 Pan Gong
Pan Gong Jiao Xue
Jiao Xue Xianru Jiao
Xianru Jiao Yuehua Zhang
Yuehua Zhang Zhixian Yang
Zhixian Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Genet. , 08 April 2021
Sec. Genetics of Common and Rare Diseases
Volume 12 - 2021 | https://doi.org/10.3389/fgene.2021.607965
Background: Recently, the electroencephalogram pattern of electrical status epilepticus during sleep (ESES) had been reported in some genetic disorders, and most of them were noted with developmental and epileptic encephalopathy (DEE) or epileptic encephalopathy (EE). This study aimed to determine the genetic etiologies and clinical characteristics of ESES in DEE/EE.
Methods: We performed a cohort study in cases of DEE or EE with ESES. Tio-based genetic testing was performed in 74 cases and was analyzed to identify underlying variants.
Results: Pathogenic or likely pathogenic variants were identified in 17/74 cases, including KCNQ2 (n = 6), KCNA2 (n = 5), GRIN2A (n = 3), SLC9A6 (n = 1), HIVEP2 (n = 1), and RARS2 (n = 1). Eleven were boys. The median age at seizure onset was 6 months. ESES occurred at the mean age of 2.0 ± 1.2 years, predominant in the Rolandic region in 14 years. Twelve of 17 cases had the first stage of different epilepsy preceding ESES: 2/12 were diagnosed as Ohtahara syndrome, 2/12 were diagnosed as infantile spasms, 3/12 were diagnosed as DEE, and 5/12 were diagnosed as EE without the epileptic syndrome.
Conclusion: Monogenic variants explained over 20% of DEE/EE with ESES. ESES could be an age-related feature in genetic disorders and occurred after the first stage of different epilepsy. Both age-related factors and genetic etiology were suggested to play a role in the occurrence of ESES in genetic DEE/EE.
Electrical status epilepticus during sleep (ESES) initially described an electroencephalogram (EEG) pattern where interictal focal or multifocal spike-and-wave occupied at least 85% of the EEG tracing during non-rapid eye movement (NREM) sleep (Patry et al., 1971). It was now also used as a cutoff value ranging from 50 to 85% in the literatures (Loddenkemper et al., 2011; Sánchez Fernández et al., 2012). The widely recognized ESES-related syndromes included benign childhood epilepsy with centrotemporal spikes, atypical benign partial epilepsy (ABPE), Landau–Kleffner syndrome, and epileptic encephalopathy with continuous spike-and-wave during sleep (CSWS). The latter three were known as epileptic encephalopathy (EE) (Loddenkemper et al., 2011). A few cases and small series suggested some degree of genetic predisposition in the EEG pattern of ESES (Sánchez Fernández et al., 2012). Recently, ESES had been reported in some genetic disorders, such as KCNQ2, ZEB2, and SLC9A6 (Bonanni et al., 2017; Lee et al., 2017; Mathieu et al., 2018). Masnada et al. (2017) described an ESES-like pattern in KCNA2-related developmental and epileptic encephalopathy (DEE). In 2018, a systematic review on all reported genetic etiologies of ESES-related syndrome summarized 11 monogenic variants identified in 60 cases (Kessi et al., 2018). Most of the reported patients were noted with EE or DEE. Here, we aimed to describe the clinical and genetic characteristics of patients with ESES and EE or DEE caused by monogenic variants.
This study was approved by the Ethical Committee of Peking University First Hospital, and written informed consents were obtained from the legal guardians (parents) of the subjects for publication.
Firstly, the EEG database over the past 3 years at the Pediatric Department of Peking University First Hospital was queried for ESES. For the purpose of this study, the spike-wave index (SWI) during NREM sleep of ESES was demarcated as above 50%. Secondly, patients with ESES recognized from the first step were screened by the following clinical criteria: (a) seizures, (b) having a developmental delay from birth or suffering from remarkable developmental impairments after seizure onset, and (c) having taken a clinical or a research-based genetic assessment, including trio-based targeted gene panels testing and whole-exome sequencing, (d) before the ESES occurrence, EEG pattern at the early stage of epilepsy was especially included in our study, including burst suppression, multifocal discharges, hypsarrhythmia, and normality. Patients with acquired structural lesions and those diagnosed with Rett syndrome and autism were excluded. We reviewed history obtained from medical records and from families via phone and/or written survey, including sex, age at seizure onset, seizure types, perinatal and personal history, family history, neurological development, magnetic resonance imaging findings, treatment, and other relevant clinical data. Pediatric epileptologists reviewed series of primary EEG recordings of each patient. Reports were reviewed when original tracings were not available.
We described variants using human genome 19 (hg19) coordinates. We used inheritance pattern, in silico predictions, control database (including the Exome Aggregation Consortium, Exon Variant Server, 1000 Genomes database, and Single Nucleotide Polymorphism Database), clinical laboratory reports, and findings in the literature to assess pathogenicity. For the prediction of the pathogenicity of nonsynonymous variants, we used Polyphen21 and SIFT2. For available original data, after assessing the known epilepsy genes (Supplementary Table 1), we considered other candidate genes, among which we prioritized de novo heterozygous, compound heterozygous, or homozygous variants in genes with a plausible role in epilepsy. Variants considered pathogenic or likely pathogenic were verified by Sanger sequencing. For available genetic testing reports, the clinical significance of the identified variants was interpreted according to the guidelines set out by the American College of Medical Genetics (Richards et al., 2015). A complete description of the methods can be found in Figure 1.
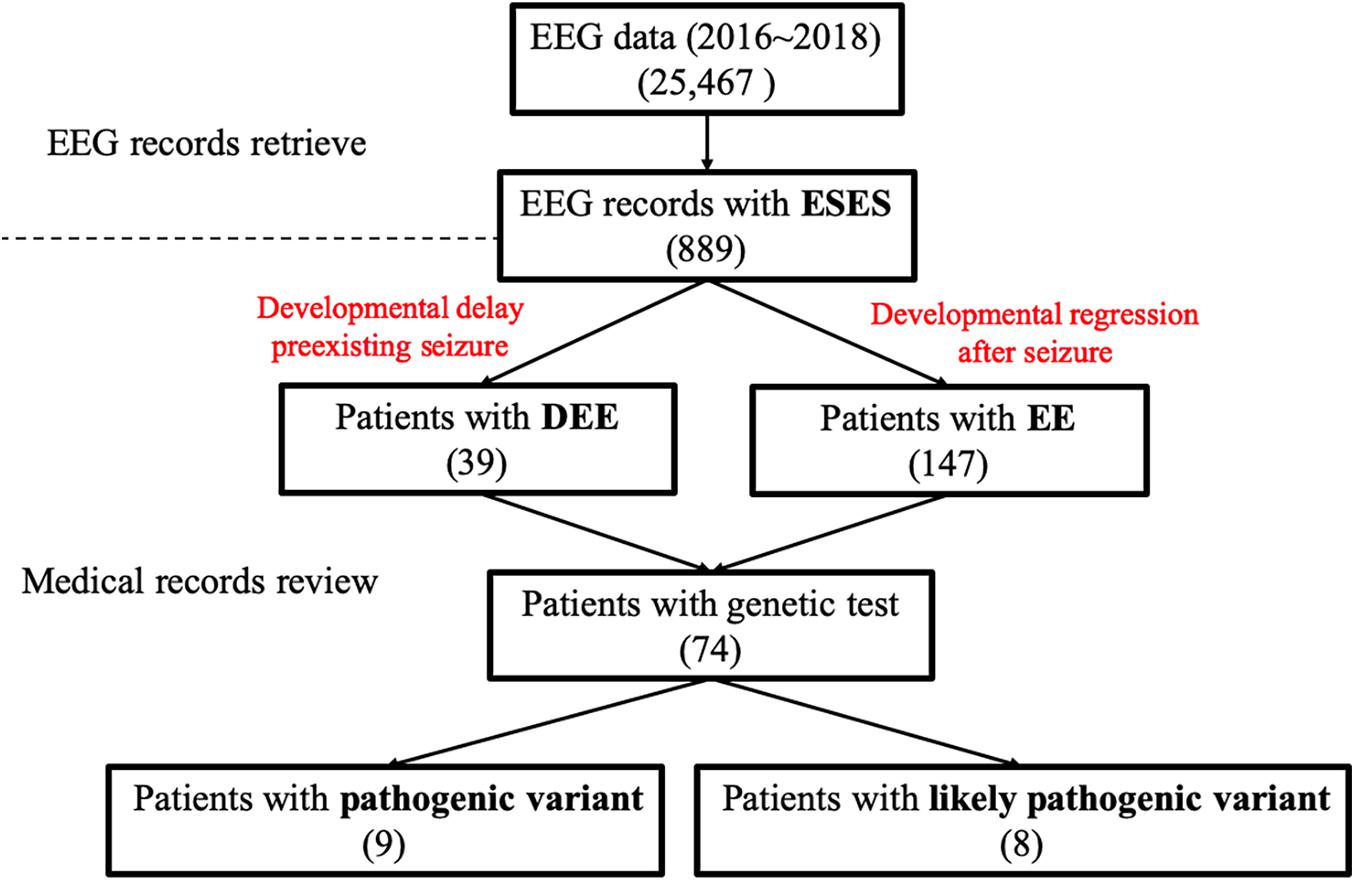
Figure 1. Data screening flow chart. EEG, electroencephalography; DEE, developmental and epileptic encephalopathy; EE, epileptic encephalopathy.
Seventy-four cases of DEE/EE with ESES had undergone genetic testing. Forty-five were boys. The median age at seizure onset was 29.4 months (range 1 day to 108 months), whereas ESES occurrence was 5.3 years (range 1.1–13 years). The median age at the last available medical records was 6.8 years (range 1.9–14 years). Eventually, a positive result was identified in 17/74 (23.0%): six with a likely pathogenic KCNQ2 variant, five with a pathogenic KCNA2 variant, three with a pathogenic or likely pathogenic GRIN2A variant, one with a pathogenic SLC9A6 variant, one with a pathogenic HIVEP2 variant, and one with likely pathogenic RARS2 variants (Table 1). Eleven of 17 were boys. The median age at seizure onset was 6 months (range 1 day to 52 months). The mean age on the occurrence of ESES was 2.0 ± 1.2 years. The SWI of ESES achieved above 85% in 13 and 50–85% in 4. Cases were followed till a mean age of 5.1 ± 2.4 years. Main clinical data are defined in Table 2.
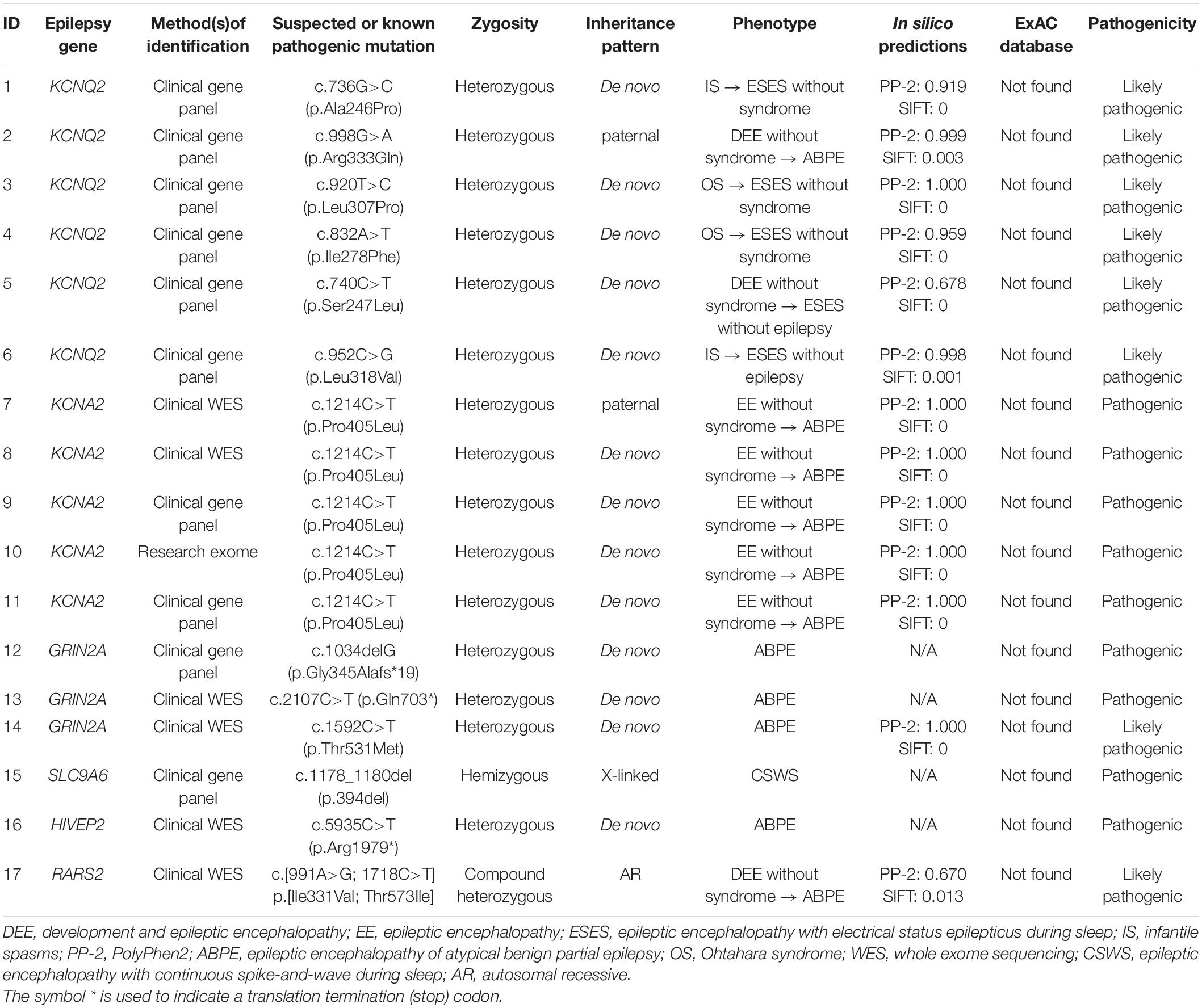
Table 1. Summary of monogenic variants for a series of 17 cases of DEE/EE with ESES.
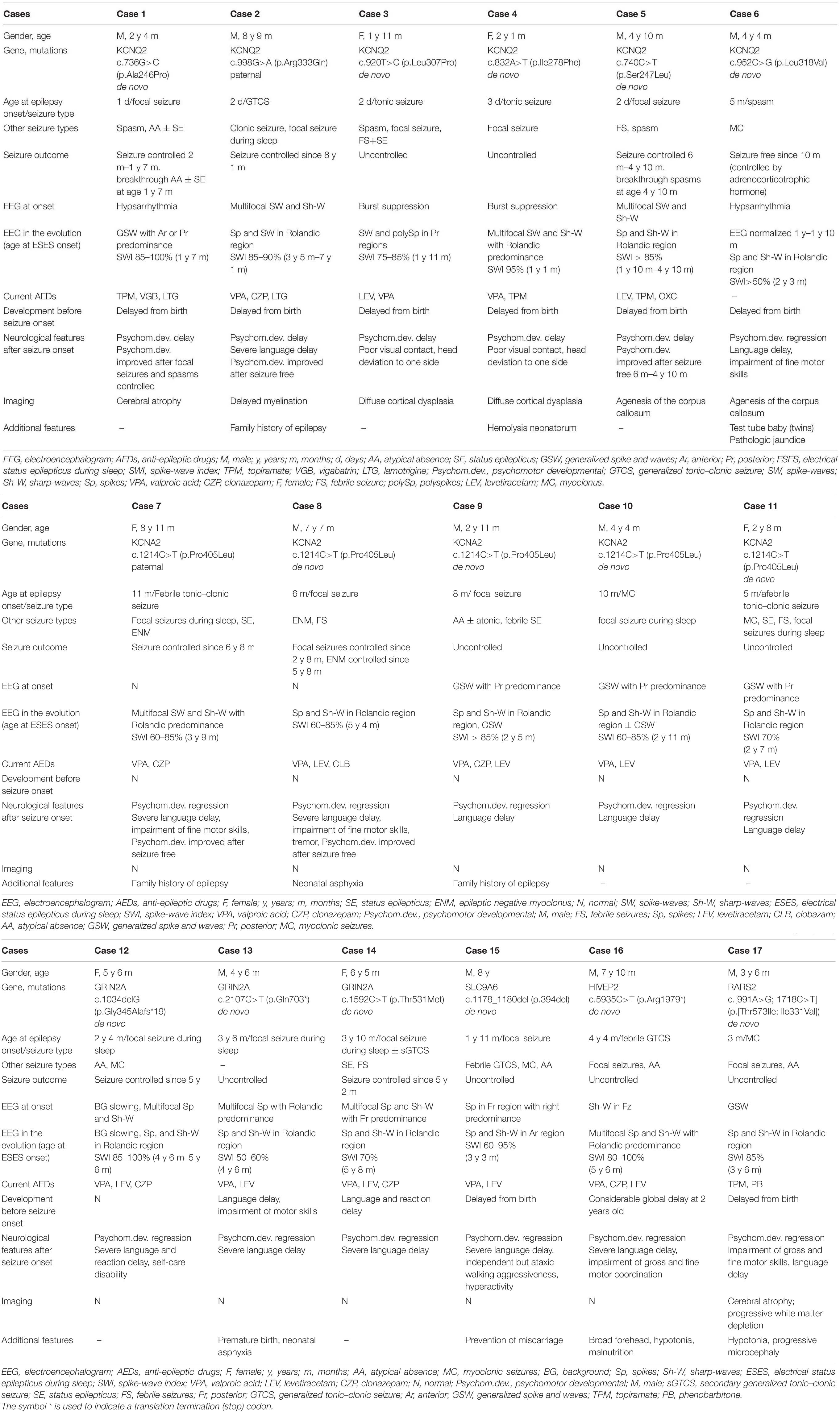
Table 2. Clinical characteristics of 17 cases of genetic DEE/EE with ESES.
Six missense variants were identified in six cases. The variants arose de novo except one. Case 2 carried a paternal variant R333Q that had been reported to be associated with benign familial neonatal seizures (BFNS) (Singh et al., 2003).
All had seizure onset within the first week except one with seizure onset at 5 months (case 6). All had a global developmental delay from birth. The first stage of epilepsy in all was inconsistent with DEE. Two had onset of tonic seizure accompanied by burst suppression, diagnosed with Ohtahara syndrome (cases 3 and 4). One had onset of a focal seizure and then epileptic spasm (case 1), and one had onset of epileptic spasm (case 6), both accompanied by hypsarrhythmia, and they were diagnosed with infantile spasms. One had onset of generalized tonic–clonic seizure (GTCS) (case 2), and one had onset of the focal seizure (case 5), both accompanied by multifocal discharges, which could not be classified into an epilepsy syndrome. The distribution of epileptiform discharges shifted, and the SWI further achieved ESES at a mean age of 2.0 ± 0.7 years, which brought the disease into the second stage of epilepsy. ESES presented with Rolandic predominance in four, posterior predominance in one, anterior or posterior predominance in one (Figure 2). In cases 1, 3, and 4, combining with seizure types, they did not fit with traditional ESES-related syndrome. Case 2 with a paternal variant had nocturnal focal seizures during the second stage, diagnosed as ABPE. He achieved ESES remission at 7.1 years old and seizure-free at 8.1 years old with a remarkably developmental improvement, especially on language. His father had seizure onset at 1 month after birth and became seizure-free at 1 year. There were additional six families affected. The age at seizure onset ranged from 2 days to 6 months after birth, and all of them became seizure-free before 1 year of age. Except for case 2, all of the affected families met the diagnosis of BFNS. Cases 5 and 6 had no seizure during the occurrence of ESES. In case 5, seizures were controlled at the age of 6 months, and ESES occurred at the age of 1.8 years. However, he had breakthrough spasms at the age of 4.8 years, accompanied by multifocal and generalized epileptiform discharges. In case 6, seizures were controlled by adrenocorticotropic hormone at 10 months, and the EEG normalized. Epileptiform discharges in the Rolandic region occurred at the age of 1.8 years and evolved into ESES at 2.3 years without a seizure. Neuroimaging was abnormal in all: cerebral atrophy in one, delayed myelination in one, diffuse cortical dysplasia in two, and agenesis of the corpus callosum in two.
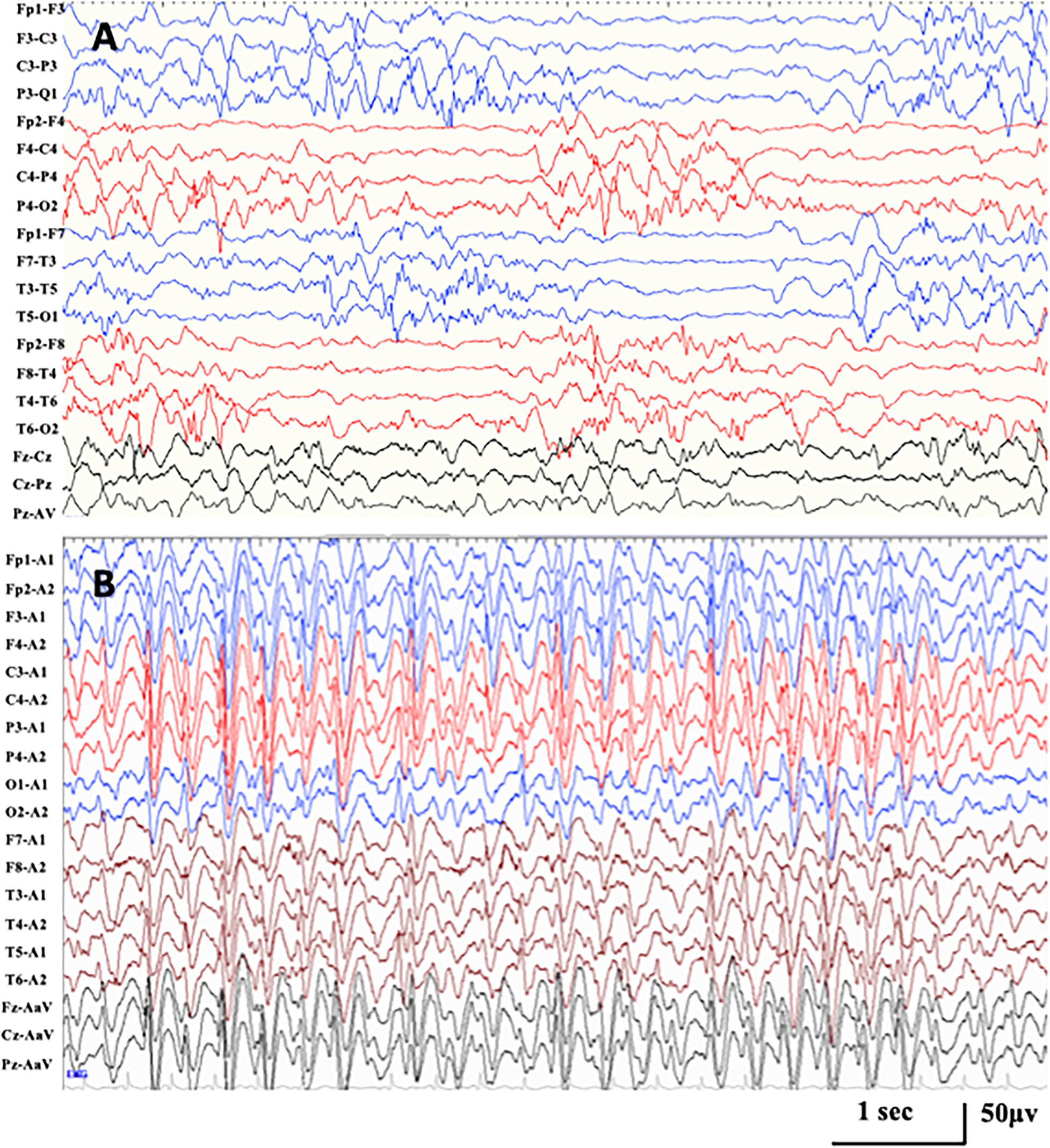
Figure 2. Representative EEG in case 1 with KCNQ2-related disorders. (A) Interictal EEG at seizure onset of 1 month after birth demonstrating hypsarrhythmia with high amplitude multifocal spikes and asynchronization between hemispheres and chaotic background. (B) Interictal EEG at the age of 1.6 years, demonstrating generalized spike-and-wave with anterior predominance during NREM sleep (SWI 85%).
All five cases carried the same missense variant of P405L that was reported to be a loss-of-function variant (Masnada et al., 2017). The variants of four arose de novo, with only the remaining one having a paternal variant (case 7).
The mean age at seizure onset was 8.0 ± 2.5 months, and prior cognitive and motor development was normal in all. Seizure semiology at onset was a focal seizure in two, myoclonic seizure in one, febrile tonic–clonic seizure in one, and afebrile tonic–clonic seizure in one. Epilepsy onset was accompanied or followed by a developmental regression in all. The EEG at onset was normal in two and showed generalized spikes and waves with posterior predominance in three. All five cases were diagnosed as EE without relative epilepsy syndrome at the first stage of epilepsy. They all had nocturnal focal seizures at a mean age of 2.5 ± 0.9 years. The distribution of epileptiform discharges transformed to Rolandic region, and ESES occurred at a mean age of 3.3 ± 1.2 years. All of them were diagnosed with ABPE at the second stage of epilepsy. A paternal variant was identified in case 7, whose father also had an epilepsy history of ABPE. The age at the last follow-up ranged from 2.7 to 8.8 years. The seizure was controlled with a remarkably developmental improvement at the age of 6.8 and 5.8 years in cases 7 and 8, respectively.
A frameshift variant, a nonsense variant, and a missense variant in GRIN2A were identified in three cases. All variants occurred de novo.
Two cases had a developmental delay from birth, with one of them delivered prematurely. The median age of focal seizure onset during sleep was 38.7 months (range 28–46 months). All had a developmental regression after seizure onset. ESES in the Rolandic region occurred at a median age of 4.9 years (range 4.5–5.7 years). All of them were diagnosed with ABPE. The age at the last follow-up ranged from 4.5 to 6.4 years. Two had seizures controlled at the age of 5 and 5.2 years. They both had developmental improvement but still presented with considerable developmental delay.
A novel hemizygous SLC9A6 variant was found in case 14. He had a delayed milestone from birth. The age of focal seizure onset during sleep was 1.9 years old with developmental regression. He developed multiple seizure types during the course, including febrile GTCS, myoclonic seizures, and atypical seizures. ESES in the anterior region was identified at 3.3 years old. He was diagnosed with CSWS. At the last follow-up of age 8 years, he presented with independent but ataxic walking, no oral speech, hyperkinetic behavior, and active seizures.
A heterozygous HIVEP2 variant was identified in case 15. The same nonsense variant had been reported (Goldsmith et al., 2019). The considerable global developmental delay became apparent at the age of 2 years. He had an onset of febrile GTCS at 4.3 years old. Then, nocturnal focal seizures and atypical absence occurred during the course. ESES with Rolandic predominance occurred at 5.6 years old (Figure 3). With a gradual developmental regression, he was diagnosed with ABPE. At the last follow-up of age 7.8 years, he had notable difficulties in gross and fine motor coordination with poor expression language skills and active seizures. Additional features include a broad forehead, hypotonia, and malnutrition.
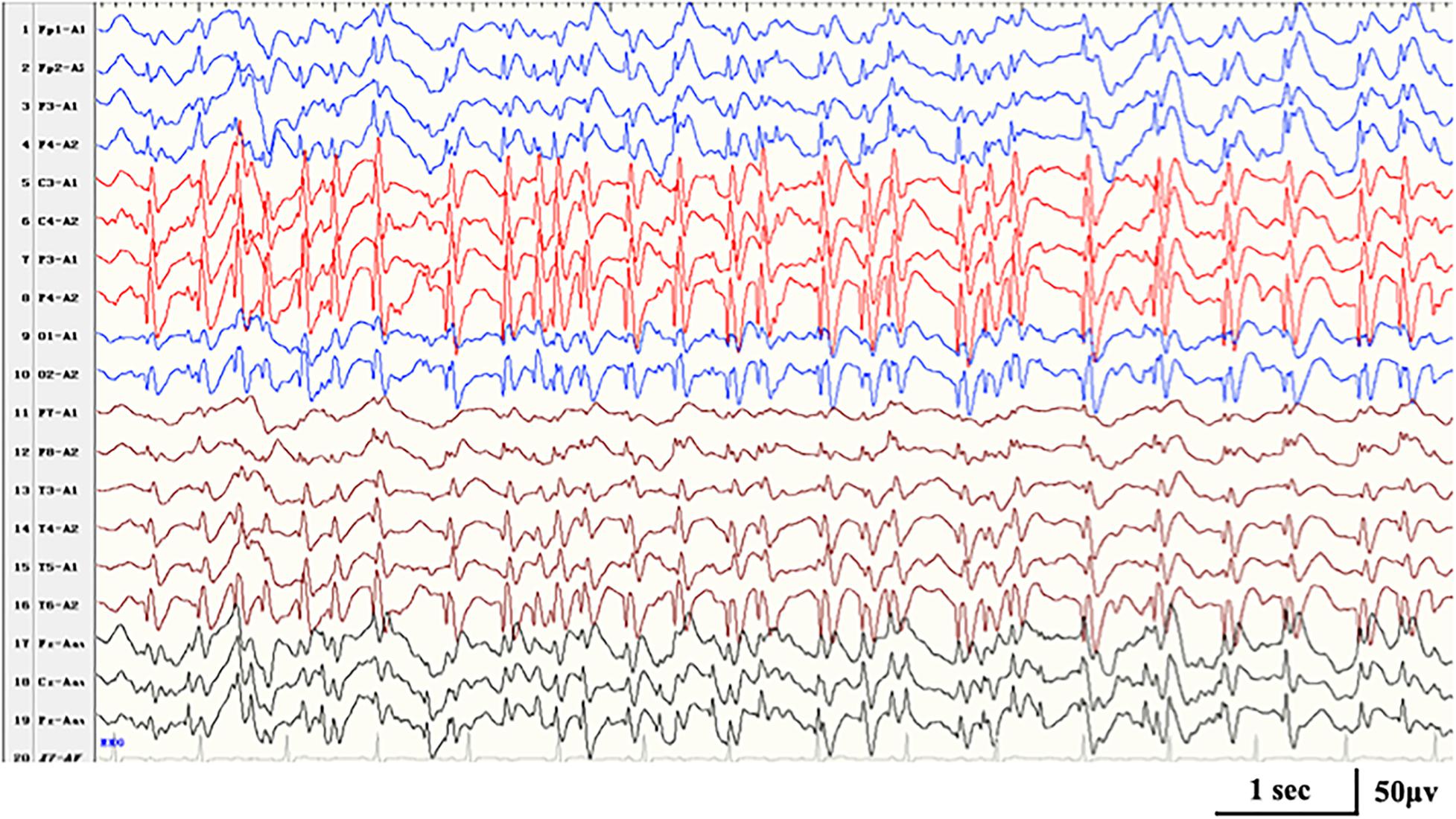
Figure 3. Interictal EEG in case 16 with HIVEP2-related disorders at the age of 7 years, demonstrating generalized spike-and-wave with Rolandic predominance during NREM sleep (SWI 90%).
Compound heterozygous variants in RARS2 were identified in case 16. The patient had a global developmental delay since birth. He gradually presented with hypotonia, with a feeding difficulty apparent from 5 months and lethargy and progressive microcephaly from 7 months. He had frequent multifocal myoclonic seizures at 3 months with developmental regression. Neuroimaging was notable for cerebral atrophy and progressive white matter depletion. He was diagnosed as DEE without relative epilepsy syndrome at the first stage of epilepsy. During the course, he had nocturnal focal seizures and atypical seizures. ESES in the Rolandic region occurred at 3.5 years old (Figure 4). He was diagnosed with ABPE at the second stage of epilepsy. At the last follow-up of age 3.5 years, he still had active seizures and severe developmental delay.
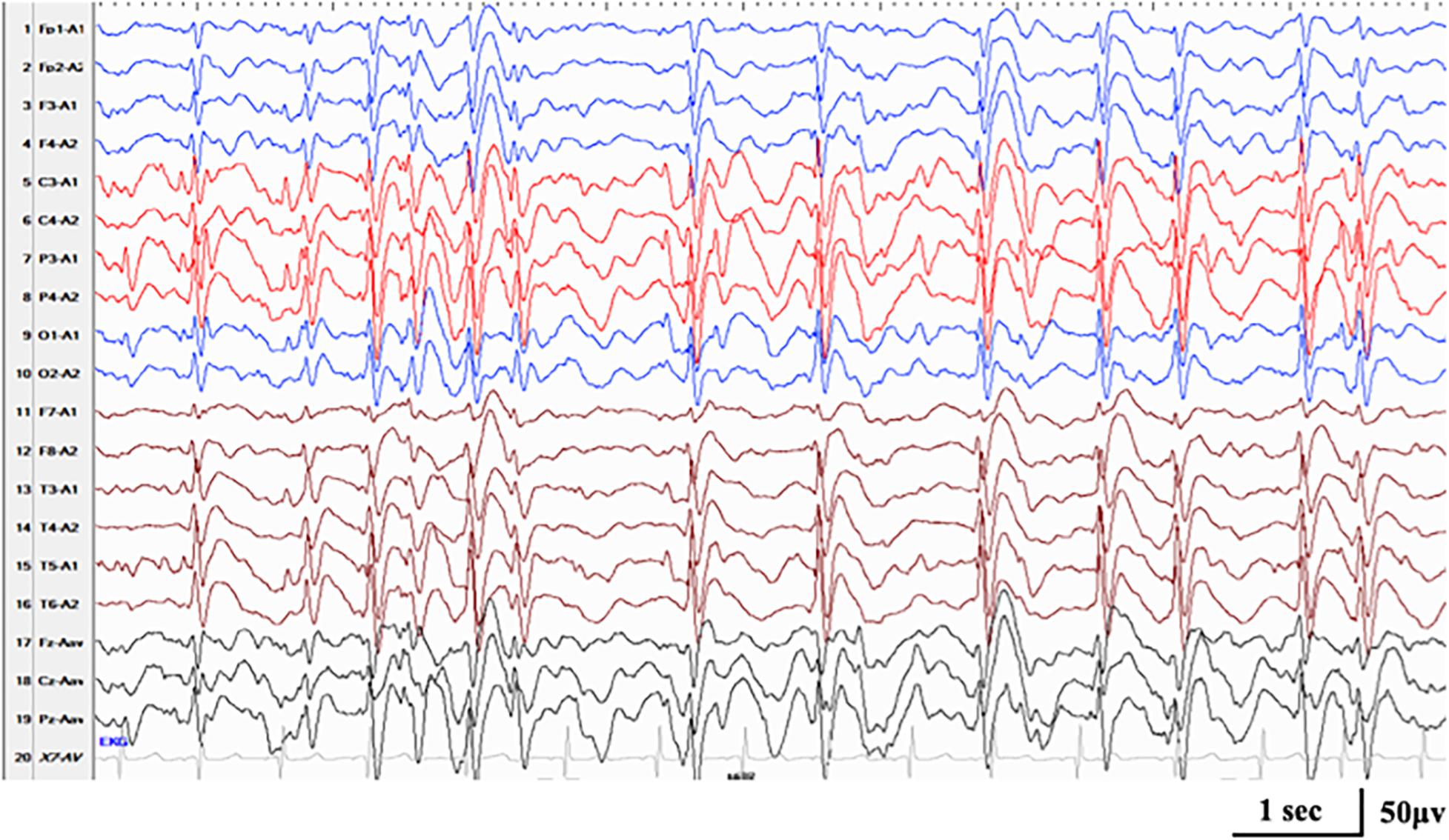
Figure 4. Interictal EEG in case 17 with RARS2-related disorders at the age of 3.5 years, demonstrating generalized spike-and-wave with Rolandic predominance during NREM sleep (SWI 85%).
The mean age at the occurrence of ESES in 17 cases was 2.0 ± 1.2 years. ESES presented with Rolandic predominance in 14, posterior predominance in one, anterior region predominance in one, and anterior or posterior predominance in one. Twelve of 17 cases had the first stage of different epilepsy preceding ESES, and 2/12 were diagnosed as Ohtahara syndrome, 2/12 as infantile spasms, 3 as DEE, and 5/12 as EE without the special epileptic syndrome. At the second stage of epilepsy with ESES, 10/17 cases were diagnosed as ABPE, 1/17 as CSWS, 4/17 as ESES without the epileptic syndrome, and 2/17 as ESES without epilepsy.
Recently, an increasing number of pieces of literature had reported the occurrence of ESES in disorders caused by monogenic variants (Coppola et al., 2003; Kobayashi et al., 2006; Bonanni et al., 2017; Lee et al., 2017; Mathieu et al., 2018). A systematic review on all reported genetic etiologies of ESES-related syndrome summarized 11 monogenic variants identified in 60 cases, including GIRN2A, SCN2A, KCNA2, KCNQ2, KCNB1, SLC6A1, SLC9A6, ATN1, SPRX2, CNKSR, and OPA3 (Kessi et al., 2018). Masnada et al. (2017) observed dramatic activation of EEG abnormalities during sleep in KCNA2-related DEE. They described it as an “ESES-like” pattern due to a different EEG pattern at the early stage of epilepsy and the lack of a longitudinal follow-up. For the purpose of this study, we still used the term “ESES,” referring to the EEG pattern. Here was the largest single-center study, which provided an insight into the genetic etiologies in DEE/EE with ESES and described the electroclinical characteristics in them. Pathogenic or likely pathogenic variants explained 23.0% of cases in our cohort.
Caused by the impaired function of potassium channels due to KCNQ2 variants, KCNQ2 encephalopathy was a clinical syndrome with a wide range from BFNS to early onset epileptic encephalopathy (Fang et al., 2019). Several affected cases with BFNS harboring KCNQ2 variants later developing benign childhood epilepsy with centrotemporal spikes had been reported (Maihara et al., 1999; Coppola et al., 2003; Ishii et al., 2012). In our cohort, all six cases had a first stage of epilepsy with DEE and developed to the second stage with the occurrence of ESES. One with a paternal variant of R333Q was diagnosed as ABPE at the second stage. He achieved ESES remission at 7.1 years old and seizure-free at 8.1 years old with a remarkably developmental improvement, further supporting a diagnosis of ABPE. The R333Q variant had been previously reported in a family with BFNS (Singh et al., 2003). In our cohort, except for this case, the other seven affected families, including his father, were diagnosed with BFNS, whose seizure became free before the first year of life. It indicated that even the same variant could lead to a different phenotype and outcome, which could be explained by other unknown modifier genes and environmental factors. The remaining five cases did not fit with traditional ESES-related syndrome. Among them, ESES appeared almost before the age of 2 years, which was earlier than the typical onset age in traditional ESES-related syndrome. Currently, only two cases of KCNQ2 encephalopathy with ESES had been reported (Lee et al., 2017). With the six cases added, we proposed that ESES could appear as an age-related EEG phenomenon in KCNQ2 encephalopathy, but it might appear earlier due to genetic factors. Also, there could be a different EEG pattern at the first stage of epilepsy preceding ESES.
As a recently discovered gene associated with epilepsy disorders, more than 50 cases bearing the KCNA2 variant had been reported (Masnada et al., 2017; Sachdev et al., 2017). Based on the severity of the encephalopathy and the seizure disorder, the phenotype associated with the KCNA2 variant might be differentiated into two main groups, with the milder phenotype correlating with loss-of-function variants and more severe phenotype with gain-of-function variants (Sachdev et al., 2017). Totally, six cases bearing the same loss-of-function P405L variant had been reported, and five of them presented with ESES (Allou et al., 2017; Sachdev et al., 2017). Here, five cases all carried the P405L variant, and the second stage of epilepsy with ESES was in line with EE of ABPE. However, there was a different EE at the first stage. With the five cases added, it suggested that the specific variant itself in KCNA2 was largely responsible for specific clinical symptoms. Epilepsy disorders related to the P405L variant tended to evolve from a different EE at the first stage into EE of ABPE. It indicated that both genetic and age-related factors played a role in the occurrence of ESES. The underlying mechanisms leading to ESES were not completely understood. The genetic basis might disrupt the normal maturation of neuronal networks, leading to an age-related pattern of electroclinical expression of ESES (Sánchez Fernández et al., 2012). Actually, our group has already worked on the study of the P405L mutation causing ESES, and we generated an induced pluripotent stem cell line from an EE patient with ESES carrying the KCNA2 (p.P405L) mutation (Gong et al., 2020). The induced pluripotent stem cell line will be useful to better understand the pathogenesis of ESES and discover new targets for pharmacological intervention.
The loss-of-function variant of GRIN2A was previously recognized as the principal genetic cause of ESES, and it was reported that up to 20% of ESES-related syndrome had a pathogenic variant in GRIN2A (Pavlidis et al., 2019). Our previous study identified the GRIN2A variant in 4/77 cases with ESES syndrome in a Chinese cohort (one with Landau–Kleffner syndrome and three with ABPE) (Yang et al., 2018). Here, three new cases with ABPE were identified. Two of them had a developmental delay preexisting seizure onset with further developmental regression accompanied by ABPE. It suggested that GRIN2A variants had a close relationship with the EEG pattern of ESES, but to what extent was still uncertain. The exact cellular and molecular mechanisms linking the GRIN2A variant and continuous spiking in NREM sleep were not well understood, but the disturbed glutamatergic pathways signaling during the neurodevelopmental age window might play a role (Bishop et al., 2015). GluN2A subunit encoded by GRIN2A mainly expressed after birth, whereas other subunits encoded by other genes were at a high level at embryonic stages, modestly diminishing after birth (Monyer et al., 1994). The theory discussed earlier could explain why genes had an age-dependent effect on the development of ESES.
In 1999, the variant involved in SLC9A6 was first described to underlie Christianson syndrome, characterized by moderate-to-severe intellectual disability with absent or very limited language development, epilepsy, ataxia, hyperkinetic behavior, and acquired microcephaly (Christianson et al., 1999). So far, ESES was reported in five cases with Christianson syndrome within the critical age window of 4–8 years (Ieda et al., 2019). In one of them, seizure-free and resolution of ESES were achieved at the age of 8 years, and transient clinical recovering of up to 15 words was noticed (Coorg and Weisenberg, 2015). In our cohort, the case was predominantly manifested with developmental impairment related to genetic variants. He was diagnosed as CSWS with ESES occurrence during the course. HIVEP2 was first considered a rare cause of neurodevelopmental abnormalities in 2016 (Steinfeld et al., 2016). To date, HIVEP2 variants had been published in 14 cases with the last follow-up of age 5 to 24 years, and the clinical feature was nonspecific, including hypotonia, development delay, intellectual disability, and dysmorphic features (Park et al., 2019). Only 2 of 14 cases were reported to have definite seizures (Park et al., 2019). Here, our case also had remarkable developmental delay preceding ABPE. It constituted the first known instance of ESES occurrence in HIVEP2-related disorder. RARS2 was an identified cause of pontocerebellar hypoplasia type 6 with autosomal recessive inheritance, typically characterized by pontine atrophy, vermian hypoplasia, infantile encephalopathy, generalized hypotonia, and intractable seizures (Park et al., 2019). Up to now, a total of 26 cases with RARS2 variants had been reported with the last follow-up of age 2 months to 24 years (Zhang et al., 2018). Here, we firstly described ESES associated with RARS2. The case was diagnosed as DEE without epilepsy syndrome at the first stage and developed to ABPE at the second stage. Our findings suggested that ESES could be an age-related feature in genetic disorders related to SLC9A6, HIVEP2, and RARS2. Severe developmental impairment and encephalopathy caused by genetic variant was the main manifestation and relatively mild epilepsy might occur during the course, just like Rett syndrome. Further deterioration of the cognitive and behavioral status during the course warranted a detection of the possible occurrence of ESES.
Studies reported the evolution of the SWI over time with the onset of ESES at 4–8 years of age (Loddenkemper et al., 2011). In our cohort, the mean age at the occurrence of ESES was 2.0 ± 1.2 years, earlier than the typical onset age. It might be due to the genetic factor. In some genetic disorders, especially KCNQ2 and KCNA2, there was always the first stage of different EEG patterns preceding ESES. In most cases, ESES was predominant in the Rolandic region, and most of them were diagnosed as ABPE. We speculated that the epileptiform discharge in these genetic disorders usually localized or transformed to the Rolandic region with further evolution of ESES.
More than 20% of DEE/EE with ESES were identified with monogenic variants, including KCNQ2, KCNA2, GRIN2A, SLC9A6, HIVEP2, and RARS2. With an age-related feature in genetic disorders, ESES could follow the first stage of different EEG patterns in precedence. Further deterioration of the cognitive and behavioral status during the course warranted a detection of the possible occurrence of ESES. Both age-related and genetic factors played a role in the occurrence of ESES in genetic DEE/EE.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by the Ethical Committee of Peking University First Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
ZY conceptualized and designed the study, coordinated the study overall, and revised the manuscript. PG co-designed the study, drafted the initial manuscript, and revised the manuscript. JX and XJ helped collect and summarize data and reviewed the manuscript. YZ interpreted the data and critically reviewed the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (81771393), Beijing Municipal Science & Technology Commission (Z171100001017125), Beijing Natural Science Foundation (7202210), and Capital’s Funds for Health Improvement and Research (2020-2-4077).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to the patients and their families who participated in the study and made this work possible.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.607965/full#supplementary-material
Allou, L., Julia, S., Amsallem, D., El Chehadeh, S., Lambert, L., Thevenon, J., et al. (2017). Rett-like phenotypes: expanding the genetic heterogeneity to the KCNA2 gene and first familial case of CDKL5-related disease. Clin. Genet. 91, 431–440. doi: 10.1111/cge.12784
Bishop, H. I., Guan, D., Bocksteins, E., Parajuli, L. K., Murray, K. D., Cobb, M. M., et al. (2015). Distinct cell-and layer-specific expression patterns and independent regulation of kv2 channel subtypes in cortical pyramidal neurons. J. Neurosci. 35, 14922–14942. doi: 10.1523/JNEUROSCI.1897-15.2015
Bonanni, P., Negrin, S., Volzone, A., Zanotta, N., Epifanio, R., Zucca, C., et al. (2017). Electrical status epilepticus during sleep in Mowat-Wilson syndrome. Brain Dev. 39, 727–734. doi: 10.1016/j.braindev.2017.04.013
Christianson, A. L., Stevenson, R. E., van der Meyden, C. H., Pelser, J., Theron, F. W., van Rensburg, P. L., et al. (1999). X linked severe mental retardation, craniofacial dysmorphology, epilepsy, ophthalmoplegia, and cerebellar atrophy in a large South African kindred is localised to Xq24-q27. J. Med. Genet. 36, 759–766. doi: 10.1136/jmg.36.10.759
Coorg, R., and Weisenberg, J. L. Z. (2015). Successful treatment of electrographic status epilepticus of sleep with felbamate in a patient with SLC9A6 mutation. Pediatr. Neurol. 53, 527–531. doi: 10.1016/j.pediatrneurol.2015.07.007
Coppola, G., Castaldo, P., Miraglia del Giudice, E., Bellini, G., Galasso, F., Soldovieri, M. V., et al. (2003). A novel KCNQ2 K+ channel mutation in benign neonatal convulsions and centrotemporal spikes. Neurology 61, 131–134. doi: 10.1212/01.WNL.0000069465.53698.BD
Fang, Z. X., Zhang, M., Xie, L. L., Jiang, L., Hong, S. Q., Li, X. J., et al. (2019). KCNQ2 related early-onset epileptic encephalopathies in Chinese children. J. Neurol. 266, 2224–2232. doi: 10.1007/s00415-019-09404-y
Goldsmith, H., Wells, A., Maria, J. N. S., Williams, M., Heussler, H., Buckman, M., et al. (2019). Expanding the phenotype of intellectual disability caused by HIVEP2 variants. Am. J. Med. Genet. A 179, 1872–1877. doi: 10.1002/ajmg.a.61271
Gong, P., Jiao, X., Zhang, Y., and Yang, Z. (2020). Generation of a human iPSC line from an epileptic encephalopathy patient with electrical status epilepticus during sleep carrying KCNA2 (p.P405L) mutation. Stem Cell Res. 49:102080. doi: 10.1016/j.scr.2020.102080
Ieda, D., Hori, I., Nakamura, Y., Ohashi, K., Negishi, Y., Hattori, A., et al. (2019). A novel splicing mutation in SLC9A6 in a boy with Christianson syndrome. Hum. Genome Var. 6:15. doi: 10.1038/s41439-019-0046-x
Ishii, A., Miyajima, T., Kurahashi, H., Wang, J. W., Yasumoto, S., Kaneko, S., et al. (2012). KCNQ2 abnormality in BECTS: benign childhood epilepsy with centrotemporal spikes following benign neonatal seizures resulting from a mutation of KCNQ2. Epilepsy Res. 102, 122–125. doi: 10.1016/j.eplepsyres.2012.07.011
Kessi, M., Peng, J., Yang, L., Xiong, J., Duan, H., Pang, N., et al. (2018). Genetic etiologies of the electrical status epilepticus during slow wave sleep: systematic review. BMC Genet. 19:40. doi: 10.1186/s12863-018-0628-5
Kobayashi, K., Hata, H., Oka, M., Ito, M., Yoshinaga, H., Kashihara, K., et al. (2006). Age-related electrical status epilepticus during sleep and epileptic negative myoclonus in DRPLA. Neurology 66, 772–773. doi: 10.1212/01.wnl.0000200958.30060.36
Lee, I. C., Yang, J. J., and Li, S. Y. (2017). A KCNQ2 E515D mutation associated with benign familial neonatal seizures and continuous spike and waves during slow-wave sleep syndrome in Taiwan. J. Formos. Med. Assoc. 116, 711–719. doi: 10.1016/j.jfma.2016.11.009
Loddenkemper, T., Fernández, I. S., and Peters, J. M. (2011). Continuous spike and waves during sleep and electrical status epilepticus in sleep. J. Clin. Neurophysiol. 28, 154–164. doi: 10.1097/WNP.0b013e31821213eb
Maihara, T., Tsuji, M., Higuchi, Y., and Hattori, H. (1999). Benign familial neonatal convulsions followed by benign epilepsy with centrotemporal spikes in two siblings. Epilepsia 40, 110–113. doi: 10.1111/j.1528-1157.1999.tb01997.x
Masnada, S., Hedrich, U. B. S., Gardella, E., Schubert, J., Kaiwar, C., Klee, E. W., et al. (2017). Clinical spectrum and genotype-phenotype associations of KCNA2-related encephalopathies. Brain 140, 2337–2354. doi: 10.1093/brain/awx184
Mathieu, M. L., de Bellescize, J., Till, M., Flurin, V., Labalme, A., Chatron, N., et al. (2018). Electrical status epilepticus in sleep, a constitutive feature of Christianson syndrome? Eur. J. Paediatr. Neurol. 22, 1124–1132. doi: 10.1016/j.ejpn.2018.07.004
Monyer, H., Burnashev, N., Laurie, D. J., Sakmann, B., and Seeburg, P. H. (1994). Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12, 529–540. doi: 10.1016/0896-6273(94)90210-0
Park, J., Colombo, R., Schäferhoff, K., Janiri, L., Grimmel, M., Sturm, M., et al. (2019). Novel HIVEP2 variants in patients with intellectual disability. Mol. Syndromol. 10, 195–201. doi: 10.1159/000499060
Patry, G., Lyagoubi, S., and Tassinari, C. A. (1971). Subclinical “electrical status epilepticus” induced by sleep in children: a clinical and electroencephalographic study of six cases. Arch. Neurol. 24, 242–252. doi: 10.1001/archneur.1971.00480330070006
Pavlidis, E., Møller, R. S., Nikanorova, M., Kölmel, M. S., Stendevad, P., Beniczky, S., et al. (2019). Idiopathic encephalopathy related to status epilepticus during slow sleep (ESES) as a “pure” model of epileptic encephalopathy. An electroclinical, genetic, and follow-up study. Epilepsy Behav. 97, 244–252. doi: 10.1016/j.yebeh.2019.05.030
Richards, S., Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for molecular pathology. Genet. Med. 17, 405–424. doi: 10.1038/gim.2015.30
Sachdev, M., Gaínza-Lein, M., Tchapyjnikov, D., Jiang, Y. H., Loddenkemper, T., and Mikati, M. A. (2017). Novel clinical manifestations in patients with KCNA2 mutations. Seizure 51, 74–76. doi: 10.1016/j.seizure.2017.07.018
Sánchez Fernández, I., Loddenkemper, T., Peters, J. M., and Kothare, S. V. (2012). Electrical status epilepticus in sleep: clinical presentation and pathophysiology. Pediatr. Neurol. 47, 390–410. doi: 10.1016/j.pediatrneurol.2012.06.016
Singh, N. A., Westenskow, P., Charlier, C., Pappas, C., Leslie, J., Dillon, J., et al. (2003). KCNQ2 and KCNQ3 potassium channel genes in benign familial neonatal convulsions: expansion of the functional and mutation spectrum. Brain 126, 2726–2737. doi: 10.1093/brain/awg286
Steinfeld, H., Cho, M. T., Retterer, K., Person, R., Schaefer, G. B., Danylchuk, N., et al. (2016). Mutations in HIVEP2 are associated with developmental delay, intellectual disability, and dysmorphic features. Neurogenetics 17, 159–164. doi: 10.1007/s10048-016-0479-z
Yang, X., Qian, P., Xu, X., Liu, X., Wu, X., Zhang, Y., et al. (2018). GRIN2A mutations in epilepsy-aphasia spectrum disorders. Brain Dev. 40, 205–210. doi: 10.1016/j.braindev.2017.09.007
Keywords: electrical status epilepticus during sleep, encephalopathy, genetic, etiology, epilepsy
Citation: Gong P, Xue J, Jiao X, Zhang Y and Yang Z (2021) Genetic Etiologies in Developmental and/or Epileptic Encephalopathy With Electrical Status Epilepticus During Sleep: Cohort Study. Front. Genet. 12:607965. doi: 10.3389/fgene.2021.607965
Received: 30 September 2020; Accepted: 02 March 2021;
Published: 08 April 2021.
Edited by:
Dan Koboldt, Nationwide Children’s Hospital, United StatesReviewed by:
Heather Mefford, University of Washington, United StatesCopyright © 2021 Gong, Xue, Jiao, Zhang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhixian Yang, emhpeGlhbi55YW5nQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.