evaluations and in silico investigations. J. Child Neurol. 30, 1715–1721. doi: 10.1177/0883073815578529
Kinde, B., Gabel, H. W., Gilbert, C. S., Griffith, E. C., and Greenberg, M. E. (2015). Reading the unique DNA methylation landscape of the brain: non-CpG methylation, hydroxymethylation, and MeCP2. Proc. Natl. Acad. Sci. U.S.A. 112, 6800–6806. doi: 10.1073/pnas.1411269112
Kishi, N., Macdonald, J. L., Ye, J., Molyneaux, B. J., Azim, E., and Macklis, J. D. (2016). Reduction of aberrant NF-kappaB signalling ameliorates Rett syndrome phenotypes in Mecp2-null mice. Nat. Commun. 7:10520.
Kitazawa, R., and Kitazawa, S. (2007). Methylation status of a single CpG locus 3 bases upstream of TATA-box of receptor activator of nuclear factor-kappaB ligand (RANKL) gene promoter modulates cell- and tissue-specific RANKL expression and osteoclastogenesis. Mol. Endocrinol. 21, 148–158. doi: 10.1210/me.2006-0205
Kriaucionis, S., and Bird, A. (2003). DNA methylation and Rett syndrome. Hum. Mol. Genet. 2, R221–R227.
Kriaucionis, S., and Bird, A. (2004). The major form of MeCP2 has a novel N-terminus generated by alternative splicing. Nucleic Acids Res. 32, 1818–1823. doi: 10.1093/nar/gkh349
Krishnaraj, R., Ho, G., and Christodoulou, J. (2017). RettBASE: Rett syndrome database update. Hum. Mutat. 38, 922–931. doi: 10.1002/humu.23263
Kruusvee, V., Lyst, M. J., Taylor, C., Tarnauskaite, Z., Bird, A. P., and Cook, A. G. (2017). Structure of the MeCP2-TBLR1 complex reveals a molecular basis for Rett syndrome and related disorders. Proc. Natl. Acad. Sci. U.S.A. 114, E3243–E3250.
Kucukkal, T. G., Yang, Y., Uvarov, O., Cao, W., and Alexov, E. (2015). Impact of Rett Syndrome Mutations on MeCP2 MBD Stability. Biochemistry 54, 6357–6368. doi: 10.1021/acs.biochem.5b00790
Kudo, S., Nomura, Y., Segawa, M., Fujita, N., Nakao, M., Schanen, C., et al. (2003). Heterogeneity in residual function of MeCP2 carrying missense mutations in the methyl CpG binding domain. J. Med. Genet. 40, 487–493. doi: 10.1136/jmg.40.7.487
Kyle, S. M., Saha, P. K., Brown, H. M., Chan, L. C., and Justice, M. J. (2016). MeCP2 co-ordinates liver lipid metabolism with the NCoR1/HDAC3 corepressor complex. Hum. Mol. Genet. 25, 3029–3041.
Kyle, S. M., Vashi, N., and Justice, M. J. (2018). Rett syndrome: a neurological disorder with metabolic components. Open Biol. 8:170216. doi: 10.1098/rsob.170216
Lamonica, J. M., Kwon, D. Y., Goffin, D., Fenik, P., Johnson, B. S., Cui, Y., et al. (2017). Elevating expression of MeCP2 T158M rescues DNA binding and Rett syndrome-like phenotypes. J. Clin. Invest. 127, 1889–1904. doi: 10.1172/jci90967
Landers, C. C., Rabeler, C. A., Ferrari, E. K., Alessandro, R. L. D., Kang, D. D., Malisa, J., et al. (2020). Ectopic expression of pericentric HSATII RNA results in nuclear RNA accumulation, MeCP2 recruitment and cell division deffects. bioRxiv [Preprint]. doi: 10.1101/2020.04.30.064329v1.full
Lavery, L. A., and Zoghbi, H. Y. (2019). The distinct methylation landscape of maturing neurons and its role in Rett syndrome pathogenesis. Curr. Opin. Neurobiol. 59, 180–188. doi: 10.1016/j.conb.2019.08.001
Lee, W., Kim, J., Yun, J. M., Ohn, T., and Gong, Q. (2020). MeCP2 regulates gene expression through recognition of H3K27me3. Nat. Commun. 11:3140.
Lehman, N. L. (2009). The ubiquitin proteasome system in neuropathology. Acta Neuropathol. 118, 329–347. doi: 10.1007/s00401-009-0560-x
Lessing, D., Dial, T. O., Wei, C., Payer, B., Carrette, L. L., Kesner, B., et al. (2016). A high-throughput small molecule screen identifies synergism between DNA methylation and Aurora kinase pathways for X reactivation. Proc. Natl. Acad. Sci. U.S.A. 113, 14366–14371. doi: 10.1073/pnas.1617597113
Lewis, J. D., Meehan, R. R., Henzel, W. J., Maurer-Fogy, I., Jeppesen, P., Klein, F., et al. (1992). Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 69, 905–914. doi: 10.1016/0092-8674(92)90610-o
Li, C., Jiang, S., Liu, S. Q., Lykken, E., Zhao, L. T., Sevilla, J., et al. (2014). MeCP2 enforces Foxp3 expression to promote regulatory T cells’ resilience to inflammation. Proc. Natl. Acad. Sci. U.S.A. 111, E2807–E2816.
Li, C. H., Coffey, E. L., Dall’agnese, A., Hannett, N. M., Tang, X., Henninger, J. E., et al. (2020). MeCP2 links heterochromatin condensates and neurodevelopmental disease. Nature 5867, 440–444. doi: 10.1038/s41586-020-2574-4
Li, Q., Loh, D. H., Kudo, T., Truong, D., Derakhshesh, M., Kaswan, Z. M., et al. (2015). Circadian rhythm disruption in a mouse model of Rett syndrome circadian disruption in RTT. Neurobiol. Dis. 77, 155–164. doi: 10.1016/j.nbd.2015.03.009
Li, R., Dong, Q., Yuan, X., Zeng, X., Gao, Y., Chiao, C., et al. (2016). Misregulation of alternative splicing in a mouse model of rett syndrome. PLoS Genet. 12:e1006129. doi: 10.1371/journal.pgen.1006129
Lilja, T., Wallenborg, K., Bjorkman, K., Albage, M., Eriksson, M., Lagercrantz, H., et al. (2013). Novel alterations in the epigenetic signature of MeCP2-targeted promoters in lymphocytes of Rett syndrome patients. Epigenetics 8, 246–251. doi: 10.4161/epi.23752
Lingbeck, J. M., Trausch-Azar, J. S., Ciechanover, A., and Schwartz, A. L. (2003). Determinants of nuclear and cytoplasmic ubiquitin-mediated degradation of MyoD. J. Biol. Chem. 278, 1817–1823. doi: 10.1074/jbc.m208815200
Lister, R., Mukamel, E. A., Nery, J. R., Urich, M., Puddifoot, C. A., Johnson, N. D., et al. (2013). Global epigenomic reconfiguration during mammalian brain development. Science 341:1237905. doi: 10.1126/science.1237905
Liyanage, V. R. B., Olson, C. O., Zachariah, R. M., Davie, J. R., and Rastegar, M. (2019). DNA methylation contributes to the differential expression levels of Mecp2 in male mice neurons and astrocytes. Int. J. Mol. Sci. 20:1845. doi: 10.3390/ijms20081845
Long, S. W., Ooi, J. Y., Yau, P. M., and Jones, P. L. (2011). A brain-derived MeCP2 complex supports a role for MeCP2 in RNA processing. Biosci. Rep. 31, 333–343. doi: 10.1042/bsr20100124
Lovci, M. T., Bengtson, M. H., and Massirer, K. B. (2016). “Post-translational modifications and RNA-binding proteins,” in RNA Processing. Advances in Experimental Medicine and Biology, ed. G. E. Yeo (Cham: Springer), 297–317. doi: 10.1007/978-3-319-29073-7_12
Lyst, M. J., Ekiert, R., Ebert, D. H., Merusi, C., Nowak, J., Selfridge, J., et al. (2013). Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci. 16, 898–902. doi: 10.1038/nn.3434
Lyst, M. J., Ekiert, R., Guy, J., Selfridge, J., Koerner, M. V., Merusi, C., et al. (2018). Affinity for DNA contributes to NLS independent nuclear localization of MeCP2. Cell Rep. 24, 2213–2220. doi: 10.1016/j.celrep.2018.07.099
Mann, J., Chu, D. C., Maxwell, A., Oakley, F., Zhu, N. L., Tsukamoto, H., et al. (2010). MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology 138, 705–714. doi: 10.1053/j.gastro.2009.10.002
Marano, D., Fioriniello, S., Fiorillo, F., Gibbons, R. J., D’esposito, M., and Della Ragione, F. (2019). ATRX contributes to MeCP2-mediated pericentric heterochromatin organization during neural differentiation. Int. J. Mol. Sci. 20:5371. doi: 10.3390/ijms20215371
Marmorstein, R., and Zhou, M. M. (2014). Writers and readers of histone acetylation: structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 6:a018762. doi: 10.1101/cshperspect.a018762
Martínez de Paz, A., Khajavi, L., Martin, H., Claveria-Gimeno, R., Tom Dieck, S., Cheema, M. S., et al. (2019). MeCP2-E1 isoform is a dynamically expressed, weakly DNA-bound protein with different protein and DNA interactions compared to MeCP2-E2. Epigenet. Chromatin 12:63.
Martinez de Paz, A., Vicente Sanchez-Mut, J., Samitier-Marti, M., Petazzi, P., Saez, M., Szczesna, K., et al. (2015). Circadian cycle-dependent MeCP2 and brain chromatin changes. PLoS One 10:e0123693. doi: 10.1371/journal.pone.0123693
Martinowich, K., Hattori, D., Wu, H., Fouse, S., He, F., Hu, Y., et al. (2003). DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 302, 890–893. doi: 10.1126/science.1090842
Maunakea, A. K., Chepelev, I., Cui, K., and Zhao, K. (2013). Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 23, 1256–1269. doi: 10.1038/cr.2013.110
Maxwell, S. S., Pelka, G. J., Tam, P. P., and El-Osta, A. (2013). Chromatin context and ncRNA highlight targets of MeCP2 in brain. RNA Biol. 10, 1741–1757. doi: 10.4161/rna.26921
McArthur, A. J., and Budden, S. S. (1998). Sleep dysfunction in Rett syndrome: a trial of exogenous melatonin treatment. Dev. Med. Child Neurol. 40, 186–192. doi: 10.1111/j.1469-8749.1998.tb15445.x
McCarthy, S. E., Gillis, J., Kramer, M., Lihm, J., Yoon, S., Berstein, Y., et al. (2014). De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol. Psychiatry 19, 652–658. doi: 10.1038/mp.2014.29
Mendoza-Viveros, L., Chiang, C. K., Ong, J. L. K., Hegazi, S., Cheng, A. H., Bouchard-Cannon, P., et al. (2017). miR-132/212 modulates seasonal adaptation and dendritic morphology of the central circadian clock. Cell Rep. 19, 505–520. doi: 10.1016/j.celrep.2017.03.057
Merritt, J. K., Collins, B. E., Erickson, K. R., Dong, H., and Neul, J. L. (2020). Pharmacological read-through of R294X Mecp2 in a novel mouse model of Rett syndrome. Hum. Mol. Genet. 29, 2461–2470. doi: 10.1093/hmg/ddaa102
Miao, C. G., Yang, Y. Y., He, X., and Li, J. (2013). New advances of DNA methylation and histone modifications in rheumatoid arthritis, with special emphasis on MeCP2. Cell. Signal. 25, 875–882. doi: 10.1016/j.cellsig.2012.12.017
Minkovsky, A., Sahakyan, A., Bonora, G., Damoiseaux, R., Dimitrova, E., Rubbi, L., et al. (2015). A high-throughput screen of inactive X chromosome reactivation identifies the enhancement of DNA demethylation by 5-aza-2’-dC upon inhibition of ribonucleotide reductase. Epigenet. Chromatin 8:42.
Mnatzakanian, G. N., Lohi, H., Munteanu, I., Alfred, S. E., Yamada, T., Macleod, P. J., et al. (2004). A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nat. Genet. 36, 339–341. doi: 10.1038/ng1327
Moncla, A., Kpebe, A., Missirian, C., Mancini, J., and Villard, L. (2002). Polymorphisms in the C-terminal domain of MECP2 in mentally handicapped boys: implications for genetic counselling. Eur. J. Hum. Genet. 10, 86–89. doi: 10.1038/sj.ejhg.5200761
Motil, K. J., Caeg, E., Barrish, J. O., Geerts, S., Lane, J. B., Percy, A. K., et al. (2012). Gastrointestinal and nutritional problems occur frequently throughout life in girls and women with Rett syndrome. J. Pediatr. Gastroenterol. Nutr. 55, 292–298. doi: 10.1097/mpg.0b013e31824b6159
Nan, X., Campoy, F. J., and Bird, A. (1997). MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell 88, 471–481. doi: 10.1016/s0092-8674(00)81887-5
Nan, X., Hou, J., Maclean, A., Nasir, J., Lafuente, M. J., Shu, X., et al. (2007). Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc. Natl. Acad. Sci. U.S.A. 104, 2709–2714. doi: 10.1073/pnas.0608056104
Nan, X., Ng, H. H., Johnson, C. A., Laherty, C. D., Turner, B. M., Eisenman, R. N., et al. (1998). Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393, 386–389. doi: 10.1038/30764
Neul, J. L., Benke, T. A., Marsh, E. D., Skinner, S. A., Merritt, J., Lieberman, D. N., et al. (2019). The array of clinical phenotypes of males with mutations in Methyl-CpG binding protein 2. Am. J. Med. Genet. B Neuropsychiatr. Genet. 180, 55–67.
Neul, J. L., Fang, P., Barrish, J., Lane, J., Caeg, E. B., Smith, E. O., et al. (2008). Specific mutations in methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology 70, 1313–1321. doi: 10.1212/01.wnl.0000291011.54508.aa
Neul, J. L., Lane, J. B., Lee, H. S., Geerts, S., Barrish, J. O., Annese, F., et al. (2014). Developmental delay in Rett syndrome: data from the natural history study. J. Neurodev. Disord. 6, 20. doi: 10.1186/1866-1955-6-20
Neupane, M., Clark, A. P., Landini, S., Birkbak, N. J., Eklund, A. C., Lim, E., et al. (2015). MECP2 is a frequently amplified oncogene with a novel epigenetic mechanism that mimics the role of activated RAS in malignancy. Cancer Discov. 6, 45–58. doi: 10.1158/2159-8290.cd-15-0341
Nikitina, T., Ghosh, R. P., Horowitz-Scherer, R. A., Hansen, J. C., Grigoryev, S. A., and Woodcock, C. L. (2007a). MeCP2-chromatin interactions include the formation of chromatosome-like structures and are altered in mutations causing Rett syndrome. J. Biol. Chem. 282, 28237–28245. doi: 10.1074/jbc.m704304200
Nikitina, T., Shi, X., Ghosh, R. P., Horowitz-Scherer, R. A., Hansen, J. C., and Woodcock, C. L. (2007b). Multiple modes of interaction between the methylated DNA binding protein MeCP2 and chromatin. Mol. Cell. Biol. 27, 864–877. doi: 10.1128/mcb.01593-06
O’Connor, R. D., Zayzafoon, M., Farach-Carson, M. C., and Schanen, N. C. (2009). Mecp2 deficiency decreases bone formation and reduces bone volume in a rodent model of Rett syndrome. Bone 45, 346–356. doi: 10.1016/j.bone.2009.04.251
Olson, C. O., Pejhan, S., Kroft, D., Sheikholeslami, K., Fuss, D., Buist, M., et al. (2018). MECP2 mutation interrupts nucleolin-mTOR-P70S6K signaling in rett syndrome patients. Front. Genet. 9:635. doi: 10.3389/fgene.2018.00635
Olson, C. O., Zachariah, R. M., Ezeonwuka, C. D., Liyanage, V. R., and Rastegar, M. (2014). Brain region-specific expression of MeCP2 isoforms correlates with DNA methylation within Mecp2 regulatory elements. PLoS One 9:e90645. doi: 10.1371/journal.pone.0090645
Osenberg, S., Karten, A., Sun, J., Li, J., Charkowick, S., Felice, C. A., et al. (2018). Activity-dependent aberrations in gene expression and alternative splicing in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 115, E5363–E5372.
Pandey, S., Simmons, G. E. Jr., Malyarchuk, S., Calhoun, T. N., and Pruitt, K. (2015). A novel MeCP2 acetylation site regulates interaction with ATRX and HDAC1. Genes Cancer 6, 408–421. doi: 10.18632/genesandcancer.84
Pazin, M. J., and Kadonaga, J. T. (1997). SWI2/SNF2 and related proteins: ATP-driven motors that disrupt protein-DNA interactions? Cell 88, 737–740. doi: 10.1016/s0092-8674(00)81918-2
Pecorelli, A., Leoni, G., Cervellati, F., Canali, R., Signorini, C., Leoncini, S., et al. (2013). Genes related to mitochondrial functions, protein degradation, and chromatin folding are differentially expressed in lymphomonocytes of Rett syndrome patients. Mediators Inflamm. 2013:137629.
Pehjan, S., Del Bigio, M. R., and Rastegar, M. (2020). The MeCP2E1/E2-BDNF-miR132 homeostasis regulatory network is region-dependent in the human brain and is impaired in rett syndrome patients. Front. Cell Dev. Biol. 8:763. doi: 10.3389/fcell.2020.00763
Petazzi, P., Sandoval, J., Szczesna, K., Jorge, O. C., Roa, L., Sayols, S., et al. (2013). Dysregulation of the long non-coding RNA transcriptome in a Rett syndrome mouse model. RNA Biol. 10, 1197–1203. doi: 10.4161/rna.24286
Pillion, J. P., Rawool, V. W., Bibat, G., and Naidu, S. (2003). Prevalence of hearing loss in Rett syndrome. Dev. Med. Child Neurol. 45, 338–343. doi: 10.1111/j.1469-8749.2003.tb00405.x
Quinodoz, S. A., Bhat, P., Ollikainen, N., Jachowicz, J. W., Banerjee, A. K., Peter, C., et al. (2020). RNA promotes the formation of spatial compartments in the nucleus. BioRxiv [Preprint]. doi: 10.1101/2020.1108.1125.267435
Rechsteiner, M., and Rogers, S. W. (1996). PEST sequences and regulation by proteolysis. Trends Biochem. Sci. 21, 267–271. doi: 10.1016/s0968-0004(96)10031-1
Reeves, R. (2001). Molecular biology of HMGA proteins: hubs of nuclear function. Gene 277, 63–81. doi: 10.1016/s0378-1119(01)00689-8
Roberts, T. C. (2014). The MicroRNA biology of the mammalian nucleus. Mol. Ther. Nucleic Acids 3:e188. doi: 10.1038/mtna.2014.40
Roberts, T. C., Morris, K. V., and Wood, M. J. (2014). The role of long non-coding RNAs in neurodevelopment, brain function and neurological disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130507. doi: 10.1098/rstb.2013.0507
Rodrigues, D. C., Kim, D. S., Yang, G., Zaslavsky, K., Ha, K. C., Mok, R. S., et al. (2016). MECP2 Is Post-transcriptionally Regulated during Human Neurodevelopment by Combinatorial Action of RNA-Binding Proteins and miRNAs. Cell. Rep. 17, 720–734. doi: 10.1016/j.celrep.2016.09.049
Rodrigues, D. C., Mufteev, M., and Ellis, J. (2020). Regulation, diversity and function of MECP2 Exon and 3’UTR isoforms. Hum. Mol. Genet. 29, R89–R99.
Rogers, S., Wells, R., and Rechsteiner, M. (1986). Amino acid sequences common to rapidly degraded proteins: the PEST hypothesis. Science 234, 364–368. doi: 10.1126/science.2876518
Rose, S. A., Wass, S., Jankowski, J. J., Feldman, J. F., and Djukic, A. (2019). Impaired visual search in children with rett syndrome. Pediatr. Neurol. 92, 26–31. doi: 10.1016/j.pediatrneurol.2018.10.002
Sakaguchi, Y., Uehara, T., Suzuki, H., Sakamoto, Y., Fujiwara, M., Kosaki, K., et al. (2018). Haploinsufficiency of NCOR1 associated with autism spectrum disorder, scoliosis, and abnormal palatogenesis. Am. J. Med. Genet. A 176, 2466–2469. doi: 10.1002/ajmg.a.40354
Sanuki, R., Onishi, A., Koike, C., Muramatsu, R., Watanabe, S., Muranishi, Y., et al. (2011). miR-124a is required for hippocampal axogenesis and retinal cone survival through Lhx2 suppression. Nat. Neurosci. 14, 1125–1134. doi: 10.1038/nn.2897
Saunders, C. J., Minassian, B. E., Chow, E. W., Zhao, W., and Vincent, J. B. (2009). Novel exon 1 mutations in MECP2 implicate isoform MeCP2_e1 in classical Rett syndrome. Am. J. Med. Genet. A 149A, 1019–1023. doi: 10.1002/ajmg.a.32776
Saxena, A., De Lagarde, D., Leonard, H., Williamson, S. L., Vasudevan, V., Christodoulou, J., et al. (2006). Lost in translation: translational interference from a recurrent mutation in exon 1 of MECP2. J. Med. Genet. 43, 470–477. doi: 10.1136/jmg.2005.036244
Schanen, C., Houwink, E. J., Dorrani, N., Lane, J., Everett, R., Feng, A., et al. (2004). Phenotypic manifestations of MECP2 mutations in classical and atypical Rett syndrome. Am. J. Med. Genet. A 126A, 129–140. doi: 10.1002/ajmg.a.20571
Schule, B., Li, H. H., Fisch-Kohl, C., Purmann, C., and Francke, U. (2007). DLX5 and DLX6 expression is biallelic and not modulated by MeCP2 deficiency. Am. J. Hum. Genet. 81, 492–506. doi: 10.1086/520063
Seto, E., and Yoshida, M. (2014). Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 6:a018713. doi: 10.1101/cshperspect.a018713
Shah, R. R., and Bird, A. P. (2017). MeCP2 mutations: progress towards understanding and treating Rett syndrome. Genome Med. 9:17.
Shahbazian, M., Young, J., Yuva-Paylor, L., Spencer, C., Antalffy, B., Noebels, J., et al. (2002). Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron 35, 243–254. doi: 10.1016/s0896-6273(02)00768-7
Shahbazian, M. D., Antalffy, B., Armstrong, D. L., and Zoghbi, H. Y. (2002). Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 11, 115–124. doi: 10.1093/hmg/11.2.115
Shahbazian, M. D., and Zoghbi, H. Y. (2002). Rett syndrome and MeCP2: linking epigenetics and neuronal function. Am. J. Hum. Genet. 71, 1259–1272. doi: 10.1086/345360
Sheikh, T. I., Ausio, J., Faghfoury, H., Silver, J., Lane, J. B., Eubanks, J. H., et al. (2016). From Function to Phenotype: Impaired DNA Binding and Clustering Correlates with Clinical Severity in Males with Missense Mutations in MECP2. Sci. Rep. 6:38590.
Sheikh, T. I., De Paz, A. M., Akhtar, S., Ausio, J., and Vincent, J. B. (2017). MeCP2_E1 N-terminal modifications affect its degradation rate and are disrupted by the Ala2Val Rett mutation. Hum. Mol. Genet. 26, 4132–4141. doi: 10.1093/hmg/ddx300
Sheikh, T. I., Harripaul, R., Ayub, M., and Vincent, J. B. (2018). MeCP2 AT-Hook1 mutations in patients with intellectual disability and/or schizophrenia disrupt DNA binding and chromatin compaction in vitro. Hum. Mutat. 39, 717–728. doi: 10.1002/humu.23409
Sheikh, T. I., Mittal, K., Willis, M. J., and Vincent, J. B. (2013). A synonymous change, p.Gly16Gly in MECP2 Exon 1, causes a cryptic splice event in a Rett syndrome patient. Orphanet. J. Rare Dis. 8:108. doi: 10.1186/1750-1172-8-108
Sheinerman, K., Djukic, A., Tsivinsky, V. G., and Umansky, S. R. (2019). Brain-enriched microRNAs circulating in plasma as novel biomarkers for Rett syndrome. PLoS One 14:e0218623. doi: 10.1371/journal.pone.0218623
Sinnamon, J. R., Kim, S. Y., Fisk, J. R., Song, Z., Nakai, H., Jeng, S., et al. (2020). In vivo repair of a protein underlying a neurological disorder by programmable RNA editing. Cell Rep. 32:107878. doi: 10.1016/j.celrep.2020.107878
Sperlazza, M. J., Bilinovich, S. M., Sinanan, L. M., and Javier, F. R. (2017). Structural Basis of MeCP2 Distribution on Non-CpG Methylated and Hydroxymethylated DNA. J. Mol. Biol. 429, 1581–1594. doi: 10.1016/j.jmb.2017.04.009
Spiga, O., Gardini, S., Rossi, N., Cicaloni, V., Pettini, F., Niccolai, N., et al. (2019). Structural investigation of Rett-inducing MeCP2 mutations. Genes Dis. 6, 31–34. doi: 10.1016/j.gendis.2018.09.005
Squillaro, T., Alessio, N., Capasso, S., Di Bernardo, G., Melone, M. A. B., Peluso, G., et al. (2019). Senescence phenomena and metabolic alteration in mesenchymal stromal cells from a mouse model of rett syndrome. Int. J. Mol. Sci. 20:2508. doi: 10.3390/ijms20102508
Sripathy, S., Leko, V., Adrianse, R. L., Loe, T., Foss, E. J., Dalrymple, E., et al. (2017). Screen for reactivation of MeCP2 on the inactive X chromosome identifies the BMP/TGF-beta superfamily as a regulator of XIST expression. Proc. Natl. Acad. Sci. U.S.A. 114, 1619–1624. doi: 10.1073/pnas.1621356114
Strati, F., Calabro, A., Donati, C., De Felice, C., Hayek, J., Jousson, O., et al. (2018). Intestinal Candida parapsilosis isolates from Rett syndrome subjects bear potential virulent traits and capacity to persist within the host. BMC Gastroenterol. 18:57. doi: 10.1186/s12876-018-0785-z
Strati, F., Cavalieri, D., Albanese, D., De Felice, C., Donati, C., Hayek, J., et al. (2016). Altered gut microbiota in Rett syndrome. Microbiome 4:41.
Stuss, D. P., Cheema, M., Ng, M. K., Martinez de Paz, A., Williamson, B., Missiaen, K., et al. (2013). Impaired in vivo binding of MeCP2 to chromatin in the absence of its DNA methyl-binding domain. Nucleic Acids Res. 41, 4888–4900. doi: 10.1093/nar/gkt213
Subbanna, S., Nagre, N. N., Shivakumar, M., Umapathy, N. S., Psychoyos, D., and Basavarajappa, B. S. (2014). Ethanol induced acetylation of histone at G9a exon1 and G9a-mediated histone H3 dimethylation leads to neurodegeneration in neonatal mice. Neuroscience 258, 422–432. doi: 10.1016/j.neuroscience.2013.11.043
Takeguchi, R., Takahashi, S., Kuroda, M., Tanaka, R., Suzuki, N., Tomonoh, Y., et al. (2020). MeCP2_e2 partially compensates for lack of MeCP2_e1: a male case of Rett syndrome. Mol. Genet. Genomic Med. 8:e1088.
Takeuchi, A., Schmitt, D., Chapple, C., Babaylova, E., Karpova, G., Guigo, R., et al. (2009). A short motif in Drosophila SECIS Binding Protein 2 provides differential binding affinity to SECIS RNA hairpins. Nucleic Acids Res. 37, 2126–2141. doi: 10.1093/nar/gkp078
Tan, Q., and Zoghbi, H. Y. (2019). Mouse models as a tool for discovering new neurological diseases. Neurobiol. Learn. Mem. 165:106902. doi: 10.1016/j.nlm.2018.07.006
Tang, X., Drotar, J., Li, K., Clairmont, C. D., Brumm, A. S., Sullins, A. J., et al. (2019). Pharmacological enhancement of KCC2 gene expression exerts therapeutic effects on human Rett syndrome neurons and Mecp2 mutant mice. Sci. Transl. Med. 11:eaau0164. doi: 10.1126/scitranslmed.aau0164
Thakur, J., Fang, H., Lagas, T., Disteche, C. M., and Henikoff, S. (2020). Architectural RNA isequired for heterochromatin organization. bioRxiv 3:784835.
Thambirajah, A. A., Eubanks, J. H., and Ausio, J. (2009). MeCP2 post-translational regulation through PEST domains: two novel hypotheses: potential relevance and implications for Rett syndrome. Bioessays 31, 561–569. doi: 10.1002/bies.200800220
Thambirajah, A. A., Ng, M. K., Frehlick, L. J., Li, A., Serpa, J. J., Petrotchenko, E. V., et al. (2012). MeCP2 binds to nucleosome free (linker DNA) regions and to H3K9/H3K27 methylated nucleosomes in the brain. Nucleic Acids Res. 40, 2884–2897. doi: 10.1093/nar/gkr1066
Thandapani, P., O’Connor, T. R., Bailey, T. L., and Richard, S. (2013). Defining the RGG/RG motif. Mol. Cell. 50, 613–623. doi: 10.1016/j.molcel.2013.05.021
Thatcher, K. N., and Lasalle, J. M. (2006). Dynamic changes in Histone H3 lysine 9 acetylation localization patterns during neuronal maturation require MeCP2. Epigenetics 1, 24–31.
Tillotson, R., and Bird, A. (2019). The molecular basis of MeCP2 function in the brain. J. Mol. Biol. [Epub ahead of print].
Tillotson, R., Selfridge, J., Koerner, M. V., Gadalla, K. K. E., Guy, J., De Sousa, D., et al. (2017). Radically truncated MeCP2 rescues Rett syndrome-like neurological defects. Nature 550, 398–401. doi: 10.1038/nature24058
Tomatsu, S., Orii, K. O., Bi, Y., Gutierrez, M. A., Nishioka, T., Yamaguchi, S., et al. (2004). General implications for CpG hot spot mutations: methylation patterns of the human iduronate-2-sulfatase gene locus. Hum. Mutat. 23, 590–598. doi: 10.1002/humu.20046
Trappe, R., Laccone, F., Cobilanschi, J., Meins, M., Huppke, P., Hanefeld, F., et al. (2001). MECP2 mutations in sporadic cases of Rett syndrome are almost exclusively of paternal origin. Am. J. Hum. Genet. 68, 1093–1101. doi: 10.1086/320109
Trendel, J., Schwarzl, T., Horos, R., Prakash, A., Bateman, A., Hentze, M. W., et al. (2019). The human RNA-binding proteome and its dynamics during translational arrest. Cell 176:e319.
Tsuchiya, Y., Minami, Y., Umemura, Y., Watanabe, H., Ono, D., Nakamura, W., et al. (2015). Disruption of MeCP2 attenuates circadian rhythm in CRISPR/Cas9-based Rett syndrome model mouse. Genes Cells 20, 992–1005. doi: 10.1111/gtc.12305
Tsujimura, K., Irie, K., Nakashima, H., Egashira, Y., Fukao, Y., Fujiwara, M., et al. (2015). miR-199a Links MeCP2 with mTOR signaling and its dysregulation leads to rett syndrome phenotypes. Cell Rep. 12, 1887–1901. doi: 10.1016/j.celrep.2015.08.028
Tyssowski, K., Kishi, Y., and Gotoh, Y. (2014). Chromatin regulators of neural development. Neuroscience 264, 4–16. doi: 10.1016/j.neuroscience.2013.10.008
Urdinguio, R. G., Pino, I., Ropero, S., Fraga, M. F., and Esteller, M. (2007). Histone H3 and H4 modification profiles in a Rett syndrome mouse model. Epigenetics 2, 11–14. doi: 10.4161/epi.2.1.3698
Vashi, N., and Justice, M. J. (2019). Treating Rett syndrome: from mouse models to human therapies. Mamm. Genome 30, 90–110. doi: 10.1007/s00335-019-09793-5
Villard, L. (2007). MECP2 mutations in males. J. Med. Genet. 44, 417–423. doi: 10.1136/jmg.2007.049452
Wan, M., Lee, S. S., Zhang, X., Houwink-Manville, I., Song, H. R., Amir, R. E., et al. (1999). Rett syndrome and beyond: recurrent spontaneous and familial MECP2 mutations at CpG hotspots. Am. J. Hum. Genet. 65, 1520–1529. doi: 10.1086/302690
Wan, M., Zhao, K., Lee, S. S., and Francke, U. (2001). MECP2 truncating mutations cause histone H4 hyperacetylation in Rett syndrome. Hum. Mol. Genet. 10, 1085–1092. doi: 10.1093/hmg/10.10.1085
Wang, C., Wang, F., Cao, Q., Li, Z., Huang, L., and Chen, S. (2018). The effect of Mecp2 on heart failure. Cell Physiol. Biochem. 47, 2380–2387. doi: 10.1159/000491610
Wang, L., Hu, M., Zuo, M. Q., Zhao, J., Wu, D., Huang, L., et al. (2020). Rett syndrome-causing mutations compromise MeCP2-mediated liquid-liquid phase separation of chromatin. Cell Res. 30, 393–407. doi: 10.1038/s41422-020-0288-7
Weise, S. C., Arumugam, G., Villarreal, A., Videm, P., Heidrich, S., Nebel, N., et al. (2019). FOXG1 regulates PRKAR2B transcriptionally and posttranscriptionally via miR200 in the adult hippocampus. Mol. Neurobiol. 56, 5188–5201. doi: 10.1007/s12035-018-1444-7
Wen, L., Li, X., Yan, L., Tan, Y., Li, R., Zhao, Y., et al. (2014). Whole-genome analysis of 5-hydroxymethylcytosine and 5-methylcytosine at base resolution in the human brain. Genome Biol. 15:R49.
Wen, Y., Wang, J., Zhang, Q., Chen, Y., Wu, X., and Bao, X. (2020). MECP2 mutation spectrum and its clinical characteristics in a Chinese cohort. Clin. Genet. 98, 240–250. doi: 10.1111/cge.13790
Weyn-Vanhentenryck, S. M., Feng, H., Ustianenko, D., Duffie, R., Yan, Q., Jacko, M., et al. (2018). Precise temporal regulation of alternative splicing during neural development. Nat. Commun. 9:2189.
Wilson, S. A., Brown, E. C., Kingsman, A. J., and Kingsman, S. M. (1998). TRIP: a novel double stranded RNA binding protein which interacts with the leucine rich repeat of flightless I. Nucleic Acids Res. 26, 3460–3467. doi: 10.1093/nar/26.15.3460
Wong, J. J., Gao, D., Nguyen, T. V., Kwok, C. T., Van Geldermalsen, M., Middleton, R., et al. (2017). Intron retention is regulated by altered MeCP2-mediated splicing factor recruitment. Nat. Commun. 8:15134.
Wu, H., Tao, J., Chen, P. J., Shahab, A., Ge, W., Hart, R. P., et al. (2010). Genome-wide analysis reveals methyl-CpG-binding protein 2-dependent regulation of microRNAs in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 107, 18161–18166. doi: 10.1073/pnas.1005595107
Xu, Y., Wu, W., Han, Q., Wang, Y., Li, C., Zhang, P., et al. (2019). Post-translational modification control of RNA-binding protein hnRNPK function. Open Biol. 9:180239. doi: 10.1098/rsob.180239
Xue, J., Wijeratne, S. S., and Zempleni, J. (2013). Holocarboxylase synthetase synergizes with methyl CpG binding protein 2 and DNA methyltransferase 1 in the transcriptional repression of long-terminal repeats. Epigenetics 8, 504–511. doi: 10.4161/epi.24449
Yamazaki, T., Nakagawa, S., and Hirose, T. (2020). Architectural RNAs for membraneless nuclear body formation. Cold Spring Harb. Symp. Quant. Biol. 84, 227–237. doi: 10.1101/sqb.2019.84.039404
Yang, F., Deng, X., Ma, W., Berletch, J. B., Rabaia, N., Wei, G., et al. (2015). The lncRNA Firre anchors the inactive X chromosome to the nucleolus by binding CTCF and maintains H3K27me3 methylation. Genome Biol. 16:52.
Yang, Y., Kucukkal, T. G., Li, J., Alexov, E., and Cao, W. (2016). Binding analysis of Methyl-CpG binding domain of MeCP2 and Rett syndrome mutations. ACS Chem. Biol. 11, 2706–2715. doi: 10.1021/acschembio.6b00450
Yasui, D. H., Peddada, S., Bieda, M. C., Vallero, R. O., Hogart, A., Nagarajan, R. P., et al. (2007). Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc. Natl. Acad. Sci. U.S.A. 104, 19416–19421. doi: 10.1073/pnas.0707442104
Yoon, S. H., Choi, J., Lee, W. J., and Do, J. T. (2020). Genetic and epigenetic etiology underlying autism spectrum disorder. J. Clin. Med. 9:966. doi: 10.3390/jcm9040966
Young, J. I., Hong, E. P., Castle, J. C., Crespo-Barreto, J., Bowman, A. B., Rose, M. F., et al. (2005). Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc. Natl. Acad. Sci. U.S.A. 102, 17551–17558. doi: 10.1073/pnas.0507856102
Yusufzai, T. M., and Wolffe, A. P. (2000). Functional consequences of Rett syndrome mutations on human MeCP2. Nucleic Acids Res. 28, 4172–4179. doi: 10.1093/nar/28.21.4172
Zachariah, R. M., Olson, C. O., Ezeonwuka, C., and Rastegar, M. (2012). Novel MeCP2 isoform-specific antibody reveals the endogenous MeCP2E1 expression in murine brain, primary neurons and astrocytes. PLoS One 7:e49763. doi: 10.1371/journal.pone.0049763
Zaghlula, M., Glaze, D. G., Enns, G. M., Potocki, L., Schwabe, A. L., and Suter, B. (2018). Current clinical evidence does not support a link between TBL1XR1 and Rett syndrome: description of one patient with Rett features and a novel mutation in TBL1XR1, and a review of TBL1XR1 phenotypes. Am. J. Med. Genet. A 176, 1683–1687. doi: 10.1002/ajmg.a.38689
Zhang, X., Wang, W., Zhu, W., Dong, J., Cheng, Y., Yin, Z., et al. (2019). Mechanisms and functions of long non-coding RNAs at multiple regulatory levels. Int. J. Mol. Sci. 20:5573. doi: 10.3390/ijms20225573
Keywords: methyl CpG binding protein 2, Rett syndrome, mutations, protein stability, RNA binding
Citation: Good KV, Vincent JB and Ausió J (2021) MeCP2: The Genetic Driver of Rett Syndrome Epigenetics. Front. Genet. 12:620859. doi: 10.3389/fgene.2021.620859
Received: 23 October 2020; Accepted: 05 January 2021;
Published: 21 January 2021.
Edited by:
Mojgan Rastegar, University of Manitoba, CanadaReviewed by:
Walter Erwin Kaufmann, Emory University, United StatesNathalie Berube, Western University, Canada
Copyright © 2021 Good, Vincent and Ausió. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Ausió, amF1c2lvQHV2aWMuY2E=