 Jing Zhang
Jing Zhang Huaying Hu
Huaying Hu Weihong Mu1
Weihong Mu1 Kai Yang
Kai Yang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Genet. , 16 February 2021
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.619948
This article is part of the Research Topic NGS Technologies of Rare Diseases Diagnosis View all 34 articles
Bruck Syndrome (BRKS) is a rare type of recessive osteogenesis imperfecta (OI) and consists of two subtypes, BRKS1 and BRKS2, which are caused by variations in FKBP10 and PLOD2 genes, respectively. In this study, a family that had experienced multiple miscarriages and recurrent fetal skeletal dysplasia was recruited for the purpose of a multiplatform laboratory investigation. Prenatal genetic testing with whole-exome sequencing (WES) identified a compound heterozygous variation in the PLOD2 gene with two variants, namely c.2038C>T (p.R680*) and c.191_201+3 delATACTGTGAAGGTA (p.Y64Cfs*12). The amino acids affected by the two variants maintained conserved across species. And the result of immunohistochemistry (IHC) indicated that the expression of PLOD2 protein in the proband's osteochondral tissue was significantly decreased. These findings in our study expanded the variation spectrum of PLOD2 gene, provided solid evidence for the family's counseling in regard to future pregnancies, strongly supported the application of WES in prenatal diagnosis, and might give insight into the understanding of PLOD2 function.
Congenital skeletal dysplasia (SD) often displays severe in utero manifestations, which provides clues and challenges for prenatal diagnosis (Offiah, 2015; Liu et al., 2019). First, they can provide evidence for the timely formation of management plans; on the other hand, due to the strong clinical heterogeneity of SDs, it is difficult to accurately judge the prognosis by these indeterminate indications alone (Konstantinidou et al., 2009; Offiah, 2015). Under such circumstances, a meticulously designed strategy of genetic detection would help overcome the setbacks in clinical differential diagnosis (Zhou et al., 2018; Yang et al., 2019).
Osteogenesis imperfecta (OI) is a series of congenital metabolic bone disorders and comprises dozens of conditions ranging from early lethality to mild manifestations (Marini, 2013; Liu et al., 2017). OI is mainly characterized by increasing bone fragility, recurrent fracture, and subsequent growth retardation (Forlino and Marini, 2016; Liu et al., 2017). OI patients also share some extraskeletal symptoms, such as blue sclera, hearing deficits, dentinogenesis imperfecta (DI), and valvular heart disease (Forlino and Marini, 2016). Bruck syndrome types 1 and 2 (BRKS1, MIM #259450; BRKS2, MIM #609220), caused by biallelic pathogenic variants in theFKBP10 (MIM *607063) and PLOD2 (MIM *601865) genes, respectively, are two rare subtypes of OI, mainly characterized by congenital joint contractures and pterygia other than the common OI manifestations (Viuoen et al., 1989). PLOD2 encodes lysyl hydroxylase 2 (LH2, EC 1.14.11.4), while FKBP10 encodes the prolyl cis-trans isomerase FKBP65, the activities of which are required in forming mature crosslinks in bone collagen (Bank et al., 1999; Alanay et al., 2010).
To our knowledge, fewer than 20 studies have reported the BRKS2 cases with respect to defects in PLOD2 (Mumm et al., 2020). Moreover, the genotype-phenotype correlation of BRKS2 is not fully established, owing to the low amount and relatively clustered distribution of constitutional variations in PLOD2, the vague structural and functional delineation of the N-terminal of LH2 protein as well as the strong phenotypic variability of BRKS2 (Tham et al., 2018; Mumm et al., 2020). Furthermore, given the commonality of cell signaling pathways between BRKS2 and multiple types of cancers, the in-depth study of corresponding molecules would benefit scientific and clinical understanding in both fields (Guo et al., 2018; Wang et al., 2020).
Thus, we studied a family having experienced multiple abnormal gestations. Prenatal ultrasonography examination and genetic detections were conducted to identify the cause of fetal skeletal dysplasia in the subject family.
A 34-year-old woman was first referred to our Center in February 2015 for having experienced multiple abnormal gestations. At that point she was 8 weeks pregnant.
Based on medical records and her personal dictation, we combed through the couple's complete medical history and illustrated in a pedigree diagram (Figure 1A). They had eight previous pregnancies in total: In August 2005, missed abortion occurred at the eighth gestational week of their first pregnancy. In October 2006, it was diagnosed that the second pregnancy was an ectopic pregnancy on the left fallopian tube, whereupon transabdominal resection of the left fallopian tube was performed. In October 2007, July 2008, August 2010, and December 2012, the couple went through four missed abortions or miscarriages, all of which occurred between 8 and 16 gestational weeks. In November 2013, the fetus of their seventh pregnancy was diagnosed with thickened nuchal translucency (NT), right abdominal fissure and visceral valgus. Induced labor was performed at 15 weeks of gestation.
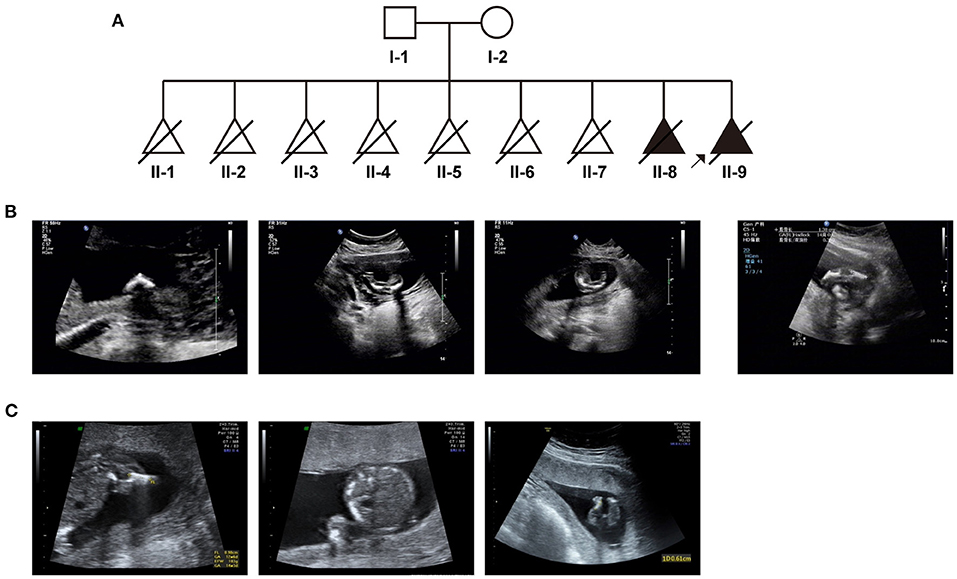
Figure 1. The clinical findings in this case: (A) Pedigree diagram of the family with fetal skeletal dysplasia and multiple miscarriages. (B) Ultrasonographic indications of the couple's eighth pregnancy. (C) Ultrasonographic indications of the couple's most recent (ninth) pregnancy.
In 2015, all indicators of their 8th pregnancy during early pregnancy were normal, including serological screening, NIPT (non-invasive prenatal testing) results. However, at 17th week of gestation, ultrasonic examination revealed that the fetus had extremely short limbs and bowing of long bones (Figure 1B). Amniocentesis and subsequent prenatal diagnosis with chromosomal karyotyping and micro array (CMA) were conducted. However, no variation with clinical significance was identified. The fetus was aborted at 20 gestational weeks.
In April 2018, we performed prenatal diagnosis for their most recent pregnancy at the 16th week of gestation. Ultrasonography indicated that bilateral lower limbs of the fetus were shortened, not excluding the presence of bending (Figure 1C). Besides regular prenatal genetic testing as described above, whole-exome sequencing was also introduced after induction.
Prenatal genetic testing including karyotyping and CMA (with Cytoscan 750k, Thermo Fisher platform) were performed on the last two adverse pregnancies in 2015 and 2018. Both results of those were normal.
Accordingly, whole-exome sequencing was performed on the skin tissue obtained from the aborted fetus of the last pregnancy. In brief, the enrichment of target-region sequences was performed by means of the Sure Select Human Exon Sequence Capture Kit (Agilent, USA). The sequencing libraries were quantified using the DNA Standards and Primer Premix Kit (Kapa Biosystems, USA), massively parallel-sequenced using the XTEN platform and then massively parallel-sequenced again (Illumina, USA). After sequencing and filtering out the low-quality readings, the high-quality readings were compared to the human genome reference sequence [(hg19/GRC h37)]. The GATK software (Genome Analysis TK3.3.0, https://software.broadinstitute.org/gatk/) and the Verita Trekker® Variants Detection system (Berry Genomics, China) were used to identify single-nucleotide polymorphisms, insertion and deletions, copy-number variants (CNVs), mitochondrial gene variants, and runs of homozygosity. Subsequently, the Enliven® Variants Annotation Interpretation (Berry Genomics, China) system was used to fulfill the annotation and interpretation progress referring to multiple databases [1000g2015aug_eas (https://www.internationalgenome.org/); ExAC_EAS (http://exac.broadinstitute.org); gnomAD_exome_EAS (http://gnomad.broadinstitute.org/); HGMD®: Human Gene Mutation Database (Professional Version 2019.4)]. The suspected pathogenic variant was validated by Sanger sequencing through use of ABI 3730 Automated Sequencer (Applied Biosystems). The mutations were identified by sequence alignment with the NCBI Reference Sequence (NG 011537.1) using Chromas 2.33. WES identified a compound heterozygous variation in the PLOD2 gene consisting of two variants, namely c.2038C>T (p.R680*) and c.191_201+3delATACTGTGAAGGTA (p.Y64Cfs*12) (NM_000935) (Figure 2A). Sanger sequencing revealed that the father carried the heterozygous c.2038C>T variant, while the mother carried c.191_201+3del (Figure 2A). The former one was reported by Lyu et al. as a pathogenic variant (Lv et al., 2017), while the latter, which would result in a premature truncated protein, was first reported in this study. According to the variant interpretation criteria by ACMG (Richards et al., 2015), it was classified as pathogenic, with the evidence of PVS1, PM2, PM3, and PP4.
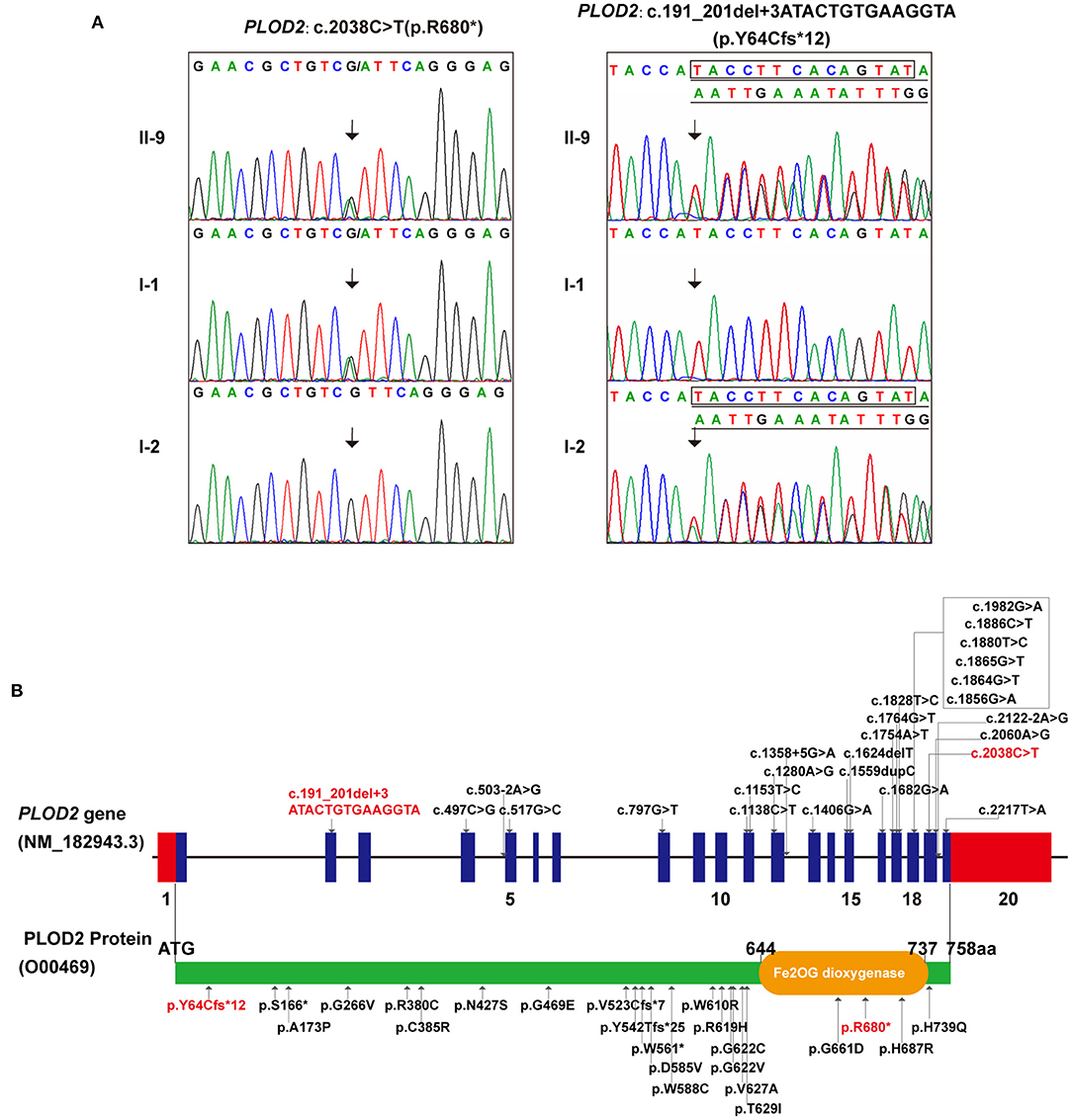
Figure 2. The genetic variations identified in this case: (A) The two variants detected in this fetus and his parents (individual no. corresponding to Figure 1A). (B) All PLOD2 variants associating with BRKS2-like phenotype in the literature and in this study, illustrated in diagrams of gene and protein. (In this study, red font represents variants).
The conservatism of all amino acids effected by detected variants was analyzed using MEGA7 (http://www.megasoftware.net/previousVersions.php) with default parameters. The results showed that the amino acids Y64 and R680 (i.e., the two variants) maintained evolutionary conservatism among species (Figure 3A).
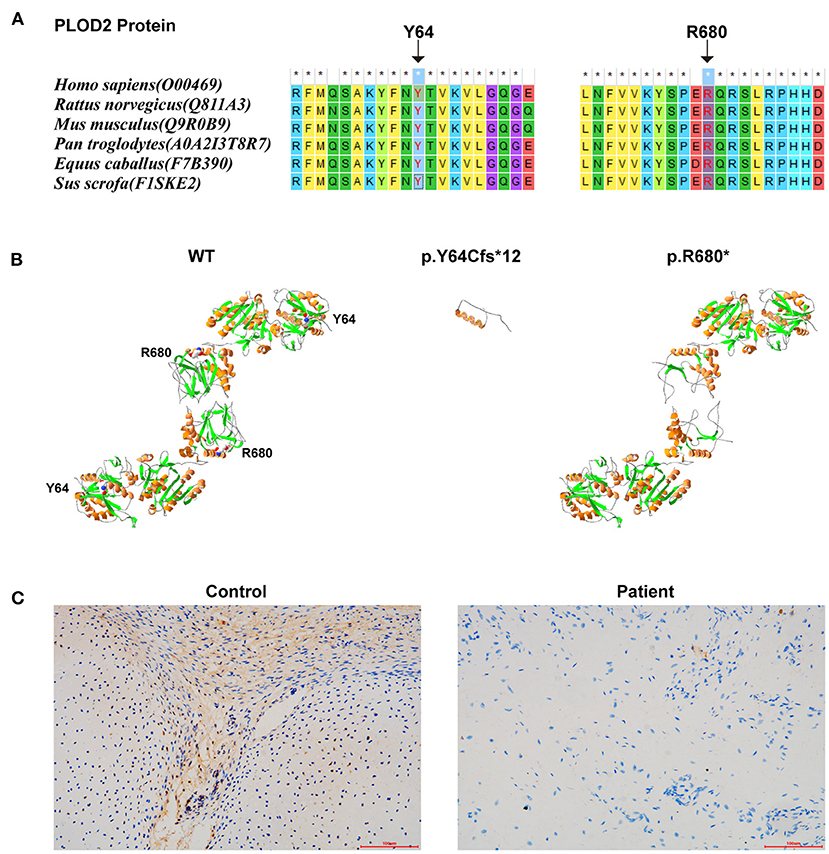
Figure 3. The results of in silico analysis and IHC experimentation: (A) The conservatism of the two amino acids effected by the two variants. (B) The protein models of WT, p.Y64Cfs*12, and p.R680* of PLOD2 protein model 6fxm.1.A. (C) PLOD2 protein expression in the osteochondrocytes of the proband and control samples, by IHC.
Protein modeling was conducted using SWISS-MODEL (https://swissmodel.expasy.org/) and Swiss-Pdb Viewer software (https://spdbv.vital-it.ch/disclaim.html#). PLOD2 protein model of wild type (WT)-6fxm.1.A (Seq Identity:59.26%/GMQE: 0.82/QMEAN: −0.67), p.Y64Cfs*12-6fxm.1.A (Seq Identity:57.80%/GMQE: 0.80/QMEAN: −0.78), and p.R680*-6fxk.1.A (Seq Identity:43.59%/GMQE:0.18/QMEAN: −0.87) were shown (Figure 3B). According to the results, these two variants would result in premature truncated peptides, which could cause severe damage to protein function. Immunohistochemistry (IHC) (with Anti-PLOD2/LH2 antibody, ab90088, Abcam, USA) was conducted on the paraffin sections of fetal auricular finger osteochondrocytes of the proband (along with a cartilage tissue sample from an aborted fetus (PLOD2 wild-type) at similar gestational age as normal control). The results indicated that the expression level of PLOD2 protein was significantly decreased in the osteochondral tissues of the proband (Figure 3C). The low protein expression of PLOD2 was possibly because c.2038C>T (p.R680*) and c.191_201+3delATACTGTGAAGGTA would be expected to induce non-sensical mediated decay.
Recessive forms only account for <10% in OI disorders, yet the list of causative genes has been growing through recent years (Eyre and Weis, 2013). Additionally, with the defects in various proteins involved in bone formation, OIs display a broad range of phenotypic variability (Forlino and Marini, 2016). This promotes the challenge in prenatal diagnosis and management on fetal SDs with similar early manifestations. Therefore, it would appear to be more and more arbitrary to consider the in utero limb shortening and bowing of long bones as a collagen-related autosomal dominant condition.
As a rare but typical example of recessive OI, most cases of BRKS are distinguished by congenital joint contractures and pterygia, albeit with several exceptions (Tham et al., 2018; Mumm et al., 2020). Currently, two BRKS subtypes, BRKS1 and BRKS2, with almost identical phenotypes but different pathogenesis are recognized (van der Slot et al., 2003; Alanay et al., 2010). To our knowledge, 25 constitutional mutations in PLOD2 associating with BRKS2-like phenotypes have been detected in fewer than 20 studies, showing significant C-terminal aggregation (Figure 2B; Supplementary Table 1) (Mumm et al., 2020). In this study, a compound heterozygous variation in thePLOD2 gene with two variants, c.2038C>T and c.191_201+3del, was identified and confirmed. The former one, c.2038C>T, was once reported in BRKS2 patient (Lv et al., 2017); while the latter one, c.191_201+3del, was a novelly detected frame-shift variant causing early termination of protein translation. This finding expanded the variation spectrum of PLOD2 gene. Additionally, Guo et al. have determined a 2Å crystal structure of the lysyl hydroxylase (LH) domain corresponding to human PLOD2 amino acids 548–758, which provided sub-molecular insight into how specific variants affect the structure of LH2 (Guo et al., 2018). The genotype-phenotype correlation of PLOD2 skeletal dysplasia is pending further elucidation along with the identification of more variations, particularly at the N-terminal (Leal et al., 2018).
LH2, cloned by Valtavaara et al. (1997), is encoded by PLOD2 gene, the defect of which is responsible for the onset of BRKS2. LH2 specifically catalyzes the hydroxylation of telopeptide lysyl residues in collagen, which is essential for the normal crosslinking of bone collagen (Bank et al., 1999; van der Slot et al., 2003). FKBP10, whose deficiency is causative for BRKS1, belongs to a group of proteins termed immunophilins and displays high binding affinity for the immunosuppressant drugs FK506 (Patterson et al., 2000). FKBP10 is thought to function as a collagen chaperone and to assist in collagen folding (Lietman et al., 2014). Subsequently, Chen et al. found that the peptidyl prolyl isomerase activity of FKBP10 positively modulated LH2 enzymatic activity and was critical for the formation of hydroxylysine-aldehyde derived intermolecular collagen crosslinks, which indirectly explained why phenotypes of BRKS1 and BRKS2 were so similar (Chen et al., 2017). LH2 has two splice forms–LH2a (short) and LH2b (long)–resulting from alternative splicing of PLOD2, with LH2b containing an additional exon 13 A encoding 21 amino acids (Gjaltema and Bank, 2017). The absence of LH2b has great impact on musculoskeletal tissues, particularly bone, as illustrated by mutations in PLOD2 (Gjaltema and Bank, 2017). LH2 activity is positively correlated with pyridinoline levels, which indicates its important role in the formation of collagen telopeptide-derived pyridinoline crosslinks (van der Slot et al., 2005; Gjaltema and Bank, 2017). In this study, the result of IHC indicated the extremely low expression of PLOD2 in cartilage tissue of the proband, which should be the result of the variation he carried. Both mutations c.2038C>T (p.R680*) and c.191_201+3delATACTGTGAAGGTA would be possibly expected to induce nonsense mediated mRNA decay (NMD), whereby the low expression level of PLOD2 could be responsible for the NMD. Further functional experiments are needed to explore the possible mechanism. However, we failed to detect the mRNA-level expression of PLOD2 due to the degradation of RNA in the fibroblast sample, which is a flaw in our study and needs to be remedied by subsequent functional studies. From another angle, the protein modeling result revealed that both mutations possibly caused the peptide chain truncation, which clearly affected the dimerization and maturation process and even the presence of the protein. Besides, the evolutionary conservatism of the amino acids affected by the two variants further supported their pathogenicity. However, further functional experiments are necessary, not only to clarify the effect of each variant on the protein itself but also to understand the role of this effect in the overall process of osteogenic differentiation.
In practical terms, diagnostic report in this study was obtained before the fetal pregnancy entered the perinatal period (after 27+6 gestational weeks), which was supportive for the application of WES in the field of prenatal diagnosis.
For any future pregnancy of the proband in this study, the recurrent risk of Bruck syndrome 2 condition would be 25%. Given such circumstances, the couple were informed of reproductive options such as prenatal testing and preimplantation genetic diagnosis (PGD). We need to follow up on the outcome of the couple's pregnancy.
In summary, this study detected a novel PLOD2 compound heterozygous variation in a 17-gestation-week fetus with BRKS2 by a multi-platform genetic approach. It clarified the cause of fetal OI in the subject family, provided guidance for any future pregnancy the couple might have and highlighted the value of WES in diagnosis of SD with unclear prenatal indications.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/, NG 011537.1; http://www.wwpdb.org/, NM_000935.
The studies involving human participants were reviewed and approved by the Research Ethics Committee of the Shijiazhuang Obstetrics and Gynecology Hospital. The patients/participants provided their written informed consent to participate in this study.
QG designed the overall research strategy and supervised the whole process. JZ recruited the case, performed the prenatal counseling, sampling, and part of the genetic experiments. HH performed the other part of the genetic experiments and data analysis. WM and MY performed IHC. WC conducted protein modeling. DM performed prenatal ultrasonic exam. JZ and HH cooperated in the preparation of this manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the patients and their families for their participation in this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.619948/full#supplementary-material
Alanay, Y., Avaygan, H., Camacho, N., Utine, G. E., Boduroglu, K., Aktas, D., et al. (2010). Mutations in the gene encoding the RER protein FKBP65 cause autosomal-recessive osteogenesisimperfecta. Am. J. Hum. Genet. 86, 551–559. doi: 10.1016/j.ajhg.2010.02.022
Bank, R. A., Robins, S. P., Wijmenga, C., Breslau-Siderius, L. J., Bardoel, A. F. J., Sluijs, H. A. V. D., et al. (1999). Defective collagen crosslinking in bone, but not in ligament or cartilage, in Bruck syndrome: indications for a bone-specific telopeptidelysyl hydroxylase on chromosome 17. Proc. Natl. Acad. Sci. U.S.A. 96, 1054–1058. doi: 10.1073/pnas.96.3.1054
Chen, Y., Terajima, M., Banerjee, P., Guo, H., Liu, X., Yu, J., et al. (2017). FKBP65-dependent peptidyl-prolylisomerase activity potentiates the lysyl hydroxylase 2-driven collagen cross-link switch. Sci. Rep. 7:46021. doi: 10.1038/srep46021
Eyre, D. R., and Weis, M. A. (2013). Bone collagen: new clues to its mineralization mechanism from recessive osteogenesisimperfecta. Calcif. Tissue Int. 93, 338–347. doi: 10.1007/s00223-013-9723-9
Forlino, A., and Marini, J. C. (2016). Osteogenesisimperfecta. Lancet 387, 1657–1671. doi: 10.1016/S0140-6736(15)00728-X
Gjaltema, R. A., and Bank, R. A. (2017). Molecular insights into prolyl and lysyl hydroxylation of fibrillar collagens in health and disease. Crit. Rev. Biochem. Mol. Biol. 52, 74–95. doi: 10.1080/10409238.2016.1269716
Guo, H. F., Tsai, C. L., Terajima, M., Tan, X., Banerjee, P., Miller, M. D., et al. (2018). Pro-metastatic collagen lysyl hydroxylase dimer assemblies stabilized by Fe(2+)-binding. Nat. Commun. 9:512. doi: 10.1038/s41467-018-02859-z
Konstantinidou, A. E., Agrogiannis, G., Sifakis, S., Karantanas, A., Harakoglou, V., Kaminopetros, P., et al. (2009). Genetic skeletal disorders of the fetus and infant: pathologic and molecular findings in a series of 41 cases. Birth Defects Res. A Clin. Mol. Teratol. 85, 811–821. doi: 10.1002/bdra.20617
Leal, G. F., Nishimura, G., Voss, U., Bertola, D. R., Astrom, E., Svensson, J., et al. (2018). Expanding the clinical spectrum of phenotypes caused by pathogenic variants in PLOD2. J. Bone Miner. Res. 33, 753–760. doi: 10.1002/jbmr.3348
Lietman, C. D., Rajagopal, A., Homan, E. P., Munivez, E., Jiang, M. M., Bertin, T. K., et al. (2014). Connective tissue alterations in Fkbp10-/- mice. Hum. Mol. Genet. 23, 4822–4831. doi: 10.1093/hmg/ddu197
Liu, Y., Asan Ma, D., Lv, F., Xu, X., Wang, J., et al. (2017). Gene mutation spectrum and genotype-phenotype correlation in a cohort of Chinese osteogenesisimperfecta patients revealed by targeted next generation sequencing. Osteoporos. Int. 28, 2985–2995. doi: 10.1007/s00198-017-4143-8
Liu, Y., Wang, L., Yang, Y. K., Liang, Y., Zhang, T. J., Liang, N., et al. (2019). Prenatal diagnosis of fetal skeletal dysplasia using targeted next-generation sequencing: an analysis of 30 cases. Diagn. Pathol. 14:76. doi: 10.1186/s13000-019-0853-x
Lv, F., Xu, X., Song, Y., Li, L., Asan Wang, J., et al. (2017). Novel Mutations in PLOD2 Cause Rare Bruck Syndrome. Calcif. Tissue Int. 102, 296–309. doi: 10.1007/s00223-017-0360-6
Marini, J. C. (2013). New genes in bone development: what's new in osteogenesis imperfecta. J Clin. Endocrinol. Metab. 98, 3095–3103. doi: 10.1210/jc.2013-1505
Mumm, S., Gottesman, G. S., Wenkert, D., Campeau, P. M., Nenninger, A., Huskey, M., et al. (2020). Bruck syndrome 2 variant lacking congenital contractures and involving a novel compound heterozygous PLOD2 mutation. Bone 130:115047. doi: 10.1016/j.bone.2019.115047
Offiah, A. C. (2015). Skeletal dysplasias: an overview. Endocr. Dev. 28, 259–276. doi: 10.1159/000381051
Patterson, C. E., Schaub, T., Coleman, E. J., and Davis, E. C. (2000). Developmental regulation of FKBP65 an ER-localized extracellular matrix binding-protein. Mol. Biol. Cell 11, 3925–3935. doi: 10.1091/mbc.11.11.3925
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Tham, E., Grigelionis, G., Hammarsjo, A., and Grigelioniene, G. (2018). Genotype-phenotype correlation of PLOD2 skeletal dysplasias using structural information. J. Bone Miner. Res. 33, 1377–1378. doi: 10.1002/jbmr.3460
Valtavaara, M., Papponen, H., Pirttila, A.-M., Helander, H., Hiltunen, K., and Myllyla, R. (1997). Cloning and characterization of a novel human lysyl hydroxylase isoform highly expressed in pancreas and muscle. J. Biol. Chem. 272, 6831–6834.
van der Slot, A. J., van Dura, E. A., de Wit, E. C., DeGroot, J., Huizinga, T. W. J., Bank, R. A., et al. (2005). Elevated formation of pyridinoline cross-links by profibrotic cytokines is associated with enhanced lysyl hydroxylase 2b levels. Biochim. Biophys. Acta 1741, 95–102. doi: 10.1016/j.bbadis.2004.09.009
van der Slot, A. J., Zuurmond, A. M., Bardoel, A. F., Wijmenga, C., Pruijs, H. E., Sillence, D. O., et al. (2003). Identification of PLOD2 as telopeptidelysyl hydroxylase, an important enzyme in fibrosis. J. Biol. Chem. 278, 40967–40972. doi: 10.1074/jbc.M307380200
Viuoen, D., Versfeld, G., and Beighton, P. (1989). Osteogenesisimperfecta with congenital joint contractures (Bruck Syndrome). Clin. Genet. 36, 122–126. doi: 10.1111/j.1399-0004.1989.tb03174.x
Wang, X., Guo, J., Dai, M., Wang, T., Yang, T., Xiao, X., et al. (2020). PLOD2 increases resistance of gastric cancer cells to 5-fluorouracil by upregulating BCRP and inhibiting apoptosis. J. Cancer 11, 3467–3475. doi: 10.7150/jca.41828
Yang, K., Shen, M., Yan, Y., Tan, Y., Zhang, J., Wu, J., et al. (2019). Genetic analysis in fetal skeletal dysplasias by trio whole-exome sequencing. Biomed. Res. Int. 2019, 1–8. doi: 10.1155/2019/2492590
Keywords: PLOD2 gene, Bruck syndrome type 2, osteogenesis imperfecta, whole-exome sequencing, immunohistochemistry detection
Citation: Zhang J, Hu H, Mu W, Yu M, Chen W, Mi D, Yang K and Guo Q (2021) Case Report: Exome Sequencing Identified a Novel Compound Heterozygous Variation in PLOD2 Causing Bruck Syndrome Type 2. Front. Genet. 12:619948. doi: 10.3389/fgene.2021.619948
Received: 21 October 2020; Accepted: 28 January 2021;
Published: 16 February 2021.
Edited by:
María L. Couce, Complejo Hospitalario Universitario de Santiago, SpainReviewed by:
Fulya Taylan, Karolinska Institutet (KI), SwedenCopyright © 2021 Zhang, Hu, Mu, Yu, Chen, Mi, Yang and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qing Guo, eWZndW9xaW5nQDE2My5jb20=; Kai Yang, eWsxOTgzMDkxOUAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.