 Igor Araujo Vieira1,2†
Igor Araujo Vieira1,2† Tiago Finger Andreis1,2†
Tiago Finger Andreis1,2† Bruna Vieira Fernandes2,3
Bruna Vieira Fernandes2,3 Maria Isabel Achatz4
Maria Isabel Achatz4 Gabriel S. Macedo2,5
Gabriel S. Macedo2,5 Daniel Schramek6,7
Daniel Schramek6,7 Patricia Ashton-Prolla1,2,5*
Patricia Ashton-Prolla1,2,5*- 1Programa de Pós-Graduação em Genética e Biologia Molecular, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
- 2Laboratório de Medicina Genômica, Centro de Pesquisa Experimental, Hospital de Clínicas de Porto Alegre, Porto Alegre, Brazil
- 3Curso de Graduação em Biomedicina, Universidade Federal de Ciências da Saúde de Porto Alegre, Porto Alegre, Brazil
- 4Departamento de Oncogenética, Hospital Sírio Libanês, São Paulo, Brazil
- 5Programa de Medicina Personalizada, Hospital de Clínicas de Porto Alegre, Porto Alegre, Brazil
- 6Centre for Molecular and Systems Biology, Lunenfeld-Tanenbaum Research Institute, Mount Sinai Hospital, Toronto, ON, Canada
- 7Department of Molecular Genetics, University of Toronto, Toronto, ON, Canada
In Southern and Southeastern Brazil, there is a germline pathogenic variant with incomplete penetrance located in the oligomerization domain of TP53, c.1010G>A (p.Arg337His). Due to a founder effect, the variant is present in 0.3% of the general population of the region. Recently, this variant was identified in 4.4 and 8.9% of two apparently unselected, single center case series of Brazilian lung adenocarcinoma (LUAD) patients from the Southeastern and Central regions of the country, respectively. In the present study, our aim was to examine TP53 c.1010G>A allele and genotype frequencies in LUAD samples obtained from patients diagnosed in Southern Brazil. A total of 586 LUAD samples (tumor DNA) recruited from multiple centers in the region were tested, and the mutant allele was identified using TaqMan® assays in seven cases (7/586, 1.2%) which were submitted to next generation sequencing analyses for confirmation. Somatic EGFR mutations were more frequent in TP53 c.1010G>A carriers than in non-carriers (57.1 vs. 17.6%, respectively). Further studies are needed to confirm if TP53 c.1010G>A is a driver in LUAD carcinogenesis and to verify if there is a combined effect of EGFR and germline TP53 c.1010G>A. Although variant frequency was higher than observed in the general population, it is less than previously reported in LUAD patients from other Brazilian regions. Additional data, producing regional allele frequency information in larger series of patients and including cost-effectiveness analyses, are necessary to determine if TP53 c.1010G>A screening in all Brazilian LUAD patients is justified.
Introduction
In Southern and Southeastern Brazil, a germline founder pathogenic variant with incomplete penetrance, c.1010G>A (rs121912664), also known as R337H or p.(Arg337His) has been detected in 0.3% of the general population (Achatz et al., 2009; Custódio et al., 2013). It is located in the oligomerization domain (exon 10) of TP53, and it is associated with Li-Fraumeni syndrome (LFS). Carriers are at a high risk for developing a wide spectrum of tumors and are prone to develop multiple primary cancers at different ages. The core tumors in LFS patients include early onset breast cancer, soft-tissue sarcomas, brain tumors and adrenocortical carcinomas (Achatz et al., 2007; Giacomazzi et al., 2013; Achatz and Zambetti, 2016). Lung cancer (LC), especially lung adenocarcinoma (LUAD) of the lepidic subtype, has been reported in LFS families and is included in the Chompret criteria for genetic testing of suspected patients, but there is limited evidence for a strong association of its occurrence with germline TP53 variants (Tinat et al., 2009; Ricordel et al., 2015; Caron et al., 2017). Although TP53 is considered one of the most commonly mutated genes in solid tumors, somatic occurrence of TP53 c.1010G>A is extremely rare. In the IARC TP53 database [International Agency for Research on Cancer TP53 database (Iarc), 2020], c.1010G>A has been described in only 4 of 28,869 solid tumors. In other public databases, its frequency in solid tumors is also very low. In COSMIC [Catalogue Of Somatic Mutations In Cancer (Cosmic), 2020], for instance, among over 20.000 samples, TP53 c.1010G>A is not present, although G > T and G > C alterations in codon 1,010 do occur. In contrast, two lung tumors harboring germline c.1010G>A have been described in the IARC TP53 database. Finally, Nogueira et al. (2015) reported a mixed acinar/bronchiolo-alveolar carcinoma in a known germline carrier of the TP53 c.1010G>A variant (Nogueira et al., 2015).
LC is the leading cause of cancer related deaths worldwide, responsible for 1.7 million deaths every year. In Brazil, the National Cancer Institute [Instituto Nacional de Câncer José Alencar Gomes da Silva (Inca), 2020] estimated 30,200 new LC cases in 2020, rendering it the second most common solid tumor in the country. In Southern Brazil, LC is the third most common cancer diagnosed in adults, with non-small cell lung cancer (NSCLC) accounting for approximately 85% of all LCs cases. Recently, Couto et al. (2017) genotyped TP53 c.1010G>A in 45 NSCLC patients from a single center in the central region of Brazil, where this variant has not been explored and its population prevalence has not been determined. They identified 4 (8.9%) heterozygotes, a surprisingly high variant frequency for a small, apparently unselected cohort (Couto et al., 2017). Importantly, a more recent single center study in Southeastern Brazil also observed a high prevalence of TP53 c.1010G>A in an unselected series of 114 EGFR-positive LUAD patients: the variant was present in 4.4 and 12.5% of samples when considering diagnosis at any age or before the age of 50 years, respectively (Barbosa et al., 2020). Furthermore, the authors assessed LUAD tumors diagnosed in known TP53 c.1010G>A carriers and observed that LUAD tumors from 8/9 (89%) TP53 c.1010G>A carriers harbored an activating EGFR variant. To our knowledge, the TP53 c.1010G>A variant has not been described in other studies assessing somatic TP53 variants in sporadic lung adenocarcinoma (LUAD) patients (Greenman et al., 2007; Ding et al., 2008; Hammerman et al., 2012; Imielinski et al., 2012; Peifer et al., 2012; Rizvi et al., 2015; Jordan et al., 2017). Thus, in the present study, our goal was to examine the TP53 c.1010G>A allele and genotype frequencies in a series of 586 LUAD samples obtained from patients diagnosed in multiple centers of the three states of Southern Brazil, a region with the highest population frequency of this particular variant observed to date, regardless of their clinical features.
Methods
Study Subjects and Ethical Aspects
A total of 586 LC samples derived from a cohort described in a previous study from our group (Andreis et al., 2019) were analyzed. Patients were originally recruited for somatic mutation testing in EGFR (exons 18–21), KRAS (exons 2 and 3), BRAF (exons 11 and 15), and NRAS (exons 2 and 3) genes from different hospitals and clinics distributed in 22 healthcare centers located in the three states of the southern region of Brazil: Rio Grande do Sul (N = 496), Santa Catarina (N = 20), and Paraná (N = 70). Patients were not selected on previous cancer history or family history of cancer. Pathologic analyses confirmed typical adenocarcinoma histology in all cases. However, histological subtype data were available for only a small proportion (72/586) of patients. The mutation status of EGFR, BRAF, KRAS, and NRAS genes was evaluated using technical procedures as previously reported (Andreis et al., 2019). Genotyping was performed in a central laboratory, a diagnostic precision medicine program established in a tertiary care hospital in southern Brazil. Before initiation of this study, age at LUAD diagnosis, EGFR/KRAS/BRAF status, and histological subtype (when available) were annotated and samples were posteriorly de-identified. A consent waiver was approved by the Institutional Review Board specifically for the TP53 c.1010G>A analysis, given that patient identification using only age at diagnosis, EGFR/KRAS/BRAF/TP53 status and LUAD histological subtype would not allow patient re-identification.
DNA Extraction and TP53 c.1010G>A Genotyping
Tumor DNA was extracted using the ReliaPrepTM FFPE gDNA Miniprep System (Promega), according to the manufacturer’s recommendations. Next, TaqMan® allelic discrimination analyses of the pathogenic variant TP53 c.1010G>A (rs121912664) were performed according to Applied Biosystems® standard protocols (Applied Biosystems, Carlsbad, CA United States), using fluorescent allele-specific probes as previously published (Fitarelli-Kiehl et al., 2016). An attempt to confirm rs121912664-positive results identified by TaqMan® by next generation sequencing (NGS) for determination of allele frequencies was made. Briefly, NGS of the TP53 entire coding region (exons 2–11) and 70 pb exon-intron boundaries was done using a custom panel (Thermo Fisher Scientific, CA, United States, reference number TP53.20140108.designed) on the Ion Torrent-Personal Genome Machine (PGM) platform. Amplicon library was prepared using the Ion AmpliSeqTM Library Kit 2.0 (Thermo Fisher Scientific, CA, United States). PCR products were then sequenced on the Ion GeneStudio S5 system (Ion Torrent Systems Inc., Gilford, NH, United States). NGS results were interpreted using the Ion Reporter software considering a minimum coverage of 100X by amplicon. Integrative Genomics Viewer (IGV) was used for visualization of the mapped reads. Human TP53 cDNA sequence corresponding to the NM_000546.5 was used as a wild-type (WT) reference.
Statistical Analyses
Genotype and allele frequencies were estimated by simple counting. Clinical and molecular features of LUAD patients were assessed using descriptive statistics. Considering the low number of mutant alleles found in our study and limitations in clinical data availability, it was not possible to perform any meaningful statistical test in our comparisons between groups of carriers and non-carriers (see more in Results section). SPSS® version 18 (SPSS® Inc., Chicago, IL, United States) was used for data handling and for all descriptive analyses.
Results
A total of 586 LUAD samples were included in this study. Clinical and molecular data are summarized in Table 1. Histopathological subclassifications were available only for 72 cases (12.3%). The most common subtypes were acinar (65.3%) and lepidic (34.7%). Moreover, the majority of LUAD samples (502/586, 85.7%) were tested for somatic mutations in EGFR, KRAS, BRAF and NRAS driver genes. Somatic mutations were mostly identified in KRAS (29.5%) and EGFR (21.1%).
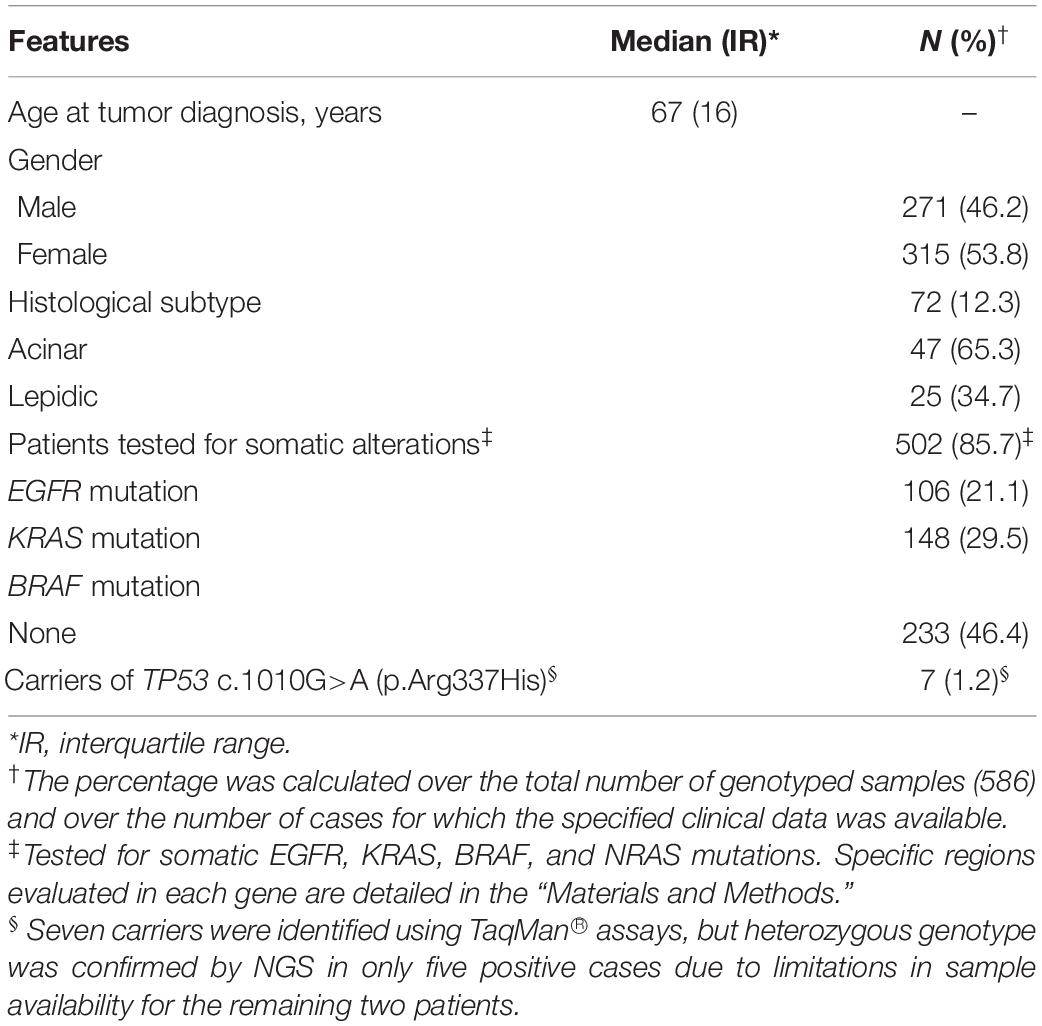
Table 1. Clinical and molecular features of 586 patients with lung adenocarcinoma (LUAD) included in this study.
Specific TP53 c.1010G>A (p.Arg337His) genotyping by TaqMan® resulted in the identification of seven heterozygotes (GA genotype). Heterozygous genotype and mutant allele (A) frequencies were thus defined at 7/586 (1.2%) and 7/1,172 (0.6%), respectively. All patients in this subgroup were diagnosed with LUAD after age 50 years, and the median age at tumor onset was 60 years. In addition, most c.1010G>A tumors also had somatic EGFR (4/7, 57.1%) variants and none of them had a KRAS variant. Importantly, heterozygous genotype was confirmed by NGS in five (p.Arg337His)-positive cases (5/586, 0.85%). In the other two positive samples, further analyses were not possible due limitations in sample availability. Mutant allele frequencies determined by NGS were close to 50% in 3/5 cases, suggesting that at least in these cases, the variant may also be present in the germline. Details on each of the heterozygous samples are summarized in Table 2. Additional TP53 alterations detected in the tumors by NGS are depicted in Supplementary Table 1.
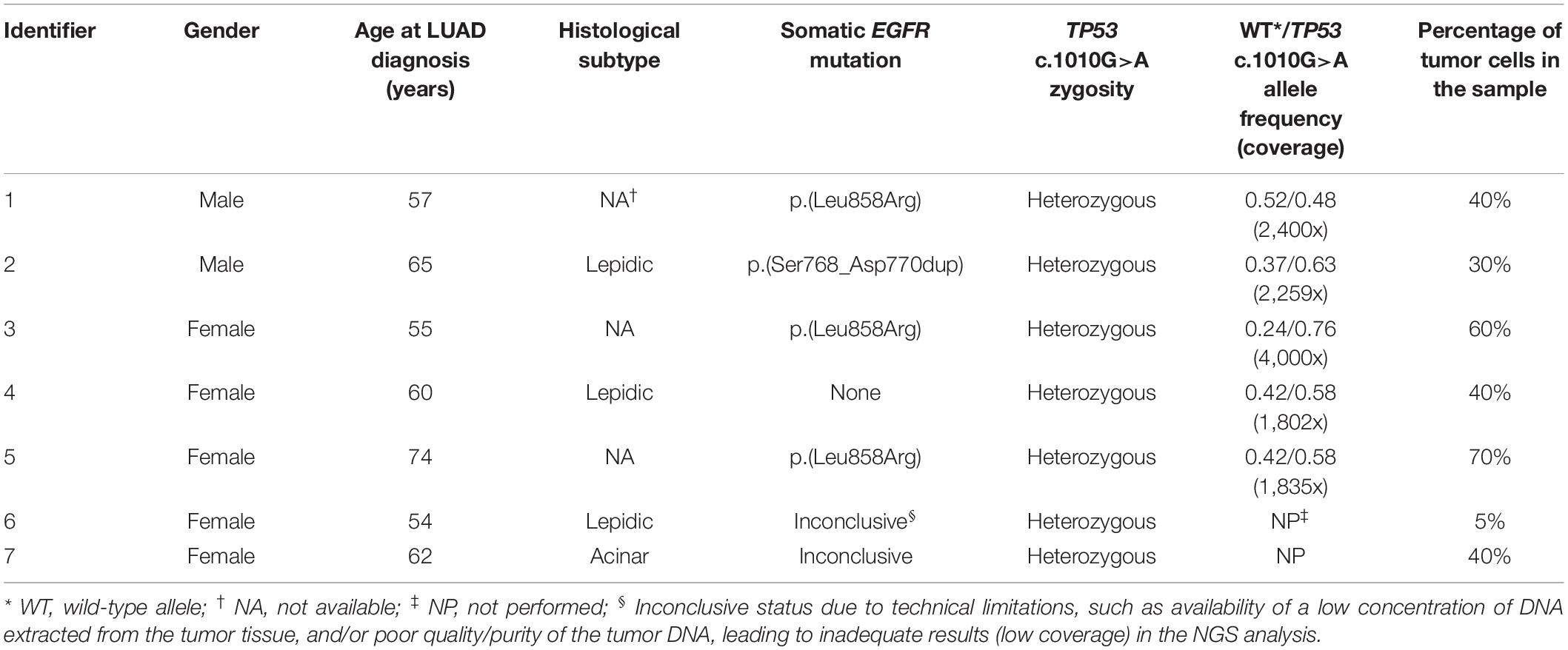
Table 2. Characterization of LUAD tumors with an identifiable TP53 c.1010G>A pathogenic variant.
Lastly, a comparison of the available clinical and molecular features between TP53 c.1010G>A carriers identified by TaqMan® and non-carriers is presented in Table 3 and Supplementary Figure 1. A difference between groups was observed for median age at cancer diagnosis and histological subtype but the number of mutant allele carriers (7/586) was too small to make meaningful comparisons. LUAD from three carriers were of the lepidic subtype (3/7, 42.8%), however, it is important to emphasize that three of the seven heterozygotes had no complete histological data, which hindered our ability to analyze a possible association between the c.1010G>A variant and occurrence of the lepidic subtype. Interestingly, the presence of somatic EGFR mutations was found in a much higher frequency in TP53 c.1010G>A carriers than in non-carriers (57.1 vs. 17.6%, respectively).
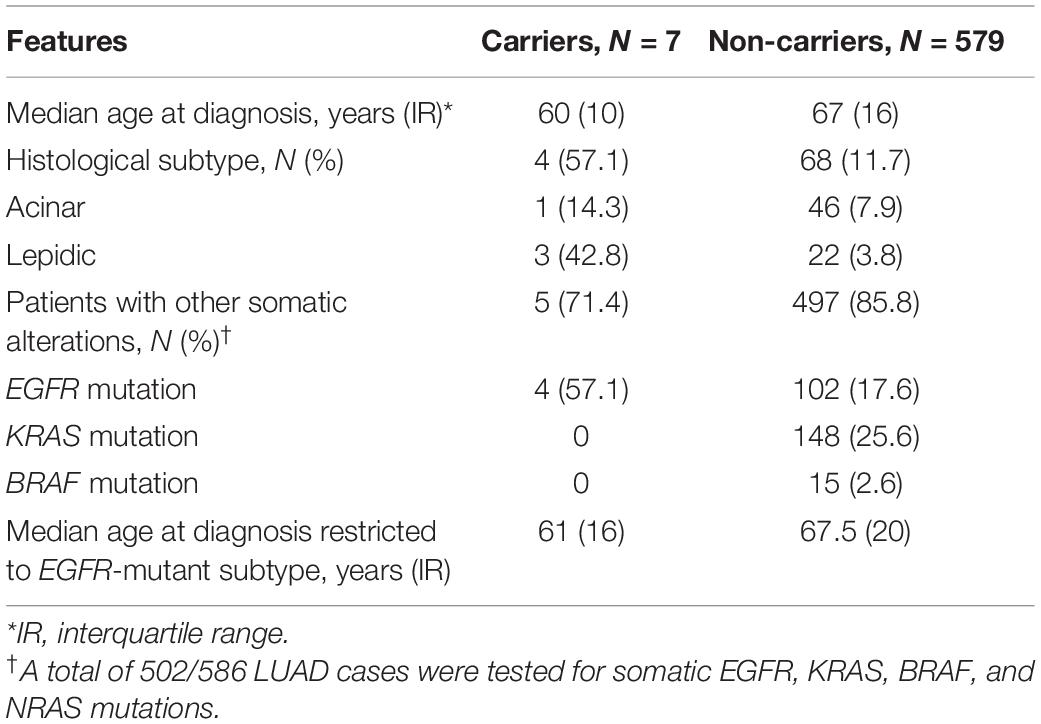
Table 3. Comparison of clinical and molecular features between LUAD samples of TP53 c.1010G>A carriers and non-carriers.
Discussion
The TP53 founder variant c.1010G>A, widely referred as R337H, is likely responsible for a significant proportion of the cancer burden in the Southern and Southeastern regions of Brazil due to its high frequency in the population (Achatz et al., 2009; Garritano et al., 2010). In a landmark study conducted in 171,000 newborns from the Southern Brazilian State of Paraná the variant was identified in 461 individuals (∼0.3%) (Custódio et al., 2013). Previous analyses performed by our research group (Palmero et al., 2008) identified a similar prevalence (2/750, ∼0,3%) in a cohort of healthy, asymptomatic women participating in a community-based breast cancer screening program in the state of Rio Grande do Sul (Southern Brazil). Furthermore, it has been well-documented that the variant exhibits incomplete, variable penetrance resulting in significant inter- and intra-familiar heterogeneity in phenotypic presentation with some carriers surviving without any cancer diagnosis to older ages and others having the typical LFS phenotype, with one or more cancers diagnosed in childhood and/or adult life (reviewed in Achatz and Zambetti, 2016). Unlike most TP53 somatic and/or germline deleterious variants involved in tumorigenesis (i.e., those located in the DNA-binding domain of p53 protein), c.1010G>A is located in the p53 oligomerization domain and has been associated with a unique intracellular pH-dependent effect on protein stability, through which the mutant protein retains some partial tumor suppressor activity (DiGiammarino et al., 2002; Zerdoumi et al., 2017). More recently, a knock-in animal model containing the homologous TP53 R337H variant (mouse R334H) demonstrated that this alteration triggers reduced formation of p53 dimers and tetramers (deficient oligomerization capacity) compared to WT p53 in mouse liver tissues after exposure to a specific carcinogen (Park et al., 2018). Interestingly, the homozygous p53 R334H mutant mice exposed to this carcinogen showed an increased liver tumor development, while mice with the same genotype that were not under exposure to the liver carcinogen developed normally without any significant difference in terms of either cancer incidence or life span compared with WT mice (Park et al., 2018, 2019).
Given its relevance, prevalence of the mutant allele (germline and/or somatic DNA analysis) has been extensively investigated in different Brazilian cohorts of patients with several tumor types, regardless of cancer family history (FH). In three studies evaluating patients with adrenocortical carcinoma (ACC), c.1010G>A was present in 90–97%, independent of cancer FH (Ribeiro et al., 2001; Seidinger et al., 2011; Mastellaro et al., 2018). Moreover, the variant was found in 69, 8.4, and 7.3% of unselected subjects diagnosed with choroid plexus carcinoma, neuroblastoma and osteosarcoma, respectively (Seidinger et al., 2011, 2015). Prevalence of the variant in breast cancer (BC) patients from southern and southeastern Brazil was also studied. In three independent case series, prevalences of 3.4% (familial BC but without a clear LFS phenotype) and 2.5–8.6% (BC patients unselected for cancer FH diagnosed in different age groups) were observed (Giacomazzi et al., 2014; Hahn et al., 2018).
In the present study, we identified seven LUAD tumors harboring the founder TP53 variant among 586 samples recruited from patients in southern Brazil, a cohort described in detail elsewhere (Andreis et al., 2019). Remarkably, all variant-positive tumors were diagnosed over age 50 years. The late onset of LC in TP53 c.1010G>A carriers is in agreement with a recent finding showing a lifelong cancer risk pattern characterized by a bimodal age distribution: one peak in the first 10 years of life associated mainly with ACC and CNS tumors, and a second peak in the fifth decade in which different tumor types occur, including LC (Mastellaro et al., 2017). Although LC (especially LUAD) is not among the core, most frequent tumors originally described in LFS, it has been described in Brazilian LFS families with the TP53 c.1010G>A variant (Barbosa et al., 2020), together with other atypical tumors such as papillary thyroid and renal cancers (Achatz and Zambetti, 2016).
Although TP53 c.1010G>A prevalence in LUAD reported in the present study (1.2%) is substantially higher than the population frequency observed in large cohorts of healthy individuals from the same Brazilian region (0.3%) (Palmero et al., 2008; Custódio et al., 2013), it is much lower than the prevalences previously described by Couto et al. (2017), and Barbosa et al. (2020). The conflicting results between our study and the study performed by Couto et al. (2017) may be explained by two main reasons: (a) small sample size along with a putative selection bias in patient recruitment in the study conducted by Couto et al. indirectly evidenced by high proportion of carriers identified with a cancer FH; and (b) employment of different variant screening approaches (PCR-RFLP vs. TaqMan®), since use of restriction endonucleases may be associated with increased false-positive rates (Uemura et al., 2004; Kang et al., 2009). The study by Barbosa et al. (2020), although performed on a larger sample size, is also a single center study developed in an institution that has been a reference center for the diagnosis and follow-up of LFS families. In addition, one cannot exclude presence of specific, regional environmental factors (carcinogenic exposure, similar to what has been observed in mouse models) which might act triggering LUAD carcinogenesis in c.1010G>A carriers and result in a variable susceptibility to LUAD in carriers from different geographic regions.
Another interesting finding of our study was the co-occurrence of TP53 c.1010G>A and activating EGFR mutations, which is in agreement with two previous reports (Barbosa et al., 2020; Mezquita et al., 2020). Studying LFS patients with a germline TP53 pathogenic variant (either DNA-binding variants or the founder c.1010G>A) and LUAD, Barbosa et al. (2020) and Mezquita et al. (2020) identified somatic EGFR alterations in 89 and 85% of the tumors, respectively. In our study, EGFR variants occurred in 57.1% of the TP53 c.1010G>A-positive tumors, a lower proportion than reported before, but our cohort was not of LFS patients. In addition, when looking at 114 LUAD patients with EGFR mutations from a non-LFS cohort, Barbosa et al. (2020) observed a TP53 codon 337 variant in 5.3%, which was further increased to 12.5% (4/32) when considering only patients diagnosed before age 50 years. In our study, TP53 c.1010G>A was present in 4/106 (3.8%) LUAD samples with activating EGFR mutations, and all patients with both alterations were diagnosed after the age of 50 years. Importantly, ethnic ancestry has major impact on the incidence of EGFR mutation status in NSCLC patients, being lower (around 10%) in regions with higher European ancestry representation (Gahr et al., 2013). The prevalence of European ancestry in the population studied here, i.e., from Southern Brazil, has been estimated at 80–90%, the highest among all Brazilian regions (Ruiz-Linares et al., 2014). In agreement with this observation, several studies evaluating the human leukocyte antigen genotypic diversity in this region also confirmed a high European ancestry and a meaningful difference from Asians or even Brazilian indigenous populations (Castro et al., 2019).
Indeed, the frequent co-occurrence of TP53 and EGFR sequence variants is striking and it might be explained by a p53/miR-193a/EGFR feedback loop mechanism previously reported as a driving force for NSCLC tumorigenesis (Wang et al., 2019). In vitro and in vivo studies demonstrated that WT p53 directly activates miR-193a transcription and, in turn, miR-193a directly targets EGFR, whereas EGFR functions as a transcriptional repressor to negatively control miR-193a expression, forming a feedback loop (Figure 1A). Considering the repeated reports of co-occurrence of TP53 and EGFR variants in LUAD, including the present study, we hypothesize that occurrence of an activating EGFR somatic event on a background of a tissue already harboring a mutant (germline) TP53 allele (such as c.1010G>A) may result in an impairment of this loop function, promoting NSCLC formation and tumor growth (Figure 1B). In parallel, it has been widely described that, upon conditions of genotoxic stress in the cellular context, presence of functional TP53 germline variants, especially hypomorphic variants (c.1010G>A), leads to a loss of p53 tumor-suppressive functions (such as DNA damage repair and cell-cycle arrest), triggering a genomic instability that, in turn, may promote the accumulation of somatic mutations at different genes (Zerdoumi et al., 2017; Park et al., 2018), which could include the hotspot regions of somatic EGFR mutations. Additionally, a previous study suggested that radiation-induced cancers are more common among LFS patients (harboring germline TP53 pathogenic variants) (Heymann et al., 2010). Although the underlying mechanism is not known, in recent reports of EGFR-mutated lung tumors in LFS patients, researchers hypothesized that the first genetic hit was a germline TP53 mutation and suggested that chemotherapy or radiotherapy (i.e., genotoxic stress-inducing conditions) promoted a second hit, which might have consisted specifically of somatic EGFR mutations (Michalarea et al., 2014; Ricordel et al., 2015). Further functional studies are required to confirm this latter hypothesis, as well as to elucidate, mechanistically, the reason why patients with TP53 germline mutations seem to harbor, more frequently, activating EGFR mutations in lung tissue.
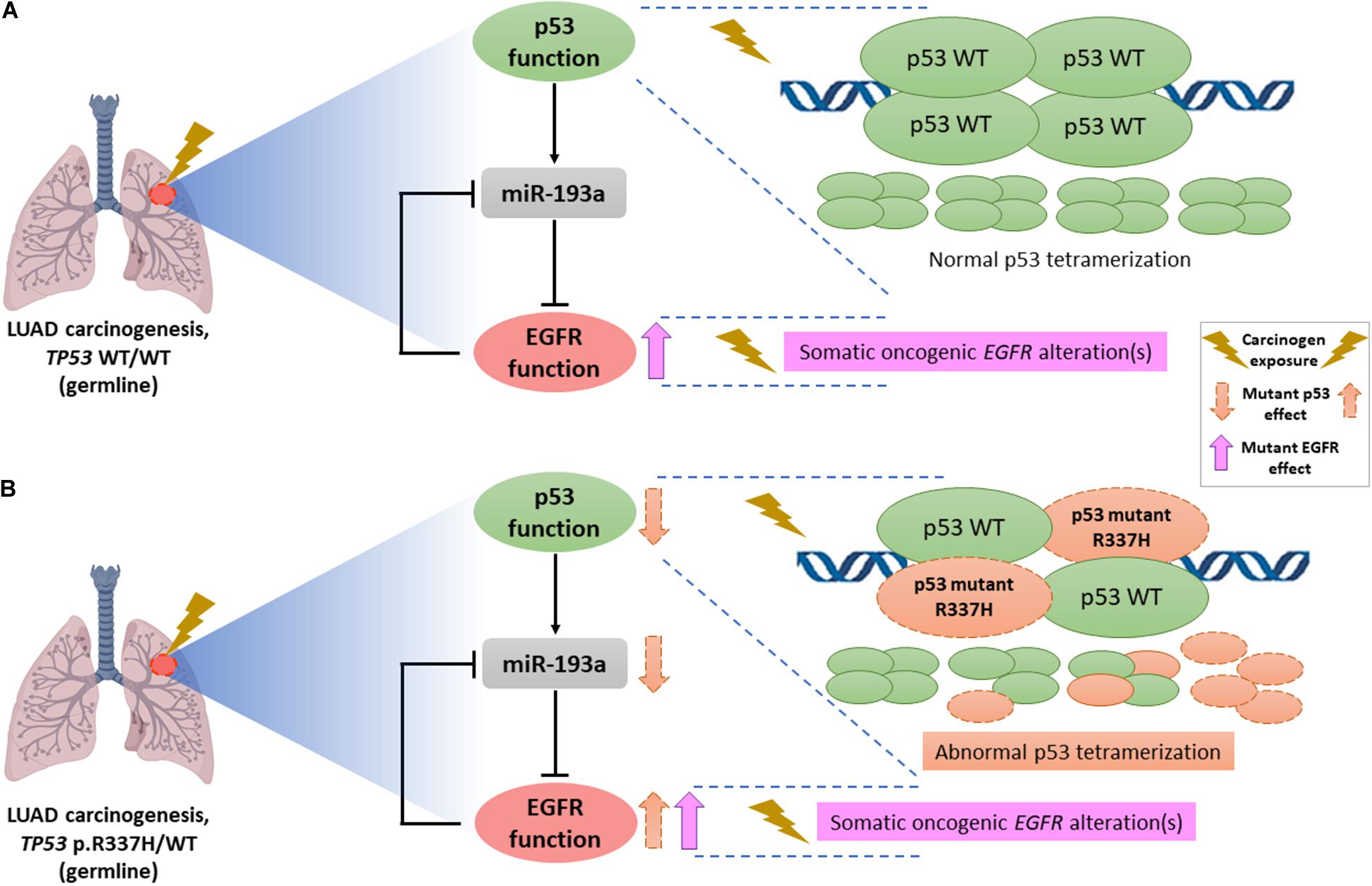
Figure 1. Schematic representation of the hypothetical interaction between mutant p53 (due to hypomorphic TP53 germline variants such as c.1010G>A, p.R337H) and mutant EGFR (due to somatic activating EGFR alterations) in NSCLC tumors. (A) Lung carcinogenesis in an individual without germline TP53 pathogenic variants, i.e., a homozygous context of wild-type (WT) p53 (TP53 WT/WT genotype) characterized by constitutive formation of normal p53 tetramers in response to genotoxic stress and/or carcinogenic exposure. Occurrence of an activating EGFR somatic event leads to an EGFR upregulation. Moreover, according to previous functional studies, there is a link between p53 and EGFR through miR-193a, forming a double-negative feedback loop that contributes to NSCLC tumorigenesis. (B) LUAD formation process in a carrier of the germline TP53 p.R337H (TP53 p.R337H/WT genotype). To explain the frequent co-occurrence of germline TP53 and somatic EGFR variants recently described in different case series, we postulate that the deficient p53 oligomerization capacity conferred by the TP53-p.R337H founder allele, specifically in the context of exposure to carcinogenic environmental factors triggers reduced tumor-suppressive p53 functions in lung tissue, representing an initial factor (first hit) in NSCLC/LUAD carcinogenesis. Subsequent mutational events at the somatic level (possibly resulting also from environmental exposure factors), including oncogenic EGFR variants (second hit) could dramatically impair the p53/miR-193a/EGFR feedback loop regulation, acting as a mechanism driving the NSCLC/LUAD development.
Finally, the results of the current study must be interpreted with caution, considering its limitations. First, LUAD specimens were obtained retrospectively from a diagnostic cohort de-identified for this study, hindering the inquiry about clinical data, such as histological subtype, smoking habit, cancer FH, and ethnic ancestry. Second, for the same reason, germline screening for the variant was not done and, thus, the study design employed here did not allow contact with R337H-positive patients or their relatives. Lastly, the limited availability of samples did not allow additional analyses of the tumor samples such as LOH testing.
In conclusion, when compared to previous studies in Brazilian patients with LUAD, the prevalence of TP53 c.1010G>A, although higher than expected for the general population, was much lower in our series from Southern Brazil, suggesting that there may be regional variations. The variability observed so far, in the absence of large prevalence studies in different regions of the country, and also without a more detailed cost-effectiveness analysis, do not allow, in our view, proposition of a general recommendation of testing all Brazilian LUAD patients for TP53 c.1010G>A. Further studies assessing presence of TP53 germline variants, or at least the founder c.1010G>A variant, in Brazilian LUAD patients, regardless of the age at tumor diagnosis and especially if they harbor activating EGFR mutations, should be undertaken in order to determine if universal screening for TP53 c.1010G>A is justified. Ultimately, predictive testing in healthy family members of variant-positive LUAD probands might be useful to assess the clinical actionability toward the occurrence of other tumor types, and future cost-effectiveness analysis should include this clinical actionability after predictive testing.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by Institutional Review Board from Hospital de Clinicas de Porto Alegre (IRB-HCPA). Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author Contributions
IV, TA, GM, and PA-P conceived the work and conception design of the brief research report. IV, TA, and PA-P designed the draft of the manuscript and carried out the statistical analyses. TA, GM, and PA-P were involved in recruitment of tumor DNA samples and retrospective search of clinical data. TA, BF, IV, GM, MA, and DS performed the genotyping and NGS analyses, as well as the interpretation of results from these experiments. IV, TA, MA, GM, DS, and PA-P helped to draft the manuscript. PA-P supervised the work. All authors revised the manuscript critically, contributed with interpretation of the findings and gave final approval of the version to be published.
Funding
This study was supported in part by funds from Fundo de Incentivo à Pesquisa do Hospital de Clínicas de Porto Alegre (FIPE-HCPA, grant number #18-0099 to GM) and from the Canada-Israel Health Research Initiative, jointly funded by the Canadian Institutes of Health Research, the Israel Science Foundation, the International Development Research Centre, Canada and the Azrieli Foundation (application #384428 2017-02-14/IDRC File #108587-001 to DS). PA-P. receives a researcher grant from Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Bruno da Silveira Corrêa, Fernanda de Paris, Bárbara Alemar, and Kendi Nishino Miyamoto for their valuable contributions and technical support.
References
Achatz, M. I., Hainaut, P., and Ashton-Prolla, P. (2009). Highly prevalent TP53 mutation predisposing to many cancers in the Brazilian population: a case for newborn screening? Lancet Oncol. 10, 920–925. doi: 10.1016/S1470-2045(09)70089-0
Achatz, M. I., Olivier, M., Le Calvez, F., Martel-Planche, G., Lopes, A., Rossi, B. M., et al. (2007). The TP53 mutation, R337H, is associated with Li-Fraumeni and Li-Fraumeni-like syndromes in Brazilian families. Cancer Lett. 245, 96–102. doi: 10.1016/j.canlet.2005.12.039
Achatz, M. I., and Zambetti, G. P. (2016). The Inherited p53 mutation in the Brazilian population. Cold Spring Harb. Perspect. Med. 6:a026195. doi: 10.1101/cshperspect.a026195
Andreis, T. F., Correa, B. S., Vianna, F. S., De-Paris, F., Siebert, M., Leistner-Segal, S., et al. (2019). Analysis of predictive biomarkers in patients with lung adenocarcinoma from southern brazil reveals a distinct profile from other regions of the country. J. Glob. Oncol. 5, 1–9. doi: 10.1200/JGO.19.00174
Barbosa, M. V. R., de Lima, V. C., Formiga, M. N., de Paula, C. A., Torrezan, G. T., and Carraro, D. M. (2020). High prevalence of egfr mutations in lung adenocarcinomas from Brazilian patients harboring the TP53 p.R337H variant. Clin. Lung Cancer 21, e37–e44. doi: 10.1016/j.cllc.2019.11.012
Caron, O., Frebourg, T., Benusiglio, P. R., Foulon, S., and Brugières, L. (2017). Lung adenocarcinoma as part of the Li-Fraumeni syndrome spectrum. JAMA Oncol. 3, 1736–1737. doi: 10.1001/jamaoncol.2017.1358
Castro, M. S., Issler, H. C., Gelmini, G. F., de Miranda, B. L. M., Calonga-Solís, V., Schmidt, A. H., et al. (2019). High-resolution characterization of 12 classical and non-classical HLA loci in Southern Brazilians. HLA 93, 80–88. doi: 10.1111/tan.13488
Catalogue Of Somatic Mutations In Cancer (Cosmic) (2020). Graphical View of Mutations Across TP53 Gene. https://cancer.sanger.ac.uk/cosmic/gene/analysis?ln=TP53 (Accessed September 15, 2020).
Couto, P. P., Bastos-Rodrigues, L., Schayek, H., Melo, F. M., Lisboa, R. G. C., Miranda, D. M., et al. (2017). Spectrum of germline mutations in smokers and non-smokers in Brazilian non-small-cell lung cancer (NSCLC) patients. Carcinogenesis 38, 1112–1118. doi: 10.1093/carcin/bgx089
Custódio, G., Parise, G. A., Kiesel Filho, N., Komechen, H., Sabbaga, C. C., Rosati, R., et al. (2013). Impact of neonatal screening and surveillance for the TP53 R337H mutation on early detection of childhood adrenocortical tumors. J. Clin. Oncol. 31, 2619–2626. doi: 10.1200/JCO.2012.46.3711
DiGiammarino, E. L., Lee, A. S., Cadwell, C., Zhang, W., Bothner, B., Ribeiro, R. C., et al. (2002). A novel mechanism of tumorigenesis involving pH-dependent destabilization of a mutant p53 tetramer. Nat. Struct. Biol. 9, 12–16. doi: 10.1038/nsb730
Ding, L., Getz, G., Wheeler, D. A., Mardis, E. R., McLellan, M. D., Cibulskis, K., et al. (2008). Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455, 1069–1075. doi: 10.1038/nature07423
Fitarelli-Kiehl, M., Macedo, G. S., Schlatter, R. P., Koehler-Santos, P., Matte, U. S., Ashton-Prolla, P., et al. (2016). Comparison of multiple genotyping methods for the identification of the cancer predisposing founder mutation p.R337H in TP53. Genet. Mol. Biol. 39, 203–209. doi: 10.1590/1678-4685-GMB-2014-0351
Gahr, S., Stoehr, R., Geissinger, E., Ficker, J. H., Brueckl, W. M., Gschwendtner, A., et al. (2013). EGFR mutational status in a large series of Caucasian European NSCLC patients: data from daily practice. Br. J. Cancer 109, 1821–1828. doi: 10.1038/bjc.2013.511
Garritano, S., Gemignani, F., Palmero, E. I., Olivier, M., Martel-Planche, G., Le Calvez-Kelm, F., et al. (2010). Detailed haplotype analysis at the TP53 locus in p.R337H mutation carriers in the population of Southern Brazil: evidence for a founder effect. Hum. Mutat. 31, 143–150. doi: 10.1002/humu.21151
Giacomazzi, J., Graudenz, M. S., Osorio, C. A., Koehler-Santos, P., Palmero, E. I., Zagonel-Oliveira, M., et al. (2014). Prevalence of the TP53 p.R337H mutation in breast cancer patients in Brazil. PLoS One 9:e99893. doi: 10.1371/journal.pone.0099893
Giacomazzi, J., Selistre, S. G., Rossi, C., Alemar, B., Santos-Silva, P., Pereira, F. S., et al. (2013). Li-Fraumeni and Li-Fraumeni-like syndrome among children diagnosed with pediatric cancer in Southern Brazil. Cancer 119, 4341–4349. doi: 10.1002/cncr.28346
Greenman, C., Stephens, P., Smith, R., Dalgliesh, G. L., Hunter, C., Bignell, G., et al. (2007). Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158. doi: 10.1038/nature05610
Hahn, E. C., Bittar, C. M., Vianna, F. S. L., Netto, C. B. O., Biazús, J. V., Cericatto, R., et al. (2018). TP53 p.Arg337His germline mutation prevalence in Southern Brazil: further evidence for mutation testing in young breast cancer patients. PLoS One 13:e0209934. doi: 10.1371/journal.pone.0209934
Hammerman, P. S., Lawrence, M. S., Voet, D., Jing, R., Cibulskis, K., Sivachenko, A., et al. (2012). Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525. doi: 10.1038/nature11404
Heymann, S., Delaloge, S., Rahal, A., Caron, O., Frebourg, T., Barreau, L., et al. (2010). Radio-induced malignancies after breast cancer postoperative radiotherapy in patients with Li-Fraumeni syndrome. Radiat. Oncol. 5:104. doi: 10.1186/1748-717X-5-104
Imielinski, M., Berger, A. H., Hammerman, P. S., Hernandez, B., Pugh, T. J., Hodis, E., et al. (2012). Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150, 1107–1120. doi: 10.1016/j.cell.2012.08.029
Instituto Nacional de Câncer José Alencar Gomes da Silva (Inca) (2020). Estimativa 2020: Incidência de Câncer no Brasil. https://www.inca.gov.br (Accessed September 15, 2020)
International Agency for Research on Cancer TP53 database (Iarc) (2020). Selected Statistics on Somatic Mutations. http://p53.iarc.fr/TP53SomaticMutations.aspx (Accessed September 15, 2020)
Jordan, E. J., Kim, H. R., Arcila, M. E., Barron, D., Chakravarty, D., Gao, J., et al. (2017). Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. 7, 596–609. doi: 10.1158/2159-8290.CD-16-1337
Kang, S. H., Cho, S. I., Chae, J. H., Chung, K. N., Ra, E. K., Kim, S. Y., et al. (2009). False homozygous deletions of SMN1 Exon 7 Using Dra I PCR-RFLP caused by a novel mutation in spinal muscular atrophy. Genet. Test. Mol. Biomark. 13, 511–513. doi: 10.1089/gtmb.2008.0158
Mastellaro, M. J., Ribeiro, R. C., Oliveira-Filho, A. G., Seidinger, A. L., Cardinalli, I. A., Miranda, E. C. M., et al. (2018). Adrenocortical tumors associated with the TP53 p.R337H germline mutation can be identified during child-care consultations. J. Pediatr. 94, 432–439. doi: 10.1016/j.jped.2017.06.009
Mastellaro, M. J., Seidinger, A. L., Kang, G., Abrahão, R., Miranda, E. C. M., Pounds, S. B., et al. (2017). Contribution of the TP53 R337H mutation to the cancer burden in southern Brazil: insights from the study of 55 families of children with adrenocortical tumors. Cancer 123, 3150–3158. doi: 10.1002/cncr.30703
Mezquita, L., Jové, M., Nadal, E., Kfoury, M., Morán, T., Ricordel, C., et al. (2020). High prevalence of somatic oncogenic driver alterations in patients With NSCLC and Li-Fraumeni Syndrome. J. Thorac. Oncol. 15, 1232–1239. doi: 10.1016/j.jtho.2020.03.005
Michalarea, V., Calcasola, M., Cane, P., Tobal, K., Izatt, L., and Spicer, J. (2014). EGFR-mutated lung cancer in Li-Fraumeni syndrome. Lung Cancer 85, 485–487. doi: 10.1016/j.lungcan.2014.06.017
Nogueira, S. T. S., Lima, E. N. P., Nóbrega, A. F., Torres, I. C. G., Cavicchioli, M., Hainaut, P., et al. (2015). 18F-FDG PET-CT for surveillance of Brazilian Patients with Li-Fraumeni syndrome. Front. Oncol. 5:38. doi: 10.3389/fonc.2015.00038
Palmero, E. I., Schüler-Faccini, L., Caleffi, M., Achatz, M. I., Olivier, M., Martel-Planche, G., et al. (2008). Detection of R337H, a germline TP53 mutation predisposing to multiple cancers, in asymptomatic women participating in a breast cancer screening program in Southern Brazil. Cancer Lett. 261, 21–25. doi: 10.1016/j.canlet.2007.10.044
Park, J. H., Li, J., Starost, M. F., Liu, C., Zhuang, J., Chen, J., et al. (2018). Mouse homolog of the human TP53 R337H mutation reveals its role in tumorigenesis. Cancer Res. 78, 5375–5383. doi: 10.1158/0008-5472.CAN-18-0016
Park, J. H., Wang, P. Y., and Hwang, P. M. (2019). Modeling the prevalent germline TP53 R337H mutation in mouse. Oncotarget 10, 631–632. doi: 10.18632/oncotarget.26603
Peifer, M., Fernández-Cuesta, L., Sos, M. L., George, J., Seidel, D., Kasper, L. H., et al. (2012). Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 44, 1104–1110. doi: 10.1038/ng.2396
Ribeiro, R. C., Sandrini, F., Figueiredo, B., Zambetti, G. P., Michalkiewicz, E., Lafferty, A. R., et al. (2001). An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc. Natl. Acad. Sci.U.S.A. 98, 9330–9335. doi: 10.1073/pnas.161479898
Ricordel, C., Labalette-Tiercin, M., Lespagnol, A., Kerjouan, M., Dugast, C., Mosser, J., et al. (2015). EFGR-mutant lung adenocarcinoma and Li-Fraumeni syndrome: report of two cases and review of the literature. Lung Cancer 87, 80–84. doi: 10.1016/j.lungcan.2014.11.005
Rizvi, N. A., Hellmann, M. D., Snyder, A., Kvistborg, P., Makarov, V., Havel, J. J., et al. (2015). Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 348, 124–128. doi: 10.1126/science.aaa1348
Ruiz-Linares, A., Adhikari, K., Acuña-Alonzo, V., Quinto-Sanchez, M., Jaramillo, C., Arias, W., et al. (2014). Admixture in Latin America: geographic structure, phenotypic diversity and self-perception of ancestry based on 7,342 individuals. PLoS Genet. 10:e1004572. doi: 10.1371/journal.pgen.1004572
Seidinger, A. L., Fortes, F. P., Mastellaro, M. J., Cardinalli, I. A., Zambaldi, L. G., Aguiar, S. S., et al. (2015). Occurrence of Neuroblastoma among TP53 p.R337H Carriers. PLoS One 10:e0140356. doi: 10.1371/journal.pone.0140356
Seidinger, A. L., Mastellaro, M. J., Paschoal Fortes, F., Godoy Assumpção, J., Aparecida Cardinalli, I., Aparecida Ganazza, M., et al. (2011). Association of the highly prevalent TP53 R337H mutation with pediatric choroid plexus carcinoma and osteosarcoma in Southeast Brazil. Cancer 117, 2228–2235. doi: 10.1002/cncr.25826
Tinat, J., Bougeard, G., Baert-Desurmont, S., Vasseur, S., Martin, C., Bouvignies, E., et al. (2009). 2009 Version of the chompret criteria for Li Fraumeni syndrome. J. Clin. Oncol. 27, e108–e109. doi: 10.1200/JCO.2009.22.7967
Uemura, T., Hibi, K., Kaneko, T., Takeda, S., Inoue, S., Okochi, O., et al. (2004). Detection of K-ras mutations in the plasma DNA of pancreatic cancer patients. J. Gastroenterol. 39, 56–60. doi: 10.1007/s00535-003-1245-1
Wang, W., Shen, X. B., Jia, W., Huang, D. B., Wang, Y., and Pan, Y. Y. (2019). The p53/miR-193a/EGFR feedback loop function as a driving force for non-small cell lung carcinoma tumorigenesis. Ther. Adv. Med. Oncol. 11:1758835919850665. doi: 10.1177/1758835919850665
Keywords: TP53 gene, p53 protein, lung adenocarcinoma, founder variant, TP53 (p.Arg337His), R337H, non-small cell lung cancer, Li-Fraumeni syndrome
Citation: Vieira IA, Andreis TF, Fernandes BV, Achatz MI, Macedo GS, Schramek D and Ashton-Prolla P (2021) Prevalence of the Brazilian TP53 Founder c.1010G>A (p.Arg337His) in Lung Adenocarcinoma: Is Genotyping Warranted in All Brazilian Patients? Front. Genet. 12:606537. doi: 10.3389/fgene.2021.606537
Received: 15 September 2020; Accepted: 11 January 2021;
Published: 02 February 2021.
Edited by:
Joan Brunet, Catalan Institute of Oncology, SpainReviewed by:
Ernest Nadal, Catalan Institute of Oncology, SpainJudith Balmaña, Vall d’Hebron University Hospital, Spain
Copyright © 2021 Vieira, Andreis, Fernandes, Achatz, Macedo, Schramek and Ashton-Prolla. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patricia Ashton-Prolla, cHByb2xsYUBoY3BhLmVkdS5icg==; cHByb2xsYUBnbWFpbC5jb20=
†These authors have contributed equally to this work