 Andressa O. de Lima1
Andressa O. de Lima1 Juliana Afonso2
Juliana Afonso2 Janette Edson3
Janette Edson3 Esteban Marcellin4
Esteban Marcellin4 Robin Palfreyman4
Robin Palfreyman4 Laercio R. Porto-Neto5
Laercio R. Porto-Neto5 Antonio Reverter5
Antonio Reverter5 Marina R. S. Fortes3*
Marina R. S. Fortes3*- 1Department of Production and Animal Health, School of Veterinary Medicine, São Paulo State University (UNESP), Araçatuba, Brazil
- 2Department of Animal Science, University of São Paulo/ESALQ, Piracicaba, Brazil
- 3School of Chemistry and Molecular Biosciences, The University of Queensland, St Lucia, QLD, Australia
- 4Australian Institute for Bioengineering and Nanotechnology (AIBN), The University of Queensland, St Lucia, QLD, Australia
- 5CSIRO Agriculture and Food, Queensland Bioscience Precinct, St. Lucia, QLD, Australia
Spermatogenesis relies on complex molecular mechanisms, essential for the genesis and differentiation of the male gamete. Germ cell differentiation starts at the testicular parenchyma and finishes in the epididymis, which has three main regions: head, body, and tail. RNA-sequencing data of the testicular parenchyma (TP), head epididymis (HE), and tail epididymis (TE) from four bulls (three biopsies per bull: 12 samples) were subjected to differential expression analyses, functional enrichment analyses, and co-expression analyses. The aim was to investigate the co-expression and infer possible regulatory roles for transcripts involved in the spermatogenesis of Bos indicus bulls. Across the three pairwise comparisons, 3,826 differentially expressed (DE) transcripts were identified, of which 384 are small RNAs. Functional enrichment analysis pointed to gene ontology (GO) terms related to ion channel activity, detoxification of copper, neuroactive receptors, and spermatogenesis. Using the regulatory impact factor (RIF) algorithm, we detected 70 DE small RNAs likely to regulate the DE transcripts considering all pairwise comparisons among tissues. The pattern of small RNA co-expression suggested that these elements are involved in spermatogenesis regulation. The 3,826 DE transcripts (mRNAs and small RNAs) were further subjected to co-expression analyses using the partial correlation and information theory (PCIT) algorithm for network prediction. Significant correlations underpinned the co-expression network, which had 2,216 transcripts connected by 158,807 predicted interactions. The larger network cluster was enriched for male gamete generation and had 15 miRNAs with significant RIF. The miRNA bta-mir-2886 showed the highest number of connections (601) and was predicted to down-regulate ELOVL3, FEZF2, and HOXA13 (negative co-expression correlations and confirmed with TargetScan). In short, we suggest that bta-mir-2886 and other small RNAs might modulate gene expression in the testis and epididymis, in Bos indicus cattle.
Introduction
Spermatozoid is the most specialized cell in mammalian organisms. Spermatogenesis, the differentiation of male germ cells, relies on a complex network of specialized molecular mechanisms that are critical to male fertility (MacLean and Wilkinson, 2005; Marengo, 2008; Hermann et al., 2018). During spermatogenesis, three sequential phases of cell proliferation and differentiation occur, where there is an extensive multiplication and proliferation of spermatogonial stem cells, followed by a meiotic division, and finally a remodeling of the nuclear and cellular components forming sperm cells (Abou-Haila and Tulsiani, 2000). Spermatogenesis starts with the multiplication of spermatogonial stem cells followed by their meiotic division into spermatids, which then differentiate into spermatozoa that are released into the lumen of seminiferous tubules in the testis (Staub and Johnson, 2018). Spermatozoa leaving the testis transit through the epididymis, where they further mature, acquiring motility and the ability to fertilize the egg (Cornwall, 2009). The epididymis is composed of caput (head), corpus (body), and cauda (tail), consisting of region-specific characteristics, including a region-specific luminal protein profile (Cornwall, 2009). The spermatozoa from the testis pass to the epididymis, which contributes to their maturation (Belleannée et al., 2012). In the epididymis, secreted luminal proteins, water, and solute balance contribute to the luminal environment necessary for sperm maturation (Huang et al., 2006). Fully formed mature sperm cells emerge from the tail epididymis and are stored until the ejaculation event in the vas deferens.
Recently, Hombach and Kretz (2016) have proposed a role for small RNAs in the testis and epididymis: they may be key regulators of gene expression in spermatogenesis, as they are in most cellular processes. RNA polymerase II transcribes small RNAs, and because of this, their expression is mostly regulated by mechanisms that regulate RNA polymerase II activity, such as the interaction of transcription factors and specific DNA sequences (Fuda et al., 2009). Some classes of small RNAs, such as micro (miRNAs), small nuclear (snRNAs), and small nucleolar (snoRNAs) RNAs, play a role in spermatogenesis by being involved in meiosis (Pradillo and Santos, 2018). Small RNAs regulate sperm maturation through mRNA-silencing mechanisms (Nixon et al., 2019), such as destabilizing mRNAs via deadenylation complexes (Bartel, 2018). In addition, miRNAs are important to maintain the epididymis homeostasis and function (Nixon et al., 2019). Small RNAs are present in epididymosomes (Sullivan, 2016) and can modulate mRNA expression in spermatozoa during the epididymal transit (Belleannée, 2015).
Considering the different roles played by the testis and epididymis, some studies investigate the pattern of gene expression of male tract reproductive tissues to shed light on the biological processes related to each specific tissue. Among these studies, there was a characterization of epididymis gene expression in humans (Thimon et al., 2007; Browne et al., 2015) and yak (Zhao et al., 2019). Guyonnet et al. (2009) have reported the differences in the expression pattern between the testis and epididymis in boar. However, knowledge of gene expression patterns in the testis or epididymis of Bos indicus bulls is lacking, and the hypothesized role of small RNAs in these tissues remains to be confirmed.
By sampling biopsies from testicular parenchyma (TP), head epididymis (HE), and tail epididymis (TE), we obtained different cell groups that are representative of spermatogenesis in three different stages. In the TP, Sertoli, Leydig, and differentiating male germ cells represent a group of cells with the DNA still bound to histones. In TE and HE, sperm cells are further along their differentiation process, and protamines instead of histones are observed, which is typical of mature sperm cells as described before (Fortes et al., 2014). Therefore, when sampling these tissues, we opened a window to investigate spermatogenesis. Our aim was to combine RNA sequencing, differential gene expression, functional enrichment, and co-expression analyses to investigate potential transcript interactions in the male reproductive system, using Bos indicus bulls as a model organism.
Studies on the testicular transcriptome, such as this one, are not only useful for understanding male fertility but also very helpful for genome annotation. Testicular tissue may be under less evolutionary pressure and this can be promoting duplication of protein-coding events and an overabundance of non-coding RNAs (ncRNAs), and not all the protein-coding genes expressed are functional (Soumillon et al., 2013). It is generally reported that the testes have higher gene expression than other tissues (Soumillon et al., 2013; Uhlén et al., 2015). The data reported on this study is available through the Functional Annotation of Animal Genomes (FAANG) Consortium for further research1.
Materials and Methods
Samples and Data
All the experimental procedures were conducted and approved by the ethics committee of the University of Queensland, Brisbane, Australia (protocol number: ANRFA/SCMB/094/16). Tissue samples were collected after euthanasia of cattle for commercial purposes, as part of normal beef industry activities. Testicular samples (n = 4) from mature Brahman bulls (approx. 2 years old) were collected shortly after slaughter and delivered to the research team, who performed the biopsies. For each bull, we performed three biopsies: testicular parenchyma (TP), head epididymis (HE), and tail epididymis (TE). Each biopsy (approximately 50 mg of tissue) was collected in Eppendorf tubes with 1 ml of RNAlater® (RNA stabilizing reagent, Ambion Inc., Austin, TX, United States). The biopsies were left to stabilize in a cold room overnight. After that, the RNAlater® fluid was pipetted out, and the tubes with tissue samples were stored in a −80°C freezer until RNA extraction.
RNA Extraction and Integrity
Biopsy samples were homogenized with Precellys 25 system with zirconium oxide beads (Bertin Technologies SAS, Montigny-le-Bretonneux, France). Following homogenization, RNA was extracted using the total RNA extraction protocol, with the RNeasy kit (QIAGEN Pty Ltd., Melbourne, VIC, Australia). After DNAse treatment, using TURBO DNAse I, each sample was purified using the Zymo Clean and Concentrator Kit as per the manufacturer’s instructions (Zymo Research, CA, United States). The RNA concentration was measured by a NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, United States). Samples without the optimal 260:280 ratio, which was between 1.8 and 2.1, were excluded from the experiment. The RNA integrity was verified by Agilent Bioanalyzer (Agilent, Santa Clara, CA, United States), and only samples with an RNA integrity number (RIN) above eight (RIN > 8) were used for RNA sequencing. When needed, RNA extraction was repeated to achieve this quality and integrity.
RNA Sequencing, Data Processing, and Quantification
Library preparation and RNA sequencing were performed following the standard Illumina protocols for the HiSeq platform (Illumina, San Diego, CA, United States). The library prep kit was the Illumina stranded total RNA kit with Ribo-Zero Gold (Illumina, San Diego, CA, United States). Pair-end 125-base pair (bp) sequencing was conducted across three lanes of an Illumina HiSeq 2000 v4 analyzer (Illumina Inc., San Diego, CA, United States) using standard protocols, generating approximately 60 to 100 million reads per sample. All the samples were run across all the lanes used, in order to avoid any lane effect on our dataset. The quality control procedure included removing adaptors and short reads. The software TrimGalore 0.4.5 was used for trimming adaptors and for the removal of short reads, where one of the pair-end reads was shorter than 20 base pairs2. Before trimming, all reads were 126-bases long, and after trimming, lengths ranged from 20 to 126. Trimming was run in paired mode to avoid unpaired reads after trimming. The quality of trimmed reads was high as evaluated with FastQC 0.11.73, and no quality cut off was required.
The sequencing reads were aligned to the Bos taurus genome assembly (UMD 3.1 assembly available in Ensembl database) using the HISAT2 v.2.1.0 (Kim et al., 2015) following the mapping evaluation by Qualimap 2.2.1 (Okonechnikov et al., 2016), reporting only known transcripts from the current bovine annotation. The “reads per kilobase per million mapped reads” (RPKM = total exon reads/mapped reads in millions × exon length in kilobase) were calculated and log2 transformed for data normalization (Mortazavi et al., 2008). To further normalize the gene expression values, we used a mixed model approach that considered the effects of library, tissue, and gene-by-tissue interaction as previously detailed (Reverter et al., 2005; Cánovas et al., 2014). In brief, the mixed model contained the sequencing library treated as a fixed effect, while the interaction of tissue, gene, and animal were fitted as random effects. Fitting this animal, gene and tissue interaction is a robust methodology, commonly used in gene expression experiments to reduce the noise. We were able to fit tissue as we had three different tissues per animal: TP, HE, and TE. The VCE6 software4 was used to solve the mixed model equations and to estimate variance components associated with random effects. The normalized gene expression values were used in all subsequent analyses, including differential gene expression.
Differential Expression Analysis
To identify differentially expressed (DE) transcripts (protein-coding and small non-coding RNAs) in specific regions of the epididymis (head and tail) and in the testis (testicular parenchyma), we carried out pairwise comparisons among the epididymis (head and tail) and testis (testicular parenchyma) tissues.
Testis and epididymis expression data comprised over 21,000 transcripts, with at least 10 counts per million reads in the data. Among expressed transcripts, 20,155 were small non-coding RNAs (miRNAs, snRNAs, and snoRNAs) and protein-coding RNAs (mRNA); for more detail, see Figure 1. Prior to differential expression analysis, transcripts with less than two RPKM in at least three samples were removed. After filtering, we considered 17,221 transcripts for differential expression and subsequent analyses, which investigated the co-expression relationships between protein-coding RNA (mRNA) and small non-coding RNAs (miRNAs and snoRNAs) in testicular and epididymis tissues. We performed differential expression analysis contrasting the three tissues sampled, in pairwise comparisons: HE vs. TE, HE vs. TP, and TE vs. TP. To identify the DE transcripts (mRNAs and small RNAs), we used the Limma package in R (Ritchie et al., 2015) to compute the moderated t-statistics, using the empirical Bayes methods (eBayes) and the default parameters. The DE transcripts with adjusted P value ≤ 0.05 (Benjamini and Hochberg, 1995) and fold change ≥ 2 were considered significant. We generated three lists of DE transcripts, one for each pairwise comparison: HE/TE, HE/TP, and TE/TP.
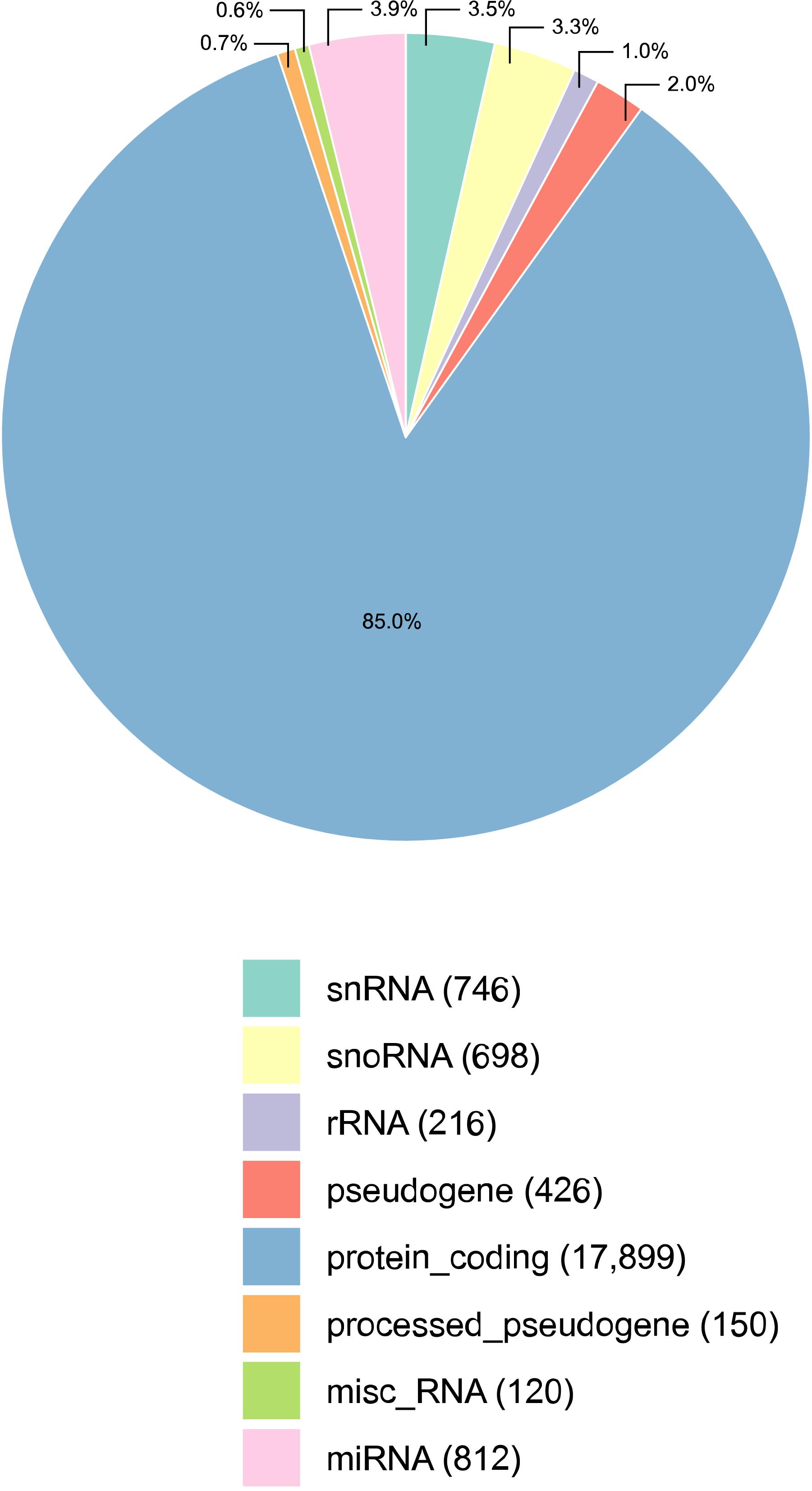
Figure 1. Percentage of transcripts detected per annotation category, in all samples of the male reproductive tract (epididymis and testis) collected from four Bos indicus bulls. The majority of the transcripts detected were protein coding mRNAs. The absolute number of transcripts detected per category is given in parenthesis, next to its classification. Three types of small RNAs were detected: micro RNAs (miRNAs), small nuclear RNAs (snRNAs), and small nucleolar RNAs (snoRNAs). Misc_RNA stands for miscellaneous types of RNAs.
Functional Enrichment Analysis of DE Transcripts
The three lists of DE transcripts were the target lists for functional enrichment analyses. We performed the enrichment analysis using the ClueGO v. 2.5.1 bioinformatics tool (Bindea et al., 2009), a plug-in of the Cytoscape software (Shannon et al., 2003). The background gene list for functional enrichment was based on the Bos taurus genome, available as a default database in ClueGO. In this analysis, we identified the gene ontology terms (GO terms) and pathways [from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database] that were over-represented in the target DE list. Redundant GO terms were clustered, considering a Kappa score = 0.4, and adjusted P values ≤ 0.05 (Bonferroni step-down method) were observed when reporting on significant GO terms or pathways. To improve the functional annotation of the DE transcripts, we cross-checked these lists with the manually curated database for bovine transcription factors (TF) (de Souza et al., 2018).
miRNA Target Genes Prediction
We predicted the target genes for the DE miRNA using the TargetScan function of R package hoardeR5. This function uses all the information stored in the database targetscan.org (release 7.2) that is available for the Bos taurus genome in terms of miRNA data. TargetScan predicts the targets of miRNAs by searching for the presence of conserved 8mer, 7mer, and 6mer sites that match the seed region of each miRNA (Lewis et al., 2005). Release 7 of TargetScan uses an improved method to predict targeting efficacy (the context + + model) (Agarwal et al., 2015), uses 3′ UTR profiles that indicate the fraction of mRNA containing each site (Nam et al., 2014), and uses updated miRNA families curated by Chiang et al. (2010) and Fromm et al. (2015). Of note, TargetScan is limited to known sites and 3′ UTR profiles and so it cannot predict all possible interactions between miRNA and target genes.
Co-expression Network Analysis
We performed a co-expression analysis with the log-normalized expression values of all transcripts (mRNAs and small RNAs that were DE) using the partial correlation and information theory (PCIT) algorithm (Reverter and Chan, 2008). Among the significant correlations according to PCIT, we prioritized the most extreme correlations (higher than 0.95 or lower than -0.95) as stronger evidence of interaction between transcripts. These significant and extreme correlations were used to construct a co-expression network, visualized with the Cytoscape software (Shannon et al., 2003). In the network, we marked as attributes the small RNAs (miRNAs, snoRNA, and snRNA), transcription factors (TFs), the tissue comparison in which the transcript was DE, and the small RNAs presenting significant regulatory impact factor (RIF) values for at least one tissue comparison (Reverter et al., 2010). Also, we pointed out hub transcripts in the network. Hubs are transcripts with higher than the average significant correlations, beyond two standard deviations (i.e., hubs are hyper-connected). In the same context, hub centrality elements are transcripts in the network with higher betweenness centrality than the average (more than two standard deviations), meaning that they tend to link different parts of the network.
Regulatory Impact Factor Analysis
The regulatory impact of each DE small RNAs over the DE genes for the same comparison analysis was estimated with the RIF algorithm (Reverter et al., 2010). The original application of the RIF algorithm was to determine the regulatory impact of TFs over selected genes (targets) related to a given trait through their expression values between contrasting groups (Reverter et al., 2010). In our experiment, for each pairwise comparison between sampled tissues, we used the RIF algorithm to determine the regulatory impact of each DE small RNA over the DE genes that were identified in the same pairwise comparison. For example, DE small RNA in the TP/HE comparison were tested as potential regulators of the DE genes identified in the TP/TE analyses. The RIF algorithm was selected for this analyses as its predicted regulatory roles have been showcased and validated in previous studies (Bottje et al., 2017; Nolte et al., 2019). A limitation of our analyses is that in vitro validations for the predicted co-expression and regulatory relationships were beyond the project scope. To mitigate this limitation, we used RIF in combination with PCIT, TargetScan, and in silico analyses of the minimum free energy of miRNA-target hybridization.
Minimum Free Energy: miRNA and Target Hybridization
The miRNA that were DE, significant according to RIF, and had potential targets with negative co-expression correlations were hypothesized as down-regulators of their targets. When the hypothesis was supported by the identification of binding sites confirmed with TargetScan, the miRNA were subjected to a final analysis: we estimated the minimum free energy (mfe) of the hybridization between the selected miRNA and their confirmed target genes, using RNAhybrid tool (Rehmsmeier et al., 2004). For this, we retrieved the miRNA mature sequence from the miRBase sequence database6 and the cDNA sequences of the genes from BioMart (Durinck et al., 2009). A transcript with a mfe less than -20 kcal/mol can be considered a potential target for the miRNA in question (Yen et al., 2019).
Results
Samples from the head and tail epididymis (HE and TE) and the testicular parenchyma (TP) of Bos indicus bulls were used for RNA sequencing. A total of 3,826 DE transcripts (mRNAs and small RNAs) were identified across the tissues in three pairwise comparisons: HE/TE, HE/TP, and TE/TP. A co-expression network was predicted and analyzed, with emphasis on investigating potential regulators of DE genes in these tissues. The network was enriched for male gamete generation and so we infer that the potential regulators of the identified DE genes might contribute to spermatogenesis.
Transcript Expression Patterns in Male Reproductive Tissues
Reads from RNA-sequencing of HE, TE, and TP were mapped to the genome, and the expression data was summarized per transcript category (Figure 1). All samples considered, the RNA sequencing data comprised of 85.0% mRNAs (17,899) and 10.7% small RNAs, including 812 miRNAs, 746 snRNAs, and 698 snoRNAs. In the bovine reference genome, approx. 13% of all transcripts are small RNAs, and so this is not too far from the 10% identified here. Ribosomal RNA (rRNA) were not well represented as expected in view of the library preparation methods. The library preparation allowed quantifying the expression of mRNAs and small RNAs, but it is also a limitation of this study since it did not enrich for small RNAs and no discovery of small RNAs was conducted. Mitochondrial RNA is not included in Figure 1 because they were less than 1% of the distribution. After the quality control, we kept 17,221 transcripts expressed that were quantified across tissues for all subsequent analyses. The expression pattern of TP samples was different from the epididymis samples (both HE and TE) according to the principal component analysis (PCA) performed, see Supplementary Figure 1.
Differentially Expressed Transcripts and Functional Enrichment Analysis
The number of DE transcripts identified (FDR ≤ 0.05 and log2 fold-change > 2) in each pairwise comparison between HE, TE, and TP are reported in Table 1. The full details on all DE transcripts are provided in Supplementary Table 1. In Supplementary Table 1, positive and negative signals of the log-transformed fold change indicate if the transcript is up- or down-regulated for the first tissue in each comparison (for HE/TE and HE/TP comparisons, a positive fold change represents up-regulation in HE; in TE/TP comparison, a positive fold-change means the transcript was up-regulated in TE). Figure 2 showcases the transcript expression patterns as volcano plots with the fold change plotted against the significance for each transcript, in each of the comparisons. We identified 40 DE transcripts that were in common for all the comparisons (Figure 2D and Supplementary Table 2). Our DE analysis identified a total of 3,826 transcripts that were DE in at least one of the three comparisons.
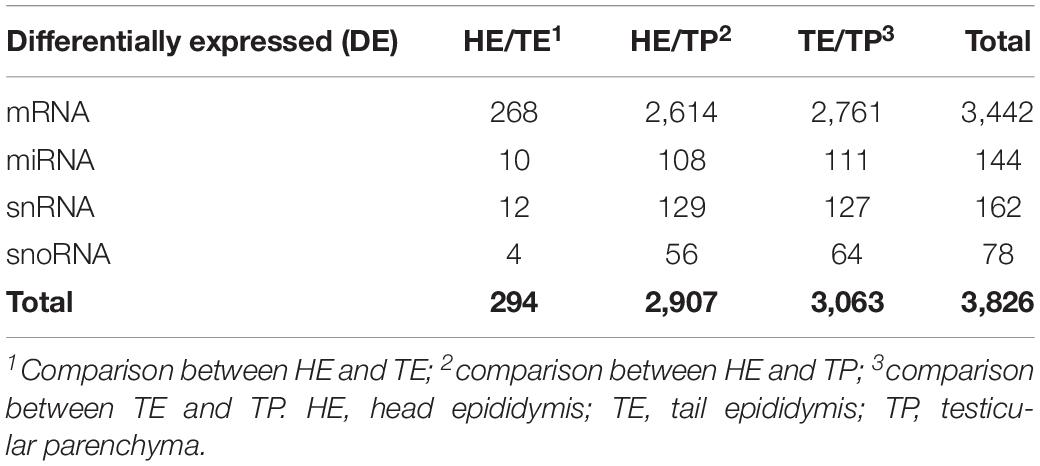
Table 1. Summarized differentially expressed (DE) genes and small RNAs in each comparison of male reproductive (epididymis and testis) tissues of Bos indicus bulls.
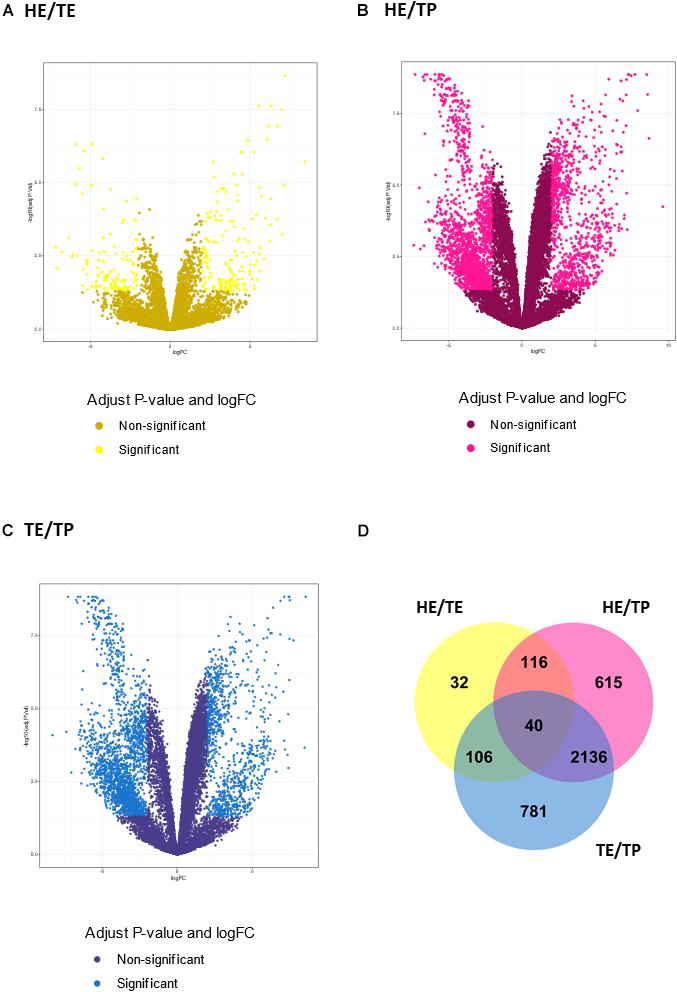
Figure 2. Transcript expression patterns displayed as volcano plots with the log2 fold change in the x-axis and the –log10 (P value) in the y-axis for three pairwise comparisons: (A) quantified transcripts in the head versus tail epididymis comparison (HE/TE, in yellow). (B) quantified transcripts in the head epididymis versus testicular parenchyma comparison (HE/TP, in pink). (C) quantified transcripts in the tail epididymis versus testicular parenchyma comparison (TE/TP, in blue). In (A–C), the light shade dots represent the significantly different genes, while the dark shade dots are not significant. In (D), a Venn diagram summarizes the significantly different transcripts identified in each comparison, including their overlaps.
Enriched GO terms and KEGG pathways for the total of 3,826 DE transcripts are shown in Figure 3 (details in Supplementary Table 3). The DE genes identified between HE and TE formed a target list that was enriched for nine GO terms and four KEGG pathways. The most significant GO term in the HE/TE comparison was detoxification of copper ion (corrected P = 2.62 × 10–7). DE genes identified between HE and TP were enriched for 46 GO terms and one KEGG pathway. In the third comparison, TE/TP, the DE genes were enriched for 36 GO terms and two KEGG pathways. When TP was compared to the epididymis regions, some of the most significant GO terms were gated channel activity, cellular protein modification, male gamete generation, neuroactive ligand-receptor interaction, spermatogenesis, and acrosomal vesicle (Supplementary Table 3).
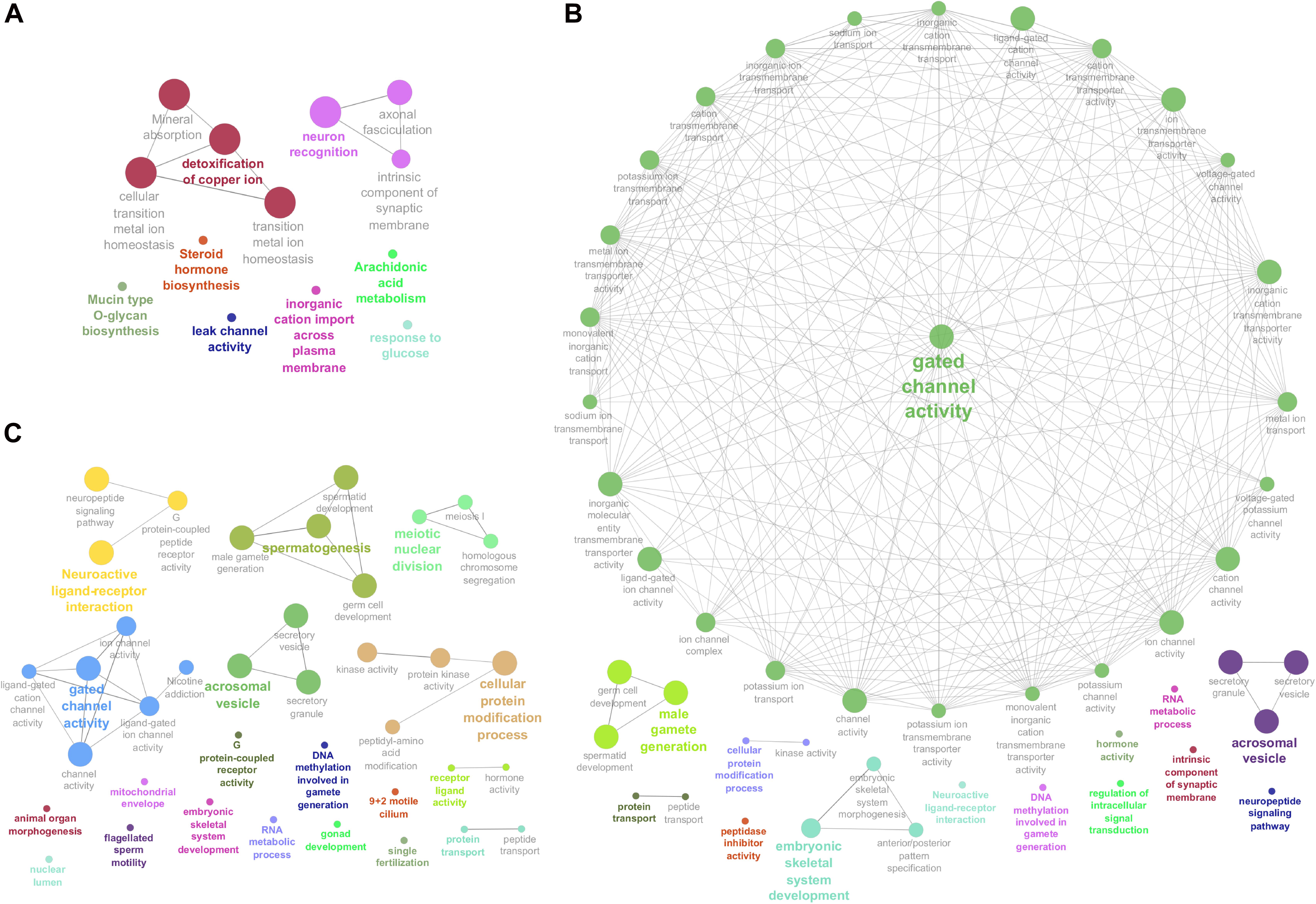
Figure 3. Enriched Genome Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways identified by the ClueGO software for each comparison of differentially expressed (DE) analysis between the male reproductive tissues (HE/TE: head vs. tail; HE/TP: head vs. testicular parenchyma; TE/TP: tail vs. testicular parenchyma) of Bos indicus bulls. (A) Enriched GO terms and KEGG pathways of HE/TE comparison. (B) Enriched GO terms and KEGG pathways of HE/TP comparison. (C) Enriched GO terms and KEGG pathways of TE/TP comparison. GO terms and KEGG pathways are represented by circles.
Small RNAs With Regulatory Potential and Co-expression Networks
Among the 3,826 DE transcripts, 384 were small RNAs and 3,442 were mRNA genes. We identified 71 small RNAs that might modulate the DE genes, according to the significant RIF score (RIF 1 or 2 higher than | 1.96|; Supplementary Table 4). The expression pattern of these 71 small RNAs with regulatory potential differed between samples, across the male reproductive tract (Figure 4). Overall, we observed that small RNAs showed an expression pattern in the testis that was different from their epididymis expression. The difference between the head and tail epididymis was less pronounced, and this is similar to the PCA results for all transcripts.
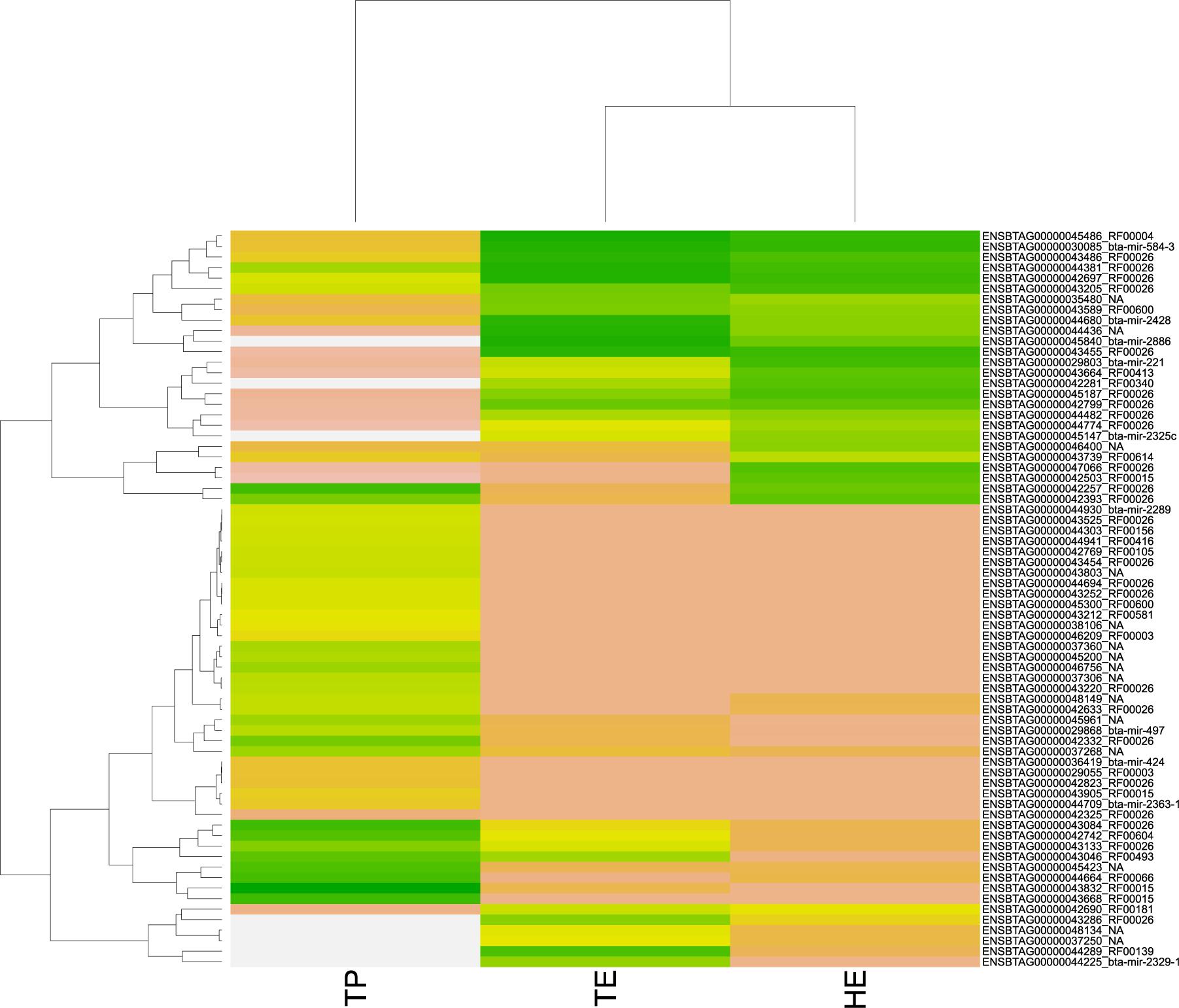
Figure 4. Heatmap of pattern gene expression of small RNAs (snRNAs and miRNAs) with regulatory potential, according to significant values in the regulatory impact factor (RIF) metric. Small RNAs are shown on the Y-axis and the total of expression per biopsies of epididymis head (HE), epididymis tail (TE), and testicular parenchyma (TP). The colors correspond to the intensity of expression per tissue. The intensity is scaled by the colors white, yellow, and green. The low expression is represented by white, followed by salmon, yellow, and green (high expression).
The co-expression network was inferred using significant correlations (> |0.95|). This meant that 3,639 transcripts were nodes linked by 175,052 edges in the network, which is available as a Cytoscape file (Supplementary Material, cys file). The co-expression network was formed by multiple clusters, not all connected to each other (Supplementary Figures 2, 3). A larger cluster with 2,216 transcripts connected through 158,807 edges was the prominent feature in the network. This large cluster was functionally enriched for male gamete generation, germ cell development, and sperm capacitation, among other GO terms (Figure 5 and Supplementary Table 5).
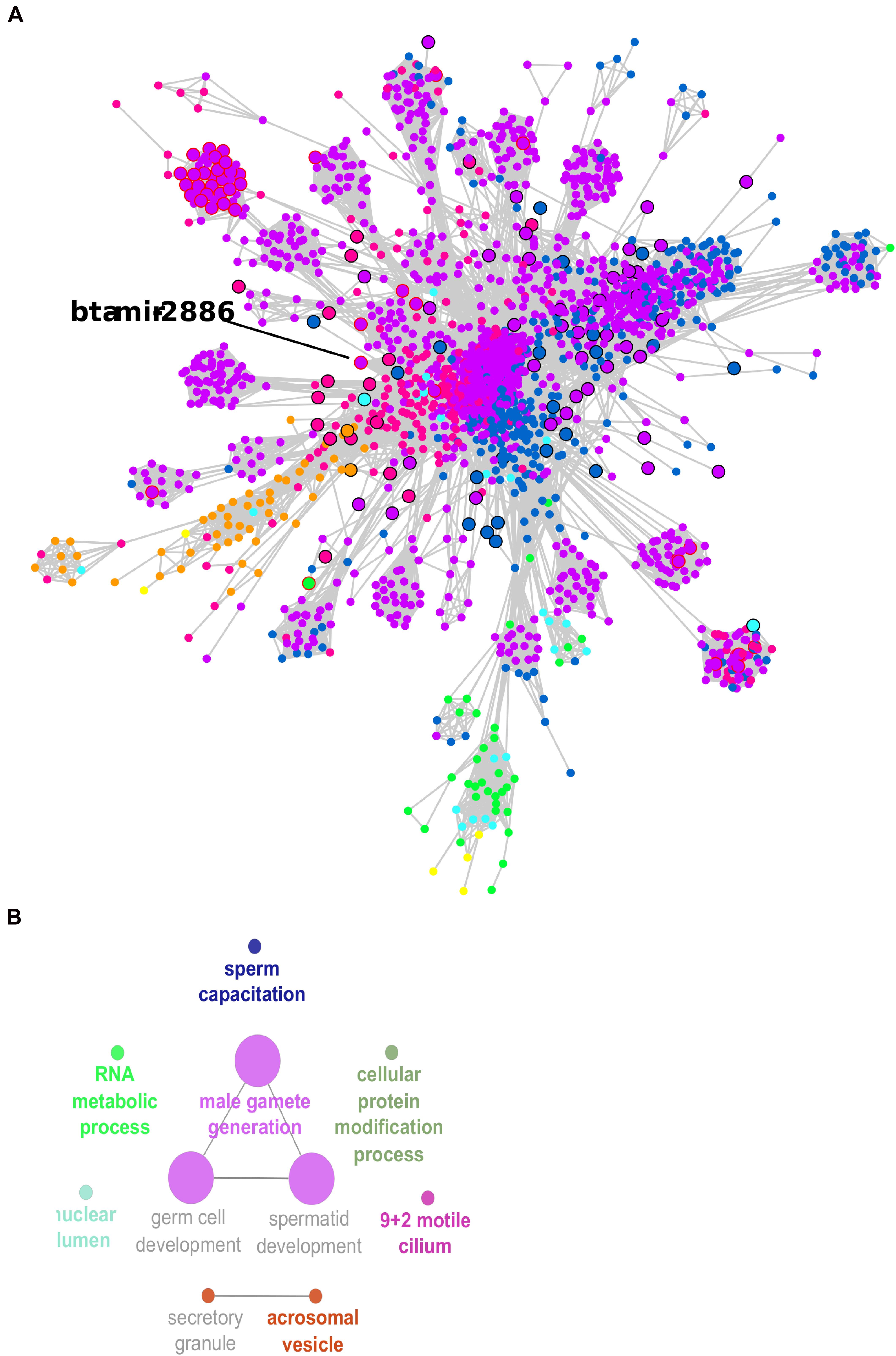
Figure 5. Co-expression network large cluster of differentially expressed (DE) transcripts enriched for male gamete generation. (A) Node sizes are proportional to the number of connections (highly connected nodes appear bigger). The nodes are the DE transcripts and the edges are the significant correlations (partial correlation and information theory, PCIT > | 0.95|). Black borders represent hub transcripts. Red borders represent small RNAs with significant regulatory impactor factor values. Node color: yellow for DE only in the head and tail epididymis comparison (HE/TE); pink for DE only in the head epididymis and testicular parenchyma comparison (HE/TP); blue for DE only in the tail epididymis and testicular parenchyma comparison (HE/TP); orange for DE in the HE/TE and the HE/TP comparisons; green for DE in the HE/TE and the TE/TP comparisons; purple for DE in the HE/TP and the TE/TP comparisons; and turquoise for DE in all comparisons. (B) Enriched GO terms and KEGG pathways for the cluster represented in (A).
In the large cluster of the co-expression network, the RIF significant small RNA bta-mir-2886 was the hub. Significant correlations suggested 601 co-expressed transcripts for bta-mir-2886, including genes and other small RNA. This is the highest number of connections for a RIF regulator in the network (Supplementary Table 4). Considering the connections between these 601 potential targets and their first neighbors, we observed a total of 1,035 transcripts that were directly or indirectly linked to bta-mir-2886 in the network (Supplementary Table 6). Among the 601 directly co-expressed transcripts, we identified one isoform of U4 spliceosomal RNA with significant RIF value and 38 transcription factors (TF).
Considering the first neighbors of bta-mir-2886 in the network, we identified a total of 241 negative correlations and 360 positive correlations. Among the negative correlations, 204 were mRNA genes. Only three of these potential targets were confirmed to have a site for hybridization with bta-mir-2886 according to TargetScan. The confirmed targets were ELOVL3, FEZF2, and HOXA13. All three had a mfe that is further evidence for bta-mir-2886 acting as their down-regulator: −28.5 kcal/mol for ELOVL3, -34.95 kcal/mol for FEZF2, and -35.75 kcal/mol for HOXA13. All TargetScan results for bta-mir-2886 are provided in Supplementary Table 7. TargetScan analyses of all DE miRNAs were performed and provided evidence for 1,846 DE genes that can be proposed as targets of miRNA regulation in the male reproductive tract. However, TargetScan analyses could not explain all the co-expression observed between miRNA and genes. This result is expected, since co-expression is not necessarily caused by direct hybridization and regulation, as there are many – and complex – molecular mechanisms that can lead to co-expression (Fionda, 2019).
Discussion
Transcript Expression Patterns in Male Reproductive Tissues
In this study, we identified 17,221 transcripts quantified in bovine samples of the head and tail epididymis and the testicular parenchyma (HE, TE, and TP). This amounts to 64% of the genome (or 17,221 of 26,740 transcripts). The expression of a relatively large number of genes and small RNAs confirmed previous reports that suggest the testis as a good sample for functional genome annotation. In humans, the testis expressed a larger number of genes in comparison to other tissues (Uhlén et al., 2015). Harhay et al. (2010) has shown a cluster of genes exclusively expressed in the testis of bovine. The data we reported on is available through the Functional Annotation of Animal Genomes (FAANG) Consortium for future reference and further use (see text footnote 1). Herein, we focus on differential expression analyses, functional enrichment, co-expression network analyses, and regulatory impact metrics (RIF and additional in silico tests) that point to potential modulators of transcription in male reproductive tissues.
We identified 71 small RNAs with significant RIF values, interpreted as potential contributors to the modulation of over 3,000 DE transcripts. The expression patterns of these 71 potential regulators were similar to the overall pattern observed, with the testis expression contrasting with the epididymis expression. The expression patterns in all epididymis samples were relatively similar. These expression patterns might reflect the specific function and distinguished cell populations of the studied samples. In boar samples, Guyonnet et al. (2009) has observed different patterns of gene expression in the testis and epididymis. A different role for small regulatory RNAs has been proposed, specific for each region of the male reproductive system, associated with regional function. For example, Guyonnet et al. (2009) has found that genes related to spermatogenesis are more prominent in the testis, compared to the epididymis. Their observation corroborates our findings. In the HE/TE comparison, only two of the 71 small RNAs detected with RIF were DE and relatively fewer genes were DE. Still, the epididymis regions have different roles in the biological processes involved in sperm maturation and transit, and this has been linked to the regionalization of gene expression patterns (Guyonnet et al., 2009; Belleannée et al., 2012; Browne et al., 2015). Our results indicated that specific small RNAs might play regulatory roles that contribute to the regionalization of gene expression in the reproductive system of Bos indicus bulls.
Detoxification of Copper Ion in the Head Epididymis
The regionalization of gene expression in the reproductive system can be discussed in light of the enriched GO terms and pathways associated with the DE transcripts. The most significant GO term in the HE/TE comparison was detoxification of copper ion. Genes associated with detoxification of copper ion were up-regulated in the head, including three members of the metallothionein family: MT1A, MT2A, and MT1E (Wong et al., 2017). These genes are involved in metal homeostasis and metal detoxification (Schulkens et al., 2014) and can protect cells from oxidative stress (Sutherland and Stillman, 2011). The expression of MT1A, MT2A, and MT1E is up-regulated in the presence of copper in adult human prostatic cell lines (Cu treatment vs. untreated) (Bigagli et al., 2010). In dogs with hepatitis, MT1A and MT2A expression levels decrease together with a copper concentration in hepatic cells (Dirksen et al., 2017). Protection from oxidative stress is important for sperm cells; in fact, the molecular environment of the epididymis is crucial for sperm maturation and capacitation (Belleannée et al., 2012). Increased dietary copper is associated to improved spermatozoa mobility and quality in bulls (Hidiroglou, 1979). However, high levels of copper can affect cell homeostasis and be detrimental to sperm quality and its fertilization capacity (Roblero, 1996). High levels of copper can disturb the integrity of the epididymis and affect sperm maturation (Xu et al., 1985). In this context, our results point to MT1A, MT2A, and MT1E as genes that may assist with copper homeostasis in the head epididymis and might have a role in sperm maturation.
The regionalization of gene expression in the male reproductive system is likely a consequence of regulatory mechanisms, including small RNAs that target genes post-transcriptionally. Bta-mir-362 had a negative co-expression correlation with MT1E and MT1A (lower than −0.95) and might down-regulate these genes. Bta-mir-362 is reported to contribute to spermatogenesis processes in pigs too (Ran et al., 2018). In short, this miRNA might modulate genes involved with detoxification of copper ion in the epididymis, and as a consequence, it might affect sperm quality.
Gated Channel Activity, Ions, and Water Transport
In our study, the GO term gated channel activity and other terms related to ion transport and channel activity were significant for the comparisons between the epididymis and testis. In both the HE/TP and the TE/TP comparisons, DE genes suggest that ion channels are relevant to spermatogenesis. This result is in agreement with previous knowledge, because ions such as Ca2+ and Na+ contribute to the acrosomal reaction, hyper-activation, sperm capacitation, and sperm quality (Mirnamniha et al., 2019).
Differentially expressed and enrichment analyses suggest regionalization of expression patterns in the male reproductive system for genes that code proteins related to ion channels. Specific proteins related to ion channels and solute transporters are responsible for the epididymis homeostasis and for the luminal environment that is adequate to sperm maturation (Belleannée et al., 2012). In short, ion channels, anions, cations, and water transport molecules (i.e., AQPs) are involved in the control of the luminal fluid (Browne et al., 2015).
The DE genes ATP6V0A4, ATP6V0D2, ATP6V1G3, CFTR, SCNN1G, and SCNN4A are related to ion channel activity and were up-regulated in the epididymis when compared to the testis. The ATP6V0A4, ATP6V0D2, and ATP6V1G3 genes were up-regulated in both HE/TP and TE/TP comparisons. They code for proteins that compose the subunit of vacuolar H + –ATPase (V-ATPase) (Wagner et al., 2004). The V-ATPase is a multi-subunit ATP-driven proton pump (Pamarthy et al., 2018), with influence in the acidification of luminal fluid (Brown et al., 1992; Roy et al., 2013) that may help sperm maturation (Pamarthy et al., 2018). Failure in luminal fluid acidification can result in poor sperm maturation and infertility (Breton et al., 1996). In this context, a higher expression of these genes in the epididymis suggests they are relevant to sperm maturation in Bos indicus bulls.
The cystic fibrosis transmembrane conductance regulator (CFTR) gene was up-regulated in the head epididymis when compared to the testis. The CFTR channel is involved in sperm capacitation (Touré, 2019) and can contribute to the Cl– and bicarbonate fluxes (Touré, 2019). The DE gene SLC26A3, up-regulated in the epididymis, is essential to bicarbonate fluxes and interacts with the CFTR channel (Touré, 2019). The SLC26A3 knockout mice present lesions in the epididymis and sperm reduction (El Khouri et al., 2018). CFTR and epithelial Na+ channel (ENaC) contribute to sperm capacitation (Visconti et al., 2011; Sharma and Hanukoglu, 2019). ENaC is probably involved in the uptake of Na+ ions from the epididymal lumen into the cells. Like CFTR channels, ENaC channels are observed in patterns along the length of the mouse and rat epididymis (Sharma and Hanukoglu, 2019). Two genes that code for ENaC proteins, SCNN1G and SCNN4A, were up-regulated in the epididymis when compared to the testis. SCNN1G was up-regulated in the head (HE/TP) while SCNN4A was up-regulated in both the head (HE/TP) and tail (TE/TP). The regional regulation of genes coding for ENaC proteins might be an evidence of their role in sperm maturation. The bovine DE patterns discussed here conform to the expectations from studies in other species and might reveal mechanisms that are relevant to male fertility across mammals.
Another gene that was up-regulated in the head and tail epididymis when compared to the testis was AQP9, an aquaporin. Aquaporins (AQPs) are channels of proteins that facilitate the movement of water across the plasma membrane and contribute to epididymal sperm concentration (Belleannée et al., 2012; Schimming et al., 2017). AQP9 has been previously reported as expressed in the epididymis and related to water resorption in the proximal epididymis (Belleannée et al., 2012). Gene expression patterns of AQPs have been related to disorders of the male reproductive system in mammals (Huang et al., 2006). Therefore, we hypothesize that AQP9 might affect water transport in the epididymis in Bos indicus.
Small RNAs might modulate DE genes involved in gated channel activity, ion channels, and water transport. We predicted interactions between 24 small RNAs (15 miRNAs and 9 snRNA) and the DE genes discussed above. For example, bta-mir-2886 was co-expressed with AQP9 and CFTR. The snRNA RF00026 (U6 spliceosomal RNA) was co-expressed with ATP6V0D2, AQP9, CFTR, and SLC26A3. It is possible then that bta-mir-2886 and RF00026 among other small RNAs modulate gated channel activity, ions, and water transport, affecting epididymis function.
Neuroactive Ligand-Receptors and Spermatogenesis
Among the DE transcripts, we identified genes related to prolactin, GABA, and muscarinic acetylcholine receptors, all part of the enriched neuroactive ligand-receptor interaction pathway. Overall, this pathway seems more important to testicular function than to the epididymis, with a few exceptions as discussed below.
Prolactin is a peptide hormone that acts via its transmembrane receptor (Raut et al., 2019). In our study, nine DE genes related prolactin signaling – PRL, PRLH, PRP1, PRP14, PRP2, PRP4, PRP6, PRP8, and PRP9 – were up-regulated in the testis when compared to the epididymis. Prolactin receptors are expressed in the testis of humans (Jabbour and Kelly, 1997) and bulls (Pratt et al., 2015). Prolactin signaling affects the male reproduction system (Hermanns and Hafez, 1981) as it interferers with steroidogenesis and spermatogenesis (Jabbour and Kelly, 1997). Also, prolactin signaling relates to testosterone concentration (Franchimont, 1983). In summary, the DE analyses recapitulated some known biology of testicular function and suggested prolactin genes that were regulated in male tissues.
GABA receptors were previously reported in the testis and sperm of mice (He et al., 2001, 2003). We identified 10 GABA receptors as DE genes in the HE/TP and TE/TP comparisons: GABBR2, GABRA2, GABRA3, GABRA4, GABRA5, GABRB1, GABRB3, GABRG2, GABRP, and GABRQ. Three receptors (GABRG2, GABRB1, and GABRA5) had higher expression in the epididymis, while the other seven were up-regulated in the testis. In the testis, GABA receptors affect the Leydig cell function, influence germ cell maturation (Geigerseder et al., 2003), and regulate spermatogenesis (Geigerseder et al., 2003; Kanbara et al., 2005; Du et al., 2013). The function of these DE GABA receptors, with expression that is specific to each region of the male system examined, requires further research.
Three DE genes, CHRM1, CHRM2, and CHRM3, are receptors related to muscarinic acetylcholine signaling. Muscarinic acetylcholine receptors (or MAChRs) are part of the regulatory mechanisms in the male reproductive system (Borges et al., 2001; Avellar et al., 2010). MAChRs regulate testicular cell function (Borges et al., 2001) and can influence the luminal fluid composition (Avellar et al., 2010). In our study, CHRM1, CHRM2, and CHRM3 were up-regulated in the testis compared to the epididymis, and so we suggested that MAChRs might contribute to testicular function in Bos indicus bulls.
Among DE genes of the neuroactive ligand-receptor interaction pathway, 10 were connected to the larger network cluster and directly or indirectly linked to bta-mir-2886. The prolactin signaling genes PR2, PRP14, and PRP9 were predicted to interact directly with bta-mir-2886, while GABRA5 was a second neighbor of this same miRNA. In short, it is possible that bta-mir-2886 and other small RNAs regulate genes in the neuroactive ligand-receptor interaction pathway that might affect spermatogenesis.
Co-expression Network, Small RNAs, and Male Gamete Generation
Differentially expressed transcripts associated with the GO term male gamete generation were enriched in the comparisons between the epididymis and testis, being the third most significant term in both HE/TP and TE/TP comparisons. This same GO term was significant for transcripts in the larger cluster of the co-expression network. Four well-known regulators of spermatogenesis were among the DE transcripts associated with male gamete generation: RFX2, HORMAD1, CCDC36, and DAZL. The gene RFX2 is an essential transcription factor in the regulation of spermatogenesis (Kistler et al., 2015; Pandey et al., 2019), which is expressed in spermatocytes and spermatids in mice (Pandey et al., 2019). The RFX2-deficient mice have completely blocked spermatogenesis (Kistler et al., 2015). HORMADA1 is key during the meiosis and it possibly interacts with CCDC36 (Stanzione et al., 2016). DAZL stands for “deleted in azoospermia like,” and it codes for a RNA-binding protein that is localized to the nucleus of spermatogonia, but relocates to the cytoplasm during meiosis, where it persists in spermatids and spermatozoa. DAZL is highly expressed in the testis of sheep with sexual maturity (Yuan et al., 2020) and may have a role in sex differentiation (Rossitto et al., 2015). All four genes were up-regulated in the testis when compared to the epididymis, which is expected since male gamete generation is a characteristic of the testis. Two of these well-known regulators, RFX2 and CCDC36, were also nodes in the larger network cluster.
In the larger network cluster, five out of 10 significant GO terms were very specific to spermatogenesis: male gamete generation, spermatid development, germ cell development, acrosomal vesicle, and sperm capacitation. Therefore, the small RNAs that were identified as potential regulators of this cluster of highly connected DE genes might be proposed as potential regulators of spermatogenesis. We identified 228 small RNAs in the larger network cluster and 43 of these had significant RIF values; they were 20 snRNAs, 8 snoRNAs, and 15 miRNAs. One of the significant miRNAs was bta-mir-2886, up-regulated in the epididymis for two pairwise comparisons (HE/TP and TE/TP), with a high fold change in both (approx. 7). We propose that bta-mir-2886, through its 601 co-expressed transcripts and additional 434 first neighbors, might affect spermatogenesis in Bos indicus bulls.
In our study, most of the correlations between bta-mir-2886 and predicted targets were positive, including for AQP9 and CFTR. We speculated that this miRNA might be indirectly regulating the expression of co-expressed transcripts, with which it presents positive correlations, by inhibiting their negative regulators. This indirect mechanism was suggested previously by Ritchie et al. (2009). We observed 204 predicted negative correlations with bta-mir-2886. Among negative correlations, we identified three genes that may be down-regulated by bta-mir-2886, which were confirmed by TargetScan and had mfe below -20 kcal/mol. They were ELOVL3, FEZF2, and HOXA13.
HOXA13 was among 23 genes of the Hox family that were DE in our study. Hox family transcription factors are expressed in the male reproductive tract (Lindsey and Wilkinson, 1996), including the human testis (Zhu et al., 2016) and mice epididymis (Bomgardner et al., 2003; Raines et al., 2013). Hox genes act in spermatogenesis and sperm maturation (Lindsey and Wilkinson, 1996). Zhu et al. (2016) have reported Hox genes as regulators of meiosis in the human testis. Mutations in HOXA13 were associated to male infertility in mice (Post and Innis, 1999). Further studies could investigate the role of HOXA13 in bull fertility.
Conclusion
Our results indicate that bta-mir-2886, among other small RNAs, are co-expressed with DE genes that may contribute to spermatogenesis and sperm maturation in the testis and epididymis of Bos indicus bulls. Although our study predicts potential regulators of gene expression in the testis and the epididymis of Bos indicus bulls, further work is necessary to confirm our findings and detail the roles played by small RNAs in spermatogenesis.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://data.faang.org/organism, SAMEA104495807.
Ethics Statement
The animal study was reviewed and approved by the Ethics Committee of the University of Queensland, Brisbane, Australia (protocol number: ANRFA/SCMB/094/16).
Author Contributions
MF conceived the idea of this study. MF, EM, and LP-N performed field and laboratory work. JE prepared and sequenced the RNA libraries. AdL, RP, JA, and AR performed the bioinformatics and data analysis. AdL, JA, and MF drafted the manuscript. All authors contributed to the interpretation of results, discussion, review of the concepts in this manuscript, and agreed to be responsible for the content of this study.
Funding
This work was supported by the University of Queensland provided an Early Career Research grant to MF.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the genomics facility of the University of Queensland for preparing and sequencing the cDNA libraries used in this study. We acknowledge Marina Naval Sanchez for her assistance with submitting the RNA-sequencing data to the FAANG Consortium. Elements of this research used the Queensland node of Metabolomics Australia.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.610116/full#supplementary-material
Footnotes
- ^ https://data.faang.org/home
- ^ https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/
- ^ https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- ^ https://www.openagrar.de/servlets/MCRFileNodeServlet/openagrar_derivate_00022208/
- ^ https://cran.r-project.org/web/packages/hoardeR/index.html
- ^ http://www.mirbase.org/
References
Abou-Haila, A., and Tulsiani, D. R. (2000). Mammalian sperm acrosome: formation, contents, and f Function. Arch. Biochem. Biophys. 379, 173–182. doi: 10.1006/abbi.2000.1880
Agarwal, V., Bell, G. W., Nam, J. W., and Bartel, D. P. (2015). Predicting effective microRNA target sites in mammalian mRNAs. Elife 4, 1–38. doi: 10.7554/eLife.05005
Avellar, M. C. W., Siu, E. R., Yasuhara, F., Maróstica, E., and Porto, C. S. (2010). Muscarinic acetylcholine receptor subtypes in the male reproductive tract: expression and function in rat efferent ductules and epididymis. J. Mol. Neurosci. 40, 127–134. doi: 10.1007/s12031-009-9268-6
Belleannée, C. (2015). Extracellular microRNAs from the epididymis as potential mediators of cell-to-cell communication. Asian J. Androl. 17, 730–736. doi: 10.4103/1008-682X.155532
Belleannée, C., Thimon, V., and Sullivan, R. (2012). Region-specific gene expression in the epididymis. Cell Tissue Res. 349, 717–731. doi: 10.1007/s00441-012-1381-0
Benjamini, Y., and Hochberg, Y. (1995). Controlling the False discovery rate – a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. 57, 289–300. doi: 10.2307/2346101
Bigagli, E., Luceri, C., Bernardini, S., Dei, A., and Dolara, P. (2010). Extremely low copper concentrations affect gene expression profiles of human prostate epithelial cell lines. Chem. Biol. Interact. 188, 214–219. doi: 10.1016/j.cbi.2010.06.009
Bindea, G., Mlecnik, B., Hackl, H., Charoentong, P., Tosolini, M., Kirilovsky, A., et al. (2009). ClueGO: a cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinform 25, 1091–1093. doi: 10.1093/bioinformatics/btp101
Bomgardner, D., Hinton, B. T., and Turner, T. T. (2003). 5′ Hox Genes and Meis 1, a Hox-DNA binding cofactor, are expressed in the adult mouse epididymis1. Biol. Reprod. 68, 644–650. doi: 10.1095/biolreprod.102.009324
Borges, M. O. R., Abreu, M. L. C., Porto, C. S., and Avellar, M. C. W. (2001). Characterization of muscarinic acetylcholine receptor in rat sertoli cells. Endocrinology 142, 4701–4710. doi: 10.1210/endo.142.11.8465
Bottje, W., Kong, B. W., Reverter, A., Waardenberg, A. J., Lassiter, K., and Hudson, N. J. (2017). Progesterone signalling in broiler skeletal muscle is associated with divergent feed efficiency. BMC Syst. Biol. 11:396.
Breton, S., Smith, P. J. S., Lui, B., and Brown, D. (1996). Acidification of the male reproductive tract by a proton pumping (H+)-ATPase. Nat. Med. 2, 470–472. doi: 10.1038/nm0496-470
Brown, D., Lui, B., Gluck, S., and Sabolic, I. (1992). A plasma membrane proton ATPase in specialized cells of rat epididymis. Am. J. Physiol. Cell Physiol. 263, 913–916. doi: 10.1152/ajpcell.1992.263.4.c913
Browne, J. A., Yang, R., Leir, S. H., Eggener, S. E., and Harris, A. (2015). Expression profiles of human epididymis epithelial cells reveal the functional diversity of caput, corpus and cauda regions. Mol. Hum. Reprod. 22, 69–82. doi: 10.1093/molehr/gav066
Cánovas, A., Reverter, A., DeAtley, K. L., Ashley, R. L., Colgrave, M. L., Fortes, M. R. S., et al. (2014). Multi-tissue omics analyses reveal molecular regulatory networks for puberty in composite beef cattle. PLoS One 9:e0102551. doi: 10.1371/journal.pone.0102551
Chiang, H. R., Schoenfeld, L. W., Ruby, J. G., Auyeung, V. C., Spies, N., Baek, D., et al. (2010). Mammalian microRNAs: experimental evaluation of novel and previously annotated genes. Genes Dev. 24, 992–1009. doi: 10.1101/gad.1884710
Cornwall, G. A. (2009). New insights into epididymal biology and function. Hum. Reprod. 15, 213–227. doi: 10.1093/humupd/dmn055
de Souza, M. M., Zerlotini, A., Geistlinger, L., Tizioto, P. C., Taylor, J. F., Rocha, M. I. P., et al. (2018). A comprehensive manually-curated compendium of bovine transcription factors. Sci. Rep. 8, 1–12. doi: 10.1038/s41598-018-32146-2
Dirksen, K., Spee, B., Penning, L. C., Van Den Ingh, T. S. G. A. M., Burgener, I. A., Watson, A. L., et al. (2017). Gene expression patterns in the progression of canine copper-associated chronic hepatitis. PLoS One 12:e0176826. doi: 10.1371/journal.pone.0176826
Du, Y., Du, Z., Zheng, H., Wang, D., Li, S., Yan, Y., et al. (2013). GABA exists as a negative regulator of cell proliferation in spermaogonial stem cells. Cell. Mol. Biol. Lett. 18, 149–162. doi: 10.2478/s11658-013-0081-4
Durinck, S., Spellman, P. T., Birney, E., and Huber, W. (2009). Mapping identifiers for the integration of genomic datasets with the R/bioconductor package biomaRt steffen. Nat. Protoc. 4, 1184–1191. doi: 10.1038/nprot.2009.97.Mapping
El Khouri, E., Whitfield, M., Stouvenel, L., Kini, A., Riederer, B., Lores, P., et al. (2018). Slc26a3 deficiency is associated with epididymis dysplasia and impaired sperm fertilization potential in the mouse. Mol. Reprod. Dev. 85, 682–695. doi: 10.1002/mrd.23055
Fionda, V. (2019). “Networks in biology,” in Encyclopedia of Bioinformatics and Computational Biology, eds S. Ranganathan, M. Gribskov, K. Nakai, and C. Schönbach (Oxford: Academic Press), 915–921.
Fortes, M. R. S., Satake, N., Corbet, D. H., Corbet, N. J., Burns, B. M., Moore, S. S., et al. (2014). Sperm protamine deficiency correlates with sperm DNA damage in Bos indicus bulls. Andrology 2, 370–378. doi: 10.1111/j.2047-2927.2014.00196.x
Franchimont, P. (1983). Regulation of gonadal androgen secretion. Horm. Res. Paediatr. 18, 7–17. doi: 10.1159/000179774
Fromm, B., Billipp, T., Peck, L. E., Johansen, M., Tarver, J. E., King, B. L., et al. (2015). A uniform system for the annotation of human microRNA genes and the evolution of the human microRNAome bastian. Annu. Rev. Genet. 49, 213–242. doi: 10.1146/annurev-genet-120213-092023.A
Fuda, N. J., Ardehali, M. B., and Lis, J. T. (2009). Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature 461, 186–192. doi: 10.1038/nature08449
Geigerseder, C., Doepner, R., Thalhammer, A., Frungieri, M. B., Gamel-Didelon, K., Calandra, R. S., et al. (2003). Evidence for a GABAergic system in rodent and human testis: local GABA production and GABA receptors. Neuroendocrinology 77, 314–323. doi: 10.1159/000070897
Guyonnet, B., Marot, G., Dacheux, J. L., Mercat, M. J., Schwob, S., Jaffrézic, F., et al. (2009). The adult boar testicular and epididymal transcriptomes. BMC Genom. 10:369. doi: 10.1186/1471-2164-10-369
Harhay, G. P., Smith, T. P. L., Alexander, L. J., Haudenschild, C. D., Keele, J. W., Matukumalli, L. K., et al. (2010). An atlas of bovine gene expression reveals novel distinctive tissue characteristics and evidence for improving genome annotation. Genome Biol. 11:102. doi: 10.1186/gb-2010-11-10-r102
He, X. B., Hu, J. H., Wu, Q., Yan, Y. C., and Koide, S. S. (2001). Identification of GABAB receptor in rat testis and sperm. Biochem. Biophys. Res. Commun. 283, 243–247. doi: 10.1006/bbrc.2001.4732
He, X., Zhang, Y., Yan, Y., Li, Y., and Koide, S. S. (2003). Identification of GABABR2 in rat testis and sperm. J. Reprod. Dev. 49, 397–402. doi: 10.1262/jrd.49.397
Hermann, B. P., Cheng, K., Singh, A., Roa-De, La Cruz, L., Mutoji, K. N., et al. (2018). The mammalian spermatogenesis single-cell transcriptome, from spermatogonial stem cells to spermatids. Cell Rep. 25, 1650–1667. doi: 10.1016/j.celrep.2018.10.026
Hermanns, U., and Hafez, E. S. E. (1981). Prolactin and male reproduction. Syst. Biol. Reprod. Med. 6, 95–125. doi: 10.3109/01485018108987351
Hidiroglou, M. (1979). Trace element deficiencies and fertility in ruminants: a review. J. Dairy Sci. 62, 1195–1206. doi: 10.3168/jds.S0022-0302(79)83400-1
Hombach, S., and Kretz, M. (2016). “Non-coding RNAs: classification, biology and functioning,” in Non-Coding RNAs in Colorectal Cancer, eds O. Slaby and G. A. Calin (Cham: Springer), 3–17. doi: 10.1007/978-3-319-42059-2
Huang, H. F., He, R. H., Sun, C. C., Zhang, Y., Meng, Q. X., and Ma, Y. Y. (2006). Function of aquaporins in female and male reproductive systems. Hum. Reprod. 12, 785–795. doi: 10.1093/humupd/dml035
Jabbour, H. N., and Kelly, P. A. (1997). Prolactin receptor subtypes: a possible mode of tissue specific regulation of prolactin function. Rev. Reprod. 2, 14–18. doi: 10.1530/ror.0.0020014
Kanbara, K., Okamoto, K., Nomura, S., Kaneko, T., Shigemoto, R., Azuma, H., et al. (2005). Cellular localization of GABA and GABAB receptor subunit proteins during spermiogenesis in rat testis. J. Androl. 26, 485–493. doi: 10.2164/jandrol.04185
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Kistler, W. S., Baas, D., Lemeille, S., Paschaki, M., Seguin-Estevez, Q., Barras, E., et al. (2015). RFX2 is a major transcriptional regulator of spermiogenesis. PLoS Genet. 11:e1005368. doi: 10.1371/journal.pgen.1005368
Lewis, B. P., Burge, C. B., and Bartel, D. P. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20. doi: 10.1016/j.cell.2004.12.035
Lindsey, S., and Wilkinson, M. F. (1996). Homeobox genes and male reproductive development. J. Assist. Reprod. Genet. 13, 182–192. doi: 10.1007/BF02072542
MacLean, J. A., and Wilkinson, M. F. (2005). Gene regulation in spermatogenesis. Curr. Top. Dev. Biol. 5, 131–197. doi: 10.1016/S0070-2153(05)71005-X
Marengo, S. R. (2008). Maturing the sperm: unique mechanisms for modifying integral proteins in the sperm plasma membrane. Anim. Reprod. Sci. 105, 52–63. doi: 10.1016/j.anireprosci.2007.11.018
Mirnamniha, M., Faroughi, F., Tahmasbpour, E., Ebrahimi, P., and Beigi Harchegani, A. (2019). An overview on role of some trace elements in human reproductive health, sperm function and fertilization process. Rev. Environ. Health 19, 1–10. doi: 10.1515/reveh-2019-0008
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Nam, J., Rissland, O. S., Koppstein, D., Abreu-goodger, C., Jan, C. H., Agarwal, V., et al. (2014). Global analyses of the effect of different cellular contexts on microRNA targeting. Mol. Cell 53, 1031–1043. doi: 10.1016/j.molcel.2014.02.013.Global
Nixon, B., De Iuliis, G. N., Dun, M. D., Zhou, W., Trigg, N. A., and Eamens, A. L. (2019). Profiling of epididymal small non-protein-coding RNAs. Andrology 7, 669–680. doi: 10.1111/andr.12640
Nolte, W., Weikard, R., Brunner, R. M., Albrecht, E., Hammon, H. M., Reverter, A., et al. (2019). Biological network approach for the identification of regulatory long Non-coding RNAs associated with metabolic efficiency in cattle. Front. Genet. 10:1130. doi: 10.3389/fgene.2019.01130
Okonechnikov, K., Conesa, A., and García-Alcalde, F. (2016). Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294. doi: 10.1093/bioinformatics/btv566
Pamarthy, S., Kulshrestha, A., Katara, G. K., and Beaman, K. D. (2018). The curious case of vacuolar ATPase: regulation of signaling pathways. Mol. Cancer 17, 1–9. doi: 10.1186/s12943-018-0811-3
Pandey, A., Yadav, S. K., Vishvkarma, R., Singh, B., Maikhuri, J. P., Rajender, S., et al. (2019). The dynamics of gene expression during and post meiosis sets the sperm agenda. Mol. Reprod. Dev. 86, 1921–1939. doi: 10.1002/mrd.23278
Post, L. C., and Innis, J. W. (1999). Infertility in adult hypodactyly mice is associated with hypoplasia of distal reproductive structures1. Biol. Reprod. 61, 1402–1408. doi: 10.1095/biolreprod61.6.1402
Pradillo, M., and Santos, J. L. (2018). Genes involved in miRNA biogenesis affect meiosis and fertility. Chromosom. Res. 26, 233–241. doi: 10.1007/s10577-018-9588-x
Pratt, S. L., Calcatera, S. M., Stowe, H. M., Dimmick, M. A., Schrick, F. N., Duckett, S. K., et al. (2015). Identification of bovine prolactin in seminal fluid, and expression and localization of the prolactin receptor and prolactin-inducible protein in the testis and epididymis of bulls exposed to ergot alkaloids. Theriogenology 83, 662–669. doi: 10.1016/j.theriogenology.2014.10.031
Raines, A. M., Adam, M., Magella, B., Meyer, S. E., Grimes, H. L., Dey, S. K., et al. (2013). Recombineering-based dissection of flanking and paralogous Hox gene functions in mouse reproductive tracts. Development 140, 2942–2952. doi: 10.1242/dev.092569
Ran, M., Weng, B., Cao, R., Li, Z., Peng, F., Luo, H., et al. (2018). miR-362 regulates the proliferation and apoptosis of porcine immature Sertoli cells through targeting the ZNF644 gene. Yi Chuan 40, 572–584. doi: 10.16288/j.yczz.18-028
Raut, S., Deshpande, S., and Balasinor, N. H. (2019). Unveiling the role of prolactin and its receptor in male reproduction. Horm. Metab. Res. 51, 215–219. doi: 10.1055/a-0859-1144
Rehmsmeier, M., Steffen, P., Ho, M., and Giegerich, R. (2004). Fast and effective prediction of microRNA/target duplexes. Bioinformatics 10, 1507–1517. doi: 10.1261/rna.5248604.and
Reverter, A., and Chan, E. K. F. (2008). Combining partial correlation and an information theory approach to the reversed engineering of gene co-expression networks. Bioinformatics 24, 2491–2497. doi: 10.1093/bioinformatics/btn482
Reverter, A., Barris, W., McWilliam, S., Byrne, K. A., Wang, Y. H., Tan, S. H., et al. (2005). Validation of alternative methods of data normalization in gene co-expression studies. Bioinformatics 21, 1112–1120. doi: 10.1093/bioinformatics/bti124
Reverter, A., Hudson, N. J., Nagaraj, S. H., Pérez-Enciso, M., and Dalrymple, B. P. (2010). Regulatory impact factors: unraveling the transcriptional regulation of complex traits from expression data. Bioinformatics 26, 896–904. doi: 10.1093/bioinformatics/btq051
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., et al. (2015). Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43:e47. doi: 10.1093/nar/gkv007
Ritchie, W., Rajasekhar, M., Flamant, S., and Rasko, J. E. J. (2009). Conserved expression patterns predict microRNA targets. PLoS Comput. Biol. 5:513. doi: 10.1371/journal.pcbi.1000513
Roblero, L. (1996). Effect of copper ion on the motility, viability, acrosome reaction and fertilizing capacity of human spermatozoa in vitro. Reprod. Fertil. Dev. 8, 871–874. doi: 10.1071/RD9960871
Rossitto, M., Philibert, P., Poulat, F., and Boizet-Bonhoure, B. (2015). Molecular events and signalling pathways of male germ cell differentiation in mouse. Semin. Cell Dev. Biol. 45, 84–93. doi: 10.1016/j.semcdb.2015.09.014
Roy, J. W., Hill, E., Ruan, Y. C., Vedovelli, L., Păunescu, T. G., Brown, D., et al. (2013). Circulating aldosterone induces the apical accumulation of the proton pumping V-ATPase and increases proton secretion in clear cells in the caput epididymis. Am. J. Physiol. Cell Physiol. 305, 436–446. doi: 10.1152/ajpcell.00410.2012
Schimming, B. C., Baumam, C. A. E., Pinheiro, P. F. F., de Matteis, R., and Domeniconi, R. F. (2017). Aquaporin 9 is expressed in the epididymis of immature and mature pigs. Reprod. Domest. Anim. 52, 617–624. doi: 10.1111/rda.12957
Schulkens, I. A., Castricum, K. C. M., Weijers, E. M., Koolwijk, P., Griffioen, A. W., and Thijssen, V. L. (2014). Expression, regulation and function of human metallothioneins in endothelial cells. J. Vasc. Res. 51, 231–238. doi: 10.1159/000365550
Shannon, P., Markiel, A., Owen Ozier Nitin, S., Baliga Jonathan, T., Wang, D. R., Amin, N., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13:6. doi: 10.1101/gr.1239303.metabolite
Sharma, S., and Hanukoglu, I. (2019). Mapping the sites of localization of epithelial sodium channel (ENaC) and CFTR in segments of the mammalian epididymis. J. Mol. Histol. 50, 141–154. doi: 10.1007/s10735-019-09813-3
Soumillon, M., Necsulea, A., Weier, M., Brawand, D., Zhang, X., Gu, H., et al. (2013). Cellular source and mechanisms of high transcriptome complexity in the mammalian testis. Cell Rep. 3, 2179–2190. doi: 10.1016/j.celrep.2013.05.031
Stanzione, M., Marek, B., Frantzeskod, P., Dereli, I., Julian, L., Ramlal, A., et al. (2016). Meiotic DNA break formation requires the unsynapsed chromosome axis-binding protein IHO1 (CCDC36) in mice. Nat. Cell Biol. 18, 1208–1220. doi: 10.1038/ncb3417
Staub, C., and Johnson, L. (2018). Review: Spermatogenesis in the bull. Animal 12, s27–s35. doi: 10.1017/S1751731118000435
Sullivan, R. (2016). Epididymosomes: role of extracellular microvesicles in sperm maturation. Front. Biosci. Sch. 8:106–114. doi: 10.2741/S450
Sutherland, D. E. K., and Stillman, M. J. (2011). The “magic numbers” of metallothionein. Metallomics 3, 444–463. doi: 10.1039/c0mt00102c
Thimon, V., Koukoui, O., Calvo, E., and Sullivan, R. (2007). Region-specific gene expression profiling along the human epididymis. Mol. Hum. Reprod. 13, 691–704. doi: 10.1093/molehr/gam051
Touré, A. (2019). Importance of slc26 transmembrane anion exchangers in sperm post-testicular maturation and fertilization potential. Front. Cell Dev. Biol. 7:230. doi: 10.3389/fcell.2019.00230
Uhlén, M., Fagerberg, L., Hallström, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A., et al. (2015). Tissue-based map of the human proteome. Science 347:1260419. doi: 10.1126/science.1260419
Visconti, P. E., Krapf, D., De La Vega-Beltrán, J. L., Acevedo, J. J., and Darszon, A. (2011). Ion channels, phosphorylation and mammalian sperm capacitation. Asian J. Androl. 13, 395–405. doi: 10.1038/aja.2010.69
Wagner, C. A., Finberg, K. E., Breton, S., Marshansky, V., Brown, D., and Geibel, J. P. (2004). Renal vacuolar H+-ATPase. Physiol. Rev. 84, 1263–1314. doi: 10.1152/physrev.00045.2003
Wong, D., Merrifield-MacRae, M., and Stillman, M. (2017). “9. Lead(II) Binding in Metallothioneins,” in Lead: Its Effects on Environment and Health, eds A. Sigel, H. Sigel, and R. Sigel (Berlin: De Gruyter), 241–270. doi: 10.1515/9783110434330-009
Xu, Y., Xiao, F. -L., Xu, N., and Qian, S. -Z. (1985). Effect of intra-epididymal injection of copper particles on fertility, spermatogenesis, and tissue copper levels in rats. Int. J. Androl. 8, 168–174. doi: 10.1111/j.1365-2605.1985.tb00830.x
Yen, P. S., Chen, C. H., Sreenu, V., Kohl, A., and Failloux, A. B. (2019). Assessing the potential interactions between cellular mirna and arboviral genomic RNA in the yellow fever mosquito, aedes aegypti. Viruses 11:540. doi: 10.3390/v11060540
Yuan, Z., Luo, J., Wang, L., Li, F., Li, W., and Yue, X. (2020). Expression of DAZL gene in selected tissues and association of its polymorphisms with testicular size in Hu sheep. Animals 10, 1–12. doi: 10.3390/ani10040740
Zhao, W., Mengal, K., Yuan, M., Quansah, E., Li, P., Wu, S., et al. (2019). Comparative RNA-Seq analysis of differentially expressed genes in the epididymides of Yak and cattleyak. Curr. Genom. 20, 293–305. doi: 10.2174/1389202920666190809092819
Keywords: bovine, RNA-sequencing, systems biology, spermatozoa, miRNA, bta-mir-2886, small RNAs, spermatogenesis
Citation: de Lima AO, Afonso J, Edson J, Marcellin E, Palfreyman R, Porto-Neto LR, Reverter A and Fortes MRS (2021) Network Analyses Predict Small RNAs That Might Modulate Gene Expression in the Testis and Epididymis of Bos indicus Bulls. Front. Genet. 12:610116. doi: 10.3389/fgene.2021.610116
Received: 25 September 2020; Accepted: 19 March 2021;
Published: 30 April 2021.
Edited by:
Christopher K. Tuggle, Iowa State University, United StatesReviewed by:
Terje Raudsepp, Texas A&M University, United StatesWansheng Liu, Pennsylvania State University (PSU), United States
Gregorio Miguel Ferreira De Camargo, Federal University of Bahia, Brazil
Copyright © 2021 de Lima, Afonso, Edson, Marcellin, Palfreyman, Porto-Neto, Reverter and Fortes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marina R. S. Fortes, bS5mb3J0ZXNAdXEuZWR1LmF1