 Yanjie Zhao
Yanjie Zhao Heng Zhang
Heng Zhang Qiang Ju
Qiang Ju Xinmei Li
Xinmei Li Yuxin Zheng
Yuxin Zheng- 1School of Public Health, Qingdao University, Qingdao, China
- 2Department of Blood Transfusion, The Affiliated Hospital of Qingdao University, Qingdao University, Qingdao, China
To analyze and construct a survival-related endogenous RNA (ceRNA) network in gastric cancer (GC) with lymph node metastasis, we obtained expression profiles of long non-coding RNAs (lncRNAs), mRNAs, and microRNAs (miRNAs) in GC from The Cancer Genome Atlas database. The edgeR package was used to screen differentially expressed lncRNAs, mRNAs, and miRNAs between GC patients with lymphatic metastasis and those without lymphatic metastasis. Then, we used univariate Cox regression analysis to identify survival-related differentially expressed RNAs. In addition, we used multivariate Cox regression analysis to screen lncRNAs, miRNAs, and mRNAs for use in the prognostic prediction models. The results showed that 2,247 lncRNAs, 155 miRNAs, and 1,253 mRNAs were differentially expressed between the two patient groups. Using univariate Cox regression analysis, we found that 395 lncRNAs, eight miRNAs, and 180 mRNAs were significantly related to the survival time of GC patients. We next created a survival-related network consisting of 59 lncRNAs, seven miRNAs, and 36 mRNAs. In addition, we identified eight RNAs associated with prognosis by multivariate Cox regression analysis, comprising three lncRNAs (AC094104.2, AC010457.1, and AC091832.1), two miRNAs (miR-653-5p and miR-3923), and three mRNAs (C5orf46, EPHA8, and HPR); these were used to construct the prognostic prediction models, and their risk scores could be used to assess GC patients’ prognosis. In conclusion, this study provides new insights into ceRNA networks in GC and the screening of prognostic biomarkers for GC.
Introduction
Gastric cancer (GC) is the third leading cause of cancer-related mortality worldwide (Venerito et al., 2018). A recent study demonstrated that lymph node metastasis is common among patients with GC (Bravo Neto et al., 2014). Lymphatic metastasis is the leading cause of mortality and a significant prognostic factor in GC (Zhang et al., 2014). Despite many studies of the progression of GC, the biomarkers and underlying molecular mechanisms of lymph node metastasis in GC remain unclear. Long non-coding RNAs (lncRNAs), originally were thought to be have no coding ability, but recent studies have shown that some of the RNAs, considered as non-coding, are translated into proteins (Matsumoto et al., 2017; Rion and Rüegg, 2017; Cardon et al., 2020). Previous studies have suggested that lncRNAs are engaged in the many biological processes of diseases including cancer. For instance, PVT1 promotes cancer progression in gallbladder cancer (Chen et al., 2019); NORAD promotes cell proliferation, invasion, and migration and inhibits apoptosis in lung cancer (Li et al., 2020); and MFI2-AS1 promotes cancer progression and metastasis in hepatocellular carcinoma (Wei et al., 2020). MicroRNAs (miRNAs) are a type of noncoding RNA of approximately 22 nucleotides in length. They can target the 3' untranslated regions (3' UTRs) of mRNAs, thereby silencing RNA expression at the post-transcriptional level (Kim et al., 2018; Zhang et al., 2018b). The famous competing endogenous RNA (ceRNA) hypothesis proposes that lncRNAs can act as ceRNAs to compete for miRNA response elements, thus regulating target mRNA expression (Salmena et al., 2011). The function of such a ceRNA network has been verified in many cancers, including Wilms’ tumor (Zheng et al., 2020) and cervical cancer (Chen et al., 2020). However, the survival-related ceRNA network in GC needs further study.
In this study, we obtained GC RNA sequencing data from The Cancer Genome Atlas (TCGA) to identify differentially expressed RNAs (DE-RNAs) between patients with lymphatic metastasis and those without lymphatic metastasis. Based on these DE-RNAs and the clinical information of GC patients, we screened survival-related DE-RNAs to construct a survival-related ceRNA network. In addition, we used three lncRNAs (AC094104.2, AC010457.1, and AC091832.1), two miRNAs (miR-653-5p and miR-3923), and three mRNAs (C5orf46, EPHA8, and HPR) to establish a prognostic prediction model. Figure 1 depicts the procedure used for the bioinformatics analysis, and the Table 1 shows the details of the datasets. The purpose of our study was to acquire potential biomarkers associated with prognosis of GC patients and clarify the regulatory mechanism of RNAs in GC.
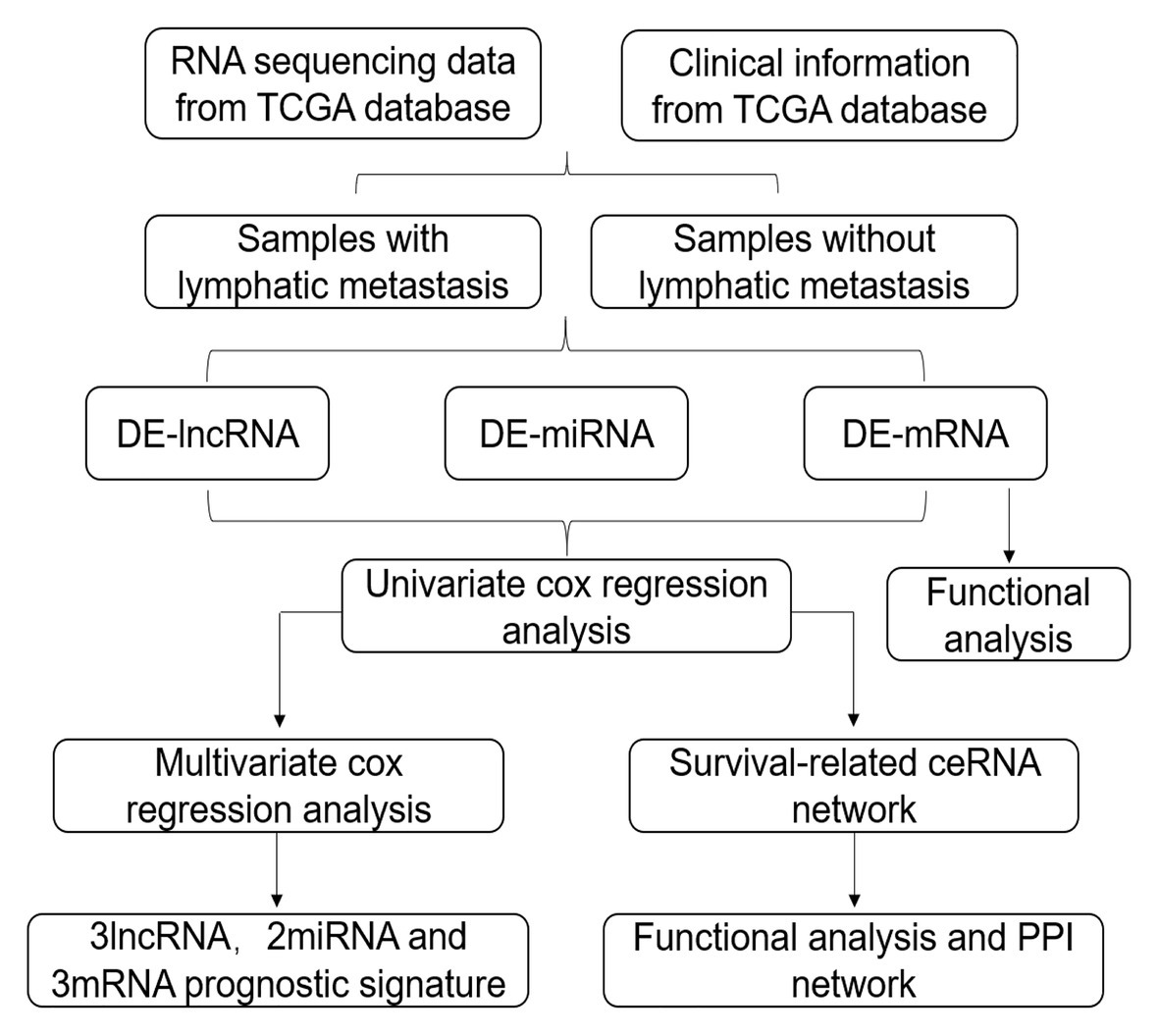
Figure 1. Flow chart of the analysis procedure. The chart shows the steps involved in screening the prognostic risk score model.
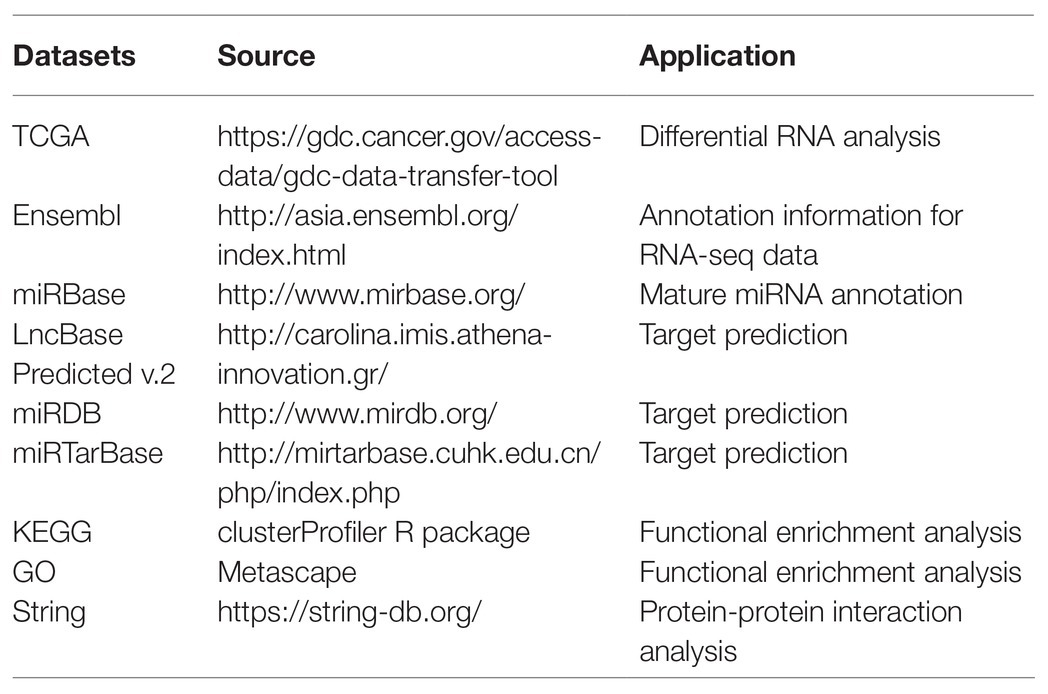
Table 1. Information of datasets.
Materials and Methods
Data Collection and Preprocessing
Clinical data for GC patients, mature miRNA datasets (the Count platform), and RNA expression profiles from TCGA were obtained using the Genomics Data Commons data transfer tool.1 Annotated RNA sequencing datasets were downloaded from the Ensembl database,2 and mature annotated miRNA datasets were downloaded from the miRBase database.3
Identification of DE-RNAs
According to the clinical information and RNA expression profiles of cancer patients, a total of 209 samples from patients with lymphatic metastasis (N1, N2, N3, and N4) and 93 samples from patients without lymphatic metastasis (N0) were obtained. Similarly, based on clinical information and mature miRNA datasets, a total of 223 samples from patients with lymphatic metastasis and 104 samples from patients without lymphatic metastasis were identified. The DE-RNAs between samples from the two patient groups were determined using the edgeR package of the R software. For screening criteria, the thresholds were set as |log2(fold change)| > 0.5 and p < 0.05. DE-RNAs were visualized using a volcano plot. The raw count was further converted into log2 values (normalized value + 1) after normalization by edgeR for use in subsequent analysis.
Univariate Cox Regression Analysis
Based on clinical data, the survival study included patients who were followed-up from 90 to 3,650 days. Univariate Cox regression analysis was used to determine correlations between the expression levels of RNAs and overall survival. RNAs with p < 0.05 were regarded as candidate prognostic markers and were used for establishing the ceRNA network. Hazard ratios (HR) for the top 10 differentially expressed lncRNAs (DE-lncRNAs), all the differentially expressed miRNAs (DE-miRNAs), and the top 10 DE-mRNAs are illustrated in forest plots.
Establishment of Survival-Related ceRNA Regulatory Network
The ceRNA network was constructed using the survival-related RNAs. The LncBase Predicted v.2 database was used to predict relationships between DE-lncRNAs and DE-miRNAs.4 The miRDB (version 6.0) and miRTarBase (Release 7.0) databases were used to establish relationships between DE-miRNAs and DE-mRNAs.5,6 Finally, the lncRNA-miRNA-mRNA ceRNA network was constructed using the Cytoscape software (version 3.7.2).
Functional Enrichment Analysis
Functional enrichment analysis of DE-mRNAs and survival-related DE-mRNAs was used to reveal their potential functions. To analyze the DE-mRNAs at a functional level, gene ontology (GO) analysis was performed using the Metascape online tool,7 and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis was conducted using the clusterProfiler R package. The survival-related DE-mRNAs were also analyzed at the functional level using Metascape. Interactive relationships among survival-related DE-mRNAs were evaluated using the STRING (version 11.0) database; those with an interaction score greater than 0.4 were considered to be significant. A protein-protein interaction (PPI) network was constructed using the Cytoscape software.
Establishment of the Prognostic Model
Survival-related RNAs were further screened by multiple regression analysis, and those with p < 0.05 were used to establish lncRNA, miRNA, and mRNA signature scores according to the following formula: risk score = (β1*expression level of RNA1) + (β2*expression level of RNA2) + (β3*expression level of RNA3), where β is the regression coefficient obtained from the multivariate Cox regression model. The survivalROC R package was used to construct a time-dependent receiver operating characteristic (ROC) curve for 3-year survival, and the area under the curve (AUC) was calculated to assess prognostic performance.
Statistical Analysis
The statistical analyses in this study were performed in the R software (version 3.6.2). The figures were drawn using GraphPad Prism 8, and the overall survival rate was evaluated via Kaplan-Meier (K-M) analysis. p < 0.05 was considered to indicate statistical significance.
Results
Identification of DE-RNAs
We identified DE-RNAs in GC patients with and without lymphatic metastasis. As a result, 2,247 lncRNAs, 155 miRNAs, and 1,253 mRNAs were found to be differentially expressed, comprising 469 upregulated and 1,778 downregulated DE-lncRNAs (Figure 2A), 87 upregulated and 68 downregulated DE-miRNAs (Figure 2B), and 521 upregulated and 732 downregulated DE-mRNAs (Figure 2C). To further understand the functions of the DE-mRNAs, we conducted KEGG and GO analyses. The KEGG results showed that the crucial pathways included “complement and coagulation cascades,” “neuroactive ligand-receptor interaction,” and “PPAR signaling pathway” (Supplementary Figure S1A). Significant GO categories identified using the Metascape tool included “developmental process,” “biological regulation,” and “multicellular organismal process” (Supplementary Figure S1B).
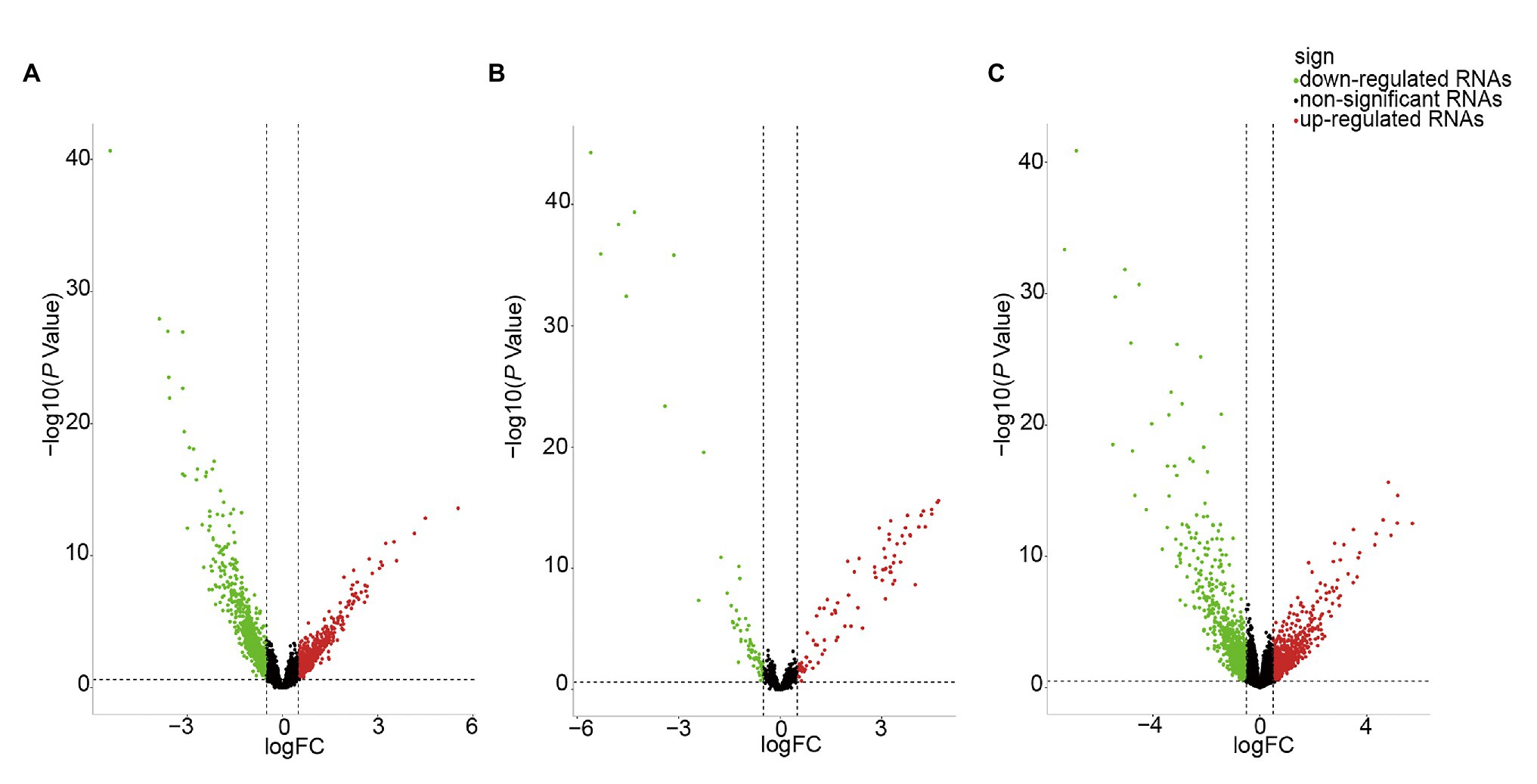
Figure 2. Volcano plot of differentially expressed RNAs (DE-RNAs) in lymph node metastatic gastric cancer (GC) samples and lymph node nonmetastatic GC samples. (A) DE-lncRNAs. (B) DE-miRNAs. (C) DE-mRNAs. Red dots indicate upregulated RNAs, green dots indicate downregulated RNAs, and black dots indicate non-differentially expressed RNAs.
Screening of Survival-Related DE-RNAs
We screened survival-related DE-RNAs by univariate Cox regression analysis. As a result, 395 lncRNAs, eight miRNAs, and 180 mRNAs were found to be significantly related to patients’ overall survival time. The top 10 survival-related lncRNAs, all survival-related miRNAs, and the top 10 survival-related mRNAs are shown in Figure 3. Functional enrichment analysis using the Metascape online tool showed that these survival-related DE-mRNAs were significantly enriched in “neuropeptide signaling pathway,” “NABA matrisome associated,” and “protein-lipid complex remodeling” (Figure 4A). In addition, a PPI network of the survival-related DE-mRNAs was constructed using STRING to show the relationships among proteins. Based on the number of protein nodes forming interactions, we identified significant RNAs including HP, CRH, GCG, APOA5, and PENK (Figure 4B).
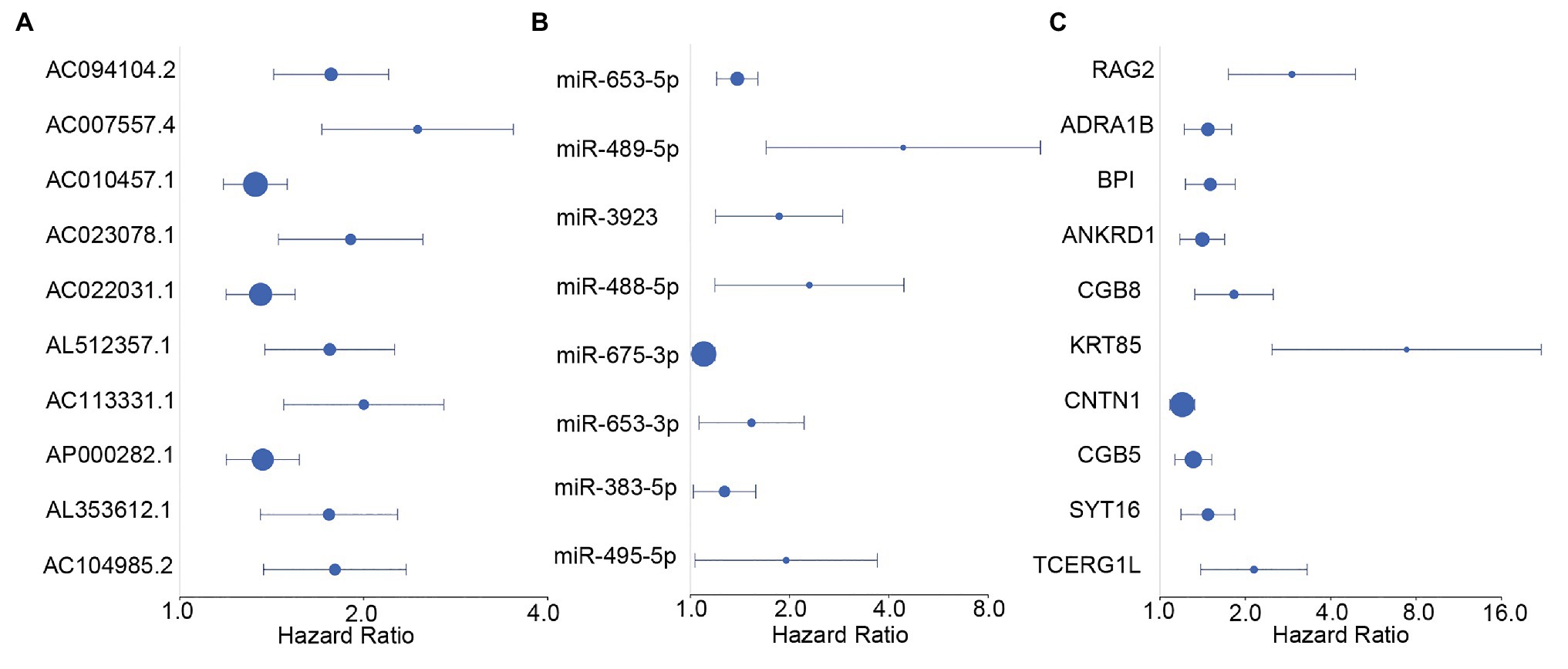
Figure 3. Forest plots of hazard ratios (HRs) of survival-related RNAs. (A) HRs of top 10 survival-related long non-coding RNAs (lncRNAs). (B) HRs of all significant survival-related microRNAs (miRNAs). (C) HRs of top 10 survival-related mRNAs.
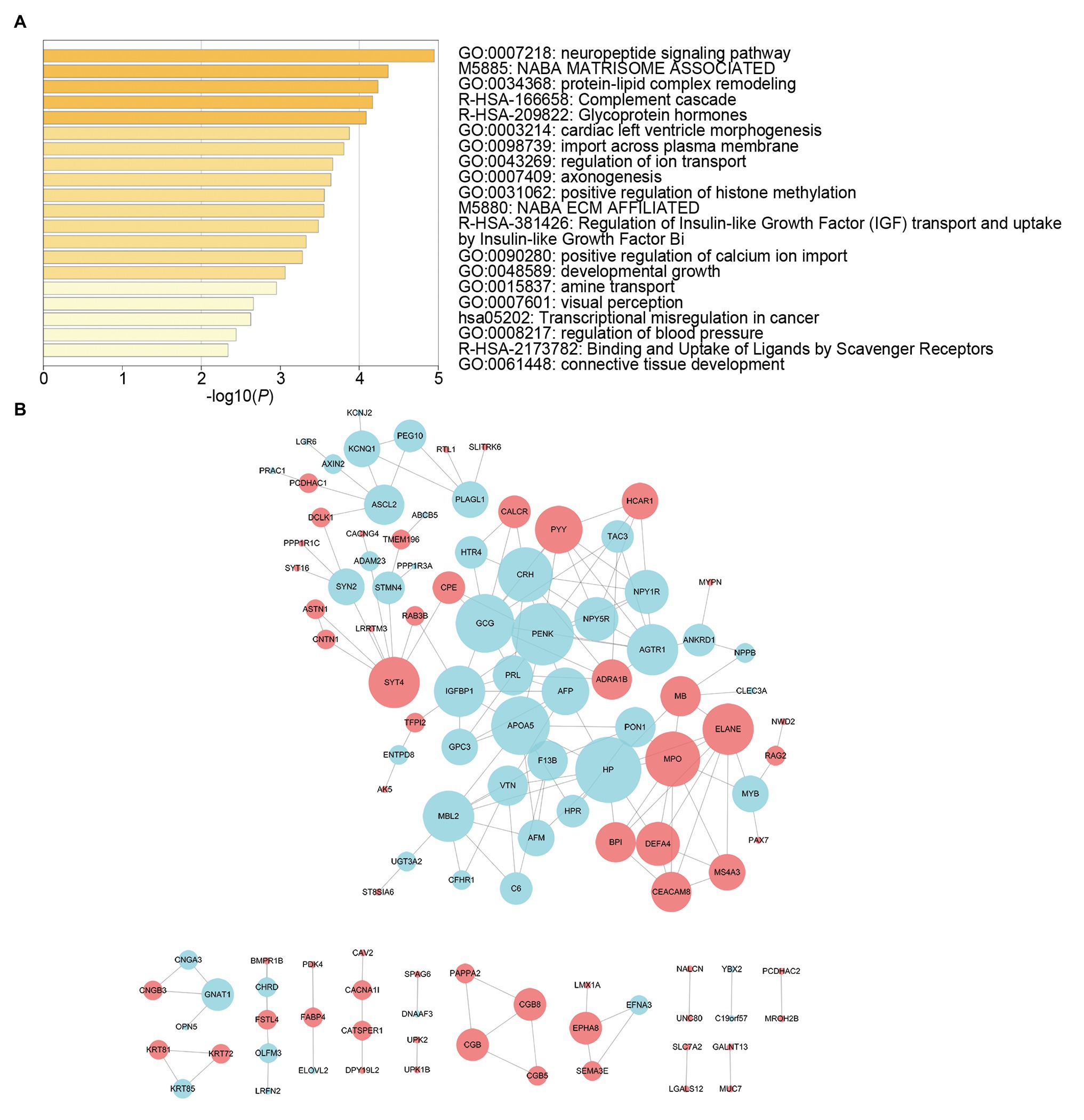
Figure 4. Functional enrichment analysis and protein-protein interaction (PPI) network of survival-related mRNAs. (A) Functional enrichment analysis of survival-related mRNAs. (B) PPI network of survival-related mRNAs. Red represents upregulation and blue represents downregulation. Node size is proportional to the degree of relationships among genes.
Construction of a Survival-Related ceRNA Network
To further study the correlations among the survival-related DE-RNAs (lncRNAs, miRNAs, and mRNAs), we established a ceRNA network and generated a visual representation using the Cytoscape software. There were 111 interactions and 102 molecules in the network, including 59 DE-lncRNAs, seven DE-miRNAs, and 36 DE-mRNAs (Figure 5A). Furthermore, considering the significant role of lncRNAs in the ceRNA network, we selected three representative lncRNAs according to the gene expression data and determined their value in predicting prognosis using K-M analysis. Of these three DE-lncRNAs, two upregulated DE-lncRNAs (ZFPM2-AS1 and APCDD1L-DT) and one downregulated DE-lncRNA (PVT1) were associated with poor survival (Figures 5B–D).
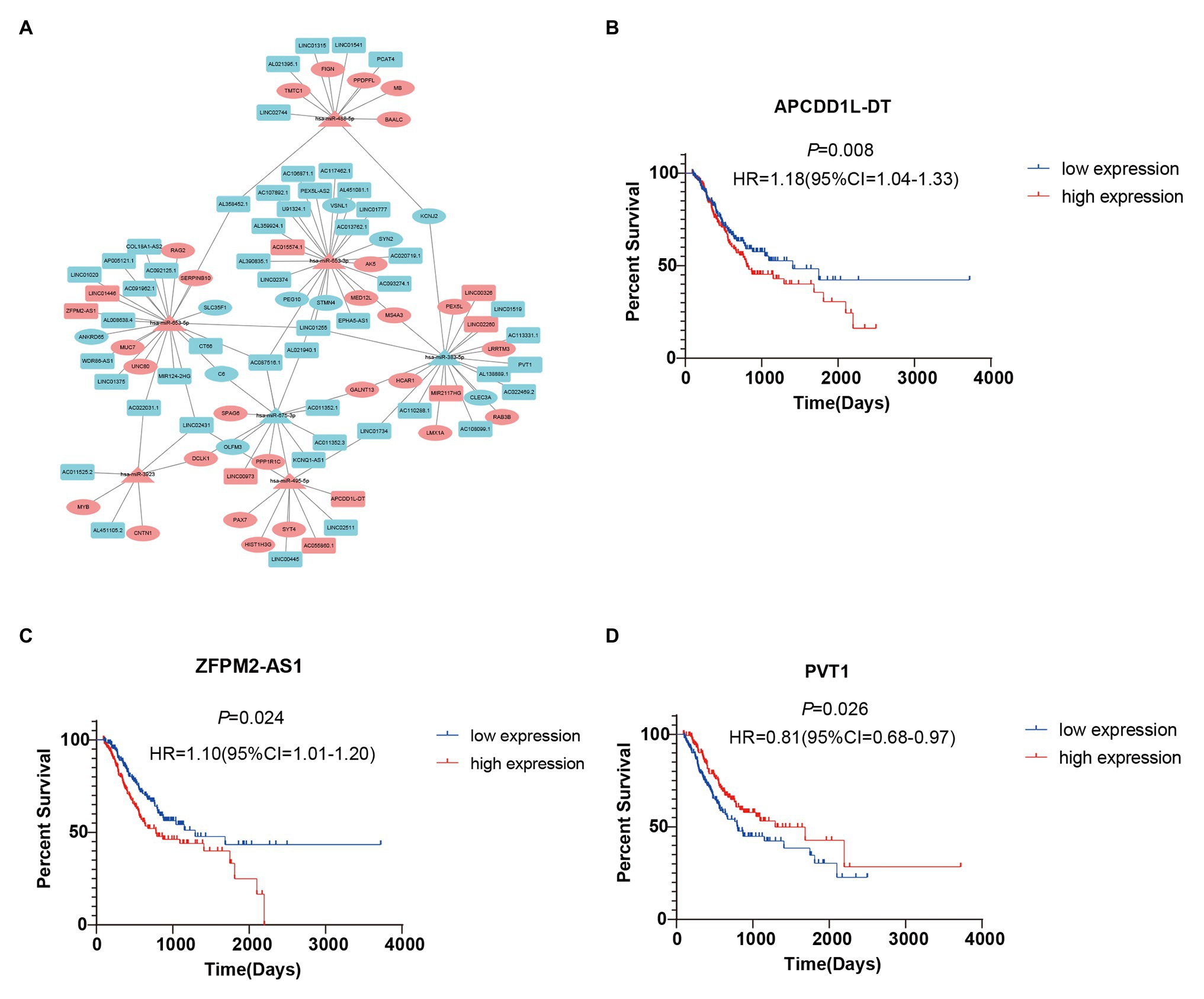
Figure 5. Survival-related competing endogenous RNA (ceRNA) network and Kaplan-Meier (K-M) curves for three DE-lncRNAs. (A) Survival-related ceRNA network in GC. Red represents upregulation and blue represents downregulation. The rectangle, triangle, and ellipse denote survival-related lncRNAs, miRNAs, and mRNAs, respectively. (B–D) Overall survival analysis for three DE-lncRNAs in the ceRNA network.
Construction of Prognostic Prediction Model
We used multivariate Cox regression analysis to further identify prognosis-related RNAs. Using p < 0.05 as the threshold for significance, five significant lncRNAs (Table 2), two significant miRNAs (Table 3), and nine significant mRNAs (Table 4) were found to have independent predictive value. Based on their values of p, the top three lncRNAs (AC094104.2, AC010457.1, and AC091832.1), all the miRNAs (miR-653-5p and miR-3923), and the top three mRNAs (C5orf46, EPHA8, and HPR) were used to establish the prognostic models. Risk score analysis was performed for the three lncRNAs, two miRNAs, and three mRNAs in each sample, and patients were divided into low- and high-risk groups according to their risk scores. K-M analysis showed that the high-risk group had worse prognoses (Figures 6A–C). The efficiencies of the above predictive models were assessed using ROC curves. The AUC values for the three lncRNAs, two miRNAs, and three mRNAs were 0.656, 0.640, and 0.628, respectively (Figures 6D–F). Thus, the risk scores could be used to evaluate the patients’ prognosis (Supplementary Figure S2).
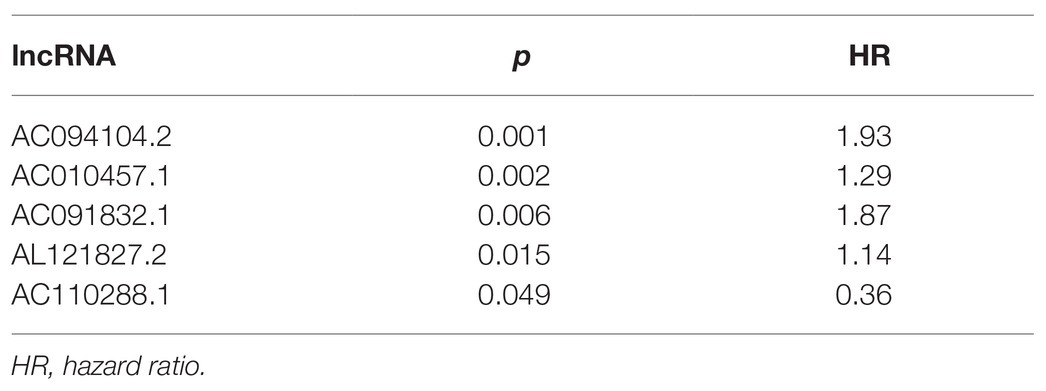
Table 2. Prognosis-related lncRNAs by multivariate Cox regression analysis.
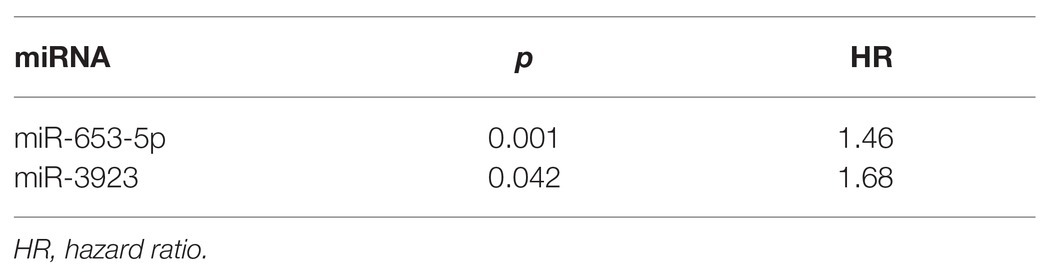
Table 3. Prognosis-related miRNAs by multivariate Cox regression analysis.
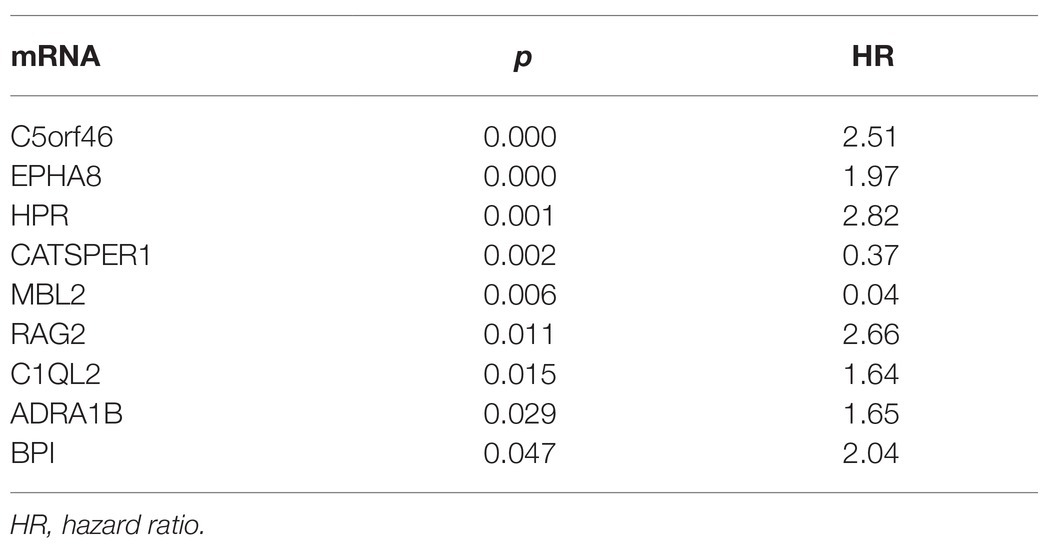
Table 4. Prognosis-related mRNAs by multivariate Cox regression analysis.
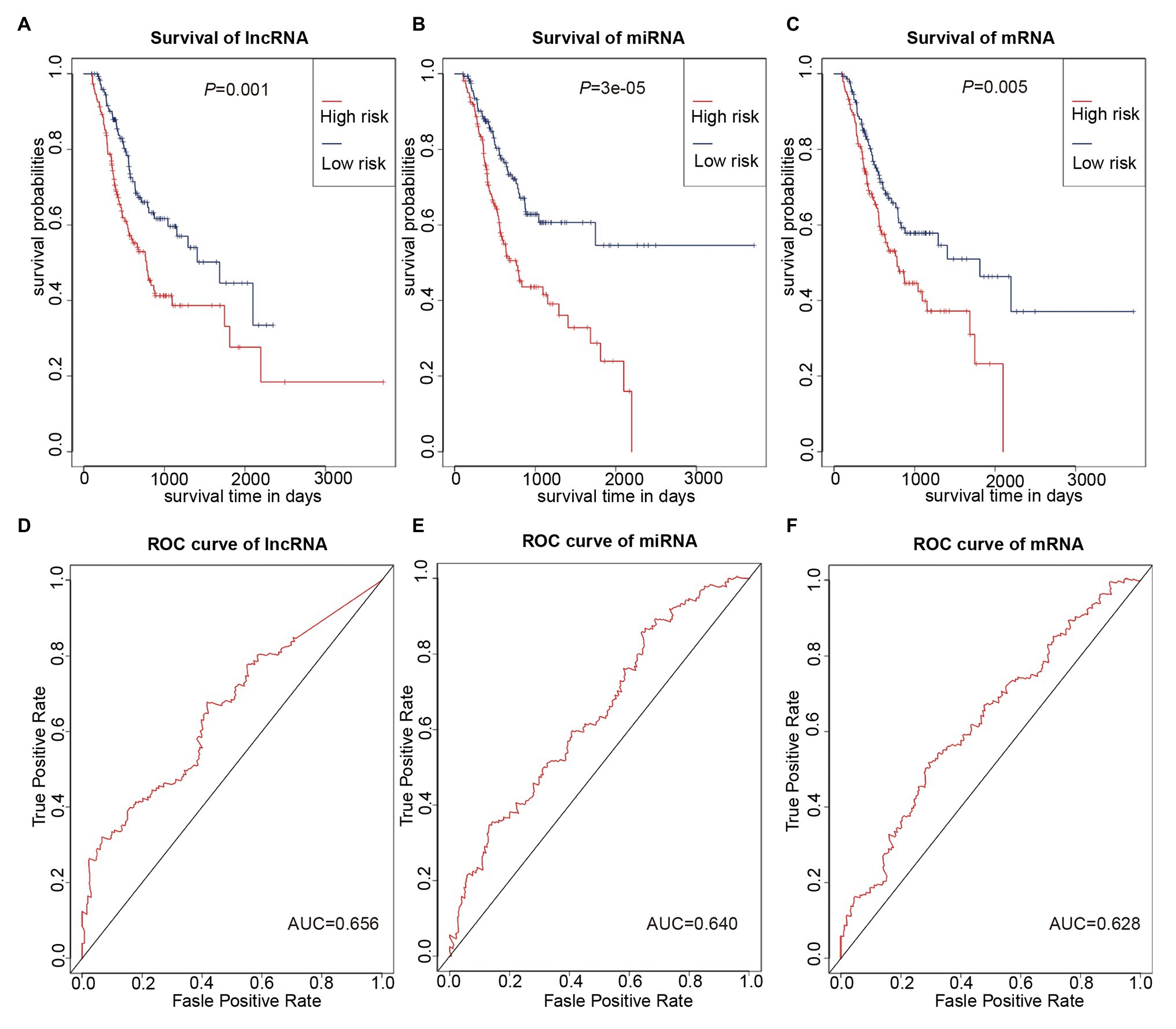
Figure 6. Prognostic prediction model based on three lncRNAs, two miRNAs, and three mRNAs. (A–C) K-M curves to assess overall survival according to high- or low-risk group based on risk scores for the three lncRNAs (A), two miRNAs (B), three mRNAs (C). (D–F) Receiver operating characteristic (ROC) curves for prognostic prediction according to the risk scores of the three lncRNAs (D), two miRNAs (E), and three mRNAs (F).
As multiple types of RNAs are thought to be involved in the occurrence and progression of cancer, we calculated risk scores using the above-mentioned eight RNAs to improve the accuracy of the prediction. The results of the K-M analysis showed that the high-risk group had worse prognoses (Figure 7A). The AUC value for the eight-RNA signature was 0.742 (Figure 7B).
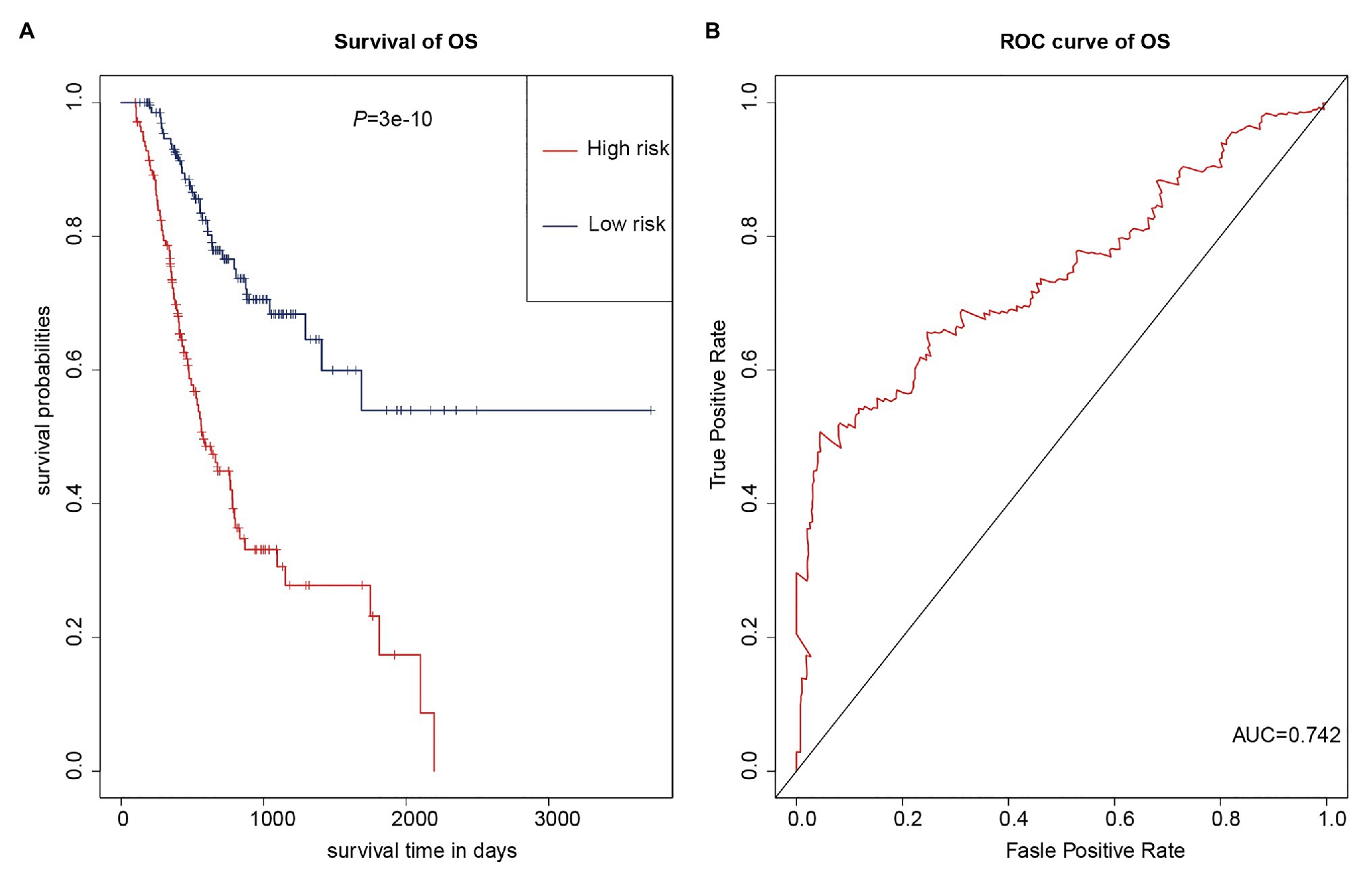
Figure 7. Eight-RNA prediction model. (A) K-M curves to assess overall survival in high- and low-risk groups according the risk scores of the eight RNAs. (B) ROC curves for prognostic prediction according the risk scores of the eight RNAs.
Discussion
A growing number of studies suggest that many diseases, particularly cancers, are controlled by complex gene regulation (Nibbe et al., 2011). In this study, in order to reveal the potential molecular mechanism of GC prognosis, we screened DE-lncRNAs, DE-miRNAs, and DE-mRNAs between GC patients with and without lymph node metastasis by bioinformatics analysis. Survival-related DE-RNAs were also obtained and used to construct a ceRNA network. Moreover, we used eight DE-RNAs to establish a prognostic prediction model. These results provide new insights for further research on ceRNAs in GC.
Recently, the ceRNA hypothesis was proposed as a new way for lncRNAs to regulate mRNAs by binding miRNAs competitively (Thomson and Dinger, 2016). To date, many studies have shown that lncRNAs can act as ceRNAs to regulate the development of GC; these lncRNAs include MT1JP (Zhang et al., 2018a), TMPO-AS1 (Sun and Han, 2020), and TRPM2-AS (Xiao et al., 2020). On the other hand, many miRNAs have also been shown to have important roles in the progression of GC, including miR-103a-3p (Hu et al., 2018a), miR-194 (Peng et al., 2017), and miR-4317 (Hu et al., 2018b). However, there has been limited research into the role of RNAs in lymph node metastasis in GC. In contrast with previous studies, we screened DE-RNAs between GC patients with lymphatic metastasis and those without lymphatic metastasis, rather than between the GC group and the normal group. As lymphatic metastasis may affect the prognosis of patients, DE-RNAs obtained in this way will be more representative as prognostic biomarkers for GC patients. Moreover, survival analysis of DE-RNAs was performed and, based on the identified survival-related DE-RNAs and the ceRNA theory, we established a survival-related ceRNA network containing 59 DE-lncRNAs, seven DE-miRNAs, and 36 DE-mRNAs. Some of these had been previously reported in GC, for instance, miR-383-5p was reported to be downregulated in GC tissues and cell lines (Xu et al., 2019). Moreover, downregulation of miR-383-5p promotes the development of GC and is associated with poor prognosis (Wei and Gao, 2019); miR-383-5p was also downregulated in our survival-related ceRNA network. Based on the ceRNA network, we found that C6 and DCLK1 bound to two miRNAs, respectively, and they were involved in the progression of GC. The expression level of C6 can be used to distinguish GC patients with good or poor prognosis (Yang et al., 2019), and high expression of DCLK1 predicts worse clinical outcomes in GC (Wu et al., 2020). In addition, we found that some of the RNAs had been reported in other cancers. For example, DCLK1 is not only associated with GC but also with colorectal cancer, where it can promote the epithelial-mesenchymal transition process (Liu et al., 2018b). MS4A3, which was found to bind to two miRNAs in our ceRNA network, was shown in a previous study to be a target of EVI1, and its suppression has an important role in cancer (Heller et al., 2015). However, there are still some novel RNAs that have not been reported in previous studies but were shown in our study to be related to prognosis in GC patients. Overall, we showed here for the first time that the above RNAs might be involved in regulation of the progression of GC.
To further understand the potential functions of survival-related DE-mRNAs, we performed functional enrichment analysis and constructed a PPI network. On the one hand, the enrichment analysis showed that the most significant pathway was “neuropeptide signaling pathway.” Studies have shown that the neuropeptide Y (NPY) family and its associated receptors have crucial roles in the progression of cancers (Ruscica et al., 2007). In addition, gastrin serves physiological functions as hormones in the gastrointestinal tract and as neuropeptides in the nervous system (Heasley, 2001). The gastrin-mediated signaling pathways, such as Indian Hedgehog signaling, may be related to the development of GC (Hayakawa et al., 2016). Moreover, some other pathways were associated with cancer, including “complement cascade” and “Matrisome associated.” Recent studies have shown that complement system promotes cancer development and progression (Rutkowski et al., 2010). And the complement activation enhances tumor growth and increases metastasis in the tumor microenvironment (Afshar-Kharghan, 2017). Otherwise, as is known to all, the extracellular matrix (ECM) consists of 1,100 core-matrisome and matrisome-associated proteins and of glycosaminoglycans (Ricard-Blum and Vallet, 2016). Studies have shown that ECM plays an important role in the development of gastric cancer (Moreira et al., 2020). On the other hand, we identified several hub RNAs based on the PPI network, including HP, CRH, GCG, APOA5, and PENK. Some of these genes are involved in the development of cancer, for instance, PENK is downregulated in osteosarcoma and can activate the PI3K/Akt signaling pathway to restrain osteosarcoma cell migration (Zhang et al., 2020). Notably, PENK was also downregulated in our PPI network. Moreover, APOA5 on 11q23.3 is significantly related to prostate cancer risk (Major et al., 2014). In brief, these results provide clues for further study of the mechanisms underlying GC.
In the present study, based on multivariate Cox regression analysis, we used three lncRNAs, two miRNAs, and three mRNAs to construct the prognostic prediction models. These biomarkers have rarely been reported in GC, but they have been widely studied in other cancers. For instance, miR-653-5p overexpression was found to inhibit tumor growth in melanoma (Liu et al., 2020) but to promote cell proliferation and invasion in prostate cancer (Fu et al., 2019). In pancreatic cancer, miR-3923 was shown to inhibit cell viability (Li et al., 2016), and the expression of miR-3923 was related to lymph node metastasis in breast cancer (Wang et al., 2014). EphA8 was identified as a prognostic biomarker for epithelial ovarian cancer and oral tongue squamous cell carcinoma (Liu et al., 2016, 2018a). Most importantly, EphA8 contributed to poor prognosis via regulation of ADAM10 in GC (Chi et al., 2019). Our models also included AC094104.2, AC010457.1, AC091832.1, C5orf46, and HPR; their molecular mechanisms in GC need further research. At present, there have been few studies of lncRNAs, miRNAs, and mRNAs related to lymph node metastasis in GC. However, considering the important role of lymph node metastasis and ceRNA regulatory networks in cancer, the RNAs identified in this study may become novel diagnostic biomarkers and therapeutic targets for GC, and provide new insights for further in-depth research on the diagnosis and treatment of GC.
In conclusion, in the present study, a survival-related ceRNA network was constructed and prognostic biomarker prediction models were established. These results provide new insights into the ceRNA network in GC and novel clues for the screening of prognostic biomarkers of GC. However, this study had several limitations: first, the results of the analysis need to be verified by other experiments; and second, the biological roles of these biomarkers in GC need to be further studied.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, and further inquiries can be directed to the corresponding author.
Author Contributions
YZe and YZa designed and supervised the study. HZ and QJ analyzed the data. HZ wrote the original draft of the paper. XL edited the draft. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by The National Science Foundation for Young Scientists of China (grant no. 81802415 to YZa) and Shandong Provincial Natural Science Foundation (grant no. ZR2018PH025 to YZa).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We sincerely thank all participants involved in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.610501/full#supplementary-material
Footnotes
1. https://gdc.cancer.gov/access-data/gdc-data-transfer-tool
2. http://asia.ensembl.org/index.html
4. http://carolina.imis.athena-innovation.gr/
References
Afshar-Kharghan, V. (2017). The role of the complement system in cancer. J. Clin. Invest. 127, 780–789. doi: 10.1172/JCI90962
Bravo Neto, G. P., dos Santos, E. G., Victer, F. C., and Carvalho, C. E. (2014). Lymph node metastasis in early gastric cancer. Rev. Col. Bras. Cir. 41, 11–17. doi: 10.1590/S0100-69912014000100004
Cardon, T., Franck, J., Coyaud, E., Laurent, E. M. N., Damato, M., Maffia, M., et al. (2020). Alternative proteins are functional regulators in cell reprogramming by PKA activation. Nucleic Acids Res. 48, 7864–7882. doi: 10.1093/nar/gkaa277
Chen, J., Yu, Y., Li, H., Hu, Q., Chen, X., He, Y., et al. (2019). Long non-coding RNA PVT1 promotes tumor progression by regulating the miR-143/HK2 axis in gallbladder cancer. Mol. Cancer 18:33. doi: 10.1186/s12943-019-0947-9
Chen, P., Zhang, W., Chen, Y., Zheng, X., and Yang, D. (2020). Comprehensive analysis of aberrantly expressed long non-coding RNAs, microRNAs, and mRNAs associated with the competitive endogenous RNA network in cervical cancer. Mol. Med. Rep. 22, 405–415. doi: 10.3892/mmr.2020.11120
Chi, Y., Wang, D., Wang, J., Yu, W., and Yang, J. (2019). Long non-coding RNA in the pathogenesis of cancers. Cells 8:1015. doi: 10.3390/cells8091015
Fu, Q., Sun, Z., Yang, F., Mao, T., Gao, Y., and Wang, H. (2019). SOX30, a target gene of miR-653-5p, represses the proliferation and invasion of prostate cancer cells through inhibition of Wnt/β-catenin signaling. Cell. Mol. Biol. Lett. 24:71. doi: 10.1186/s11658-019-0195-4
Hayakawa, Y., Chang, W., Jin, G., and Wang, T. C. (2016). Gastrin and upper GI cancers. Curr. Opin. Pharmacol. 31, 31–37. doi: 10.1016/j.coph.2016.08.013
Heasley, L. E. (2001). Autocrine and paracrine signaling through neuropeptide receptors in human cancer. Oncogene 20, 1563–1569. doi: 10.1038/sj.onc.1204183
Heller, G., Rommer, A., Steinleitner, K., Etzler, J., Hackl, H., Heffeter, P., et al. (2015). EVI1 promotes tumor growth via transcriptional repression of MS4A3. J. Hematol. Oncol. 8:28. doi: 10.1186/s13045-015-0124-6
Hu, X., Miao, J., Zhang, M., Wang, X., Wang, Z., Han, J., et al. (2018a). miRNA-103a-3p promotes human gastric cancer cell proliferation by targeting and suppressing ATF7 in vitro. Mol. Cell 41, 390–400. doi: 10.14348/molcells.2018.2078
Hu, X., Zhang, M., Miao, J., Wang, X., and Huang, C. (2018b). miRNA-4317 suppresses human gastric cancer cell proliferation by targeting ZNF322. Cell Biol. Int. 42, 923–930. doi: 10.1002/cbin.10870
Kim, J., Yao, F., Xiao, Z., Sun, Y., and Ma, L. (2018). MicroRNAs and metastasis: small RNAs play big roles. Cancer Metastasis Rev. 37, 5–15. doi: 10.1007/s10555-017-9712-y
Li, X., Deng, S. J., Zhu, S., Jin, Y., Cui, S. P., Chen, J. Y., et al. (2016). Hypoxia-induced lncRNA-NUTF2P3-001 contributes to tumorigenesis of pancreatic cancer by derepressing the miR-3923/KRAS pathway. Oncotarget 7, 6000–6014. doi: 10.18632/oncotarget.6830
Li, J., Xu, X., Wei, C., Liu, L., and Wang, T. (2020). Long noncoding RNA NORAD regulates lung cancer cell proliferation, apoptosis, migration, and invasion by the miR-30a-5p/ADAM19 axis. Int. J. Clin. Exp. Pathol. 13, 1–13.
Liu, F., Hu, L., Pei, Y., Zheng, K., Wang, W., Li, S., et al. (2020). Long non-coding RNA AFAP1-AS1 accelerates the progression of melanoma by targeting miR-653-5p/RAI14 axis. BMC Cancer 20:258. doi: 10.1186/s12885-020-6665-2
Liu, L., Wang, X., and Ge, W. (2018a). EphA8 is a prognostic factor for oral tongue squamous cell carcinoma. Med. Sci. Monit. 24, 7213–7222. doi: 10.12659/MSM.910909
Liu, W., Wang, S., Sun, Q., Yang, Z., Liu, M., and Tang, H. (2018b). DCLK1 promotes epithelial-mesenchymal transition via the PI3K/Akt/NF-κB pathway in colorectal cancer. Int. J. Cancer 142, 2068–2079. doi: 10.1002/ijc.31232
Liu, X., Xu, Y., Jin, Q., Wang, W., Zhang, S., Wang, X., et al. (2016). EphA8 is a prognostic marker for epithelial ovarian cancer. Oncotarget 7, 20801–20809. doi: 10.18632/oncotarget.8018
Major, J. M., Yu, K., Weinstein, S. J., Berndt, S. I., Hyland, P. L., Yeager, M., et al. (2014). Genetic variants reflecting higher vitamin e status in men are associated with reduced risk of prostate cancer. J. Nutr. 144, 729–733. doi: 10.3945/jn.113.189928
Matsumoto, A., Pasut, A., Matsumoto, M., Yamashita, R., Fung, J., Monteleone, E., et al. (2017). mTORC1 and muscle regeneration are regulated by the LINC00961-encoded SPAR polypeptide. Nature 541, 228–232. doi: 10.1038/nature21034
Moreira, A. M., Pereira, J., Melo, S., Fernandes, M. S., Carneiro, P., Seruca, R., et al. (2020). The extracellular matrix: an accomplice in gastric cancer development and progression. Cells 9:394. doi: 10.3390/cells9020394
Nibbe, R. K., Chowdhury, S. A., Koyutürk, M., Ewing, R., and Chance, M. R. (2011). Protein-protein interaction networks and subnetworks in the biology of disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 3, 357–367. doi: 10.1002/wsbm.121
Peng, Y., Zhang, X., Ma, Q., Yan, R., Qin, Y., Zhao, Y., et al. (2017). MiRNA-194 activates the Wnt/β-catenin signaling pathway in gastric cancer by targeting the negative Wnt regulator, SUFU. Cancer Lett. 385, 117–127. doi: 10.1016/j.canlet.2016.10.035
Ricard-Blum, S., and Vallet, S. D. (2016). Proteases decode the extracellular matrix cryptome. Biochimie 122, 300–313. doi: 10.1016/j.biochi.2015.09.016
Rion, N., and Rüegg, M. A. (2017). LncRNA-encoded peptides: more than translational noise? Cell Res. 27, 604–605. doi: 10.1038/cr.2017.35
Ruscica, M., Dozio, E., Motta, M., and Magni, P. (2007). Relevance of the neuropeptide Y system in the biology of cancer progression. Curr. Top. Med. Chem. 7, 1682–1691. doi: 10.2174/156802607782341019
Rutkowski, M. J., Sughrue, M. E., Kane, A. J., Mills, S. A., and Parsa, A. T. (2010). Cancer and the complement cascade. Mol. Cancer Res. 8, 1453–1465. doi: 10.1158/1541-7786.MCR-10-0225
Salmena, L., Poliseno, L., Tay, Y., Kats, L., and Pandolfi, P. P. (2011). A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 146, 353–358. doi: 10.1016/j.cell.2011.07.014
Sun, Y., and Han, C. (2020). Long non-coding RNA TMPO-AS1 promotes cell migration and invasion by sponging miR-140-5p and inducing SOX4-mediated EMT in gastric cancer. Cancer Manag. Res. 12, 1261–1268. doi: 10.2147/cmar.S235898
Thomson, D. W., and Dinger, M. E. (2016). Endogenous microRNA sponges: evidence and controversy. Nat. Rev. Genet. 17, 272–283. doi: 10.1038/nrg.2016.20
Venerito, M., Vasapolli, R., Rokkas, T., and Malfertheiner, P. (2018). Gastric cancer: epidemiology, prevention, and therapy. Helicobacter 23 (Suppl. 1): e12518. doi: 10.1111/hel.12518
Wang, B., Li, J., Sun, M., Sun, L., and Zhang, X. (2014). miRNA expression in breast cancer varies with lymph node metastasis and other clinicopathologic features. IUBMB Life 66, 371–377. doi: 10.1002/iub.1273
Wei, C., and Gao, J. J. (2019). Downregulated miR-383-5p contributes to the proliferation and migration of gastric cancer cells and is associated with poor prognosis. PeerJ 7:e7882. doi: 10.7717/peerj.7882
Wei, Y., Wang, Z., Zong, Y., Deng, D., Chen, P., and Lu, J. (2020). LncRNA MFI2-AS1 promotes HCC progression and metastasis by acting as a competing endogenous RNA of miR-134 to upregulate FOXM1 expression. Biomed. Pharmacother. 125:109890. doi: 10.1016/j.biopha.2020.109890
Wu, X., Qu, D., Weygant, N., Peng, J., and Houchen, C. W. (2020). Cancer stem cell marker DCLK1 correlates with tumorigenic immune infiltrates in the colon and gastric adenocarcinoma microenvironments. Cancer 12:274. doi: 10.3390/cancers12020274
Xiao, J., Lin, L., Luo, D., Shi, L., Chen, W., Fan, H., et al. (2020). Long noncoding RNA TRPM2-AS acts as a microRNA sponge of miR-612 to promote gastric cancer progression and radioresistance. Oncogene 9:29. doi: 10.1038/s41389-020-0215-2
Xu, G., Li, N., Zhang, Y., Zhang, J., Xu, R., and Wu, Y. (2019). MicroRNA-383-5p inhibits the progression of gastric carcinoma via targeting HDAC9 expression. Braz. J. Med. Biol. Res. 52:e8341. doi: 10.1590/1414-431x20198341
Yang, W., Lai, Z., Li, Y., Mu, J., Yang, M., Xie, J., et al. (2019). Immune signature profiling identified prognostic factors for gastric cancer. Chin. J. Cancer Res. 31, 463–470. doi: 10.21147/j.issn.1000-9604.2019.03.08
Zhang, G., Li, S., Lu, J., Ge, Y., Wang, Q., Ma, G., et al. (2018a). LncRNA MT1JP functions as a ceRNA in regulating FBXW7 through competitively binding to miR-92a-3p in gastric cancer. Mol. Cancer 17:87. doi: 10.1186/s12943-018-0829-6
Zhang, H. P., Yu, Z. L., Wu, B. B., and Sun, F. R. (2020). PENK inhibits osteosarcoma cell migration by activating the PI3K/Akt signaling pathway. J. Orthop. Surg. Res. 15:162. doi: 10.1186/s13018-020-01679-6
Zhang, K., Zhang, X., Cai, Z., Zhou, J., Cao, R., Zhao, Y., et al. (2018b). A novel class of microRNA-recognition elements that function only within open reading frames. Nat. Struct. Mol. Biol. 25, 1019–1027. doi: 10.1038/s41594-018-0136-3
Zhang, J., Zhu, Z., Sun, Z., Sun, X., Wang, Z., and Xu, H. (2014). Survivin gene expression increases gastric cancer cell lymphatic metastasis by upregulating vascular endothelial growth factor-C expression levels. Mol. Med. Rep. 9, 600–606. doi: 10.3892/mmr.2013.1858
Keywords: gastric cancer, lncRNA, miRNA, mRNA, ceRNA, prognosis
Citation: Zhao Y, Zhang H, Ju Q, Li X and Zheng Y (2021) Comprehensive Analysis of Survival-Related lncRNAs, miRNAs, and mRNAs Forming a Competing Endogenous RNA Network in Gastric Cancer. Front. Genet. 12:610501. doi: 10.3389/fgene.2021.610501
Edited by:
Daniele Vergara, University of Salento, ItalyReviewed by:
Kyoung-Mee Kim, Sungkyunkwan University, South KoreaMarina Damato, University of Salento, Italy
Copyright © 2021 Zhao, Zhang, Ju, Li and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuxin Zheng, eXh6aGVuZ0BxZHUuZWR1LmNu
†These authors have contributed equally to this work