 Xiaolei Yu
Xiaolei Yu Wenxiu Wang1
Wenxiu Wang1 Dan Wang
Dan Wang Xiaoxuan Tian
Xiaoxuan Tian- 1State Key Laboratory of Component-based Chinese Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin, China
- 2Key Laboratory of Pharmacology of Traditional Chinese Medical Formulae, Tianjin University of Traditional Chinese Medicine, Tianjin, China
Vincetoxicum versicolor (Bunge) Decne is the original plant species of the Chinese herbal medicine Cynanchi Atrati Radix et Rhizoma. The lack of information on the transcriptome and chloroplast genome of V. versicolor hinders its evolutionary and taxonomic studies. Here, the V. versicolor transcriptome and chloroplast genome were assembled and functionally annotated. In addition, the comparative chloroplast genome analysis was conducted between the genera Vincetoxicum and Cynanchum. A total of 49,801 transcripts were generated, and 20,943 unigenes were obtained from V. versicolor. One thousand thirty-two unigenes from V. versicolor were classified into 73 functional transcription factor families. The transcription factors bHLH and AP2/ERF were the most significantly abundant, indicating that they should be analyzed carefully in the V. versicolor ecological adaptation studies. The chloroplast genomes of Vincetoxicum and Cynanchum exhibited a typical quadripartite structure with highly conserved gene order and gene content. They shared an analogous codon bias pattern in which the codons of protein-coding genes had a preference for A/U endings. The natural selection pressure predominantly influenced the chloroplast genes. A total of 35 RNA editing sites were detected in the V. versicolor chloroplast genome by RNA sequencing (RNA-Seq) data, and one of them restored the start codon in the chloroplast ndhD of V. versicolor. Phylogenetic trees constructed with protein-coding genes supported the view that Vincetoxicum and Cynanchum were two distinct genera.
Introduction
Apocynaceae is a large family of plants distributed globally, which contains around 4,500 species in approximately 370 genera (Endress et al., 2014; Fishbein et al., 2018). Vincetoxicum versicolor (also known as Cynanchum versicolor in Flora of China) belongs to the Apocynaceae family and is the original plant species of the Chinese herbal medicine Cynanchi Atrati Radix et Rhizoma (Chinese Pharmacopoeia Commission, 2015). However, the genus of this plant has not been unified due to the controversial phylogenetic relationship between the genera Vincetoxicum and Cynanchum, which may affect the Cynanchi Atrati Radix et Rhizoma application in the world. The phylogenetic relationship between Vincetoxicum and Cynanchum has been controversial since the first transfer of Vincetoxicum hirundinaria and several other Eurasian Vincetoxicum species to the genus Cynanchum by Persoon in 1805 (Persoon, 1805). Some researchers have suggested that Vincetoxicum should be grouped into the genus Cynanchum based on the corona structure similarity (Jiang and Li, 1977; Gilbert et al., 1996). On the other hand, these two genera were considered to be distinct, and Vincetoxicum was regarded as an independent genus based on molecular data and chemical substances (Qiu et al., 1989; Liede-Schumann, 2000). Besides, the second opinion is supported by studies based on some regions of the nuclear and chloroplast DNA (Yamashiro et al., 2004; Fishbein et al., 2018). Although Vincetoxicum is generally considered an independent genus in Apocynaceae taxonomy around the world (Goyder et al., 2012; Endress et al., 2014; Liede-Schumann et al., 2016; Liede-Schumann and Meve, 2018), the concept of Vincetoxicum as a section of the genus Cynanchum is still reflected in the taxonomy of modern flora in China (Feng et al., 2012; Li et al., 2012; Yang J. et al., 2018). Therefore, more evidence should be provided to promote the unification of the phylogenetic relationship between Vincetoxicum and Cynanchum.
Chloroplasts originated from ancient endosymbiotic cyanobacteria and are active metabolic centers that sustain life on Earth by converting solar energy into carbohydrates via the photosynthesis process and oxygen release (Leister, 2003; Daniell et al., 2016). Chloroplasts carry their own genomes and genetic systems. The typical angiosperm chloroplast genome has a quadripartite structure, with a genome size of 107–218 kb and gene content of 120–130 genes (Daniell et al., 2016; Kim et al., 2019). The chloroplast genome has the characteristics of uniparental inheritance, moderate nucleotide substitution rate, haploid status, and no homologous recombination (Shaw et al., 2005; Hansen et al., 2007; Yang et al., 2019b). These features make it a suitable tool for molecular identification of species and genetic diversity studies (Zhang et al., 2017; Chen et al., 2018). Moreover, the entire chloroplast genome contains more informative sites than chloroplast DNA fragments, which can provide a higher resolution of the phylogenetic relationship at multiple taxonomic levels (Yang X.-Y. et al., 2018). The development of next-generation sequencing technology has led to more and more angiosperm chloroplast genomes available, making comparative chloroplast genomics a convenient and efficient method for phylogenetic and evolutionary studies (Ge et al., 2018; Gu et al., 2019).
Next-generation sequencing not only greatly improves our ability to obtain genomic resources in non-model species but also facilitates the development of the RNA-Seq technique. RNA-Seq is an efficient technology for large scale transcriptome investigations, which provides a convenient way to obtain information from expressed genomic regions quickly and offers an opportunity to solve comparative transcriptomic-level problems for non-model organisms (Logacheva et al., 2011; Zhang et al., 2013). Transcriptome analysis provides an effective way for novel gene discovery (Emrich et al., 2007) and expression profile construction (Fox et al., 2014), as well as for molecular marker development (Zhang et al., 2013) and analysis of adaptive evolution (Jia et al., 2017). As a non-model species, V. versicolor lacks transcriptome analysis, delaying molecular studies at the transcriptional level.
RNA editing, which is identified primarily by the RNA-Seq technique, is a repair mechanism derived by species in response to abnormal DNA mutations during evolution. RNA editing is a post-transcriptional process in which the nucleotide in the transcript differs from the encoded DNA sequence by nucleotide insertion, deletion, or conversion (Takenaka et al., 2013). Most RNA editing events occur in internal codons, resulting in amino-acid substitutions. However, in some cases, the ACG codon is restored to the AUG start codon because of the C-to-U RNA editing, contributing to the conservation of the translation start signals at the gene level, which is essential for protein synthesis (Hirose and Sugiura, 1997). This editing-restored start codon has been reported in the chloroplast transcripts from maize (rpl2), tobacco (psbL), but especially in the ndhD transcript of several species, including Arabidopsis, Betula, tobacco, spinach, and snapdragon (Neckermann et al., 1994; Wang et al., 2018).
Here, we de novo assembled the transcriptome and chloroplast genome of V. versicolor and performed a comparative chloroplast genome analysis between species of the genera Vincetoxicum and Cynanchum. The aims of this study were (1) to characterize the transcriptome and chloroplast genome of V. versicolor, (2) to explore the V. versicolor molecular evolution, and (3) to provide insights into the phylogenetic relationship between the genera Vincetoxicum and Cynanchum.
Materials and Methods
Plant Materials Collection and DNA and RNA Extraction
The young fresh leaves of a single plant of V. versicolor were collected in August 2019 from Tianjin University of Traditional Chinese Medicine (117.06°E, 38.96°N), Tianjin City, China. The voucher specimens were deposited at Tianjin State Key Laboratory of Modern Chinese Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin, China (voucher number 2019bsbq). The collected leaves were snap-frozen in liquid nitrogen and then stored at -80°C until DNA and RNA extraction. The total DNA was extracted using the extract Plant DNA kit (QIAGEN, Germany) following the manufacturer’s instructions. Total RNA was extracted using the QIAGEN RNeasy Plant Mini Kit (QIAGEN, Germany) following the manufacturer’s instructions. The purity and concentration of DNA and RNA were checked using NanoPhotometer®spectrophotometer (IMPLEN, CA, United States) and Qubit® DNA Assay Kit in Qubit® 2.0 Fluorometer (Life Technologies, CA, United States), respectively.
DNA and RNA Sequencing, Assembly, and Annotation of Chloroplast Genome and Transcriptome
The DNA-Seq library with an average insert size of 350 bp was constructed using the Truseq Nano DNA HT Sample Preparation Kit (Illumina United States). The strand-specific RNA-Seq library was constructed using the protocol described by Zhong et al. (2011). Then, the RNA-Seq library was sequenced on the Illumina HiSeqTM 2,500 platform. Subsequently, clean DNA and RNA data were obtained by removing adaptors and low-quality reads from the raw data. The V. versicolor chloroplast genome was de novo assembled using NOVOPlasty3.7.2 (Dierckxsens et al., 2017). To validate the reads coverage of the assembled chloroplast genome, clean data were mapped to the V. versicolor chloroplast genome using bowtie 2 (Langmead and Salzberg, 2012), and the average reads coverage was 2,418×. The V. versicolor chloroplast genome was annotated using GeSeq (Tillich et al., 2017), coupled with manual corrections for the start and stop codons. Finally, the V. versicolor chloroplast genome was deposited in the National Center for Biotechnology Information (NCBI) GenBank under accession number MT558564. For the transcriptome assembly, high-quality RNA-Seq data were de novo assembled into transcripts using Trinity (Grabherr et al., 2011) with min_kmer_cov set to two and other parameters set to default. The trinity-obtained contigs were then linked into transcripts. To remove redundant transcripts and obtain the primary representative of each gene locus, only the longest transcript in each cluster was selected as the unigene for subsequent analysis. Finally, the obtained unigenes were annotated using a BLAST search against the following databases, namely KOG (euKaryotic Ortholog Groups), GO (Gene Ontology), KO (KEGG Ortholog), Swiss-Prot (a manually annotated and reviewed protein sequence database), Nr (NCBI non-redundant protein sequences), Nt (NCBI non-redundant nucleotide sequences), and Pfam (protein family).
Annotation of Functional Genes, Prediction of Biochemical Pathways, and Detection of Transcription Factors
Gene Ontology functional analysis was implemented using blast2go tool (Götz et al., 2008). The KAAS software (Moriya et al., 2007) was used to predict the biochemical pathways of the V. versicolor unigenes based on the KO database. The transcription factors were detected using the iTAK program (Zheng et al., 2016).
Identification of RNA Editing Sites
RNA-Seq reads were mapped to the chloroplast genome of V. versicolor using bowtie 2 (Langmead and Salzberg, 2012). Then, samtools was applied to call single nucleotide polymorphisms to recognize editing sites in the V. versicolor chloroplast genome.
Codon Usage Calculation
The number of codons and the relative synonymous codon usage (RSCU) were calculated using Mega X (Kumar et al., 2018). The effective number of codons (ENc) values against GC content in the third position of synonymously variable codons (GC3s) values of protein-coding genes of chloroplast genome were calculated using CodonW v1.4.4 (Peden, 1999). Then, the relationships between ENc and GC3s were analyzed using the R script.
Phylogenetic Analyses
A total of 20 chloroplast genomes (Supplementary Table 1) of 18 Apocynaceae species and two Gentianaceae species available in GenBank were collected to reconstruct phylogenetic trees. Besides, another Vincetoxicum species (V. rossicum) was added to phylogenetic analysis. Although the full-length chloroplast genome of V. rossicum was not available, its raw reads were present in NCBI Sequence Read Archive under accession number SRR934046 (Straub et al., 2013). So, a draft chloroplast genome of V. rossicum was assembled using NOVOPlasty3.7.2. The draft chloroplast genome was incomplete and contained many degenerate bases in the intergenic regions, but its protein-coding genes were complete and could be used for phylogenetic analysis. The protein-coding genes from 21 chloroplast genomes were extracted, aligned separately, and recombined to construct a matrix using PhyloSuite_v1.1.15 (Zhang et al., 2020). The generated matrix was used to conduct the Bayesian inference (BI) and Maximum likelihood (ML) phylogenies. The BI phylogenies were inferred using MrBayes 3.2.6 (Ronquist et al., 2012) under JC + I + G model, which was determined from the ModelFinder (Kalyaanamoorthy et al., 2017). The ML phylogenies were inferred using IQ-TREE (Nguyen et al., 2015) under an edge-linked partition model for 5,000 ultrafast (Minh et al., 2013) bootstraps, as well as the Shimodaira–Hasegawa-like approximate likelihood-ratio test (Guindon et al., 2010).
Results and Discussion
Transcriptome Features
Illumina pair-end sequencing produced 52,502,062 raw reads for V. versicolor, and 51,764,112 clean reads were obtained after removing adaptors and low-quality data (Table 1). The base quality value Q20 and Q30 reached 97.51 and 93.01%, respectively, which indicated that the produced data could be used for further analysis. A total of 49,801 transcripts were generated in V. versicolor, of which 20,943 unigenes (N50 = 2,128 bp, average length = 1,491 bp) were identified. Most transcripts and unigenes were 1,001–2,000 bp, and the number of transcripts and unigenes over 2,000 bp were 14,787 and 5,443, respectively (Supplementary Figure 1). There were 16,895 unigenes (80.60%) for V. versicolor with at least one significant match to the databases discussed earlier and 3,177 unigenes (15.17%) with all significant matches to the databases mentioned earlier (Table 1).
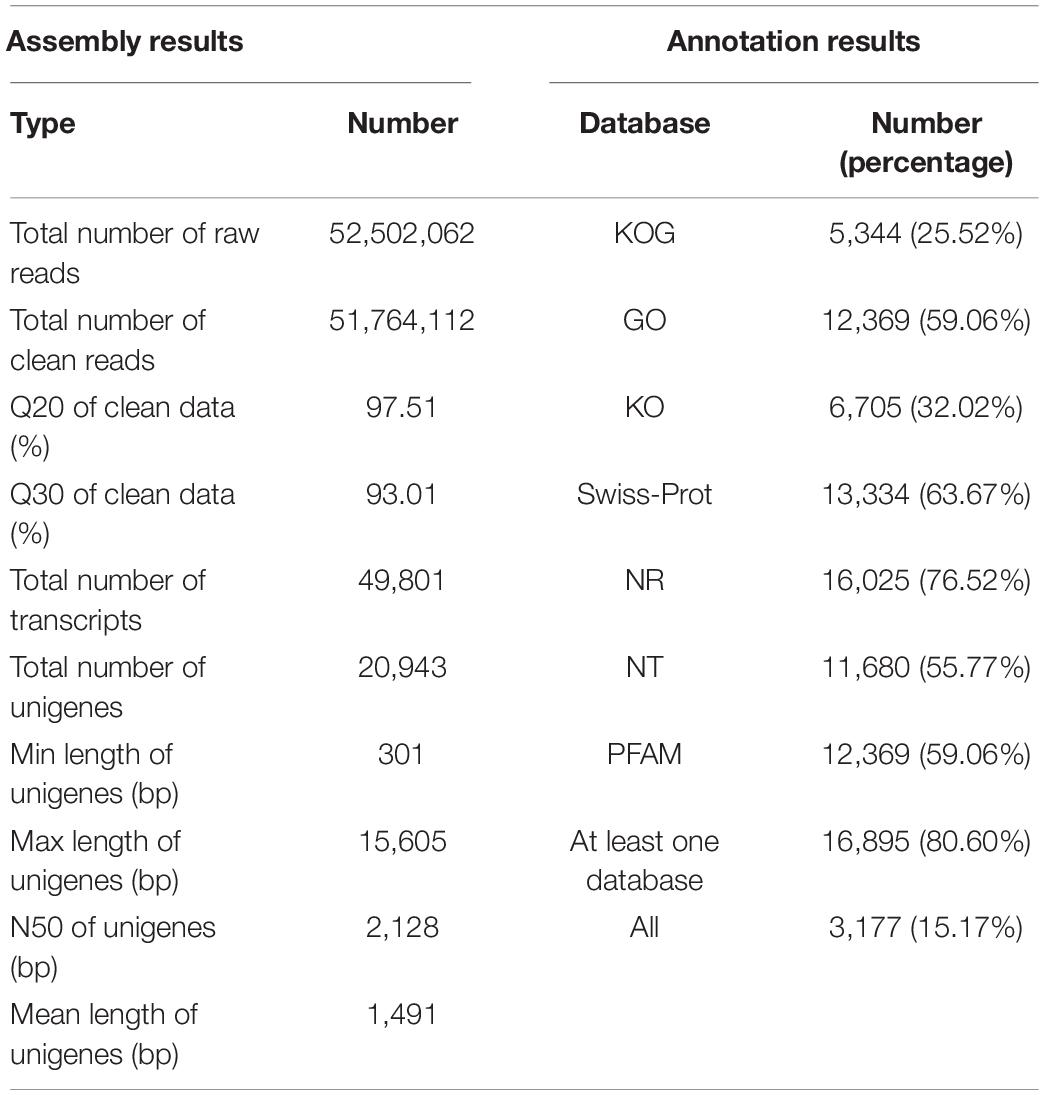
Table 1. Summary of statistics for the transcriptomes of V. versicolor.
Gene Ontology and Biochemical Pathways Prediction
The GO concept aims to use a common vocabulary to annotate homologous genes and protein sequences in various organisms in a flexible and dynamic way. Thus, scientists can query and retrieve genes and protein sequences based on their shared biology (Ashburner et al., 2000). The functional classification of unigenes in the GO database was assigned into three categories: biological processes, cellular components, and molecular functions (Figure 1). A total of 12,369 unigenes were assigned to the GO classification groups. In the “Biological processes” group, “Cellular process” (7,374) was the most abundant term. Regarding “Cellular components,” “Cell” (4,159), and “Cell part” (4,159) were the dominant items. In the “Molecular functions” category, “Binding” (7,101) was the largest cluster. Interestingly, the most abundant terms in the corresponding GO categories in V. versicolor were highly similar to other angiosperm transcriptomes, such as Raphanus (Mei et al., 2016), Glycyrrhiza (Jiang et al., 2020), and Dipteronia (Zhou et al., 2016). These data suggested that these gene groups are highly expressed and have functional importance in angiosperms.
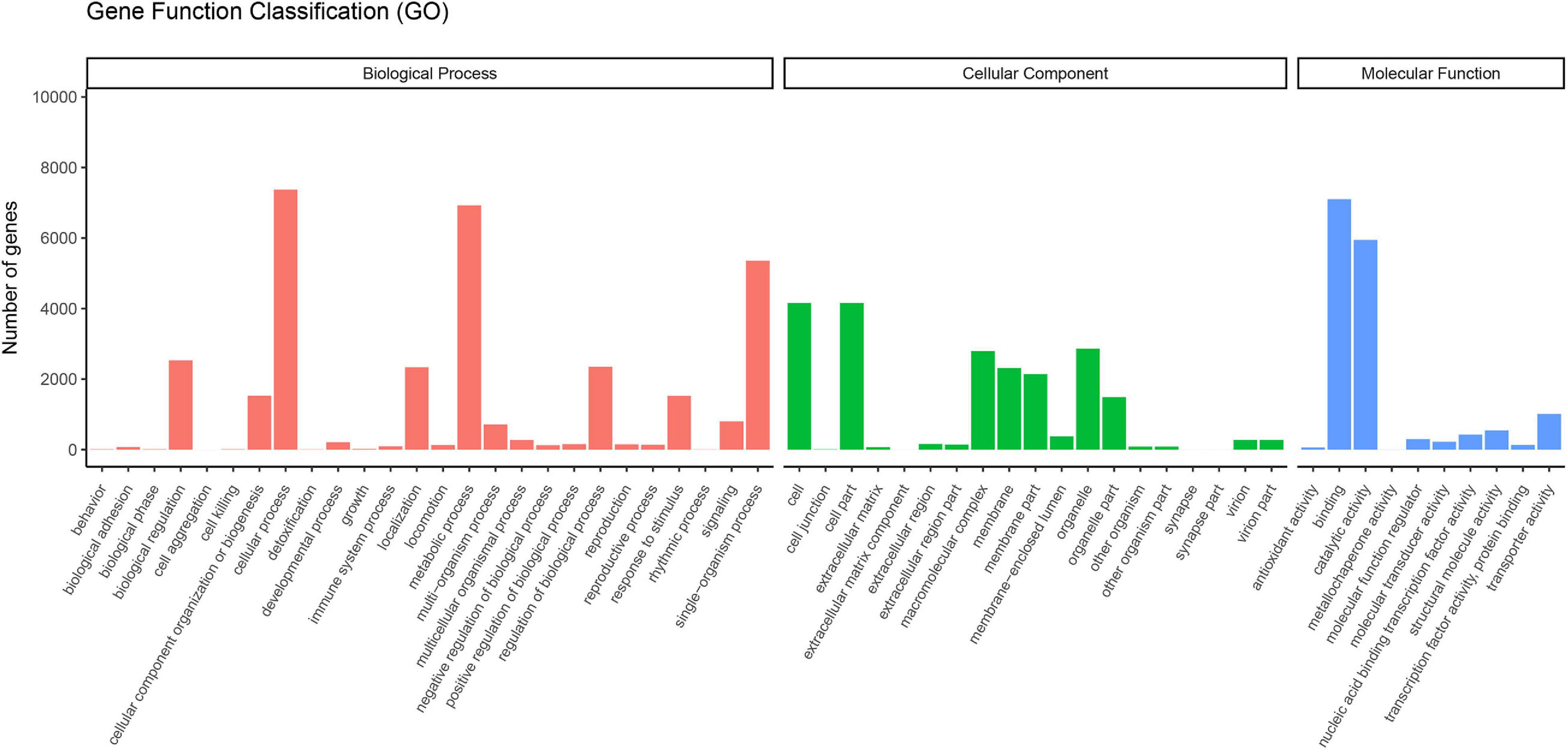
Figure 1. Functional classification of unigenes of V. versicolor in GO database. GO terms were annotated according to three main categories “biological processes,” “cellular components,” and “molecular functions.”
The KO database is an integrated database resource composed of genes, protein, small molecules, reactions, pathways, diseases, drugs, organisms, and viruses, as well as more conceptual objects, aiming to assign functional meanings to genes and genomes, both at the molecular and higher levels (Kanehisa et al., 2017). For the biochemical pathways prediction in the KO database, a total of 6,705 unigenes were assigned to the KO pathways (Figure 2). The cluster for “Translation” (799) represented the largest group, followed by “Carbohydrate metabolism” (542) and “Folding, sorting and degradation” (477), which indicated that these pathways might be crucial for the V. versicolor development.
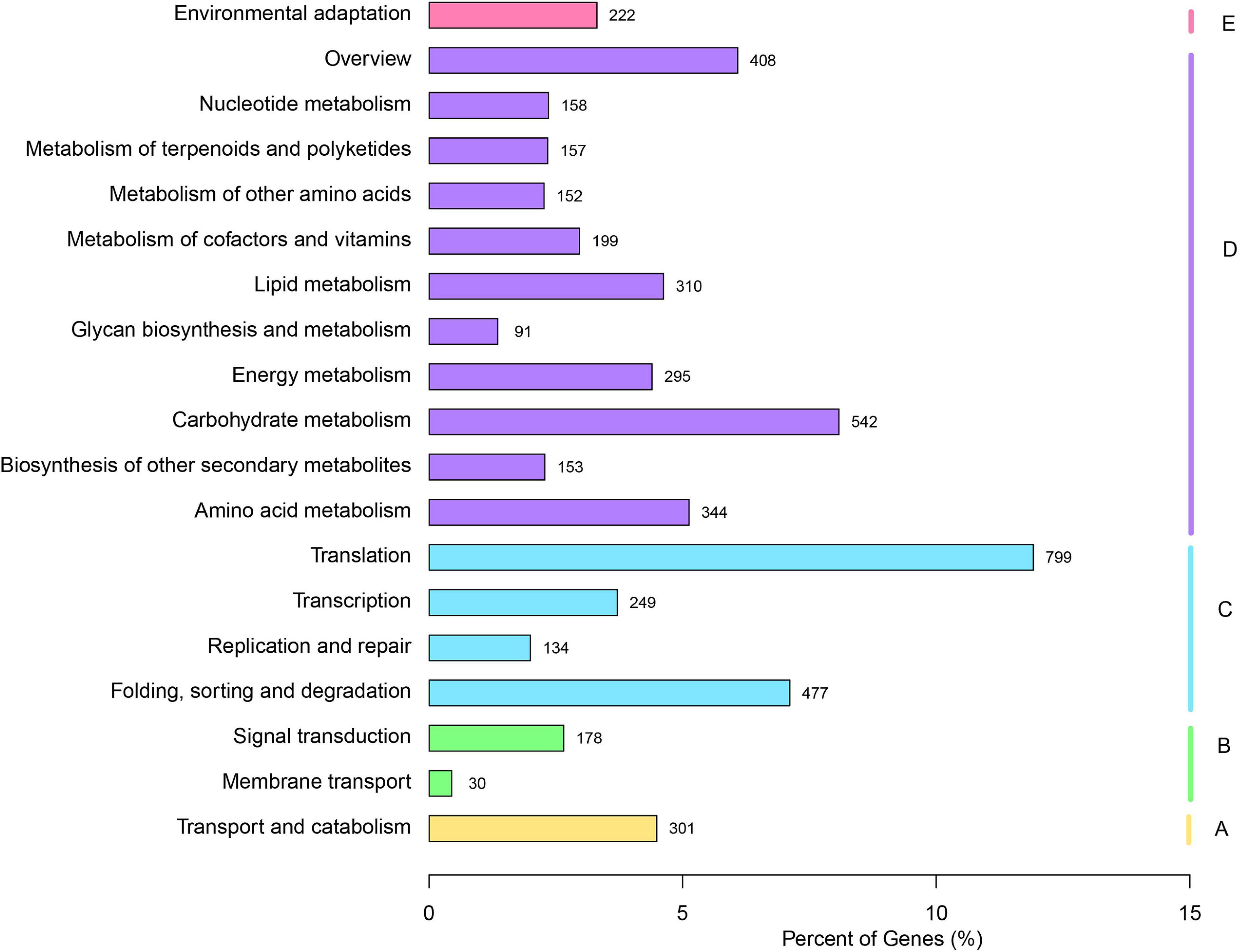
Figure 2. Annotation of unigenes of V. versicolor in KO database. “A” presents “Cellular processes,” “B” presents “Environmental information processing,” “C” presents “Genetic information processing,” “D” presents “Metabolism,” and “E” presents “Organismal Systems.”
Detection of Transcription Factors
Transcription factors play pivotal roles in complex biological processes under multiple environmental signals by regulating the gene transcription through binding to specific DNA sequences in the target gene promoters (Honys and Twell, 2004). Transcription factors are generally classified into different families based on their DNA-binding domains (Jin et al., 2014). A total of 1,032 unigenes V. versicolor were classified into 73 functional families (Table 2). Among these families, bHLH transcription factors were the most abundant (57), followed by AP2/ERF (56). It is worth paying attention to these two transcription factor families in the ecological adaptation studies of V. versicolor, as they play essential roles in resistance to abiotic stress in plants (Chinnusamy et al., 2003; Yang et al., 2016; Tripathi et al., 2017).
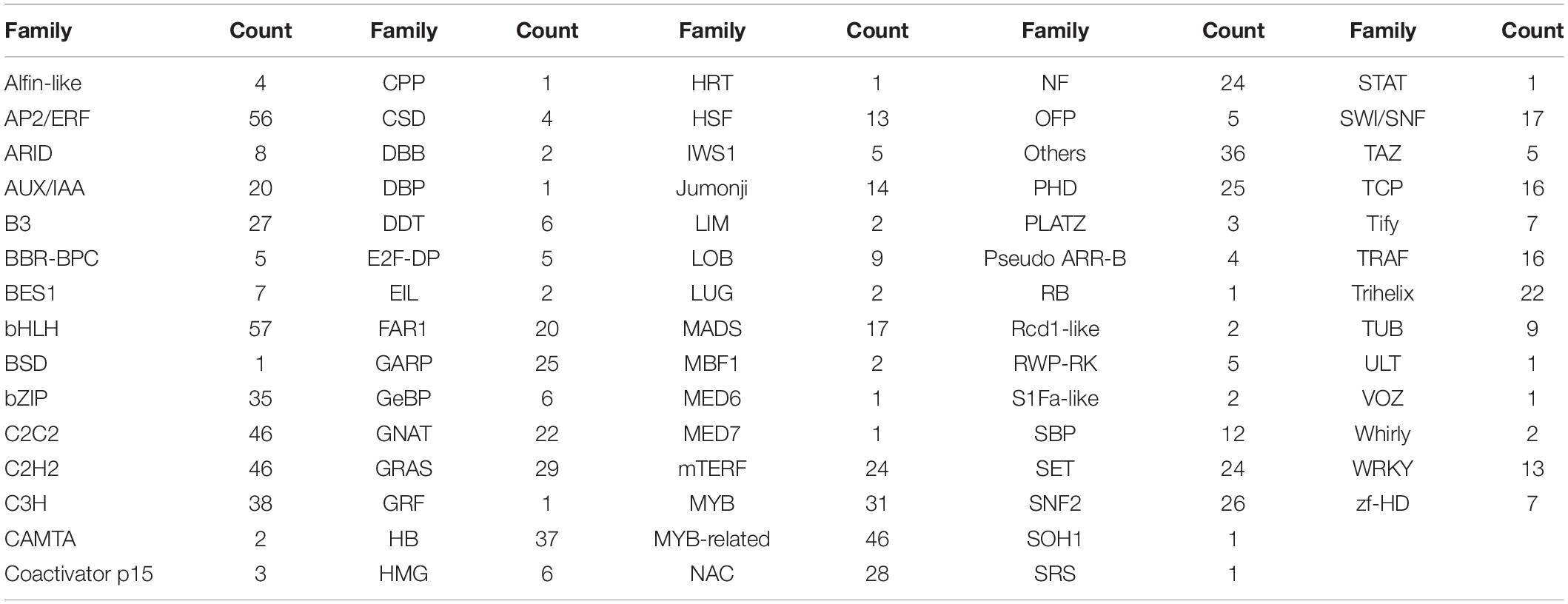
Table 2. Transcription factor families and corresponding unigenes number identified in V. versicolor.
Chloroplast Genome Features
The complete chloroplast genome of V. versicolor was 159,907 bp in length, including a pair of 24,971-bp IRs separated by 19,456-bp SSC and 90,509-bp LSC regions (Figure 3). This quadripartite structure was a typical feature of the chloroplast genome of most angiosperms (Yu et al., 2019, 2020; Tan et al., 2020). The AT content of the V. versicolor chloroplast genome was 62.2%, whereas the AT contents of the LSC, SSC, and IR regions were 63.9, 68.8, and 56.8%, respectively. These data showed that the chloroplast genome exhibited an obvious AT preference and such preference was most evident in the SSC region. The chloroplast genome of V. versicolor contained 133 genes, of which 88 were protein-coding genes, 37 were tRNA genes, and 8 were rRNA genes (Table 3). Among these genes, 19 were duplicated in the IR regions, including eight protein-coding genes (rpl2, rpl23, ycf2, ndhB, rps7, rps12, ycf15, and ycf1), seven tRNA genes (trnR-ACG, trnL-CAA, trnV-GAC, trnI-CAU, trnI-GAU, trnA-UGC, and trnN-GUU), and four rRNA genes (rrn23, rrn16, rrn5, and rrn4.5). There were 21 genes with introns, and 19 of which (atpF, petB, petD, ndhA, ndhB × 2, rpoC1, rps16, rpl16, rpl2 × 2, trnK-UUU, trnL-UAA, trnG-GCC, trnV-UAC, trnA-UGC × 2, trnI-GAU × 2) contained one intron, while two genes (clpP, ycf3) contained two introns. Although the chloroplast genome sizes of V. versicolor, Vincetoxicum shaanxiense (NCBI accession number MH210646), Cynanchum wilfordii (NC_029459), and Cynanchum auriculatum (NC_029460) ranged from 159,907 to 161,241 bp, the gene order, gene content, intron content, and AT content of these genomes were similar (Table 3 and Supplementary Table 2).
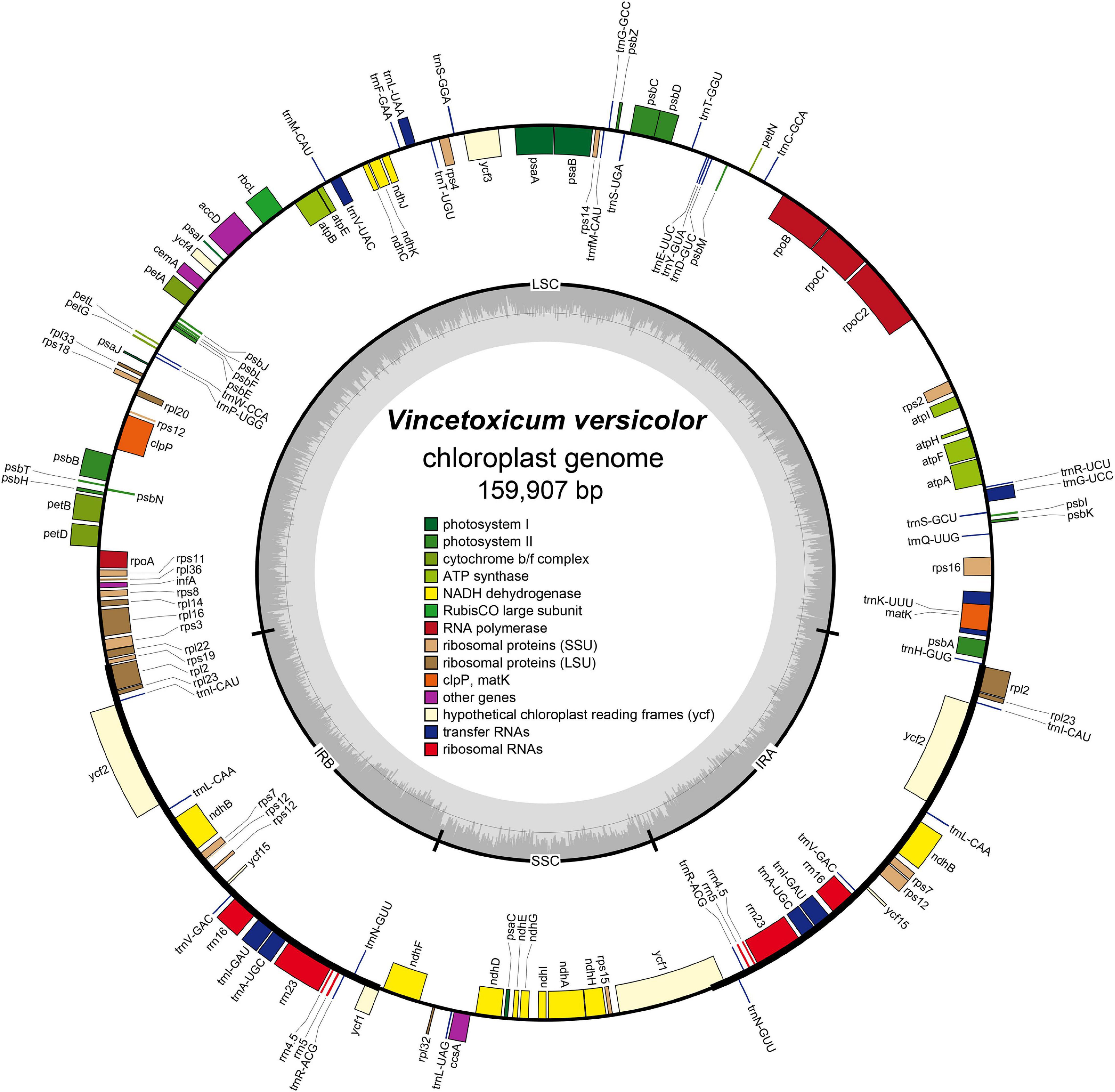
Figure 3. Chloroplast genome map of V. versicolor. Genes inside the circle are transcribed clockwise, whereas those on the outside are transcribed counterclockwise. Genes belonging to different functional groups are color-coded. Darker gray in the inner circle represents the GC content, whereas the lighter gray represents the AT content.

Table 3. Summary statistics of chloroplast genomes of Vincetoxicum and Cynanthum species.
Detection of Chloroplast RNA Editing Sites
The RNA editing sites in the V. versicolor chloroplast genome were identified based on RNA-Seq data. The type and position of the editing sites are shown in Table 4. All RNA editing sites identified were C-to-U. A total of 35 RNA editing sites were detected in the V. versicolor chloroplast genome, of which 33 were located in the protein-coding region, and the remaining two were located in the tRNA region (trnN-GUU). All identified RNA editing sites occurred at the first and second positions of the codon, resulting in amino acid changes at the transcription level. Among these changes, the change from serine (S) to leucine (L) was the most abundant.
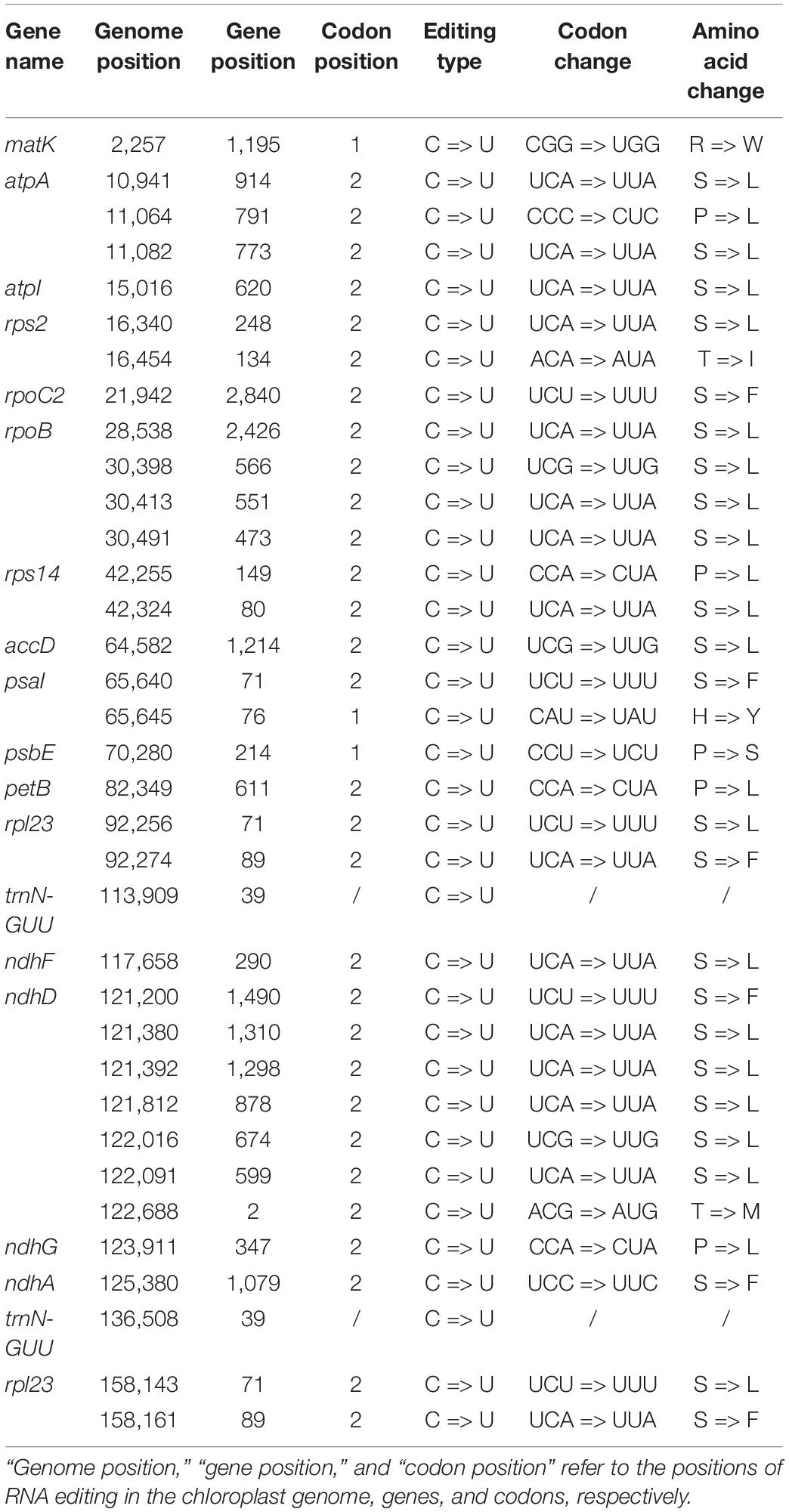
Table 4. RNA editing sites in the V. versicolor chloroplast genome detected by RNA-Seq data.
We found an interesting phenomenon when checking the annotated genes, in which the chloroplast ndhD of V. versicolor did not seem to have a start codon at the genome level (the sequence was validated by a polymerase chain reaction and Sanger sequencing data). A further comparison of the chloroplast ndhD between species of the genera Vincetoxicum and Cynanchum showed that only the C. auriculatum ndhD started with the standard AUG. In contrast, the ndhD of V. versicolor, V. shaanxiense, and C. wilfordii exhibited ACG instead of AUG at the corresponding codon position (Figure 4). Therefore, we speculated that RNA editing restored the start codon AUG in V. versicolor, V. shaanxiense, and C. wilfordii, as observed in Arabidopsis, tobacco, spinach, Betula, and snapdragon (Neckermann et al., 1994; Wang et al., 2018). Examination of V. versicolor transcripts revealed seven RNA editing sites in ndhD, one of which appeared on the ndhD first codon, causing the codon change from ACG to AUG (this editing site was validated by a reverse transcription-polymerase chain reaction, Supplementary Figure 2). This confirmation of the editing-restored ndhD start codon in V. versicolor strongly supported our hypothesis despite lacking the transcripts from the other two species.
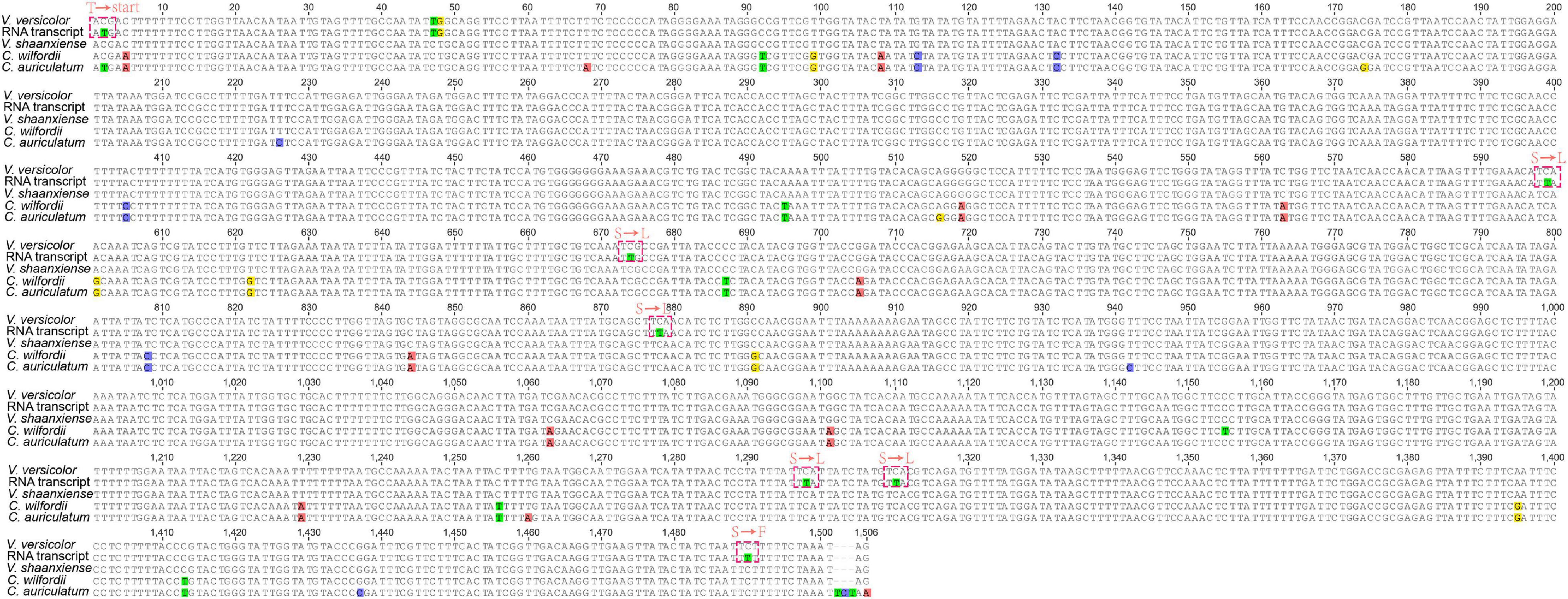
Figure 4. Comparison of chloroplast ndhD in Vincetoxicum and Cynanchum species. Red dotted box represents the amino acid changes at the transcription level. “Start” represents the start codon, whereas “T,” “S,” “L,” and “F” represent threonine, serine, leucine, and phenylalanine, respectively.
To further verify whether the editing-restored ndhD start codon was a common phenomenon in Apocynaceae, the ndhD of 17 Apocynaceae species was compared (Supplementary Table 3). The results showed that almost all of the examined Apocynaceae species exhibited ACG in the first ndhD codon (except for C. auriculatum), suggesting that the editing-restored ndhD start codon was prevalent in Apocynaceae. This kind of editing-restored ndhD start codon had also been reported in other angiosperms, especially in dicots (López-Serrano et al., 2001; Tsudzuki et al., 2001). In Apocynaceae, only C. auriculatum showed the appropriate AUG start codon in ndhD, suggesting that the mutation in this species corrected the start codon of ndhD at the genomic level after the interspecific differentiation in Cynanchum, as implied by previous studies on Liliaceae and Aloaceae (López-Serrano et al., 2001).
Codon Usage Analyses
As an important evolutionary feature, the codon usage pattern has been widely investigated in plant chloroplast genomes (Gao et al., 2018; Somaratne et al., 2019; Yang et al., 2019a). To explore the codon usage pattern in the chloroplast genomes of the Vincetoxicum and Cynanchum species, we calculated the number of codons and RSCU of protein-coding genes in the four chloroplast genomes using Mega X (Supplementary Table 4). The 88 shared protein-coding genes were encoded by 26729, 26671, 26716, and 26586 codons in the chloroplast genomes of V. versicolor, V. shaanxiense, C. wilfordii, and C. auriculatum, respectively. AAA encoding lysine was the most commonly used codon in the chloroplast genome of V. versicolor, whereas AUU encoding isoleucine was the most abundant codon in the chloroplast genomes of V. shaanxiense, C. wilfordii, and C. auriculatum. In the four chloroplast genomes, the A/U content in the third codon position was 68.70–69.11%, showing the preference for A/U-ending codons. Codon bias contributes to the efficiency of gene expression and, therefore, is generated and maintained by selection pressure (Hershberg and Petrov, 2008). The bias toward A/U in the third codon position is commonly observed in the angiosperm chloroplast genomes (Cui et al., 2019; Mehmood et al., 2020). This reflects the strong selection pressure that affects the codon usage of the chloroplast genome, thus regulating the chloroplast gene expressions. Additionally, except for UUG, all preferred synonymous codons (RSCU > 1) ended with A/U. The usage of the initial codon AUG and tryptophan UGG had no bias (RSCU = 1), as observed in other angiosperms (Li et al., 2019).
The plot of the ENc values against the GC3 values is a useful indicator to explore the factors that affect the codon usage. The predicted values are in the expected curve when the codon usage of a gene is constrained only by the G + C mutation bias. Moreover, the predicted values are much lower than the expected curve when natural selection played a major role in optimizing codon usage bias (Wright, 1990). The four chloroplast genomes shared the analogous codon bias pattern (Figure 5). A small number of protein-coding genes followed the standard curve, suggesting that the codon bias of these genes was caused mainly by the nucleotide composition bias in the third codon position. In particular, more than half of the genes were below the curve, indicating that natural selection predominantly influenced these genes. The photosynthesis-related genes represent most of them, revealing their importance so that strong selection pressure is necessary to keep these genes conserved. However, not all photosynthesis-related genes were below the curve. These photosynthesis-related genes exhibited discrete distribution, which implies that other factors such as gene expression level can also affect codon bias (Hershberg and Petrov, 2008).
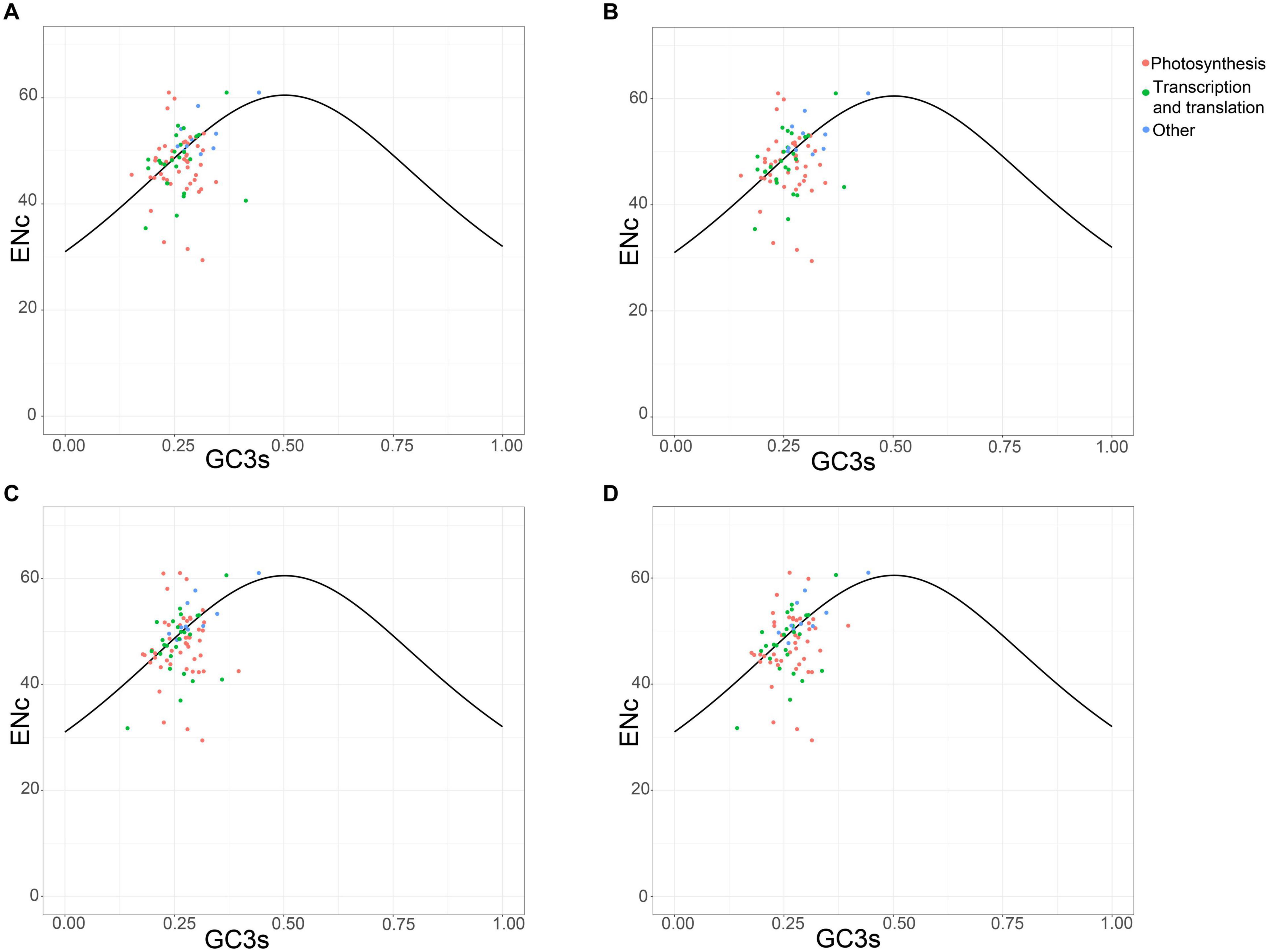
Figure 5. ENc plotted against GC3s based on protein-coding genes of chloroplast genomes of Vincetoxicum and Cynanchum species. (A) V. versicolor; (B) V. shaanxiense; (C) C. wilfordii; (D) C. auriculatum.
Phylogenetic Analysis
Complete chloroplast genomes can provide abundant genetic information for understanding the phylogenetic relationships at various taxonomic levels (Huang et al., 2019; Yang et al., 2019b). To explore the phylogenetic relationship between the genera Vincetoxicum and Cynanchum in the Apocynaceae family, the phylogenetic analysis was conducted based on protein-coding genes of chloroplast genomes of 19 Apocynaceae species (Figure 6). ML and BI trees had a highly similar typology at most branches, except that the position of Vincetoxicum hainanense between ML and BI trees was inconsistent. In the ML and BI trees, four Vincetoxicum species (V. versicolor, V. shaanxiense, V. hainanense, and V. rossicum) were clustered into a monophyletic branch (bootstrap proportions = 100, posterior probabilities = 1), whereas two Cynanchum species formed another monophyletic branch (bootstrap proportions = 100, posterior probabilities = 1). Phylogeny between Vincetoxicum and Cynanchum was described as {Cynanchum + [Vincetoxicum + (Asclepias + Calotropis)]}, which strongly supports the previous view (Liede-Schumann, 2000; Yamashiro et al., 2004; Alessandro et al., 2007) that there was no close phylogenetic relationship between the genera Vincetoxicum and Cynanchum.
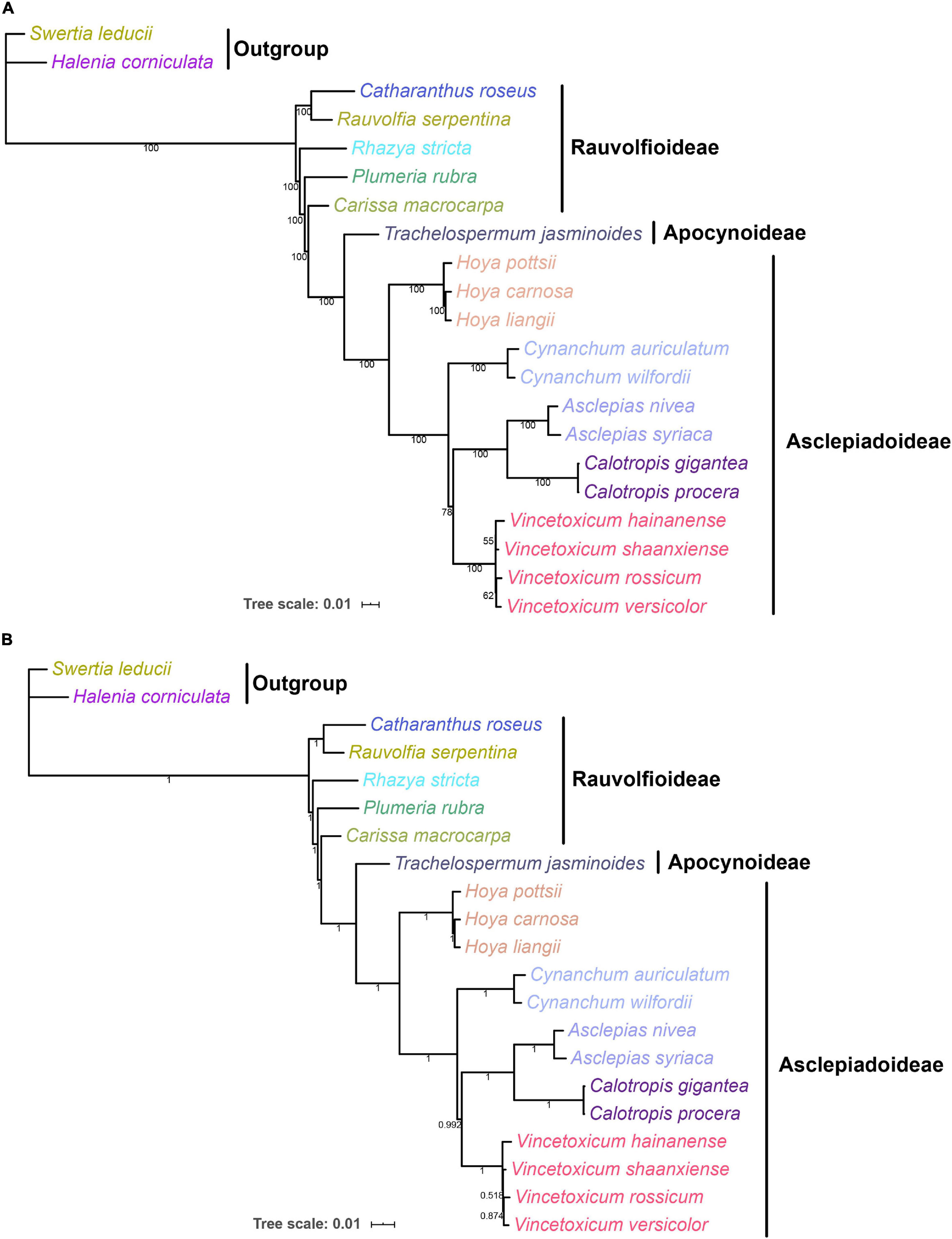
Figure 6. ML and BI phylogenetic trees of Apocynaceae based on 88 protein-coding genes in the chloroplast genome. Numbers below the lines represented ML bootstrap proportions and BI posterior probabilities. Halenia corniculata and Swertia leducii were set as the outgroups. (A) ML tree; (B) BI tree.
Conclusion
This study was the first effort to characterize the transcriptome and chloroplast genome of V. versicolor. A total of 49,801 transcripts were generated, and 20,943 unigenes were obtained from V. versicolor. The GO classification showed that “Cellular process,” “Cell,” “Cell part,” and “Binding” were the most abundant terms in the corresponding categories. KO pathway prediction indicated that the “Translation” cluster represented the largest group. A total of 1,032 unigenes from V. versicolor were classified into 73 functional transcription factor families. The bHLH and AP2/ERF transcription factors were significantly abundant, suggesting that they should be carefully evaluated in the V. versicolor ecological adaptation studies. The comparative analysis showed that the Vincetoxicum and Cynanchum chloroplast genomes were highly conserved in terms of gene order, gene content, and AT content. They shared an analogous codon bias pattern in which their protein-coding genes exhibited a preference for A/U-ending codons. More than half of the chloroplast genes were predominantly influenced by natural selection pressure, and photosynthesis-related genes accounted for most of them. The RNA-Seq data revealed 35 editing sites in the chloroplast genome of V. versicolor, and one of which restored the ndhD start codon in V. versicolor. Phylogenetic analysis based on ML and BI trees strongly supported the view that Vincetoxicum and Cynanchum were two distinct genera. Thus, Vincetoxicum should be regarded as an independent genus in the Apocynaceae family. Overall, this study provided valuable insights into the evolution and phylogeny of V. versicolor.
Data Availability Statement
The dataset generated for this study can be found in NCBI Sequence Read Archive (SRA) under the accession numbers SRR10838756 (DNA) and SRR10838799 (RNA). The assembled chloroplast genome of V. versicolor can be found in GenBank under the accession number MT558564.
Author Contributions
XT and DW designed the study and revised the manuscript. XY assembled, annotated, analyzed the chloroplast genome and transcriptome, and drafted the manuscript. XY and WW performed the experiment. XY, HY, and XZ analyzed the data. All authors contributed to the article and approved the submitted version.
Funding
This work is supported by grants from the State Key Laboratory of Component-based Chinese Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin, 300193, China.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fgene.2021.602528/full#supplementary-material
Supplementary Figure 1 | Number and length of transcripts and unigenes of the V. versicolor transcriptome.
References
Alessandro, R., Cássio van den, B., and Sigrid, L.-S. (2007). Diversification of Asclepiadoideae (Apocynaceae) in the new world. Ann. Missouri Bot. Gard. 94, 407–422.
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Chen, X., Zhou, J., Cui, Y., Wang, Y., Duan, B., and Yao, H. (2018). Identification of Ligularia herbs using the complete chloroplast genome as a super-barcode. Front. Pharmacol. 9:695. doi: 10.3389/fphar.2018.00695
Chinese Pharmacopoeia Commission (2015). Pharmacopoeia of the People’s Republic of China. Beijing: The Medicine Science and Technology Press.
Chinnusamy, V., Ohta, M., Kanrar, S., Lee, B. H., Hong, X., Agarwal, M., et al. (2003). ICE1: a regulator of cold-induced transcriptome and freezing tolerance in Arabidopsis. Genes Dev. 17, 1043–1054. doi: 10.1101/gad.1077503
Cui, Y., Nie, L., Sun, W., Xu, Z., Wang, Y., Yu, J., et al. (2019). Comparative and phylogenetic analyses of ginger (Zingiber officinale) in the family Zingiberaceae based on the complete chloroplast genome. Plants 8:283. doi: 10.3390/plants8080283
Daniell, H., Lin, C. S., Yu, M., and Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17:134. doi: 10.1186/s13059-016-1004-2
Dierckxsens, N., Mardulyn, P., and Smits, G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18. doi: 10.1093/nar/gkw955
Emrich, S. J., Barbazuk, W. B., Li, L., and Schnable, P. S. (2007). Gene discovery and annotation using LCM-454 transcriptome sequencing. Genome Res. 17, 69–73. doi: 10.1101/gr.5145806
Endress, M., Liede-Schumann, S., and Meve, U. (2014). An updated classification for Apocynaceae. Phytotaxa 159:115. doi: 10.5167/uzh-93115
Feng, Y. Q., Qin, X. S., Huang, J. L., and Hong, W. J. (2012). Pollinarium morphology of 17 species and 1 subspecies of Cynanchum (Asclepiadaceae). Acta Bot. Boreali Occident. Sin. 32, 1758–1762.
Fishbein, M., Livshultz, T., Straub, S. C. K., Simões, A. O., Boutte, J., McDonnell, A., et al. (2018). Evolution on the backbone: Apocynaceae phylogenomics and new perspectives on growth forms, flowers, and fruits. Am. J. Bot. 105, 495–513. doi: 10.1002/ajb2.1067
Fox, S. E., Geniza, M., Hanumappa, M., Naithani, S., Sullivan, C., Preece, J., et al. (2014). De novo transcriptome assembly and analyses of gene expression during photomorphogenesis in diploid wheat Triticum monococcum. PLoS One 9:e96855. doi: 10.1371/journal.pone.0096855
Gao, X., Zhang, X., Meng, H., Li, J., Zhang, D., and Liu, C. (2018). Comparative chloroplast genomes of Paris Sect. Marmorata: insights into repeat regions and evolutionary implications. BMC Genom. 19, (Suppl. 10):878. doi: 10.1186/s12864-018-5281-x
Ge, J., Cai, L., Bi, G. Q., Chen, G., and Sun, W. (2018). Characterization of the complete chloroplast genomes of Buddleja colvilei and B. sessilifolia: implications for the taxonomy of Buddleja L. Molecules 23, 1248. doi: 10.3390/molecules23061248
Gilbert, M., Stevens, W., and Ping-tao, L. (1996). Notes on the Asclepiadaceae of China. Novon 5:1. doi: 10.2307/3391820
Götz, S., García-Gómez, J. M., Terol, J., Williams, T. D., Nagaraj, S. H., Nueda, M. J., et al. (2008). High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 36, 3420–3435. doi: 10.1093/nar/gkn176
Goyder, D., Harris, T., Siro, M., Ulrich, M., and Johan, V. (2012). Apocynaceae (Part 2). Flora of Tropical East Africa. England: CRC Press.
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Gu, C., Ma, L., Wu, Z., Chen, K., and Wang, Y. (2019). Comparative analyses of chloroplast genomes from 22 Lythraceae species: inferences for phylogenetic relationships and genome evolution within Myrtales. BMC Plant. Biol. 19:281. doi: 10.1186/s12870-019-1870-3
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Hansen, D. R., Dastidar, S. G., Cai, Z., Penaflor, C., Kuehl, J. V., Boore, J. L., et al. (2007). Phylogenetic and evolutionary implications of complete chloroplast genome sequences of four early-diverging angiosperms: Buxus (Buxaceae), Chloranthus (Chloranthaceae), Dioscorea (Dioscoreaceae), and Illicium (Schisandraceae). Mol. Phylogenet. Evol. 45, 547–563. doi: 10.1016/j.ympev.2007.06.004
Hershberg, R., and Petrov, D. A. (2008). Selection on codon bias. Annu. Rev. Genet. 42, 287–299. doi: 10.1146/annurev.genet.42.110807.091442
Hirose, T., and Sugiura, M. (1997). Both RNA editing and RNA cleavage are required for translation of tobacco chloroplast ndhD mRNA: a possible regulatory mechanism for the expression of a chloroplast operon consisting of functionally unrelated genes. EMBO J. 16, 6804–6811. doi: 10.1093/emboj/16.22.6804
Honys, D., and Twell, D. (2004). Transcriptome analysis of haploid male gametophyte development in Arabidopsis. Genome Biol. 5:R85. doi: 10.1186/gb-2004-5-11-r85
Huang, Y., Yang, Z., Huang, S., An, W., Li, J., and Zheng, X. (2019). Comprehensive analysis of Rhodomyrtus tomentosa chloroplast genome. Plants 8:89. doi: 10.3390/plants8040089
Jia, Y., Liu, M. L., Yue, M., Zhao, Z., Zhao, G. F., and Li, Z. H. (2017). Comparative transcriptome analysis reveals adaptive evolution of Notopterygium incisum and Notopterygium franchetii, two high-alpine herbal species endemic to China. Molecules 22:1158. doi: 10.3390/molecules22071158
Jiang, W., Tan, W., Gao, H., Yu, X., Zhang, H., Bian, Y., et al. (2020). Transcriptome and complete chloroplast genome of Glycyrrhiza inflata and comparative analyses with the other two licorice species. Genomics 112, 4179–4188. doi: 10.1016/j.ygeno.2020.07.007
Jin, J., Zhang, H., Kong, L., Gao, G., and Luo, J. (2014). PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 42, D1182–D1187. doi: 10.1093/nar/gkt1016
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K. F., von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y., and Morishima, K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. doi: 10.1093/nar/gkw1092
Kim, S. H., Yang, J., Park, J., Yamada, T., Maki, M., and Kim, S. C. (2019). Comparison of whole plastome sequences between thermogenic skunk cabbage Symplocarpus renifolius and Nonthermogenic S. nipponicus (Orontioideae; Araceae) in East Asia. Int. J. Mol. Sci. 20:4678. doi: 10.3390/ijms20194678
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Leister, D. (2003). Chloroplast research in the genomic age. Trends Genet. 19, 47–56. doi: 10.1016/s0168-9525(02)00003-3
Li, X., Tan, W., Sun, J., Du, J., Zheng, C., Tian, X., et al. (2019). Comparison of four complete chloroplast genomes of medicinal and ornamental Meconopsis species: genome organization and species discrimination. Sci. Rep. 9:10567. doi: 10.1038/s41598-019-47008-8
Li, Y., Qin, X., and Feng, Y. (2012). Pollinarium morphology of 12 species of Cynanchum (Asclepiadaceae). Bull. Bot. Res. 32, 137–142.
Liede-Schumann, S. (2000). Subtribe Astephaninae (Apocynaceae-Asclepiadoideae) reconsidered: new evidence based on cpDNA spacers. Ann. Missouri Bot. Gard. 88:657. doi: 10.2307/3298638
Liede-Schumann, S., Khanum, R., Mumtaz, A. S., Gherghel, I., and Pahlevani, A. (2016). Going west - A subtropical lineage (Vincetoxicum, Apocynaceae: Asclepiadoideae) expanding into Europe. Mol. Phylogenet. Evol. 94(Pt A), 436–446. doi: 10.1016/j.ympev.2015.09.021
Liede-Schumann, S., and Meve, U. (2018). Vincetoxicum (Apocynaceae—Asclepiadoideae) expanded to include Tylophora and allies. Phytotaxa 369:129. doi: 10.11646/phytotaxa.369.3.1
Logacheva, M. D., Kasianov, A. S., Vinogradov, D. V., Samigullin, T. H., Gelfand, M. S., Makeev, V. J., et al. (2011). De novo sequencing and characterization of floral transcriptome in two species of buckwheat (Fagopyrum). BMC Genom. 12:30. doi: 10.1186/1471-2164-12-30
López-Serrano, M., Del Campo, E. M., Sabater, B., and Martín, M. (2001). Primary transcripts of ndhD of Liliaceae and Aloaceae require editing of the start and 20th codons. J. Exp. Bot. 52, 179–180.
Mao, X., Cai, T., Olyarchuk, J. G., and Wei, L. (2005). Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21, 3787–3793. doi: 10.1093/bioinformatics/bti430
Mehmood, F., Abdullah, S. I., Ahmed, I., Waheed, M. T., and Mirza, B. (2020). Characterization of Withania somnifera chloroplast genome and its comparison with other selected species of Solanaceae. Genomics 112, 1522–1530. doi: 10.1016/j.ygeno.2019.08.024
Mei, S., Liu, T., and Wang, Z. (2016). Comparative transcriptome profile of the cytoplasmic male sterile and fertile floral buds of radish (Raphanus sativus L.). Int. J. Mol. Sci. 17:42. doi: 10.3390/ijms17010042
Minh, B. Q., Nguyen, M. A., and von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. doi: 10.1093/molbev/mst024
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W1185. doi: 10.1093/nar/gkm321
Neckermann, K., Zeltz, P., Igloi, G. L., Kössel, H., and Maier, R. M. (1994). The role of RNA editing in conservation of start codons in chloroplast genomes. Gene 146, 177–182. doi: 10.1016/0378-1119(94)90290-9
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Persoon, C. H. (1805). Synopsis Plantarum: Seu Enchiridium Botanicum, Complectens Enumerationem Systematicam Specierum Hucusque Cognitarum. Paris: apud CF Cramerum, 456.
Qiu, S. X., Li, D. Z., Zhang, Z. X., Zhou, J., and Wu, Y. Z. (1989). Chemotaxonomy of Cynanchum and its allied genera with notes on the genetic characteristics of Vincetoxicum. Acta Bot. Yunnan. 11, 41–50.
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Shaw, J., Lickey, E. B., Beck, J. T., Farmer, S. B., Liu, W., Miller, J., et al. (2005). The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 92, 142–166. doi: 10.3732/ajb.92.1.142
Somaratne, Y., Guan, D. L., Wang, W. Q., Zhao, L., and Xu, S. Q. (2019). The complete chloroplast genomes of two Lespedeza species: insights into codon usage Bias, RNA editing sites, and phylogenetic relationships in Desmodieae (Fabaceae: Papilionoideae). Plants 9:51. doi: 10.3390/plants9010051
Straub, S. C., Cronn, R. C., Edwards, C., Fishbein, M., and Liston, A. (2013). Horizontal transfer of DNA from the mitochondrial to the plastid genome and its subsequent evolution in milkweeds (Apocynaceae). Genome Biol. Evol. 5, 1872–1885. doi: 10.1093/gbe/evt140
Takenaka, M., Zehrmann, A., Verbitskiy, D., Härtel, B., and Brennicke, A. (2013). RNA editing in plants and its evolution. Annu. Rev. Genet. 47, 335–352. doi: 10.1146/annurev-genet-111212-133519
Tan, W., Gao, H., Zhang, H., Yu, X., Tian, X., Jiang, W., et al. (2020). The complete chloroplast genome of Chinese medicine (Psoralea corylifolia): molecular structures, barcoding and phylogenetic analysis. Plant Gene 21:100216. doi: 10.1016/j.plgene.2019.100216
Tillich, M., Lehwark, P., Pellizzer, T., Ulbricht-Jones, E. S., Fischer, A., Bock, R., et al. (2017). GeSeq - versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Tripathi, S., Sangwan, R. S., Narnoliya, L. K., Srivastava, Y., Mishra, B., and Sangwan, N. S. (2017). Transcription factor repertoire in Ashwagandha (Withania somnifera) through analytics of transcriptomic resources: insights into regulation of development and withanolide metabolism. Sci. Rep. 7:16649. doi: 10.1038/s41598-017-14657-6
Tsudzuki, T., Wakasugi, T., and Sugiura, M. (2001). Comparative analysis of RNA editing sites in higher plant chloroplasts. J. Mol. Evol. 53, 327–332. doi: 10.1007/s002390010222
Wang, S., Yang, C., Zhao, X., Chen, S., and Qu, G. Z. (2018). Complete chloroplast genome sequence of Betula platyphylla: gene organization, RNA editing, and comparative and phylogenetic analyses. BMC Genom. 19:950. doi: 10.1186/s12864-018-5346-x
Wright, F. (1990). The ‘effective number of codons’ used in a gene. Gene 87, 23–29. doi: 10.1016/0378-1119(90)90491-9
Yamashiro, T., Fukuda, T., Yokoyama, J., and Maki, M. (2004). Molecular phylogeny of Vincetoxicum (Apocynaceae-Asclepiadoideae) based on the nucleotide sequences of cpDNA and nrDNA. Mol. Phylogenet. Evol. 31, 689–700. doi: 10.1016/j.ympev.2003.08.016
Yang, J., Qing, Z., Wang, D., and Yang, H. (2018). Distribution of medicinal plant resources of Cynanchum in Guizhou. Guizhou Sci. 36, 50–52.
Yang, T., Yao, S., Hao, L., Zhao, Y., Lu, W., and Xiao, K. (2016). Wheat bHLH-type transcription factor gene TabHLH1 is crucial in mediating osmotic stresses tolerance through modulating largely the ABA-associated pathway. Plant Cell Rep. 35, 2309–2323. doi: 10.1007/s00299-016-2036-5
Yang, X.-Y., Wang, Z., Luo, W.-C., Guo, X., Zhang, C.-H., Liu, J.-Q., et al. (2018). Plastomes of Betulaceae and phylogenetic implications. J. Syst. Evol. 57, 508–518. doi: 10.1111/jse.12479
Yang, Z., Huang, Y., An, W., Zheng, X., Huang, S., and Liang, L. (2019a). Sequencing and structural analysis of the complete chloroplast genome of the medicinal plant Lycium chinense Mill. Plants 8:87. doi: 10.3390/plants8040087
Yang, Z., Wang, G., Ma, Q., Ma, W., Liang, L., and Zhao, T. (2019b). The complete chloroplast genomes of three Betulaceae species: implications for molecular phylogeny and historical biogeography. PeerJ 7:e6320. doi: 10.7717/peerj.6320
Young, M. D., Wakefield, M. J., Smyth, G. K., and Oshlack, A. (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11:R14. doi: 10.1186/gb-2010-11-2-r14
Yu, X., Jiang, W., Tan, W., Zhang, X., and Tian, X. (2020). Deciphering the organelle genomes and transcriptomes of a common ornamental plant Ligustrum quihoui reveals multiple fragments of transposable elements in the mitogenome. Int. J. Biol. Macromol. 165(Pt B), 1988–1999. doi: 10.1016/j.ijbiomac.2020.10.075
Yu, X., Tan, W., Zhang, H., Gao, H., Wang, W., and Tian, X. (2019). Complete chloroplast genomes of Ampelopsis humulifolia and Ampelopsis japonica: molecular structure, comparative analysis, and phylogenetic analysis. Plants 8:410. doi: 10.3390/plants8100410
Zhang, D., Gao, F., Jakovlić, I., Zou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Zhang, L., Yan, H. F., Wu, W., Yu, H., and Ge, X. J. (2013). Comparative transcriptome analysis and marker development of two closely related Primrose species (Primula poissonii and Primula wilsonii). BMC Genom. 14:329. doi: 10.1186/1471-2164-14-329
Zhang, S. D., Jin, J. J., Chen, S. Y., Chase, M. W., Soltis, D. E., Li, H. T., et al. (2017). Diversification of Rosaceae since the late cretaceous based on plastid phylogenomics. New Phytol. 214, 1355–1367. doi: 10.1111/nph.14461
Zheng, Y., Jiao, C., Sun, H., Rosli, H. G., Pombo, M. A., Zhang, P., et al. (2016). iTAK: a program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein Kinases. Mol. Plant 9, 1667–1670. doi: 10.1016/j.molp.2016.09.014
Zhong, S., Joung, J. G., Zheng, Y., Chen, Y. R., Liu, B., Shao, Y., et al. (2011). High-throughput illumina strand-specific RNA sequencing library preparation. Cold Spring Harb. Protoc. 2011, 940–949. doi: 10.1101/pdb.prot5652
Keywords: Vincetoxicum versicolor (Bunge) Decne, transcriptome, chloroplast genome, comparative analysis, phylogeny
Citation: Yu X, Wang W, Yang H, Zhang X, Wang D and Tian X (2021) Transcriptome and Comparative Chloroplast Genome Analysis of Vincetoxicum versicolor: Insights Into Molecular Evolution and Phylogenetic Implication. Front. Genet. 12:602528. doi: 10.3389/fgene.2021.602528
Received: 03 September 2020; Accepted: 25 January 2021;
Published: 04 March 2021.
Edited by:
Abdelfattah Badr, Helwan University, EgyptReviewed by:
Ibrar Ahmed, Alpha Genomics Private Limited, PakistanXiaojun Nie, Northwest A and F University, China
Copyright © 2021 Yu, Wang, Yang, Zhang, Wang and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dan Wang, bmt3YW5nZGFuQDE2My5jb20=; Xiaoxuan Tian, dGlhbl94aWFveHVhbkB0anV0Y20uZWR1LmNu